
Abstract
Pediatric diffuse intrinsic pontine glioma (DIPG) represents approximately 20% of all pediatric CNS tumors. However, disease outcomes are dismal with a median survival of less than 1 year and a 2-year overall survival rate of less than 10%. Despite extensive efforts to improve survival outcomes, progress towards clinical improvement has been largely stagnant throughout the last 4 decades. Focal radiotherapy remains the standard of care with no promising single-agent alternatives and no evidence for improvement with the addition of a long list of systemic therapies. A better understanding of the biology of DIPG, though not easy due to obstacles in obtaining pathological material to study, is promising for the development of specific individualized treatment for this fatal disease. Recent studies have found epigenetic mutations to be successful predictors and prognostic factors for developing future management policies. The aim of this review is to give a global overview about the epidemiology, diagnosis, and treatment of DIPG. We further examine the controversial biopsy and autopsy issue that is unique to DIPG and assess the subsequent impact this issue has on the research efforts and clinical management of DIPG.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
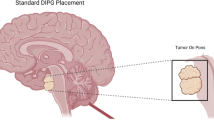
Brain tumors are common cancers in childhood and represent about 20–25% of all childhood tumors [1]. Although 15% of brain tumors affect the brain stem, 80% of these cases are diagnosed as diffuse intrinsic pontine glioma (DIPG) [2]. DIPG is a subtype of advanced grade gliomas that originates in the pons and spreads to other parts of the brainstem. After the establishment of magnetic resonance technology in the 1990s, DIPG was classified separately from other focally growing tumors with unique prognosis [3]. Before 2012, most of the epidemiological data about DIPG was mainly available from limited countries [4, 5]. The aim of this review is to give a global overview about the epidemiology, diagnosis, and treatment of DIPG. Furthermore, it highlights the controversy biopsy and autopsy issue with its subsequent progress and impact on the biology of DIPG.
2 Epidemiology
Although DIPG is a rare tumor, it is the most lethal type of pediatric brain tumor with 90% deaths within 2 years of diagnosis [6]. DIPG has a bimodal peak; it is apparent specifically in children with median age of 6–7 years at diagnosis [2, 7] and between 20 and 50 years in adults with a median age of 34 years [8]. In the USA, around 100–150 children were diagnosed as DIPG annually [4]. Its incidence is equal between males and females (1:1) [9]. Unlike other pediatric tumors, the survival of DIPG has not improved over the last 40 years. Despite the extensive continuous research to improve DIPG’s outcome, the median overall survival (OS) of DIPG patients remains poor at 8–11 months [10]. Moreover, greater than 90% of patients die within 2 years of diagnosis [6].
3 Presentation and diagnosis of DIPG pateint
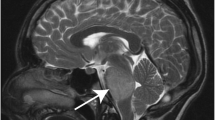
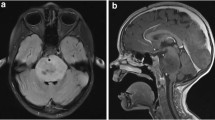
The presentation of DIPG is characterized by neurological signs and symptoms that were developed rapidly over 1–6 months prior to presentation. The classical signs and symptoms may involve long tract signs (clonus, hyperreflexia, weakness, positive Babinski reflex, and increased tone) and cranial palsy (due to tumor infiltration or mass effect on the sixth and seventh cranial nerves with uni- or bi-lateral deficits), in addition to cerebellar affection signs (ataxia, coordination deficit, and dysmetria) [11,12,13]. Around 30% of DIPG patients suffer from increased intracranial pressure, due to expansion of the pons. This expansion together with fourth ventricular narrowing consequently leads to obstructive hydrocephalus [13].
The standard and widely accepted criteria for DIPG diagnosis include symptoms that last greater than 6 months; the presence of at least 2 symptoms of the 3 brainstem dysfunctions (pyramidal tract affection, cranial nerve deficit, and coordination dysfunction); and infiltration of more than 50–66% of the pons [6, 13, 14]. A post-mortem assessment of DIPG patients revealed that around 20% of the brainstem tumors had a histologic diagnosis of brainstem primitive neuroectodermal tumors (PNET) that mimic DIPGs radiologically [13, 15]. Thus oncologists must exercise caution when making a DIPG diagnosis for a mass lesion in the brainstem.
In general, tissue biopsy from brain tumor lesions is usually required for proper diagnosis and appropriate treatment selection. However, most neurosurgeons do not perform tissue biopsy as it impacts both the quality of life and function of DIPG patients. Since the 1990s, most studies recommended using MRI as an alternative option for obtaining tissue biopsy to diagnose brainstem gliomas [3, 16]. In 2007, Schumacher et al. proved that the diagnosis of brainstem gliomas can be effectively made through precise imaging criteria, laboratory data, and clinical history and symptoms. Diagnostic biopsies are mainly reserved for tumors with atypical clinical presentation [17]. Currently, the diagnosis of DIPG depends on both the clinical symptoms and MRI. A tirade of symptoms including ataxia, pyramidal tract dysfunction, and cranial nerve palsies are the common presentations. These often have a rapid clinical onset (less than 3 months). DIPG also exhibits characteristic hypertense T2 or FLAIR MR sequence involving more than 50% of the ventral pons and no postcontrast enhancement on T1 images.
4 Autopsy, biopsy, and liquid biopsy: “Areas of Controversy”
A major obstacle that hindered progress in identification of DIPG biology and consequently hindered developing a specific treatment is the neuroanatomical location of DIPG in addition to the deficiency of in vivo models of DIPG. Generally, the two main sources for obtaining DIPG tissues are autopsies and biopsies. Autopsy, also called “post-mortem material,” is a radiological-guided process. This process depends mainly on the selfless gift and co-operation of DIPG patients and their families of donating their post-mortem tumor. Samples obtained by this process were large enough to carry out many in vivo tumor model studies for drug testing [18,19,20] and DIPG genomic profiling [21]. Many efforts were exerted to develop protocols and standardize procedures for specimen acquisition [22, 23]. However, the fact of obtaining cells and tissues exposed to many treatment modalities and evolution changes during the course of the disease is the major drawback and limitation for the absolute value of such procedure.
The indication for DIPG biopsy is a highly controversial issue [24]. Biopsy is widely considered to be a risky procedure with no direct patient benefits and was thus restricted to DIPG patients of unusual or atypical imaging or presentation. Increasing the need for improving the prognosis of DIPG and achieving progress in understanding DIPG biology lead to reconsideration of biopsy in DIPG. Many centers showed the safety and feasibility of DIPG biopsy with low morbidity and mortality [25,26,27,28]. A meta-analysis of 13 studies performing stereotactic biopsy of brain stem lesions in 381 children and adults established a diagnostic yield of 96%, notably with one incidence of death. The rate of permanent and transient deficits was 4 and 1%, respectively [29]. Another meta-analysis on 192 children revealed a similar diagnostic yield of 94.9%, 0.7% mortality, and 4.9% morbidity [30]. There is an ongoing national clinical trial in the USA that obtains upfront biopsy from newly diagnosed DIPG pediatric patients, called The DIPG Biology and Treatment Study (DIPG-BAT clinical trial) [31]. Puget et al. also put guidelines for performing biopsies in DIPG patients including the choice of the proper patient, the target, and the procedure [24]. However, dynamic monitoring of DIPG treatment using biopsy remains a major clinical challenge. On the other hand, liquid biopsy or liquid biome avoids the most troublesome aspects of biopsy. This is a non-invasive technique that has recently garnered attention in many cancer types, and involves using biofluids (e.g., serum, cerebrospinal fluids, urine, or saliva) to detect certain analytes such as cell-free tumor DNA (ctDNA), circulating tumor cells (CTC), and tumor-derived extracellular vesicles [32]. Liquid biopsy has a wide range of applications both at initial diagnosis and for post-surgical or post-therapy monitoring. There is an additional role for liquid biopsies in cancer screening, but this remains of limited application [32, 33].
Huang et al. conducted the first study on the liquid biopsy approach in pediatric midline gliomas including DIPG [34]. They used both Sanger sequencing and mutation-specific PCR to detect H3 mutations in CSF-derived tumor DNA. While the process was feasible, the Sanger sequencing method had two limitations that hurdle the clinical utility of this method: the inability to calculate allele frequency and the lack of sensitivity. Droplet digital PCR is another approach used to overcome these limitations and to detect major mutations instead of single variants [35]. Recently, molecular profiling using deep sequencing technique of CSF-derived tumor DNA in brain tumors involving DIPG patients was successful. In addition to its technique feasibility, CSF-derived tumor DNA has better sensitivity (100%) compared with plasma circulating tumor [36]. Generally, liquid biopsy is considered a promising non-invasive approach for the future diagnosis and monitoring of DIPGs. It helps at diagnosis particularly in the situation of unfeasible biopsy. Furthermore, it offers a non-invasive dynamic monitoring of DIPG including molecular-based longitudinal characteristics.
5 Biology of DIPG
A better understanding of DIPG biology will positively impact disease prognosis and overall patient outcomes. Great advances in neurosurgical techniques with the advancement of operative microscopes, intraoperative MRI, and stereotactic technology have effectively decreasing the risks associated with obtaining a surgical biopsy. In addition, using small samples for genomic profiling and drug efficacy testing has enabled researchers to perform whole genome sequencing (WGS) on DIPG tumor tissues to understand DIPG biology. They successfully revealed many oncogenic pathways and drivers involved in DIPG tumorigenesis [37,38,39,40]. DIPG biology is complex and involves epigenetic aberrations and genetic changes that affects many pathways of cellular proliferation and cell cycle machinery regulations (Table 1). Pontine precursor-like cells (PPC) are thought to be the cells of origin for DIPG. During infancy, they are available in all ventral brainstem structures and wane by the age of 2 years. At the age of 6 years, they have another peak which is corresponding to the median age of DIPG presentation [41].
5.1 Epigenetic aberrations
Epigenetic aberrations identified in DIPG involve both histone H3 mutations and polycomb repressive complex (PRC) abnormalities.
5.1.1 H3 mutations
Histone protein help in packaging DNA as a chain of nucleosome. Each nucleosome composed of 147 base pair (bp) wrapping around 2 copies of histones [H2A, H2B, H3, and H4] [42, 43]. Both lysine (K) and arginine (R) residues are the N-terminal end of histones. These terminals are transcriptionally modified by acetylation or methylation and play a role in DNA regulation, replication, and transcription. The histone H3 family is composed of functionally and structurally related proteins (H3.1, H3.2, and H3.3) encoded by different genes. Both the H3F3A and H3F3B genes encode the H3.3 protein, while H3.1 protein is encoded by multiple genes including the HIST1H3B gene. H3K27M mutation results in a lysine-to-methionine substitution at K27 and has been reported in greater than 80% of DIPG patients. This missense mutation is mostly related to the H3.3 protein (H3F3A gene) and to a lesser extent the H3.1 protein (HIST1H3B and HIST1H3C genes) [44]. Both H3.1 and H3.3 tumor mutations behave deferentially regarding location, aggressiveness, overall survival, and the response to radiotherapy (RTH) treatment [45]. G34R/V mutations represent another small proportion of H3.3 mutations (H3F3A gene). It involves a substitution of glycine (G) 34 with arginine (R) or valine (V) [39]. Additionally, the H3K27I mutation is a novel H3F3A gene mutation that results in lysine 27 to isoleucine substitution. Furthermore, mutations in chromatin-modifying enzymes have been reported in DIPG. Both the H3K27M and G34R/V mutations are associated with other mutations in DIPG, e.g., TP53, ATRX (alpha thalassemia/mental retardation syndrome X-linked), and PDGFRA mutations [38, 39, 46].
5.1.2 Polycomb Repressive Complex
The polycomb repressive complex is another relevant group of proteins (PRC1 and PRC2) in the context of DIPG that remodel chromatin and enable epigenetic gene silencing. High expression of B cell–specific Moloney murine leukemia virus integration site 1 (BMI-1) as a component of PRC1 has been detected in DIPG [47].
5.2 Gene aberrations
This includes aberrations in both cellular proliferation pathways and cell cycle regulation pathways.
5.2.1 Cellular Proliferation Pathways aberrations
ACVR1 somatic mutations
The ACVR1 or ALK2 gene encodes activin receptor type 1A, also known as the bone morphogenetic protein (BMP) type 1 receptor. It belongs to transforming growth factor beta (TGF-β) signalling family. Activation of ACVR1 by binding to its ligand activates SMAD transcription factor. Receptor-activated SMADs activates subsequently growth-promoting genes. Six somatic ACVR1 mutations have been detected in 21–25% of DIPG patients, while germline ACVR1 mutation leads to the congenital malformation syndrome fibrodysplasia ossificans progressive (FOP). Both mutations affect the same residues (R206H, Q207E, R258G, G328E, G328V, G328W, and G356D) [48,49,50]. ACVR1 tumor mutations upregulate expression of BMP1-TGF-β signalling pathway targets, e.g., members of the ID gene family [51]. Recently, cooperation between both ACVR1 (R206H) and H3.1K27M mutations in promoting DIPG tumorigenesis has been reported [52]. Also, development of ACVR1 or ALK2 inhibitors with better potency and selectivity in addition to valuable pharmacokinetic profile in vitro and in vivo has been reported to help as a targeted therapy for DIPG patients [44].
Receptor tyrosine kinase pathways
There are 58 receptor tyrosine kinase (RTK) genes in humans categorized into 20 subfamilies. RTKs are transmembrane protein receptors that are activated upon ligand binding followed by signal transduction to nucleus then protein transcription. These signalling pathways play critical role in cell proliferation, differentiation, and survival [53]. Recurrent amplification of platelet-derived growth factor subunit A (PDGFA) protein and platelet-derived growth factor receptor (PDGFR) alpha overexpression have been identified by molecular profiling of DIPG samples [54]. Also, somatic-activating mutations have been reported in DIPG samples that include missense mutations, in-frame deletions, and insertions [55, 56]. Also, these PDGR oncogenic mutations have been observed with concurrent amplification in DIPG [56]. PDGFR alpha is expressed by precursor cells that are derived from PCC, the DIPG cell of origin. In many DIPG model systems, PDGF signalling pathway is a potent driver of brainstem gliomagenesis and a potential therapeutic target. Two small molecule inhibitors targeting PDGFRA (imatinib and dasatinib) have been evaluated in phase I clinical trials for pediatric DIPG patients but both trials did not show significant prolonged survival [57,58,59]. Epidermal growth factor receptor (EGFR) aberrations have also been detected in DIPG samples, either as an immune positivity in 27% of patients [54] or as a gene amplification in 7–9% of patients [60].
The MYC-N pathway
The MYC-N protein is a proto-oncogene encoded by the MYC-N gene. It is involved in both cell proliferation and differentiation [61]. MYC-N protein amplification has been reported in DIPG samples along with genetic, epigenetic, and histological aberrations [2, 38, 62]. This amplification plays an important role as a transcriptional regulator on the epigenetic landscape causing upregulation of gene expression. The MYC-N proto-oncogene pathway can be activated through H3 mutations [38, 46, 63] and independent of them [2].
5.2.2 Cell cycle regulation pathways
The TP53 pathway
The TP53 pathway is composed of a network of genes that play a critical role in the cellular homeostasis as they respond to internal and external stress signals. The stress signals of this network activate p53 protein leading to programmed cell death or apoptosis. TP53 gene mutation is common in numerous types of cancer [46]. In DIPG, the incidence of TP53 gene mutation is between 9 and 77% [38, 64]. This showed up in glioma grade III and IV tumors, while grade II DIPG samples and those carrying wild-type TP53 represent 50% of DIPG cases and are characterized with the presence of PPM1D oncogenic mutation. This oncogene codes for the wild-type p53-induced phosphatase 1D (WIP1) that inactivates p53 protein [65]. In brief, both TP53 and PPM1D mutations lead to the abnormal function of TP53 pathway in the tumorigenesis of DIPG. Many small molecule compounds are under investigations in clinical trials [66, 67].
The RB pathway
The Rb protein plays a critical role in the negative control of the cell cycle machinery as well as in tumor progression. The cell cycle machinery is controlled by cyclins and cyclin-dependent kinases (cyclin/cdks) proteins [68]. In DIPG, abnormalities in cyclins/CDKs involved in the Rb pathway that regulate the G1/S transition of the cell cycle have been identified. These abnormalities include deletions of both CDK2A and CDK2B and amplifications of CDK4, CDK6, and CCND1 (encodes cyclin D1) [60, 69, 70]. Cdk4/6 inhibition was recently recognized as a therapeutic target. Many cdk4/6 inhibitors, e.g., abemaciclib, ribociclib, and palbociclib, have entered clinical trials for the treatment of DIPG patients [71].
The Aurora kinase signalling pathway
Aurora kinase is a serine/threonine kinase complex composed of the three kinases: AURKA, AURKB, and AURKC. They are responsible for regulation of mitosis phase in the cell cycle machinery. Recently, their role in many types of cancer has been discovered and many Aurora kinase inhibitors have been investigated in several clinical trials [72, 73]. Overexpression of AURKB has been detected in 70% of DIPG samples. Consequently, it can be a potential therapeutic target for DIPG patients [74].
The WEE1 kinase pathway
WEE1 kinase regulates both the G2 checkpoint of the cell cycle as well as irradiation-induced DNA damage repair immediately preceding mitotic entry [75]. Overexpression of WEE1 protein has been detected in DIPG samples [76]. In a preclinical study, AZD1775, a selective WEE1 inhibitor, showed promising results in DIPG cell culture and murine models when combined with chemotherapy [76,77,78]. The safety and efficacy of AZD1775 (also known as adavosertib) in combination with RTH for the treatment DIPG patients are under investigation in a phase I clinical trial (NCT01922076).
Poly (ADP-ribose) polymerase (PARP)-1 overexpression
PARP1/2 are nuclear proteins responsible for activating DNA repair proteins that facilitate apoptosis evasion [79]. Overexpression of PARP1 has been detected in DIPG samples. This overexpression facilitates repair of DNA damage post-RTH or temozolomide treatment leading to resistance [80]. In phase I and II trials, combination of veliparib/ABT-888 (PARP1/2 inhibitor), RTH, and temozolomide in DIPG patients was well-tolerated but did not show benefit compared with previous studies [81, 82].
5.3 Immune checkpoint abnormalities
B7-H3 (CD276) is a type I transmembrane glycoprotein and is a part of B7-CD28 family. B7-H3 overexpression has been detected in DIPG samples [83]. It is recognized by monoclonal antibody 8H9 with high selectivity to neuroepithelial tumors only. This selectivity along with its safety and efficacy resulted in the use of intrathecal 131I-8H9 as a salvage therapy in stage IV neuroblastoma [84]. Recently, B7-H3 CAR T therapy showed its role as a therapeutic option in many relapsed and refractory pediatric malignancies and it can be tried in DIPG patients [85, 86].
6 Treatment strategies
Early retrospective studies showed similar outcome of treating whole brain radiation or more limited approach in pediatric brainstem glioma [87, 88]. Therefore, the standard of care for children with DIPG remains along the last 40 years, focal external beam RTH. It is well proven that RTH increases the overall survival by around 3–6 months; as without RTH the overall survival is only 5 months [29]. Conformal radiation is most commonly delivered up to a dose of 54 Gy in 1.8-Gy daily fractions over 6 weeks. The timing of RTH was investigated in 95 DIPG patients treated at St. Jude Children’s Research Hospital, with or without chemotherapy. They found that time to start RTH as a continuous variable did not affect overall survival (OS) or event-free survival (EFS). The dichotomization of time at 14 days in the same cohort showed poorer outcomes for those starting treatment earlier (HR = 1.7 (95% CI 1.11–2.59, p = 0.014)). This has to be interpreted with caution as this better survival is probably due to the slower tumor proliferating capability of the patients treated later [89].
In the early 1990s, the Children’s Cancer Group (CCG) and Pediatric Oncology Group (POG) tested hyper-fractionated RTH in DIPG at 78 and 75.6 Gy, respectively. Both demonstrated no benefit over conventional RTH at 54 Gy [90, 91]. Delineating the gross target volume (GTV) is usually performed after fusion and registration of the CT simulation with T2w or Flair MR studies safety margin of 1.0–1.5 cm to GTV after adjusting for bony structures and tentorium. Larger volume did not lead to better outcome. More recently, Zaghloul et al. randomized 71 DIPG patients to 39 Gy in 13 fractions (2.6 weeks) vs the conventional RTH of 54 Gy in 30 fractions (6 weeks). The hazard ratio for OS was 1.14 (95% CI 0.70–1.89, p = 0.59). The 18-month OS was 10.9 (95% CI 5.1–16.7%) for the hypofractionated arm and 13.1 (95% CI 7.1–19.1%) for the conventional RTH arm. The OS for the hypofractionated arm was non-inferior to that of the conventional arm. The same group underwent a larger randomized controlled study (NCT01878266) including 245 children. They used 2 hypofractionated regimens: 39 Gy in 13 fractions and 45 Gy in 15 fractions compared with the conventional 54 Gy in 30 fractions. This study confirmed their previous study results of non-inferiority of both hypofractionated regimens. More importantly, there was no difference in RTH-related side effects between arms in the 2 randomized studies. The authors concluded that hypofractionated RTH has an advantage over conventional RTH of decreasing the burden on the child, his family, and the health care provider reducing the overall treatment time with similar effect on survival without any additional immediate or late side effects [92].
6.1 Re-irradiation
The concept of re-irradiation (reRT) at first DIPG progression has been emerged as a novel intervention with the potential to improve clinical course and survival in pediatric brain tumors [93]. Re-irradiation in DIPG may be justified and medically accepted, as local relapse is extremely high, the clinical end results are very poor, the prolonged survival is rare for late side effects to manifest, and the symptoms upon relapse are intense. The application of reRT to the DIPG is increasing, with all most studies reporting long follow-up periods showed good symptomatic response and minimal risk of serious toxicity when reRT is administered appropriately in well-selected patients. The late toxicity ranged between 2 and 4% [94].
7 Combination chemotherapy
The role of chemotherapy has been extensively studied in DIPG. Specifically, the use of chemotherapy as a radiosensitizer, neoadjuvant prior to radiation therapy, or adjuvant following radiation therapy, was generally tolerated but had no added benefit. In a phase III trial, the CCSG randomized 74 brainstem tumors patients between 1977 and 1980, to either RTH alone (50–60 Gy) or RTH followed by PCV (prednisone, lomustine, and vincristine). The 5-year OS rate achieved was 17% vs 23% (p = 0.56), respectively. The 5-year progression-free survival was 17% for both arms. This trial predated the use of MRIs in the diagnosis and follow-up of brainstem gliomas, so one could not exclude the inclusion of focal tumors having better prognosis. Notably, the authors reported an increased risk of infection in the PCV arm (16/37 vs 5/30) [95].
Pre-irradiation chemotherapy was also studied in the 1990s. CCG-9941 randomized participants to either carboplatin, etoposide, and vincristine (n = 32) or cisplatin, etoposide, cyclophosphamide, and vincristine (n = 31). Both arms then proceeded to receive hyperfractionated RTH of 72 Gy. There was no difference in EFS, OS, or response rate between the two regimens (p > 0.05) despite the greater chemotherapeutic intensity of the second arm. The 1-year EFS was 17% (95% CI 7.2–26.8%). Interestingly, based on the Goldie-Coldman hypothesis which implied that large tumor volume would predict resistance to therapy and hence survival, the authors reported that this seems to not apply to gliomas intrinsic to the brainstem according to their trial, although this data was not shown and the trial was not powered for such a hypothesis [96]. Allen et al. measured the cumulative maximum tolerated dose (MTD) for carboplatin when given with RTH (72 Gy) for DIPG patients in a phase I/II trial, which was 1540 mg/m2; they also reported a median OS of 12 months, although the trial was not designed to measure such an outcome. This figure, however, was still “typical” of other experiences [97].
In a multicenter Brazilian trial, the BGCG group used tamoxifen 200 mg/m2 alongside conventional RTH (median dose 52 Gy) and then for 52 weeks after RTH. Eleven DIPG cases out of 22 had an objective radiologic response, and only 3 completed the entire protocol without progression or significant toxicity. One-year OS was 37% and the median survival was, again, 10.3 months. The group recommended testing alternative treatments [98].
In 2004, COG enrolled high-grade glioma (HGG) patients, including 63 DIPG cases, in a phase II study entailed RTH plus temozolomide (TMZ) (90 mg/m2/day for 42 days), and then maintenance with TMZ (10 cycles of 200 mg/m2/day for 5 days, every 28 days) (ACNS0126). Temozolomide is an alkylating agent, especially effective in O6-methylguanine DNA methyltransferase (MGMT) promoter methylated tumors that are proven to be sensitive to the drug and show a prolonged survival compared with MGMT-unmethylated HGGs. The comparison of TMZ vs other chemotherapeutic agents in CCG-9941 did not show any benefit (p = 0.96) with median time to progression and death of 6.1 and 9.6 months, respectively [99]. This result was also confirmed in a single-arm trial (CNS 2007/4 from the UK) which studied the effect of concurrent and adjuvant TMZ in 43 cases, resulting in a median survival of 9.5 months (95% CI 7.5–11.4). Interestingly, there were five 2-year survivors with an age range between 9 and 16, which is older than the typical DIPG patient. It has been previously shown that patients < 3 years old might have better prognosis due to the presence of different oncogenic mutations. The investigators of this trial raised the possibility of another prognostic group of older adolescents [100]. These trials demonstrate that there is no progress whatsoever in the survival of DIPG patients treated with any of the chemotherapeutic agents used in other gliomas.
7.1 Novel therapeutic approaches and ongoing trials
Different approaches to the management of pediatric DIPG have recently been explored, but have unfortunately found to be similarly ineffective thus far. These include intra-arterial chemotherapy, intra-nasal chemotherapy, convection-enhanced drug delivery (CED), and targeted therapy.
CED is a surgical technique used to bypass the BBB and increase the bioavailability of the drug, which is delivered through a pump, directly into the tumor mass by a catheter. The cannula is inserted via the transfrontal extraventricular approach, and the drug is delivered to the pons in one session. The safety of drug delivery by CED into the brainstem has been demonstrated in preclinical studies [101,102,103].
Directing the focus towards the delivery of specific macromolecules, like monoclonal antibodies, the first trial of CED in DIPG patients started in 2012 (NCT01502917). It is an open-label dose-escalation safety trial of 124I-8H9 alongside standard RTH. 8H9 is a monoclonal antibody that is specific to B7-H3 protein, which is expressed by HGG of the pons but not by normal brain tissue.
Another phase I trial (NCT03086616) started in 2017 to recruit DIPG patients after RTH, to study the efficacy of irinotecan liposome injection (nal-IRI). The primary endpoint of this study is the number of treatment-related adverse events, and the secondary endpoint is the 1-year OS. More recently, a trial studying nanoparticle formulation of panobinostat MTX110, a histone deacetylase inhibitor, via CED is underway (NCT03566199). As discussed earlier, the availability of DIPG tissue encouraged the identification of targeted agents. For example, the use of HDAC inhibitors in H3K27M-mutant gliomas was studied. These studies showed an effect of HDAC inhibitors in K27M-mutant DIPG cell lines and mouse xenografts [104], although a later study showed that the activity is similar in K27M wild-type cell lines, and almost no effect in vivo [105].
Valproic acid (VPA), another HDAC inhibitor, was retrospectively studied in a single center that was administering VPA as a prophylactic anticonvulsant. Patients receiving RTH+CTH (carboplatin and VCR) were compared with RTH+CTH+VPA. They reported a statistically significant advantage in median OS and PFS for those who received VPA. Median OS was 7.8 in the control (n = 6) vs. 13.4 months in treated patients (n = 13) (HR 0.60 (95% CI 0.37–0.98, p = 0.05)) [106]. However, considering the small number of participants and the retrospective nature of this study, results should be viewed skeptically.
The Pediatric Brain Tumor Consortium (PBTC) studied gefitinib, an EGFR tyrosine kinase inhibitor, in 43 DIPG patients. The 1- and 2-year OS were 56.4% (95% CI 48.8–64%) and 19.6% (95% CI 13.7–25.5%), respectively [107]. PDGFRA and FGFR can be targeted using TKIs, such as dasatinib, ponatinib, imatinib, pazopanib, and sunitinib. The Biological Medicine for DIPG Eradication trial (BIOMEDE) is a phase II trial (NCT02233049) randomizing DIPG patients to three oral targeted therapies—erlotinib 125 mg/m2/day, everolimus 5 mg/m2/day, and dasatinib 170 mg/m2/day—based on three biomarkers—EGFR, PTEN, and PDGFRA.
CUDC-907 is a dual HDAC and PI3K inhibitor in relapsed solid tumors, including DIPG, and lymphomas (NCT02909777). The NCI-COG Pediatric MATCH (Molecular Analysis for Therapy Choice) Screening trial (NCT03155620) is an umbrella trial recruiting relapsed pediatric patients to different targeted treatments, including, for example, LY3023414, a PI3K/mTOR pathway inhibitor, in PI3K-activating tumors (NCT03213678). Finally, to date, there is no effective treatment for DIPG.
8 International collaboration
An invaluable collaboration was accomplished upon establishment of the International Diffuse Intrinsic Pontine Glioma Registry (IDIPGR) in April 2012. This research infrastructure registry collects demographics, clinical, imaging, pathological, and molecular data from many DIPG patients located in many countries: Canada, the USA, New Zealand, Australia, and many other countries including Egypt. According to a recent report, this registry recruited more than 1006 DIPG patients [108]. In addition, this enabled many interested centers in DIPG research to work closely for better outcome, and it is translated in the 9 projects initiated by 13 principal investigators using IDIPGR. Now, it is considered a central resource for many families, oncology clinicians, and patients in many DIPG aspects including literature review, clinical trials, and consultations, in addition to educational materials. It has a future plan to expand collaboration to others around the world and improve awareness about DIPG among medical staff and families through education. In addition, it may act as a good platform for multi-institutional DIPG clinical trials. This not only represents pronounced progress in DIPG registry but also jumps in DIPG understanding.
The European Society for Pediatric Oncology (SIOPE) DIPG Network is another example of collaborative effort to develop registry and imaging repository in DIPG [109]. It was established as a part of SIOPE Brain Tumour Group. SIOPE DIPG Network and registry includes 27 countries. Fortunately, SIOPE DIPG Network and registry works in close collaboration with IDIPGR to advance DIPG research outcome [109, 110].
A recent systematic review about registered clinical trials on ClinicalTrials.gov on DIPG as a primary investigation is recently published [111]. Out of 95 registered DIPG clinical trials, there are 13 trials coordinated between multicenter-based USA and sponsored by the US NCI. The USA ranks the highest in number (67 trials) as a contributing country. Outside the USA, there are three clinical trials coordinated by the Children’s Cancer and Leukaemia Group (UK). Also, they reported 49 trials that involve a single center. The rarity and lethality of DIPG disease mandate collaboration in different aspects (basic and clinical) to yield a transnational impact touched in practice in near future.
References
Hassan, H., Pinches, A., Picton, S. V., & Phillips, R. S. (2017). Survival rates and prognostic predictors of high grade brain stem gliomas in childhood: a systematic review and meta-analysis. Journal of Neuro-Oncology, 135, 1–8. https://doi.org/10.1007/s11060-017-2546-1.
Saratsis, A. M., Kambhampati, M., Snyder, K., Yadavilli, S., Devaney, J. M., Harmon, B., Hall, J., Raabe, E. H., An, P., Weingart, M., Rood, B. R., Magge, S. N., MacDonald, T., Packer, R. J., & Nazarian, J. (2014). Comparative multidimensional molecular analyses of pediatric diffuse intrinsic pontine glioma reveals distinct molecular subtypes. Acta Neuropathologica, 127(6), 881–895. https://doi.org/10.1007/s00401-013-1218-2.
Barkovich, A. J., Krischer, J., Kun, L. F., Packer, R., Zimmerman, R. A., Freeman, C. R., et al. (1990). Brain stem gliomas: a classification system based on magnetic resonance imaging. Pediatric Neurosurgery, 16(2), 73–83. https://doi.org/10.1159/000120511.
Bartels, U., Hawkins, C., Vézina, G., Kun, L., Souweidane, M., & Bouffet, E. (2011). Proceedings of the diffuse intrinsic pontine glioma (DIPG) Toronto Think Tank: advancing basic and translational research and cooperation in DIPG. Journal of Neuro-Oncology, 105(1), 119–125. https://doi.org/10.1007/s11060-011-0704-4.
Smith, M. A., Freidlin, B., Ries, L. A., & Simon, R. (1998). Trends in reported incidence of primary malignant brain tumors in children in the United States. Journal of the National Cancer Institute, 90(17), 1269–1277 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9731733.
Hargrave, D., Bartels, U., & Bouffet, E. (2006). Diffuse brainstem glioma in children: critical review of clinical trials. The Lancet Oncology, 7(3), 241–248. https://doi.org/10.1016/S1470-2045(06)70615-5.
Buczkowicz, P., Bartels, U., Bouffet, E., Becher, O., & Hawkins, C. (2014). Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathologica, 128(4), 573–581. https://doi.org/10.1007/s00401-014-1319-6.
Landolfi, J. C., Thaler, H. T., & DeAngelis, L. M. (1998). Adult brainstem gliomas. Neurology, 51(4), 1136–1139 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9781543.
Recinos, P. F., Sciubba, D. M., & Jallo, G. I. (2007). Brainstem tumors: where are we today? Pediatric Neurosurgery, 43(3), 192–201. https://doi.org/10.1159/000098831.
Freeman, C. R., & Farmer, J. P. (1998). Pediatric brain stem gliomas: a review. International Journal of Radiation Oncology, Biology, Physics, 40(2), 265–271. https://doi.org/10.1016/s0360-3016(97)00572-5.
Lassiter, K. R. L., Alexander, E., Davis, C. H., & Kelly, D. L. (1971). Surgical treatment of brain stem gliomas. Journal of Neurosurgery, 34(6), 719–725. https://doi.org/10.3171/jns.1971.34.6.0719.
Warren, K. E. (2012). Diffuse intrinsic pontine glioma: poised for progress. Frontiers in Oncology, 2, 205. https://doi.org/10.3389/fonc.2012.00205.
Robison, N. J., & Kieran, M. W. (2014). Diffuse intrinsic pontine glioma: a reassessment. Journal of Neuro-Oncology, 119(1), 7–15. https://doi.org/10.1007/s11060-014-1448-8.
Bradley, K. A., Zhou, T., McNall-Knapp, R. Y., Jakacki, R. I., Levy, A. S., Vezina, G., & Pollack, I. F. (2013). Motexafin-gadolinium and involved field radiation therapy for intrinsic pontine glioma of childhood: a Children’s Oncology Group phase 2 study. International Journal of Radiation Oncology, Biology, and Physics, 85(1), e55–e60. https://doi.org/10.1016/j.ijrobp.2012.09.004.
Angelini, P., Hawkins, C., Laperriere, N., Bouffet, E., & Bartels, U. (2011). Post mortem examinations in diffuse intrinsic pontine glioma: challenges and chances. Journal of Neuro-Oncology, 101(1), 75–81. https://doi.org/10.1007/s11060-010-0224-7.
Albright, A. L., Packer, R. J., Zimmerman, R., Rorke, L. B., Boyett, J., & Hammond, G. D. (1993). Magnetic resonance scans should replace biopsies for the diagnosis of diffuse brain stem gliomas: a report from the Children’s Cancer Group. Neurosurgery, 33(6), 1026–1029 discussion 1029-30. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8133987.
Schumacher, M., Schulte-Mönting, J., Stoeter, P., Warmuth-Metz, M., & Solymosi, L. (2007). Magnetic resonance imaging compared with biopsy in the diagnosis of brainstem diseases of childhood: a multicenter review. Journal of Neurosurgery: Pediatrics, 106(2), 111–119. https://doi.org/10.3171/ped.2007.106.2.111.
Plessier, A., Le Dret, L., Varlet, P., Beccaria, K., Lacombe, J., Mériaux, S., et al. (2017). New in vivo avatars of diffuse intrinsic pontine gliomas (DIPG) from stereotactic biopsies performed at diagnosis. Oncotarget, 8(32), 52543–52559. https://doi.org/10.18632/oncotarget.15002.
Sewing, A. C. P., Lagerweij, T., van Vuurden, D. G., Meel, M. H., Veringa, S. J. E., Carcaboso, A. M., et al. (2017). Preclinical evaluation of convection-enhanced delivery of liposomal doxorubicin to treat pediatric diffuse intrinsic pontine glioma and thalamic high-grade glioma. Journal of Neurosurgery: Pediatrics, 19(5), 518–530. https://doi.org/10.3171/2016.9.PEDS16152.
Misuraca, K. L., Cordero, F. J., & Becher, O. J. (2015). Pre-clinical models of diffuse intrinsic pontine glioma. Frontiers in Oncology, 5, 172. https://doi.org/10.3389/fonc.2015.00172.
Lapin, D. H., Tsoli, M., & Ziegler, D. S. (2017). Genomic insights into diffuse intrinsic pontine glioma. Frontiers in Oncology, 7, 57. https://doi.org/10.3389/fonc.2017.00057.
Kambhampati, M., Perez, J. P., Yadavilli, S., Saratsis, A. M., Hill, A. D., Ho, C.-Y., et al. (2015). A standardized autopsy procurement allows for the comprehensive study of DIPG biology. Oncotarget, 6(14), 12740–12747. https://doi.org/10.18632/oncotarget.3374.
Caretti, V., Jansen, M. H. A., van Vuurden, D. G., Lagerweij, T., Bugiani, M., Horsman, I., et al. (2013). Implementation of a multi-institutional diffuse intrinsic pontine glioma autopsy protocol and characterization of a primary cell culture. Neuropathology and Applied Neurobiology, 39(4), 426–436. https://doi.org/10.1111/j.1365-2990.2012.01294.x.
Puget, S., Beccaria, K., Blauwblomme, T., Roujeau, T., James, S., Grill, J., et al. (2015). Biopsy in a series of 130 pediatric diffuse intrinsic pontine gliomas. Child's Nervous System, 31(10), 1773–1780. https://doi.org/10.1007/s00381-015-2832-1.
Cage, T. A., Samagh, S. P., Mueller, S., Nicolaides, T., Haas-Kogan, D., Prados, M., Banerjee, A., Auguste, K. I., & Gupta, N. (2013). Feasibility, safety, and indications for surgical biopsy of intrinsic brainstem tumors in children. Child's Nervous System, 29(8), 1313–1319. https://doi.org/10.1007/s00381-013-2101-0.
Hamisch, C., Kickingereder, P., Fischer, M., Simon, T., & Ruge, M. I. (2017). Update on the diagnostic value and safety of stereotactic biopsy for pediatric brainstem tumors: a systematic review and meta-analysis of 735 cases. Journal of Neurosurgery: Pediatrics, 20(3), 261–268. https://doi.org/10.3171/2017.2.PEDS1665.
Pfaff, E., El Damaty, A., Balasubramanian, G. P., Blattner-Johnson, M., Worst, B. C., Stark, S., et al. (2019). Brainstem biopsy in pediatric diffuse intrinsic pontine glioma in the era of precision medicine: the INFORM study experience. European Journal of Cancer, 114, 27–35. https://doi.org/10.1016/j.ejca.2019.03.019.
Gupta, N., Goumnerova, L. C., Manley, P., Chi, S. N., Neuberg, D., Puligandla, M., Fangusaro, J., Goldman, S., Tomita, T., Alden, T., DiPatri, A., Rubin, J. B., Gauvain, K., Limbrick, D., Leonard, J., Geyer, J. R., Leary, S., Browd, S., Wang, Z., Sood, S., Bendel, A., Nagib, M., Gardner, S., Karajannis, M. A., Harter, D., Ayyanar, K., Gump, W., Bowers, D. C., Weprin, B., MacDonald, T., Aguilera, D., Brahma, B., Robison, N. J., Kiehna, E., Krieger, M., Sandler, E., Aldana, P., Khatib, Z., Ragheb, J., Bhatia, S., Mueller, S., Banerjee, A., Bredlau, A. L., Gururangan, S., Fuchs, H., Cohen, K. J., Jallo, G., Dorris, K., Handler, M., Comito, M., Dias, M., Nazemi, K., Baird, L., Murray, J., Lindeman, N., Hornick, J. L., Malkin, H., Sinai, C., Greenspan, L., Wright, K. D., Prados, M., Bandopadhayay, P., Ligon, K. L., & Kieran, M. W. (2018). Prospective feasibility and safety assessment of surgical biopsy for patients with newly diagnosed diffuse intrinsic pontine glioma. Neuro-Oncology, 20(11), 1547–1555. https://doi.org/10.1093/neuonc/noy070.
Samadani, U., & Judy, K. D. (2003). Stereotactic brainstem biopsy is indicated for the diagnosis of a vast array of brainstem pathology. Stereotactic and Functional Neurosurgery, 81(1–4), 5–9. https://doi.org/10.1159/000075097.
Pincus, D. W., Richter, E. O., Yachnis, A. T., Bennett, J., Bhatti, M. T., & Smith, A. (2006). Brainstem stereotactic biopsy sampling in children. Journal of Neurosurgery: Pediatrics, 104(2), 108–114. https://doi.org/10.3171/ped.2006.104.2.108.
Bandopadhayay, P., Greenwald, N. F., Wala, J., Sharpira, O., Tracy, A., Filbin, M., et al. (2017). DIPG-29. Genomic landscape of diffuse intrinsic pontine glioma: an analysis of the DIPG-BATs cohort. Neuro-Oncology, 19(suppl_4), iv11. https://doi.org/10.1093/neuonc/nox083.044.
Alix-Panabières, C., & Pantel, K. (2016). Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discovery, 6(5), 479–491. https://doi.org/10.1158/2159-8290.CD-15-1483.
Mattox, A. K., Bettegowda, C., Zhou, S., Papadopoulos, N., Kinzler, K. W., & Vogelstein, B. (2019). Applications of liquid biopsies for cancer. Science Translational Medicine, 11(507), eaay1984. https://doi.org/10.1126/scitranslmed.aay1984.
Huang, T. Y., Piunti, A., Lulla, R. R., Qi, J., Horbinski, C. M., Tomita, T., James, C. D., Shilatifard, A., & Saratsis, A. M. (2017). Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathologica Communications, 5(1), 28. https://doi.org/10.1186/s40478-017-0436-6.
Panditharatna, E., Kilburn, L. B., Aboian, M. S., Kambhampati, M., Gordish-Dressman, H., Magge, S. N., Gupta, N., Myseros, J. S., Hwang, E. I., Kline, C., Crawford, J. R., Warren, K. E., Cha, S., Liang, W. S., Berens, M. E., Packer, R. J., Resnick, A. C., Prados, M., Mueller, S., & Nazarian, J. (2018). Clinically relevant and minimally invasive tumor surveillance of pediatric diffuse midline gliomas using patient-derived liquid biopsy. Clinical Cancer Research, 24(23), 5850–5859. https://doi.org/10.1158/1078-0432.CCR-18-1345.
Pan, C., Diplas, B. H., Chen, X., Wu, Y., Xiao, X., Jiang, L., Geng, Y., Xu, C., Sun, Y., Zhang, P., Wu, W., Wang, Y., Wu, Z., Zhang, J., Jiao, Y., Yan, H., & Zhang, L. (2019). Molecular profiling of tumors of the brainstem by sequencing of CSF-derived circulating tumor DNA. Acta Neuropathologica, 137(2), 297–306. https://doi.org/10.1007/s00401-018-1936-6.
Buczkowicz, P., Hoeman, C., Rakopoulos, P., Pajovic, S., Letourneau, L., Dzamba, M., Morrison, A., Lewis, P., Bouffet, E., Bartels, U., Zuccaro, J., Agnihotri, S., Ryall, S., Barszczyk, M., Chornenkyy, Y., Bourgey, M., Bourque, G., Montpetit, A., Cordero, F., Castelo-Branco, P., Mangerel, J., Tabori, U., Ho, K. C., Huang, A., Taylor, K. R., Mackay, A., Bendel, A. E., Nazarian, J., Fangusaro, J. R., Karajannis, M. A., Zagzag, D., Foreman, N. K., Donson, A., Hegert, J. V., Smith, A., Chan, J., Lafay-Cousin, L., Dunn, S., Hukin, J., Dunham, C., Scheinemann, K., Michaud, J., Zelcer, S., Ramsay, D., Cain, J., Brennan, C., Souweidane, M. M., Jones, C., Allis, C. D., Brudno, M., Becher, O., & Hawkins, C. (2014). Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nature Genetics, 46(5), 451–456. https://doi.org/10.1038/ng.2936.
Khuong-Quang, D.-A., Buczkowicz, P., Rakopoulos, P., Liu, X.-Y., Fontebasso, A. M., Bouffet, E., Bartels, U., Albrecht, S., Schwartzentruber, J., Letourneau, L., Bourgey, M., Bourque, G., Montpetit, A., Bourret, G., Lepage, P., Fleming, A., Lichter, P., Kool, M., von Deimling, A., Sturm, D., Korshunov, A., Faury, D., Jones, D. T., Majewski, J., Pfister, S. M., Jabado, N., & Hawkins, C. (2012). K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathologica, 124(3), 439–447. https://doi.org/10.1007/s00401-012-0998-0.
Schwartzentruber, J., Korshunov, A., Liu, X.-Y., Jones, D. T. W., Pfaff, E., Jacob, K., et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature, 482(7384), 226–231. https://doi.org/10.1038/nature10833.
Wu, G., Broniscer, A., McEachron, T. A., Lu, C., Paugh, B. S., Becksfort, J., et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature Genetics, 44(3), 251–253. https://doi.org/10.1038/ng.1102.
Monje, M., Mitra, S. S., Freret, M. E., Raveh, T. B., Kim, J., Masek, M., Attema, J. L., Li, G., Haddix, T., Edwards, M. S., Fisher, P. G., Weissman, I. L., Rowitch, D. H., Vogel, H., Wong, A. J., & Beachy, P. A. (2011). Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proceedings of the National Academy of Sciences of the United States of America, 108(11), 4453–4458. https://doi.org/10.1073/pnas.1101657108.
Kepert, J. F., Tóth, K. F., Caudron, M., Mücke, N., Langowski, J., & Rippe, K. (2003). Conformation of reconstituted mononucleosomes and effect of linker histone H1 binding studied by scanning force microscopy. Biophysical Journal, 85(6), 4012–4022. https://doi.org/10.1016/S0006-3495(03)74815-2.
Mariño-Ramírez, L., Kann, M. G., