
Abstract
Simple sequence repeat (SSR) markers provide a powerful tool for genetic linkage map construction that can be applied for identification of quantitative trait loci (QTL). In this study, a total of 640 new SSR markers were developed from an enriched genomic DNA library of the cassava variety ‘Huay Bong 60’ and 1,500 novel expressed sequence tag-simple sequence repeat (EST-SSR) loci were developed from the Genbank database. To construct a genetic linkage map of cassava, a 100 F1 line mapping population was developed from the cross Huay Bong 60 by ‘Hanatee’. Polymorphism screening between the parental lines revealed that 199 SSRs and 168 EST-SSRs were identified as novel polymorphic markers. Combining with previously developed SSRs, we report a linkage map consisted of 510 markers encompassing 1,420.3 cM, distributed on 23 linkage groups with a mean distance between markers of 4.54 cM. Comparison analysis of the SSR order on the cassava linkage map and the cassava genome sequences allowed us to locate 284 scaffolds on the genetic map. Although the number of linkage groups reported here revealed that this F1 genetic linkage map is not yet a saturated map, it encompassed around 88% of the cassava genome indicating that the map was almost complete. Therefore, sufficient markers now exist to encompass most of the genomes and efficiently map traits in cassava.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cassava (Manihot esculenta Crantz) is an important crop that is commonly grown in tropical and subtropical areas. It is used as a major source of carbohydrate for over 500 million people (El-Sharkawy 2004). Cassava crop is also used to produce chip/pellets for starch production, animal feed and used as a raw material for ethanol fermentation. Cassava has an allopolyploid genome, with 2n = 36 chromosomes with the estimated size of the haploid genome of about 772 mega base pairs (Bennett et al. 1982). It has DNA content of 1.67 pg per cell nucleus (Awoleye et al. 1994).
Molecular markers are powerful tools for marker-assisted selection (MAS) in plant breeding (Collard and Mackill 2008; Ribaut and Hoisington 1998). MAS is more efficient, effective, reliable and cost-effective than conventional selection for many traits during plant breeding (Collard et al. 2005). Microsatellite or SSR markers are widely used to construct genetic maps, associate traits with underlying genomic regions and for MAS (Varshney et al. 2005). Microsatellites are found in all eukaryotic genomes. They consist of 1–6 bp of nucleotide motifs repeated in 5–20 copies (Field and Wills 1996) distributed throughout the genome both in coding and non-coding regions (Kashi et al. 1997). Moreover, SSRs are co-dominant in inheritance, reproducible, highly polymorphic, simple and cheap to use (Gupta and Varshney 2000; Song et al. 2004). The use of genomic DNA enriched for satellites to produce libraries for DNA sequencing was a common and reliable technique to develop markers in many plant species, including wheat (Song et al. 2005), maize (Sharopova et al. 2002), peanut (He et al. 2003), onion (Tsukazaki et al. 2007) and red clover (Sato et al. 2005). Alternatively, SSRs have been developed from published sequence databases of ESTs (Feng et al. 2009; Raji et al. 2009; Tangphatsornruang et al. 2008; Yu et al. 2004a, b) and/or BAC end sequence databases (La Rota et al. 2005; McCouch et al. 2002; Shultz et al. 2007; Temnykh et al. 2001). Although EST derived SSRs showed less polymorphism than genomic SSRs, they are directly linked to expressed genes (Cho et al. 2000; La Rota et al. 2005; Shultz et al. 2007). Therefore, direct association can be made between genotype and phenotype leading to identification of QTL underlying the traits of interest using EST linkage map (Rudd 2003).
