
Abstract
The aim of this study was to develop a large set of microsatellite markers based on publicly available BAC-end sequences (BESs), and to evaluate their transferability, discriminating capacity of genotypes and mapping ability in Citrus. A set of 1,281 simple sequence repeat (SSR) markers were developed from the 46,339 Citrus clementina BAC-end sequences (BES), of them 20.67% contained SSR longer than 20 bp, corresponding to roughly one perfect SSR per 2.04 kb. The most abundant motifs were di-nucleotide (16.82%) repeats. Among all repeat motifs (TA/AT)n is the most abundant (8.38%), followed by (AG/CT)n (4.51%). Most of the BES-SSR are located in the non-coding region, but 1.3% of BES-SSRs were found to be associated with transposable element (TE). A total of 400 novel SSR primer pairs were synthesized and their transferability and polymorphism tested on a set of 16 Citrus and Citrus relative’s species. Among these 333 (83.25%) were successfully amplified and 260 (65.00%) showed cross-species transferability with Poncirus trifoliata and Fortunella sp. These cross-species transferable markers could be useful for cultivar identification, for genomic study of Citrus, Poncirus and Fortunella sp. Utility of the developed SSR marker was demonstrated by identifying a set of 118 markers each for construction of linkage map of Citrus reticulata and Poncirus trifoliata. Genetic diversity and phylogenetic relationship among 40 Citrus and its related species were conducted with the aid of 25 randomly selected SSR primer pairs and results revealed that citrus genomic SSRs are superior to genic SSR for genetic diversity and germplasm characterization of Citrus spp.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microsatellites or simple sequence repeats (SSRs) are arrays of short motifs of 1–6 base pairs in length, which occur as interspersed repetitive elements in all eukaryotic genomes [1]. Variations in the number of tandem repeat units are mainly due to strand slippage during DNA replication where the repeats allow matching via excision or addition of repeats [2]. As slippage in replication is more likely than point mutations, microsatellite loci tend to be hyper-variable. Microsatellite arrays show extensive inter-individual length polymorphisms during PCR analysis of unique loci using discriminatory primer sets. SSR motifs are present both in protein coding and non-coding regions of DNA sequences [3, 4] and their presence in coding regions are less polymorphic compared to those in genomic regions [5]. Moreover, different taxa vary in abundance of different types of SSRs, and their abundance in non-coding regions are greater than in coding SSRs [4]. The advantages of using microsatellite markers in plant genetics and molecular breeding are their multi-allelic nature, co-dominance inheritance, easy assay, relative abundance, high reproducibility, extensive genome coverage and requirement of a small amount of sample DNA as template [6]. Therefore, SSR markers are extensively used in genetic diversity, population genetics, linkage map construction and gene mapping studies in plants.
Development of SSR markers through traditional methods can be quite costly, time consuming, labor intensive and also inefficient [7–9]. Over the past decade, a number of genome sequencing project have been initiated by different research groups which generated numerous publicly available cDNA and GSS sequences. Consequently, public databases are becoming valuable resources for plant genomic studies. An alternative strategy has been developed to generate SSR markers from publicly available sequences (both cDNA and GSS), by using data mining pipelines composed primarily of SSR search and primer design programs. This approach has been successfully applied in several plant species including lotus [10], Brassica [11–15], Citrus [5, 16], Wheat [17–19], ray [20], Zea maize [21], Saccharum spp. [22] and Vitis [23]. Thus, the use of such databases for marker development appears to be a promising alternative to the development of traditional ‘‘anonymous’’ SSRs following standard methods. SSR search programs such as MISA (MIcro SAtellite identification tool), SSR hunter and TROLL [24], SPUTNIK (http://abajian.net/sputnik/) and Phobos (http://www.ruhr-uni-bochum.de/spezzoo/cm/cm_phobos.htm) are available for public use. These programs can identify SSR motifs within sequences and compute an overview of the distribution and frequency of SSRs in the entire genome.
Citrus is an economically important fruit crop in many subtropical regions. Improvement of citrus cultivar through conventional methods is quite difficult, inefficient, costly and time consuming due to it’s prolonged juvenility, unusual sexual behavior and complex genetic background. Several approaches have the potential to shorten the breeding time and to reduce the cost. One of them is the marker assisted selection (MAS), which use the DNA stretch linked to the target agronomic traits to select the young hybrid progeny at early stages of growth and development. MAS are useful for citrus breeding. A perquisite for the use of MAS in citrus is to obtain suitable molecular markers. Unfortunately, till to date, the number of published molecular markers in citrus is limited for high density linkage map construction. Therefore, advances in molecular breeding of citrus are less explored than for other crops like rice, wheat and brassica. With the aim of facilitating citrus genetic improvement via MAS, it is important to develop molecular markers and then construct a highly dense genetic and physical map.
Although the abundance, characterization and usefulness of SSR markers in Citrus spp. is documented, the number of publicly available SSR markers is still limited for mapping, rapid genotype identification and genetic diversity analyses. Several projects had been conducted to develop SSR markers in citrus species including C. sinensis, P. trifoliata, C. clementina, C. limon [5, 16, 25]. The first citrus SSR marker was developed by Kijas et al. [26] in order to increase the density of Citrus RFLP and to construct an isozyme based linkage map. Seven SSR markers were publicly available for lemon. Chen et al. [16] developed and mapped about 100 EST-SSR markers from citrus EST data mining. More than 216 EST-SSR markers were publicly available, which were developed from unigene of C. sinensis [5], but no experimental tests of these markers have been reported. Jiang et al. [27] developed 25 SSR markers and among them 12 were published. Forty-one EST-SSR markers were produced from C. clementina EST sequences and studied for their transferability to other citrus species and their effectiveness for genetic mapping [25]. One hundred seventy-one SSR primers were designed from a genomic library of ‘Pera IAC’ sweet orange; among them 113 were functional [28]. Terol et al. [29] developed 46,000 BAC end sequences (BES) and primarily identified 3,800 putative SSRs. They suggested that the SSRs contained in BES could be useful resources for SSR marker development, which however remained to be verified in molecular markers related experiments. Clearly not all SSR loci are suitable for high quality SSR-primer development, due to insufficient or poor flanking regions (e.g., low GC content). In addition, the success rate of the PCR amplification of SSR primers was 60–90% reported in different studies [30]. Consequently, the practical use of SSR primers for germplasm identification, mapping and population genetic studies, in which data integration and comparison are crucial, requires each SSR marker to be validated for quality and robustness of the amplification product. Recently, great efforts have been made to develop SSR markers from publicly available BAC end sequences of many plant species [11, 12, 15]. The utilization of publicly available C. clementina sequence information to detect SSR motif provides a promising methodology for the development of a large number of useful molecular markers. Ollitrault et al. [31] characterized only 79 SSR markers from these BAC-end sequences and estimated genetic diversity in Citrus using a subset of 18 primer pairs. Our initial effort was to develop a new set of SSR markers from the publicly available BES of C. clementina [29]. Consequently, we designed 1,281 SSR primer pairs from those BES and validated them for their quality and robustness. Here we also analyzed their genomic distribution, including their occurrences in protein coding genes and transposable elements (TEs). In addition, 400 novel BAC-end derived SSR markers were experimentally evaluated for their transferability among the citrus and its closely related genera as well as their mapping ability on the F1 progeny established for genetic studies. We also evaluated their discriminating capacity, efficiency and informativeness for genetic similarity and phylogenetic studies among Citrus and related species.
Materials and methods
Retrieval and mining of GSS (or BES) for microsatellites
A total of 46,339 genome survey sequences (GSSs) were downloaded from the NCBI on 19 October 2008. These sequences (GenBank accession numbers from ET068227 to ET114565) were generated from BAC-end sequencing of three Citrus BAC libraries.
Retrieved GSSs were screened for SSRs by using the MISA (MIcro SAtellite identification tool) perl script. MISA was downloaded from http://pgrc.ipk-gatersleben.de/misa/misa.html and run on a local computer and the parameters were set for detection of perfect mono-, di-, tri-, tetra-, penta- and hexa-nucleotide motifs with a minimum of ten, six, five, five, five and five repeats, respectively. The following information was extracted from the MISA output for further analysis (*.fasta.misa and *.fasta.statistics files), i.e. sequence ID, SSR number, SSR type, SSR motif, SSR size, SSR start and end sites, total number of sequences examined, total size of examined sequences (bp), total number of identified SSRs, number of SSR containing sequences, number of sequences containing more than 1 SSR, number of SSRs present in compound formation, distribution to different repeat type classes and frequency of classified repeat types.
Removal of redundant sequences and primer design
BES sequences were assembled using the CAP3 software [32] to eliminate sequence redundancy in order to avoid designing multiple sets of primers for the same locus. The resulting contigs and singletons were used for SSR primer design. Microsatellites with repeat length ≥16 bp for di-, ≥18 for tri- and ≥20 for tetra-, penta- and hexa-nucleotides were selected for SSR marker development. The Primer3 program was used to design primers with the same parameters as described by Thiel et al. [19]. The SSR primers were synthesized by the Sangon Company, Shanghai, China.
Plant materials, DNA extraction and PCR amplification
Sixteen genotypes were used for the initial screening and transferability analysis of the SSR primers. These genotypes represented the major groups of Citrus and closely related genera (Supplementary file 1). Progenies of C. reticulata × P. trifoliata, were used to test the map-ability of the designed primers. To estimate discriminating capacity and the utility of BES-SSR marker in the phylogenetic study, randomly selected 25 BES-SSR primers and 40 citrus and its relative’s species were used. All the plant materials were collected from the National Center of Citrus Breeding, Huazhong Agricultural University, Wuhan, China. Total genomic DNA was isolated from young mature fresh leaf following the procedure previously described by Cheng et al. [33].
For the SSR analysis, PCR amplification was performed as described by Kijas et al. [26] with minor modifications. The total volume of PCR reactions was 20 μl, containing 50 ng genomic DNA, 1.5 mM MgCl2, 0.2 mM dNTPs, 1.0 U Taq DNA polymerase, corresponding 1× reaction buffer and 0.1 μM of each primer pair. PCR amplification was conducted in a MJ-PTC-200 thermal controller (MJ Research, Waltham Mass) using the following program: 94°C for 5 min, 32 cycles at 94°C for 1 min, 55°C for 30 s, 72°C for 1 min, followed by a final step at 72°C for 4 min. After PCR, 8 μl of loading buffer (98% formamide, 2% dextran blue, 0.2 mM EDTA) was added to each sample. Samples were denatured at 90°C for 5 min and then immediately placed on ice. An aliquot (4 μl) of each sample was loaded onto 6% polyacrylamide gel (60 cm × 30 cm × 0.4 cm), which had been run for 2 h and 30 min at 80 W. DNA bands were visualized with silver staining as described by Ruiz et al. [34].
Transposon element (TE) association and functional annotation of SSR containing BAC-end sequences
A customize plant TE data base was constructed in combination with plant repeats from Repbase, plant repeat database from TIGR (ftp://ftp.tigr.org/pub/data/TIGR_Plant_Repeats) and GeneBank for our initial classification of TEs. Then the customized TE database was compared with the SSR containing BAC-end data set using BLASTN analysis.
In order to assign a putative function of BES-SSR sequences, we used the Blast2GO tool. The mapping and annotation of the sequences according to gene ontology (GO) terms [35] is based on sequence similarity and therefore, sequences without BLAST hit were not annotated. For the annotation configuration the default settings were used (E value filter of 1E−10 and annotation cutoff of 55). Each sequence can have more than one GO term, either from different GO categories (Biological Process, Molecular Function and Cellular Component) or from the same category. Furthermore, in order to improve annotatability, we used InterProScan, which searched the data-bases BlastProDom, FPrintScan, HMMPIR, HMMPfam, HMMSmart, HMMTigr, ProfileScan, ScanRegExp and Super Family [36] (http://www.ebi.ac.uk/interpro/index.html) provided by the EBI [37] (http://www.ebi.ac.uk/) through Blast2GO.
Data analysis
Levels of polymorphism and discriminating capacity were analyzed following the procedure previously described by Belaj et al. [55]. PIC was estimated using the formula: \( {\text{PIC}} = \sum {p_{i}^{2} } \), where \( p_{i} \) is the frequency of ith allele at a locus. Phylogenetic analysis was performed with the NTSYS-Pc software package [38]. A similarity matrix was constructed based on Dice coefficient [39]; similarity matrix was used to construct a dendrogram using the unweighted pair grouping method arithmetic average (UPGMA) to determine genetic relationships among the germplasm studied.
Results
Development and characterization of BES-SSR marker
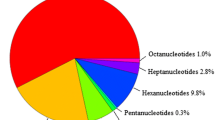
To develop a large number of citrus microsatellite markers, a total of 46,339 C. clementina BAC-end sequences (BES) were retrieved from NCBI database, representing a total length of 28.56 Mb of C. clementina genome. After SSR mining, a total of 14,009 SSRs were identified from 10,544 BES sequences and 22.61% of the BES had at least one SSR (Table 1). On average, at least one SSR was found per 2.04 kb (or 0.49 SSR/kb) in the 28.56 Mb BES sequences. Of the total SSRs identified, di- and tri-nucleotide repeat motifs were the most abundant repeat types which have a frequency of 16.82 and 9.98% respectively. The observed frequency of different repeat types comprising the SSR is summarized in supplementary file 2. In di-nucleotide repeats, the most abundant repeat motif was (TA/AT)n which accounted for 8.38%, followed by (AG/CT)n with 4.51%. The CG/GC repeats were least abundant. All of the ten possible types of tri-nucleotide repeats occurred in the citrus BES-SSR. Among the tri-nucleotide repeats the (AAT/TTA)n motif was the most common (5.54%), followed by (AAG/CTT)n (1.47%) and (AAC/GTT)n (0.66%). The GC rich tri-nucleotide repeats were the least abundant. Among the tetra-, penta- and hexa-nucleotides repeats (AAAN)n (AAAAN)n and (AAAAAN)n were more common than other combinations in C. clementina genome.
All the BES sequences (46,339) were assembled using CAP3 [37] to remove redundant sequences. As a result, 27,058 non-redundant BES sequences were identified. All microsatellites having repeat length ≥16 bp for di-, ≥18 bp for tri-, ≥20 bp for tetra- and penta- and ≥30 bp hexa-nucleotides were selected from the non-redundant BES sequences for marker development. Consequently, 1,529 sequences were selected for BES-SSR primer modeling, 1,281 non-redundant BES-SSR primers were designed, and 400 primers were evaluated for successful PCR amplification and transferability across the genera (Table 2). Among the primers, 333 (83.25%) were successfully amplified and 260 (65.00%) were transferable across genera. The efficiency of marker development was examined for each repeat motif. The success rate of PCR amplification, the transferability and map ability of the BES-SSRs markers for each SSR motif are listed in Table 2. Hexa-nucleotide motif had the highest success rate (100.00%) of PCR amplification, followed by di- (86.67%), tri- (81.29%) and penta-nucleotide (77.78%) motifs. BES-SSRs with hexa (100.00%) and penta-nucleotide (77.78%) had the highest levels of transferability, followed by tetra- (73.68%) and di-nucleotide (64.62%) repeats. Of the total number of SSRs identified in C. clementina BAC-end sequences, 1,967 (20.67%) were defined as Class I and 7,550 (79.85%) as Class II microsatellite. Class I SSRs were enriched for tri- (6%) and di-nucleotides (4%), while class II repeats were enriched in mono nucleotide repeats (65%) and di nucleotide repeats (10%), with less frequent occurrence of tetra nucleotide repeats (1%). Class I BES-SSR is on average more polymorphic than the class II microsatellite markers. More than 56% BES-SSR were in non coding sequences and the remaining 44% BES-SSR were located in the putative coding region of the C. clementina genome, while 1.35% BES-SSR were associated with TEs (Fig. 1). The abundance of mono, di and tri nucleotide motif is much higher in TEs than all other repeat motifs (Fig. 1). A total of 29 (17%) of SSR were found in copia-like and DNA transposon class TE. Most of the copia-like and gypsy-like elements contain SSRs in the 3′ end of the 5′ LTR.

Association of transposable elements (TE) with BES-SSR. a Association of TE with different repeat classes of microsatellites, b occurrences of BES-SSRs in TE classes
To determine the function of SSR containing BES, the 7,935 non-redundant BES-SSR sequences were annotated against non-redundant protein database using the Blas2GO tools. A total of 1,133 sequences (14%) matched with known, unknown, unnamed, hypothetical or expressed proteins, whereas 6,238 sequences (78%) had no blast hit (Fig. 2). Further putative functions were assigned to 1,133 BES-SSR sequences involved in molecular function, biological process and cellular component categories by GO analysis (Fig. 2c). The result revealed that a majority of the BES-SSR sequences in the molecular function category was assigned to binding (694 BRS-SSR sequences, 61%) and catalytic activity (585, BRS-SSR sequences 52%). When mapped against the biological process category, 578 (51%), 528 (47%), 130(11%) and 104 (9%) BRS-SSR sequences were involved in metabolic processes, cellular processes, localization and response to stimulus, respectively. On the other hand, when mapped against the cellular component GO terms, 640 BRS-SSR sequences (56%) were involved in cell and 375 (33%) were involved in organelle function.

Summary of the functional annotation of BES-SSR sequences; a Blast result distribution, b distribution of annotated BES-SSR sequences among the three major categories, c go level distribution of functional BES-SSR sequences, d annotation distribution among the different repeat motifs
Utility of BES-SSR marker in establishing phylogenetic relationship, genetic diversity and mapping
Level of polymorphism, informativeness and discriminating capacity were further estimated to evaluate the efficiency of designed BES-SSR primers. Twenty-five BES-SSR primer pairs were randomly selected and 40 citrus genotypes were used in this experiment. The results are shown in Table 3. A total of 118 alleles have been detected among 40 citrus genotypes. The number of alleles ranged from 4 to 13, the number of alleles/assay unit was 4.72, and the average confusion probability (C) value was negligible. Estimated average discriminating power (D) was very close to the average limit of discriminating power (D L ). Effective number of patterns/assay unit was 10.41, indicating that one BES-SSR primer set can discriminate about 10 citrus genotypes when the population size is infinite. The very low value of the effective number of alleles per locus in comparison to the average number of alleles per locus in this study may suggest the presence of many unique or less frequent alleles generated by the BES-SSR primer. The value of the marker index (MI) was very low compared to the assay efficiency index (A i ) and effective multiplex ratio (E).
In order to evaluate the ability of BES-SSR marker to be used for phylogenetic studies, a cluster analysis of genetic diversity has been conducted using 118 alleles generated by 25 BES-SSR markers (Fig. 3). Forty genotypes were clearly differentiated and the relationship between them was organized around five major groups. Acidic species such as lemon, lime, citron and sour oranges clustered together in the same group. Sweet orange species group in a single cluster. Citrus related species P. trifoliata and Fortunella sp. (Mewa kumquat and Hongkong kumquat) generate different individual cluster. Fortunella sp. is closer to the Citrus spp. than Poncirus sp.

Phylogenetic relationship among the 40 citrus genotypes determined by 118 alleles obtained from randomly selected 25 BES-SSR markers
Simultaneously, a subset of six BES-SSR markers were further used to analyze genetic diversity of 28 pummelo, 31 sweet orange and 18 wild kumquat accessions (Fig. 4). Our analysis showed that each of the six BES-SSR primers gave amplification products in all accessions and all six loci were polymorphic. The number of alleles observed at each locus ranges from four (BES-6) to nine (BES-12, BES-16) with an average of 7.2 (Table 4). Altogether, 43 alleles were generated in the set of 77 accessions. Of the total number of alleles, 61.3% were shared among the pummelo, sweet orange and wild kumquat accessions, while 12.8, 10.4 and 15.5% were unique alleles to the pummelo, sweet orange and wild kumquat accessions, respectively. Across all 77 accessions analyzed, the PIC values for individual loci ranged from 0.301 (BES-10) to 0.674 (BES-16) with an average of 0.525, which was lower than that observed in citrus and its relatives (PIC = 0.648) by Pang et al. [68] and was similar to that reported in the mandarin landraces and wild accessions (PIC = 0.5071, Chen et al. [21]). The PIC values were different among pummelo, sweet orange and wild kumquat germplasm collection. For instance it was 0.514 for wild kumquat, which was two and three fold higher than for sweet orange and pummelo, respectively. These results correlated the findings of Corazza-Nunes et al. [40], who estimated PIC value 0.2294 for pummelo and grapefruit cultivars. This is just slightly higher than our estimated PIC values (0.183) for pummelo cultivars. The discrepancy may be due to different numbers of samples and cultivar used in both studies or may even be attributed to statistical fluctuations caused by the selection of the BES-SSR markers. We also estimated genetic similarity (GS) separately among the collected germplasm (Pummelo, Sweet orange, wild kumquat; Supplementary file 1) and estimated GS were all most similar for three germplasm groups. For pummelo it was 0.50 which was slightly lower than that obtained from the same pummelo accessions with EST-SSR markers (GS = 0.67; Chai et al., unpublished). Moreover, this value is twofold higher than that for pummelo and grapefruit cultivars [40].

Electrophoretic patterns of BES-SSR markers: a 40 citrus and its relatives with primer BES-2-48; b 28 Pummelo with primer BES-10; c 31 Sweet orange with primer BES-12; d 18 wild kumquat with primer BES-6. Number of each lean represents the genotype id which is listed in supplementary file 1
The 333 BES-SSR markers which successfully amplified were further evaluated for their preliminary usability for genetic mapping; the results are summarized in Tables 2 and 5. Among the tested markers, 118 (35.44%) were heterozygous in at least one parent of the C. reticulata × P. trifoliata F1 population. Hexa-nucleotide (66.67%) repeats had the highest mapping ability, followed by penta- (57.14%), tetra- (53.57%) and tri-nucleotide (50.79%) repeats. The quality of the map-able marker was very good in that the allele bands were prominent and easy to score. In fact 42 (34.15%) BES-SSR markers amplified more than one locus in the mapping population. Non-polymorphic and non-segregating loci had been found with occurrence numbers of 45 (24.59%) and 39 (21.31%) respectively. The amount of information varied with different microsatellites. The information content was recorded in five categories from the allele segregation pattern (Table 5). The highest number of allele segregating pattern was ab-aa (33.90%), followed by ab-ac (25.42%) and ab-cd (19.49%). In this study, 118 map-able markers produced 183 scorable segregating loci, of which 129 could be mapped. For all of the map-able markers, the PIC value was calculated on the basis of observed alleles in sixteen citrus species; these markers detected 2–13 alleles with an average of 4.72 alleles per locus. Their corresponding PIC values ranged from 0.96 to 0.37 with an average of 0.69; about 90% of the PIC values were higher than 0.69. A putative gene function could be assigned to 53 (44.91%) map-able BES-SSR markers based on functional annotation. Among these markers, 38 showed homology with known proteins, 3 with putative proteins, 5 with hypothetical proteins and 7 with unknown proteins (supplementary file 3). Information about the developed map-able BES-SSR markers is listed in supplementary file 3, which includes the SSR motif, primer sequences and corresponding annealing temperature (Tm), allele number, PCR amplification profile, PIC value and BLASTX result.
Discussion
Development and characterization of BES-SSR marker
The markers developed in this study are a valuable resource for genetic analysis of citrus and related species. Here we developed SSR markers from BES data mining and experimentally validated their quality and usefulness in Citrus. BES databases have proved to be an important and useful resource for SSR marker development. BES data-bases can be screened for SSR and those SSR with suitable flanking sequences can be used for SSR marker development. This strategy has been successfully applied in several plant species [41, 42]. In this study, C. clementina BES databases have been screened for potential SSR markers. Terol et al. [29] suggested that this BES data set can be a good resource for SSR marker development, but did not experimentally verify this proposition. Previous studies suggested that SSR containing sequences are not always suitable for high quality markers development. Consequently, an individual experimental verification is required for each SSR marker in order to validate its quality and robustness. We found that 71.7% SSR containing BES sequences are suitable for BES-SSR primer development. This finding suggested that only a primary SSR analysis is not sufficient and that an empirical verification is also important for assessing their utility. The number of SSR detected in this study was twofold higher than the number reported by Terol et al. [29]. Although both studies use the same underlying data, the results obtained are significantly different. This difference is due to the SSR search parameters used in both the studies. Terol et al. [29] searched for SSRs with a minimum length of 16 bp, which discards a large number of SSR. In this study, SSR are required to have a minimum length of 11 bp. With this threshold, the SSR density was 0.49 SSR/Kbp (2.04 Kb/SSR), which is twofold higher than the result reported by Terol et al. [29]. The dependence of SSR densities on search parameters is well known [43]. Our repeat densities are comparable with those reported in Papaya [44] and Brassica rapa [41]. The density of SSRs found in this study is higher than that in barley (1/7.5 kb), maize (1/6.2 kb), rice (1/11.81 kb) and soybean (1/23.80 kb) [45, 46]. Earlier studies suggested that the density of SSRs varies strongly among species [4, 47]. The density of SSRs found in the non-coding region (BES sequences) of C. clementina is lower than that in the transcribed regions of the citrus genome (one SSR per 1.70 kb in C. sinensis, 1.30 kb in C. clementina). This finding is in good agreement with the hypothesis of SSR distribution between non coding and coding region in higher plants [48].
The number of SSRs found varies strongly with unit size. The SSRs with di-nucleotide repeats were most abundant. This finding is consistent with previous studies of citrus EST-SSR analyses, in which di-nucleotide repeats were also dominant [5]. Among di-nucleotide repeats (TA/AT)n is the most frequent di-nucleotide repeat motif, followed by (AG/CT)n repeats. This is in good agreement with the patterns observed in papaya [44], but it is different from the observations in human and Drosophila, where (AC)n are the most frequent di-nucleotide repeats [3]. (GC)n repeats are extremely rare in eukaryotic genomes and this is also the case for C. clementina [3]. Among tri-nucleotide repeats, AT-rich repeats are the most abundant in C. clementina BESs. Unfortunately, the majority of AT-rich repeats do not amplify well. Similar result are reported for rice [49], Arabidopsis and yeast [3]. This seems to be correlated with AT-rich repeats lying mostly in non-coding regions and that they are frequently associated with larger repeat elements. This conjecture however needs to be further investigated. In tetra and penta nucleotide repeats, especially (AAAT)n and (AAAAT)n are more common than other combinations, and are more common than in other plant genomes [41]. These findings suggested that the SSRs in the C. clementina genome tend to skew toward AT rich motifs.
Microsatellites can be categorized into two groups based on their total length which in turn is correlated to their potential utility as informative genetic markers: class I (≥20 bp) and class II (≥10 bp but <20 bp). Experimental evidences from human, rice and other organism [12, 49] suggests that class I SSRs are highly polymorphic. This could be verified for citrus SSRs in the present study. As a result, class I SSRs was chosen in citrus for BES-SSR markers development since they are usually more polymorphic and informative. Several previous studies showed that SSRs are randomly distributed in the genome [50, 51] but their density differed significantly among coding and non-coding regions, as was shown e.g., in the L. bicolor genome [50]. Our study confirms that BES-SSR in the protein coding sequences is less abundant than in non-coding regions. TE associated SSRs are a component of active TEs that spreading throughout the genome, act as a landing pad for TE insertion or arise other the integration of on extended and polyadenylated retro-transcript into the genome [52–54]. We found that SSRs are often extending to both the 5′ and 3′ ends of LTR retrotransposon suggesting that these SSR have arisen by a mutation followed by an expansion of the same proto-SSR and then spread through the C. clementina genome as component of an active retrotransposable element. The data on the composition and distribution of SSRs obtained in this study are useful for further research on the role of SSR in the citrus genome organization.
Out of 1,529 BES-SSR sequences, 1,281 primer pairs were generated (71.70%). The remaining sequences had insufficient flanking regions or the microsatellite or/and the sequences were inappropriate for primer design. Using these 1,281 candidates for SSR-marker development, four hundred primer pairs were assessed to detect polymorphism and transferability in the 16 citrus and their relatives. Our results demonstrate high marker transferability among citrus and its relatives and revealed a high level of sequence conservation among the citrus and its related genera. The high level of interspecific transferability of BES-SSR markers may prove useful for comparative genomic studies in citrus.
A functional characterization of BES-SSR sequences was performed with the Blast2GO annotation pipeline. A major proportion of the sequences remained unannotated, and thus may be considered as novel C. clementina sequences. Our results suggest that BES SSRs are distributed in all of the main functional categories (biological function, molecular function and cellular component) in the C. clementina genome. Functional annotation of BES-SSR sequences led to the development of functional domain markers that can provide information on functional properties of microsatellites and predicted protein domains.
Utility of BES-SSR marker in establishing phylogenetic relationship, genetic diversity and mapping
BES-SSR primers were highly polymorphic compared to previous studies of EST-SSR based polymorphism in Citrus [16, 25]. This can be explained with different levels of sequence conservation in transcribed and non-transcribed regions, with a higher level of conservation in the transcribed versus non-transcribed regions. Consequently, EST-SSRs are less polymorphic than genomic SSRs. In this respect BES-SSR markers are superior to the EST-SSRs for fingerprinting or variety identification in citrus. The value of the effective number of alleles per locus (n e ) was 1.56, which finds its reflection in lower values of the expected heterozygosity (H ep = 0.32). The low value of n e for BES-SSR markers in comparison to the number of alleles per assay unit (n u = 4.72) may suggest the presence of many unique or less frequent alleles. Confusion probability (C) and effective number of patterns per assay unit (P) provide valuable information on the evaluation of germplasm for which numerous cultivars need to be accurately characterized and identified. A low level of confusion probability was observed in this study, which again supports the utility of BES-SSRs in identification studies. The relatively high value of the effective number of patterns per assay unit (P) for BES-SSR markers revealed their discriminating capacity when handling a large number of samples. Similar result have been reported for olive [55]. In order to estimate phylogenetic relationship among 40 accessions of citrus and its relatives, a similarity matrix was calculated according to Dice coefficient [39] and dendrogram was constructed using UPGMA cluster analysis (Fig. 3). Cophenetic correlation between tree matrix and similarity matrix was found to be higher (r = 0.84, P < 0.01), indicating that the cluster analysis strongly represented the similarity matrix. The studied accessions had similarity values ranging from 0.29 to 1.00, suggesting a high level of variation exits among the accessions. The organization of genetic diversity obtained with BES-SSR is in agreement with the previously reported systematic relationship of citrus species [56]. In addition, Ollitrault et al. [31] estimated genetic diversity using 18 BES-SSR markers for 45 accessions from eight cultivated species and the papeda group and concluded that BES-SSR markers were useful for citrus germplasm identification. In our study we also performed genetic diversity analysis for 40 accessions of citrus and its relatives species (Poncirus and Fortunella sp.) and we found that BES-SSR markers are not only suitable for citrus germplasm characterization but also for the characterization of species related to citrus such as P. trifoliata and Fortunella sp. Comparing the phylogenetic relationship of citrus and its relatives obtained from EST-SSRs [25] and BES-SSRs (this study), some differences were observed, which need to be resolved in future analyses. According to Luro et al. [25] Poncirus trifoliata clustered with Citron-limes-lemon, and wild kumquat (Fortunella japonica) remained genetically distinct from other citrus species. In contrast, we found that Fortunella species (Mewia Kumquat and Hong Kong Kumquat) are closer related to the Citrus than Poncirus. A phylogenetic relation of Poncirus, Fortunella and citrus which is compatible with this study has already been reported by Barkley [56]. If this finding is also supported by future analyses, this hints at a higher resolving power of BES-SSRs compared to EST-SSR.
In view of the performance of BES-SSR markers, we conclude that these markers generate valuable information on the level of polymorphism and diversity in citrus. Consequently, we suggest a broader application of BES-SSR derived markers which seem to be more reliable compared to EST-SSR markers for the characterization of the citrus germplasm accessions.
Pummelo, sweet orange and wild kumquat germplasm are important citrus genetic resource. Knowledge of the genetic diversity of this germplasm provides an opportunity for citrus breeding programs, germplasm conservation and management strategies. In order to estimate genetic diversity of any germplasm, a set of effective molecular markers play an important role. In this study, a subset of BES-SSR markers was used to assess the efficiency of BES-SSR markers for genetic diversity analyses of the pummelo, sweet orange and wild kumquat germplasm collection. Our result showed that the genetic diversity and levels of polymorphism detected in pummelo accessions by 6 BES-SSR markers was higher than previously detected in pommelo accessions by RAPD and EST-SSR markers [57, 58]. This finding suggested that BES-SSR markers are more powerful than EST-SSR and RAPD for studying the genetic diversity of the pummelo germplasm collection. The same result was obtained for the sweet orange and wild kumquat germplasm collections. Comparing the GS (genetic similarity) obtained for the pummelo accession with EST-SSR and BES-SSRs, a significant difference was observed. Our results suggest that genomic SSR are more suitable than genic SSRs for estimate genetic diversity in the citrus germplasm collection.
SSR markers are useful for a variety of applications in plant genetics and breeding; among them mapping is one of the most important applications. Prolonged juvenility in citrus limits the probability of work on second generation hybrids. As a result, citrus genetic maps were developed on F1 progenies at interspecific or intergeneric level [59–62]. Citrus × Poncirus progenies have been extensively used for citrus genetic mapping [26, 63–66]. In this study we also used Citrus × Poncirus F1 progenies in order to evaluate the utility of BES-SSR markers for mapping studies. One hundred eighteen BES-SSRs markers were used to demonstrate the mapping ability on the Citrus × Poncirus mapping population due to ease of allele scoring. Five types of allele segregating patterns were recorded, among which the ab-aa pattern was dominant (33.90%). Similar results were observed from EST-derived microsatellites for Actinidia sp. [67]. The PIC value of markers indicates their usefulness for gene mapping, molecular breeding and germplasm evaluation [18]. In order to measure the informativeness of BES-SSR derived markers, the PIC was estimated for each of the map-able markers based on the 16 Citrus spp. The result showed that BES-SSR marker might be useful for mapping in citrus and related species. A high-density microsatellite consensus map is still lacking in citrus due to a limited number of publicly available SSR markers. Newly developed BES-SSR markers will increase the number of markers and accelerate the mapping project.
Conclusion
Our study demonstrated the utility of BES-SSR derived markers in the characterization of citrus germplasm and genetic mapping. A total of 7,935 non-redundant SSR sequences were identified from 46,339 BAC-end sequences and 1,281 BES-SSR markers were developed. Of these markers, 400 were tested in this study; 83.25% successfully amplified, 65.00% were transferable across the genera and 35.44% were potentially useful for mapping projects in the C. reticulata × P. trifoliata mapping population. These newly developed BES-SSR markers remarkably increased the number of publicly available citrus SSR markers and will certainly benefit citrus breeders and geneticists.
Abbreviations
- BES:
-
Citrus clementina BAC end sequences
- GSSs:
-
Genome survey sequences
- GO:
-
Gene ontology
- SSR:
-
Simple sequences repeat
References
Tautz D, Renz M (1984) Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res 12:4127–4138
Schlötterer C, Tautz D (1992) Slippage synthesis of simple sequence DNA. Nucleic Acids Res 20:211–215
Katti MV, Ranjekar PK, Gupta VS (2001) Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol Biol Evol 18:1161–1167
Mayer C, Leese F, Tollrian R (2010) Genome-wide analysis of tandem repeats in Daphnia pulex—a comparative approach. BMC Genomics 11:277
Shanker A, Bhargava A, Bajpai R, Singh S, Srivastava S, Sharma V (2007) Bioinformatically mined simple sequence repeats in UniGene of Citrus sinensis. Sci Hortic 113:353–361
Gupta PK, Varshney RK (2000) The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat. Euphytica 113:163–185
Chen C, Yu Q, Hou S, Li Y, Eustice M, Skelton RL, Veatch O, Herdes RE, Diebold L, Saw J, Feng Y, Qian W, Bynum L, Wang L, Moore PH, Paull RE, Alam M, Ming R (2007) Construction of a sequence-tagged high-density genetic map of papaya for comparative structural and evolutionary genomics in brassicales. Genetics 177:2481–2491
Shoemaker RC, Grant D, Olson T, Warren WC, Wing R, Yu Y, Kim H, Cregan P, Joseph B, Futrell-Griggs M, Nelson W, Davito J, Walker J, Wallis J, Kremitski C, Scheer D, Clifton SW, Graves T, Nguyen H, Wu X, Luo M, Dvorak J, Nelson R, Cannon S, Tomkins J, Schmutz J, Stacey G, Jackson S (2008) Microsatellite discovery from BAC end sequences and genetic mapping to anchor the soybean physical and genetic maps. Genome 51:294–302
Shultz JL, Kazi S, Bashir R, Afzal JA, Lightfoot DA (2007) The development of BAC-end sequence-based microsatellite markers and placement in the physical and genetic maps of soybean. Theor Appl Genet 114:1081–1090
Pan L, Xia Q, Quan Z, Liu H, Ke W, Ding Y (2010) Development of novel EST-SSRs from Sacred Lotus (Nelumbo nucifera Gaertn) and their utilization for the genetic diversity analysis of N. nucifera. J Hered 101:71–82
Batley J, Hopkins CJ, Cogan NOI, Hand M, Jewell E, Kaur J, Kaur S, Li XI, Ling AE, Love C, Mountford H, Todorovic M, Vardy M, Walkiewicz M, Spangenberg GC, Edwards D (2007) Identification and characterization of simple sequence repeat markers from Brassica napus expressed sequences. Mol Ecol Notes 7:886–889
Choi SR, Teakle GR, Plaha P, Kim JH, Allender CJ, Beynon E, Piao ZY, Soengas P, Han TH, King GJ, Barker GC, Hand P, Lydiate DJ, Batley J, Edwards D, Koo DH, Bang JW, Park BS, Lim YP (2007) The reference genetic linkage map for the multinational Brassica rapa genome sequencing project. Theor Appl Genet 115:777–792
Hopkins CJ, Cogan NOI, Hand M, Jewell E, Kaur J, Li XI, Lim GAC, Ling AE, Love C, Mountford H, Todorovic M, Vardy M, Spangenberg GC, Edwards D, Batley J (2007) Sixteen new simple sequence repeat markers from Brassica juncea expressed sequences and their cross-species amplification. Mol Ecol Notes 7:697–700
Iniguez-Luy FL, Voort AV, Osborn TC (2008) Development of a set of public SSR markers derived from genomic sequence of a rapid cycling Brassica oleracea L. genotype. Theor Appl Genet 117:977–985
Ling AE, Kaur J, Burgess B, Hand M, Hopkins CJ, Li XI, Love CG, Vardy M, Walkiewicz M, Spangenberg G, Edwards D, Batley J (2007) Characterization of simple sequence repeat markers derived in silico from Brassica rapa bacterial artificial chromosome sequences and their application in Brassica napus. Mol Ecol Notes 7:273–277
Chen C, Zhou P, Choi YA, Huang S, Gmitter FG Jr (2006) Mining and characterizing microsatellites from citrus ESTs. Theor Appl Genet 112:1248–1257
Eujayl I, Sorrells ME, Baum M, Wolters P, Powell W (2002) Isolation of EST-derived microsatellite markers for genotyping the A and B genomes of wheat. Theor Appl Genet 104:399–407
Peng J, Lapitan N (2005) Characterization of EST-derived microsatellites in the wheat genome and development of eSSR markers. Funct Integr Genomics 5:80–96
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Hackauf B, Wehling P (2002) Identification of microsatellite polymorphisms in an expressed portion of the rye genome. Plant Breed 121:17–25
Sharopova N, McMullen MD, Schultz L, Schroeder S, Sanchez-Villeda H, Gardiner J, Bergstrom D, Houchins K, Melia-Hancock S, Musket T, Duru N, Polacco M, Edwards K, Ruff T, Register JC, Brouwer C, Thompson R, Velasco R, Chin E, Lee M, Woodman-Clikeman W, Long MJ, Liscum E, Cone K, Davis G, Coe EH Jr (2002) Development and mapping of SSR markers for maize. Plant Mol Biol 48:463–481
Cordeiro GM, Casu R, McIntyre CL, Manners JM, Henry RJ (2001) Microsatellite markers from sugarcane (Saccharum spp.) ESTs cross transferable to erianthus and sorghum. Plant Sci 160:1115–1123
Scott KD, Eggler P, Seaton G, Rossetto M, Ablett EM, Lee LS, Henry RJ (2000) Analysis of SSRs derived from grape ESTs. TAG Theor Appl Genet 100:723–726
Castelo AT, Martins W, Gao GR (2002) TROLL—tandem repeat occurrence locator. Bioinformatics 18:634–636
Luro F, Costantino G, Terol J, Argout X, Allario T, Wincker P, Talon M, Ollitrault P, Morillon R (2008) Transferability of the EST-SSRs developed on Nules clementine (Citrus clementina Hort ex Tan) to other Citrus species and their effectiveness for genetic mapping. BMC Genomics 9:287
Kijas JMH, Thomas MR, Fowler JCS, Roose ML (1997) Integration of trinucleotide microsatellites into a linkage map of Citrus. TAG Theor Appl Genet 94:701–706
Jiang D, Zhong G-Y, Hong Q-B (2006) Analysis of microsatellites in citrus unigenes. Acta Genetica Sinica 33:345–353
Novelli VM, Cristofani M, Souza AA, Machado MA (2006) Development and characterization of polymorphic microsatellite markers for the sweet orange (Citrus sinensis L. Osbeck). Genet Mol Biol 29:90–96
Terol J, Naranjo MA, Ollitrault P, Talon M (2008) Development of genomic resources for Citrus clementina: characterization of three deep-coverage BAC libraries and analysis of 46, 000 BAC end sequences. BMC Genomics 9:423
Varshney RK, Graner A, Sorrells ME (2005) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23:48–55
Ollitrault F, Terol J, Pina JA, Navarro L, Talon M, Ollitrault P (2010) Development of SSR markers from Citrus clementina (Rutaceae) BAC end sequences and interspecific transferability in Citrus. Am J Bot 97:124–129
Huang X, Madan A (1999) CAP3: a DNA sequence assembly program. Genome Res 9:868–877
Cheng Y-J, Guo W–W, Yi H-L, Pang X-M, Deng X (2003) An efficient protocol for genomic DNA extraction from Citrus species. Plant Mol Biol Rep 21:177–178
Ruiz C, Paz Breto M, Asíns MJ (2000) A quick methodology to identify sexual seedlings in citrus breeding programs using SSR markers. Euphytica 112:89–94
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33:W116–W120
Labarga A, Valentin F, Anderson M, Lopez R (2007) Web services at the European bioinformatics institute. Nucleic Acids Res 35:W6–W11
Rohlf FJ (1998) NTSYSpc: numerical taxonomy and multivariate analysis system version 2.02. Exeter Software, Setauket
Dice LR (1945) Measures of the amount of ecologic association between species. Ecology 26:297–302
Corazza-Nunes MJ, Machado MA, Nunes WMC, Cristofani M, Targon MLPN (2002) Assessment of genetic variability in grapefruits (Citrus paradisi Macf.) and pummelos (C. maxima (Burm.) Merr.) using RAPD and SSR markers. Euphytica 126:169–176
Hong CP, Piao ZY, Kang TW, Batley J, Yang TJ, Hur YK, Bhak J, Park BS, Edwards D, Lim YP (2007) Genomic distribution of simple sequence repeats in Brassica rapa. Mol Cells 23:349–356
Wen M, Wang H, Xia Z, Zou M, Lu C, Wang W (2010) Developmenrt of EST-SSR and genomic-SSR markers to assess genetic diversity in Jatropha Curcas L. BMC Res Notes 3:42
Leclercq S, Rivals E, Jarne P (2007) Detecting microsatellites within genomes: significant variation among algorithms. BMC Bioinform 8:125
Lai C, Yu Q, Hou S, Skelton R, Jones M, Lewis K, Murray J, Eustice M, Guan P, Agbayani R, Moore P, Ming R, Presting G (2006) Analysis of papaya BAC end sequences reveals first insights into the organization of a fruit tree genome. Mol Genet Genomics 276:1–12
Gao L, Tang J, Li H, Jia J (2003) Analysis of microsatellites in major crops assessed by computational and experimental approaches. Mol Breed 12:245–261
Varshney RK, Thiel T, Stein N, Langridge P, Graner A (2002) In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species. Cell Mol Biol Lett 7:537–546
Tóth G, Gáspári Z, Jurka J (2000) Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res 10:967–981
Morgante M, Hanafey M, Powell W (2002) Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet 30:194–200
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:1441–1452
Labbé J, Murat C, Morin E, Le Tacon F, Martin F (2011) Survey and analysis of simple sequence repeats in the Laccaria bicolor genome, with development of microsatellite markers. Curr Genet 57:75–88
Victoria F, da Maia L, de Oliveira A (2011) In silico comparative analysis of SSR markers in plants. BMC Plant Biol 11:15
Akagi H, Yokozeki Y, Inagaki A, Mori K, Fujimura T (2001) Micron a microsatellite-targeting transposable element in the rice genome. Mol Genet Genomics 266:471–480
Ramsay L, Macaulay M, Cardle L, Morgante M, Ivanissevich SD, Maestri E, Powell W, Waugh R (1999) Intimate association of microsatellite repeats with retrotransposons and other dispersed repetitive elements in barley. Plant J 17:415–425
Tay W, Behere G, Batterham P, Heckel D (2010) Generation of microsatellite repeat families by RTE retrotransposons in lepidopteran genomes. BMC Evol Biol 10:144
Belaj A, Satovic Z, Cipriani G, Baldoni L, Testolin R, Rallo L, Trujillo I (2003) Comparative study of the discriminating capacity of RAPD, AFLP and SSR markers and of their effectiveness in establishing genetic relationships in olive. Theor Appl Genet 107:736–744
Barkley NA, Roose ML, Krueger RR, Federici CT (2006) Assessing genetic diversity and population structure in a citrus germplasm collection utilizing simple sequence repeat markers (SSRs). Theor Appl Genet 112:1519–1531
Yong L, De-Chun L, Bo W, Zhong-Hai S (2006) Genetic diversity of pummelo (Citrus grandis Osbeck) and its relatives based on simple sequence repeat markers. Chin J Agric Biotechnol 3:119–126
Zhang TP, Peng SL, Wang ZF, Ling DH, Gan LS (2001) Genetic relationships among cultivars of citrus maxima (burm.) Merr. using RAPD marker technique. J Trop Sub Trop Bot 9:322–328
Bernet GP, Margaix C, Jacas J, Carbonell EA, Asins MJ (2005) Genetic analysis of citrus leafminer susceptibility. Theor Appl Genet 110:1393–1400
Ruiz C, Asins MJ (2003) Comparison between Poncirus and Citrus genetic linkage maps. Theor Appl Genet 106:826–836
Siviero A, Cristofani M, Furtado EL, Garcia AA, Coelho AS, Machado MA (2006) Identification of QTLs associated with citrus resistance to Phytophthora gummosis. J Appl Genet 47:23–28
Weber CA, Moore GA, Deng Z, Gmitter FG Jr (2003) Mapping freeze tolerance quantitative trait loci in a Citrus grandis × Poncirus trifoliata F1 pseudo-testcross using molecular markers. J Am Soc Hortic Sci 128:508–514
Cai Q, Guy CL, Moore GA (1994) Extension of the linkage map in Citrus using random amplified polymorphic DNA (RAPD) markers and RFLP mapping of cold-acclimation-responsive loci. TAG Theor Appl Genet 89:606–614
Durham RE, Liou PC, Gmitter FG, Moore GA (1992) Linkage of restriction fragment length polymorphisms and isozymes in Citrus. TAG Theor Appl Genet 84:39–48
Jarrell DC, Roose ML, Traugh SN, Kupper RS (1992) A genetic map of citrus based on the segregation of isozymes and RFLPs in an intergeneric cross. TAG Theor Appl Genet 84:49–56
Moore GA, Tozlu I, Weber CA, Guy CL (2000) Mapping quantitative trait loci for salt tolerance and cold tolerance in Citrus grandis (L.) Osb. × Poncirus trifoliata (L.) Raf hybrid populations. Acta Hortic 535:37–45
Fraser LG, Harvey CF, Crowhurst RN, De Silva HN (2004) EST-derived microsatellites from Actinidia species and their potential for mapping. Theor Appl Genet 108:1010–1016
Pang XM, Hu CG, Deng XX (2003) Phylogenetic relationships among citrus and its relatives as reveled by SSR markers. Yi Chuan Bao 30(1):81–87
Acknowledgments
We are grateful to Dr. Yun-Jiang Cheng for discussing on the sampling strategy. This research was financially supported by the Ministry of Science and Technology of China (Nos. 2011CB100600, 2011AA100205) and the national NSF of China.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Biswas, M.K., Chai, L., Mayer, C. et al. Exploiting BAC-end sequences for the mining, characterization and utility of new short sequences repeat (SSR) markers in Citrus. Mol Biol Rep 39, 5373–5386 (2012). https://doi.org/10.1007/s11033-011-1338-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-011-1338-5