For cassava, SSRs have been developed and used in genetic linkage map construction (Fregene et al. 1997). The first genetic linkage map of cassava was constructed from F1 intra-specific cross using SSR, RFLPs, RAPDs and isoenzymes (Fregene et al. 1997). Later, more SSRs (Chavarriaga-Aguirre et al. 1998) and EST-SSRs (Raji et al. 2009; Tangphatsornruang et al. 2008) were developed for germplasm evaluation in cassava and its related species. In addition, 172 SSR markers were developed from genomic DNA-derived satellite-enriched library and mapped in an F1 population (Mba et al. 2001). In 2006, a genetic map of an F2 population was developed using SSR markers (Okogbenin et al. 2006). Kunkeaw et al. (2010a, b) presented a composite map of an F1 population that consisted of AFLP, SSR and EST markers. However, none of these maps could completely encompass the genome of cassava. A recent genetic map of cassava was constructed using F1 population (Chen et al. 2010). The map consisted of 18 linkage groups with a total length of 1,707.9 cM, however, the map was mostly based on AFLPs (65%) and SSRs, EST-SSRs and sequence-related amplified polymorphisms (SRAPs).
Here, the aim was to develop additional SSR markers from genomic DNA enriched for SSRs and the Genbank EST database from cassava and to construct genetic map using a F1 mapping population.
Methods
Plant materials
Huay Bong 60 is a commercial cassava variety for Thailand that was developed by a cross between ‘Rayong 5’ and ‘Kasetsart 50’. Huay Bong 60 is widely grown due to its high starch content and biomass yield. However, it is a bitter type with high-cyanide content. ‘Hanatee’ is a local variety with low levels of starch content, biomass and yield. It is classified as a sweet type due to low cyanide content. An F1 population consisting of 100 individuals derived from crosses of Huay Bong 60 by Hanatee was developed in 2006 and used for linkage map analysis. All F1 plant samples were propagated by stem cutting and grown at the Rayong Field Crops Research Center, Thailand. The distance between planting row was 1.5 and 1 m between individual plants. Fertilizer (15:15:15), 312.5 kg/ha, and chicken manure, 3,100 kg/ha was be applied at 1 month after planting. Pest management was applied as necessary.
SSR-enriched genomic libraries construction
An enriched genomic DNA library was constructed from Huay Bong 60 as described by Nunome et al. (2006) with some modifications. Leaf tissue was collected for DNA extraction using DNeasy Plant Mini Kit (QIAGEN, Hilden Germany). Four libraries were constructed. For the first library, 20 μg of genomic DNA was digested with two different restriction enzymes; AluI (Fermentas, Hanover, MD, USA), HaeIII (TaKaRa Bio Inc., Ohtsu-shi, Japan). The other three libraries were each made from DNA digested with three different restriction enzymes; AluI (Fermentas), HaeIII (TaKaRa Bio Inc.) and AfaI (TaKaRa Bio Inc.). Each digested DNA was purified using Wizard SV Gel and PCR Clean-Up System (Promega, Madison WI, USA) and ligated with double stranded DNA linkers (linker 1: 5′-GTTTAGCCTTGTAGCAGAAGC-3′ and linker 2: 5′-pGCTTCTGCTACAAGGCTAAACAAAA-3′). Ligation reactions were performed in 60 μl reactions containing 10 μM DNA linkers, 1× rapid ligation buffer, 24 U of T4 DNA ligase (Promega), 20 U XmnI (Promega), 10 U of each restriction enzyme in individual reaction: AluI, HaeIII and AfaI. Ligation reactions were accomplished by 25 cycles of 30 min at 16°C, and 10 min at 37°C, and incubated at 16°C overnight.
For the first library, 5 μl of linker-ligated DNA of AluI and HaeIII was pooled and hybridized with 1.5 μM of a biotinylated oligo probe of (AC)12/(CT)12. Libraries 2, 3 and 4, three different linker-ligated DNAs of AluI, HaeIII and AfaI, were pooled and hybridized with (AT)12/(CTT)8, (ACC)8/(AGC)8 and (GC)12/(GGC)8 probes, respectively.
All hybridized reactions were incubated at 95°C for 15 min followed by incubating overnight at different temperatures; 42°C for libraries 1 and 2, 50°C for library 3 and 58°C for library 4. 150 μl (1,500 μg) of Dynabeads (Dynabeads M-280 Streptavidine-DYNAL; Invitrogen Dynal AS. Smestad, Norway) was washed with B&W buffer (10 mM Tris–HCl pH 7.5, 1 mM EDTA, 2 M NaCl) followed by centrifugation and collection of magnetic beads (repeated 3 times). After adding hybridized-biotinylated oligo DNA into Dynabeads, the mixture was incubated at 43°C for 2 h, and the supernatant was removed by placing on a magnetic separation stand. The captured complexes were washed with 400 μl of 2× SSC/0.1% (w/v) SDS, twice for 5 min at room temperature followed by twice at each hybridization temperature for 5 min with 1× SSC/0.1%. After the supernatant was discarded, 120 μl of pre-heated TE buffer (at 95°C) was added and incubated for 10 min at 95°C to elute the captured DNA.
The eluted DNA was used for PCR amplifications with linker 1 primer. The PCR reactions were set-up using 1× PCR Buffer (Mg2+ plus), 0.2 mM dNTPs, 1.5 mM MgCl2, 0.8 μM linker, and 5 U Ex-Taq polymerase (TaKaRa Bio Inc.). The PCR profile was 94°C for 30 s, followed by 30 cycles of 94°C for 30 s, 60°C for 1 min, 68°C for 1 min, then 68°C for 7 min. PCR products derived from the captured DNA were separated by electrophoresis on 1% Seakem GTG agarose gel (FMC, Bioproducts, Rockland, USA) at 20 V overnight and stained with ethidium bromide. The expected DNA bands of sizes between 750 bp and 2.5 kbp was excised in a block and embedded by converting the large size of DNA fragment on the top of 0.8% Sea Plaque GTG agarose gel (FMC Bioproduct). After which, the agarose was cut into 50 ml tube and weighed to determine the appropriate volume of 1× agarose buffer (1 g of gel equal to 10 ml 1× agarose buffer). The gel was melted at 70°C for 15 min and cooled down to 45°C before β agarose (2 U/200 mg of gel) was added and incubated at 45°C for 1 h The DNA fragments were ethanol precipitated and the pellet washed in 70% (v/v) ethanol. The PCR products A-tailed by the following reaction mixture: 150–200 ng of PCR product, 1× buffer, 0.2 mM dATP, 1.5 mM of MgCl2, 2.5 U rTaq polymerase (TaKaRa Bio Inc.) and incubated at 70°C for 30 min.
The A-tailed PCR products were cloned into pGEM-T Easy vector (LigaFast Rapid DNA ligation system, Promega) according to the manufacturer’s instructions. The enriched library was transformed into the ElectroTen-Blue Electroporation competent cells (Stratagene, La Jolla, CA, USA) by electroporation. The transformants were selected on LB agar plates containing ampicillin, X-gal and IPTG (TaKaRa Bio Inc.). White colonies were used to find the insert size by PCR using T7 and SP6 primers after growth in 96-well plates. Inserts were amplified using TempliPhi 100 Amplification Kit (GE Healthcare, Piscataway, NJ) according to the manufacture’s instructions. Finally, amplified DNA was sequenced.
EST-SSR analysis
A total of 76,566 ESTs obtained from the Genbank EST database was assembled to 28,940 unique sequences (Kunkeaw et al. 2010b). The identification and localization of microsatellites were conducted as described in Kunkeaw et al. (2010b).
Primer designs
DNA sequences of clones were trimmed and analyzed to identify repeat regions. SSR primers were designed from flanking regions of SSR containing sequences using the Primer 3 program. The PCR product size was designed to be in range of 90–300 bp (Sato et al. 2005).
Analysis of SSR markers
SSR primers were analyzed with genomic DNA of cassava varieties Huay Bong 60 and Hanatee. Informative SSR markers that showed heterozygous pattern in either female or male parent were used to genotype 100 F1 progenies of the cross. Polymerase chain reaction (PCR) was performed as described in Kunkeaw et al. (2010a) in 15 μl reaction volumes containing 25 ng of genomic DNA, 0.2 μM of each primer, 200 μM dNTPs (Promega), 1× PCR buffer, 1.5 mM MgCl2, and 1 U Taq DNA polymerase (Promega). PCR was accomplished by 2 min at 94°C, followed by 45 s at 94°C, 45 s at primer annealing temperature, and 1 min at 72°C for 30 cycles and final extension for 5 min at 72°C. The PCR amplification products were visualized on 5% (w/v) denaturing polyacrylamide gel and visualized by silver staining (Benbouza et al. 2006).
Genetic linkage analysis
Genotypic data of SSR markers were scored as CP codes as described by Van Ooijen and Voorrips (2001). All informative SSR markers were used to construct a genetic linkage map using JoinMap® version 3.0 (Van Ooijen and Voorrips 2001). The genotype data were scored as CP codes (e.g. <abxcd>, <efxeg>, <lmxll>, <nnxnp> and <hkxhk>). Linkage groups were determined using an LOD threshold of 4.0. Map construction was performed, using the Kosambi mapping function with JoinMap parameter settings as follows: Rec = 0.5, LOD = 4.0 and Jump = 5 (Kosambi 1944). To compare the order of SSR loci with the cassava genome sequence, primer sequences were searched for sequence homology against the scaffolds of the cassava genome (ftp://ftp.jgi-psf.org/pub/JGI_data/phytozome/v5.0/Mesculenta) using BlastN.
Results
Ninety-six hybridizing clones from each library were selected for sequencing. The results showed that library 1 contained most SSRs that were useful for design primers. Additional clones (2,016) from the library 1 were then selected and sequenced. From 2,400 clones, 2,269 (94.5%) were successfully sequenced. Of these, 1,576 (69.4%) clones contained microsatellite regions with 1,221 (77.4%) in non-redundant sequences. This indicated that our genomic libraries are highly enriched with microsatellite regions.
In this study, 712 (58.3%) useful SSR primer pairs were designed from those sequences. The predominant microsatellite motifs were di-nucleotide repeats (73.03%), while 18.68 and 8.29% were tri-nucleotide repeats and tetra-nucleotide repeats, respectively. The majority of di-nucleotide motifs were AC/TG repeats (56.15%); the majority of tri-nucleotide repeats were AAT/TTA (21.05%) or AAG/TTC (21.05%); and the predominant tetra-nucleotide repeat was AAAT/TTTA (28.81%) as shown in Table 1.
A total of 640 of the 712 (89.89%) useful SSR primer pairs was suitable for detection of polymorphisms between the DNA of the population parents. The analysis of the population showed 439 (68.59%). SSR primer pairs that were successfully amplified in all DNA samples. Of these, 199 (31.09%) markers were found to be informative markers that showed heterozygous pattern in either or both parental lines, whereas 240 (37.50%) pairs were non-informative.
In this study, we screened 1,500 new primer pairs amplifying microsatellites in the cassava EST database. These primers were designed to amplify microsatellites with di- (1,012; 67.47%), tri- (438; 29.20%) and tetra- (50; 3.33%) nucleotide repeats, as present in Table 2. Genomic DNA of parental lines were tested with all new EST-SSR primers, the results showed that 1,222 (81.47%) primer pairs successfully amplified genomic DNA. Of these, 168 (13.75%) primer pairs showed informative polymorphism. The sequences of SSR and EST-SSR primers developed in this project are available upon request.
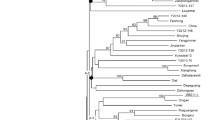
In addition to these newly synthesized primers (199 SSRs and 168 EST-SSRs), 277 of informative SSRs that were developed by CIAT and 81 informative EST-SSR markers from Kunkeaw et al. (2010b) were also included in this genetic linkage map construction. In total, 725 informative markers were successfully genotyped within the 100 F1 individuals of the mapping population and subjected to linkage map analysis. All of 725 markers were used for linkage map analysis with LOD score set at 4.0. The linkage map of the F1 population consisted of 510 SSR markers distributed on 23 linkage groups (Fig. 1). The map encompassed 1,420.3 cM. Linkage groups ranged in length from the 2.0 cM of LG23 to the 120.42 cM of LG2. The total number of markers in each linkage group varied from 2 to 71. The mean size of linkage groups was 61.8 cM containing 22.2 loci. The mean distance between linked markers was 4.54 cM, but ranged from 0.1 to 26.1 cM.


The genetic linkage map of cassava F1 population (Manihot esculenta Crantz) developed from SSR and EST-SSR markers. The map is composed of 510 SSR loci, covering 1,420.3 cM on 23 linkage groups. On the map, the SSR loci name is followed by the scaffold name(s) that each locus is located in the cassava genome project (ftp://ftp.jgi-psf.org/pub/JGI_data/phytozome/v5.0/Mesculenta)
To search for the location of SSR loci on the cassava genome sequence, primer sequences were searched for sequence homology against the scaffolds of the cassava genome (ftp://ftp.jgi-psf.org/pub/JGI_data/phytozome/v5.0/Mesculenta) using BlastN.
We were able to identify the locations of 481 (94.3%) SSR loci on the linkage map scattering on 284 scaffolds of the cassava genome (Table 3).
Discussion
Microsatellites are the most widely used DNA markers for genetic linkage map analysis (Ritschel et al. 2004). The satellite-enriched genomic library was a simple procedure used to characterize SSR markers in many plant species, including cassava (Chavarriaga-Aguirre et al. 1998; Mba et al. 2001). To characterize and develop additional SSR markers of cassava, four enriched genomic DNA libraries were constructed from cassava, and each library was enriched with a different biotinylated DNA oligomer. The most abundant microsatellite motif in plant genomes was the (GA/CT)n repeat (Li et al. 2002; Morgante et al. 2002; Saha et al. 2006). The (GA/CT) SSR motif was used to construct enriched libraries in cassava (Chavarriaga-Aguirre et al. 1998; Mba et al. 2001). Therefore, this study choose different SSR motifs (AC)12/(CT)12, (AT)12/(CTT)8, (ACC)8/(AGC)8 and (GC)12/(GGC)8 to construct enriched libraries. The results showed that AC/TG (56%) repeats were common and could be used to design primers useful for mapping. There was no evidence for clustering of repeat motifs seen in other paleopolyploid genomes (Shultz et al. 2007). In the previous study, Mba et al. (2001) constructed GA-enriched libraries with 45–60% enrichment efficiency. In this study, the highest efficiency of enriched library, 74% was found in the library using (AC)12/(CT)12. The average efficiency of other plants that ranges from 50 to 90% (Butcher et al. 2000). The enriched libraries increased the rate of SSR discovery as compared to the sequencing of genomic DNA in BAC end sequences (13–15%) (La Rota et al. 2005; McCouch et al. 2002; Shultz et al. 2007; Temnykh et al. 2001). However, the problem of SSR-enriched library for marker development is partly caused by small insert size that lead to short or redundant flanking regions not suitable for primer design (Sharopova et al. 2002).
There was concern that the F1 population may not be ideal for map development (Okogbenin et al. 2006). However, the process of population development for cassava was limited by the long growing cycle and low seed number per pollination that results in limits to developing F2 or derived populations for classical genetic studies (Kunkeaw et al. 2010a). Conversely, several genetic linkage maps of cassava (Fregene et al. 1997; Kunkeaw et al. 2010a, b; Chen et al. 2010) and other perennials, such as tea (Hackett et al. 2000) have been constructed using F1 populations. Therefore, genetic mapping populations in cassava were usually derived from crosses between heterozygous parents, F1 crosses (Fregene et al. 1997; Mba et al. 2001; Kunkeaw et al. 2010b). In the previous work on the construction of the cassava linkage map of cassava using EST markers by Kunkeaw et al. (2010b), there were more than 7,000 SSR loci identified in the Genbank ESTdb. The authors screened 425 primer pairs for polymorphism between the parental lines and found that 81 primer pairs were informative and 56 EST-SSR loci were mapped in the F1 population from crosses of Huay Bong 60 by Hanatee (Kunkeaw et al. 2010b). Based on the same mapping population as Kunkeaw et al. (2010b), 168 primer pairs were informative and used in the genetic linkage map analysis.
The linkage map in this study added 299 new microsatellite markers (181 SSR and 118 EST-SSR loci) into the previous map constructed by Kunkeaw et al. (2010b). Based on the genome size estimation method from linkage data (Hulbert et al. 1988), the size of the cassava genome was estimated to be around 1,610 cM (Fregene et al. 1997). Here, we reported the map which encompassed 1,420.3 cM or around 88% of the genome indicating that the map was almost complete. SSRs provide powerful tool for genetic linkage map construction that can be applied for identification of QTL. Importantly, the marker linked to the QTL can be further applied to MAS in cassava breeding program for selecting cassava plant that contains desirable phenotype.
In general, SSR loci in the same scaffold of the cassava genome sequence located in the same linkage group. For example, on the LG5, there were 39 loci which can be located on 18 scaffolds of the cassava genome sequences (Fig. 1). Fifteen SSR loci could be located on the scaffold03614. However, 2 out of 15 loci from the scaffold03614 were separated by markers from other scaffolds on the LG5. This information could help bringing scaffolds in the incomplete cassava genome sequences. However, the non-corresponding order of markers between genetic linkage map and the cassava genome sequence is probably due to different genetic backgrounds used in the linkage map construction and the cassava genome project. In some cases, the comparison analysis may help bridging linkage groups. For example, the scaffold02960 contained three loci (CA23, CA241 and CA258) on the LG14 and one locus (SSRY337) on the LG19. It is possible that the LG14 and LG19 are not linked on the linkage map due to a lack of bridging markers. However, more experiments are required to evaluate these hypotheses.
References
Awoleye F, Duren M, Dolezel J, Novak FJ (1994) Nuclear DNA content and in vitro induced somatic polyploidization cassava (Manihot esculenta Crantz) breeding. Euphytica 76:195–202
Benbouza H, Jacquemin J, Baudoin J, Mergeai G (2006) Optimization of a reliable, fast, cheap and sensitive silver staining method to detect SSR markers in polyacrylamide gels. Biotechnol Agron Soc Environ 10:77–81
Bennett MD, Smith JB, Heslop-Harrison JS (1982) Nuclear DNA amounts in angiosperms. Proc R Soc Lond Ser B Biol Sci 179–199
Butcher PA, Decroocq S, Gray Y, Moran GF (2000) Development, inheritance and cross-species amplification of microsatellite markers from Acacia mangium. Theor Appl Genet 101:1282–1290
Chavarriaga-Aguirre PP, Maya MM, Bonierbale MW, Kresovich S, Fregene MA, Tohme J, Kochert G (1998) Microsatellites in cassava (Manihot esculenta Crantz): discovery, inheritance and variability. Theor Appl Genet 97:493–501
Chen X, Xia Z, Fu Y, Lu C, Wang W (2010) Constructing a genetic linkage map using an F1 population of non-inbred parents in cassava (Manihot esculenta Crantz). Plant Mol Biol Report. doi:10.1007/s11105-010-0198-9
Cho YG, Ishii T, Temnykh S, Chen X, Lipovich L, McCouch SR, Park WD, Ayres N, Cartinhour S (2000) Diversity of microsatellites derived from genomic libraries and GenBank sequences in rice (Oryza sativa L.). Theor Appl Genet 100:713–722
Collard BC, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc Lond B Biol Sci 363:557–572
Collard B, Jahufer M, Brouwer J, Pang E (2005) An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142:169–196
El-Sharkawy MA (2004) Cassava biology and physiology. Plant Mol Biol 56:481–501
Feng SP, Li WG, Huang HS, Wang JY, Wu YT (2009) Development, characterization and cross-species/genera transferability of EST-SSR markers for rubber tree (Hevea brasiliensis). Mol Breed 23:85–97
Field D, Wills C (1996) Long, polymorphic microsatellites in simple organisms. Proc R Soc Lond 263:209–215
Fregene M, Angel F, Gomez R, Rodriguez F, Chavarriaga P, Roca W, Tohme J, Bonierbale M (1997) A molecular genetic map of cassava (Manihot esculenta Crantz). Theor Appl Genet 95:431–441
Gupta P, Varshney R (2000) The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat. Euphytica 113:163–185
Hackett CA, Wachira FN, Paul S, Powell W, Waugh R (2000) Construction of a genetic linkage map for Camellia sinensis (tea). Heredity 85:346–355
He G, Meng R, Newman M, Gao G, Pittman R, Prakash CS (2003) Microsatellites as DNA markers in cultivated peanut (Arachis hypogaea L.). BMC Plant Biol 3:3
Hulbert SH, Ilott TW, Legg EJ, Lincoln SE, Lander ES, Michelmore RW (1988) Genetic analysis of the fungus, Bremia lactucae, using restriction fragments length polymorphisms. Genetics 120:947–958
Kashi Y, King D, Soller M (1997) Simple sequence repeats as a source of quantitative genetic variation. Trends Genet 13:74–78
Kosambi DD (1944) The estimation of map distance from recombination values. Ann Eugen Lond 12:172–175
Kunkeaw S, Tangphatsornruang S, Smith DR, Triwitayakorn K (2010a) Genetic linkage map of cassava (Manihot esculenta Crantz) based on AFLP and SSR markers. Plant Breed 129:112–115
Kunkeaw S, Yoocha T, Sraphet S, Boonchanawiwat A, Boonseng O, Lightfoot DA, Triwitayakorn K, Tangphatsornruang S (2010b) Construction of a genetic linkage map using simple sequence repeat markers from expressed sequence tags for cassava (Manihot esculenta Crantz). Mol Breed 10. doi:10.1007/s11032-010-9414-4
La Rota M, Kantety R, Yu J-K, Sorrells M (2005) Nonrandom distribution and frequencies of genomic and EST-derived microsatellite markers in rice, wheat, and barley. BMC Genomics 6:23
Li YC, Korol AB, Fahima T, Beiles A, Nevo E (2002) Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol 11:2453–2465
Mba REC, Stephenson P, Edwards K, Melzer S, Nkumbira J, Gullberg U, Apel K, Gale M, Tohme J, Fregene MF (2001) Simple sequence repeat (SSR) markers survey of the cassava (Manihot esculenta Crantz) genome: towards an SSR-based molecular genetic map of cassava. Theor Appl Genet 102:21–31
McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, DeClerck G, Schneider D, Cartinhour S, Ware D, Stein L (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
Morgante M, Hanafey M, Powell W (2002) Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet 30:194–200
Nunome T, Negoro S, Miyatake K, Yamaguchi H, Fukuoka H (2006) A protocol for the construction of microsatellite enriched genomic library. Plant Mol Biol Rep 24:305–312
Okogbenin E, Marin J, Fregene M (2006) An SSR-based molecular genetic map of cassava. Euphytica 147:433–440
Raji AA, Anderson JV, Kolade OA, Ugwu CD, Dixon AG, Ingelbrecht IL (2009) Gene-based microsatellites for cassava (Manihot esculenta Crantz): prevalence, polymorphisms, and cross-taxa utility. BMC Plant Biol 9:118
Ribaut J-M, Hoisington D (1998) Marker-assisted selection: new tools and strategies. Trends Plant Sci 3:236–239
Ritschel PS, Lins TC, Tristan RL, Buso GS, Buso JA, Ferreira ME (2004) Development of microsatellite markers from an enriched genomic library for genetic analysis of melon (Cucumis melo L.). BMC Plant Biol 4:9
Rudd S (2003) Expressed sequence tags: alternative or complement to whole genome sequences? Trends Plant Sci 8:321–329
Saha MC, Cooper JD, Mian MA, Chekhovskiy K, May GD (2006) Tall fescue genomic SSR markers: development and transferability across multiple grass species. Theor Appl Genet 113:1449–1458
Sato S, Isobe S, Asamizu E, Ohmido N, Kataoka R, Nakamura Y, Kaneko T, Sakurai N, Okumura K, Klimenko I, Sasamoto S, Wada T, Watanabe A, Kohara M, Fujishiro T, Tabata S (2005) Comprehensive structural analysis of the genome of red clover (Trifolium pratense L.). DNA Res 12:301–364
Sharopova N, McMullen MD, Schultz L, Schroeder S, Sanchez-Villeda H, Gardiner J, Bergstrom D, Houchins K, Melia-Hancock S, Musket T, Duru N, Polacco M, Edwards K, Ruff T, Register JC, Brouwer C, Thompson R, Velasco R, Chin E, Lee M, Woodman-Clikeman W, Long MJ, Liscum E, Cone K, Davis G, Coe EH (2002) Development and mapping of SSR markers for maize. Plant Mol Biol 48:463–481
Shultz JL, Kazi S, Bashir R, Afzal JA, Lightfoot DA (2007) The development of BAC-end sequence-based microsatellite markers and placement in the physical and genetic maps of soybean. Theor Appl Genet 114:1081–1090
Song QJ, Marek LF, Shoemaker RC, Lark KG, Concibido VC, Delannay X, Specht JE, Cregan PB (2004) A new integrated genetic linkage map of the soybean. Theor Appl Genet 109:122–128
Song Q, Shi J, Singh S, Fickus E, Costa J, Lewis J, Gill B, Ward R, Cregan P (2005) Development and mapping of microsatellite (SSR) markers in wheat. Theor Appl Genet 110:550–560
Tangphatsornruang S, Sraphet S, Singh R, Okogbenin E, Fregene M, Triwitayakorn K (2008) Development of polymorphic markers from expressed sequence tags of Manihot esculenta Crantz. Mol Ecol Resour 8:682–685
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:1441–1452
Tsukazaki H, Nunome T, Fukuoka H, Kanamori H, Kono I, Yamashita K-i, Wako T, Kojima A (2007) Isolation of 1,796 SSR clones from SSR-enriched DNA libraries of bunching onion (Allium fistulosum). Euphytica 157:83–94
Van Ooijen JW, Voorrips RE (2001) JoinMap 3.0, Software for the calculation of genetic linkage maps. Plant Research International, Wageningen
Varshney RK, Graner A, Sorrells ME (2005) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23:48–55
Yu JK, Dake TM, Singh S, Benscher D, Li W, Gill B, Sorrells ME (2004a) Development and mapping of EST-derived simple sequence repeat markers for hexaploid wheat. Genome 47:805–818
Yu JK, La Rota M, Kantety RV, Sorrells ME (2004b) EST derived SSR markers for comparative mapping in wheat and rice. Mol Genet Genomics 271:742–751
Acknowledgments
This research was supported by National Center for Genetic Engineering and Biotechnology (Thailand), Commission on Higher Education, Ministry of Education (Thailand), the Thailand Research Fund and Mahidol University. Supajit Sraphet and Athipong Boonchanawiwat were supported from Thailand Graduate Institute of Science and Technology (TGIST) for PhD and MS studies, respectively. The cassava genome sequence was downloaded from “Cassava Genome Project 2009 http://www.phytozome.net/cassava”.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by A. Bervillé.
Rights and permissions
About this article
Cite this article
Sraphet, S., Boonchanawiwat, A., Thanyasiriwat, T. et al. SSR and EST-SSR-based genetic linkage map of cassava (Manihot esculenta Crantz). Theor Appl Genet 122, 1161–1170 (2011). https://doi.org/10.1007/s00122-010-1520-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1520-5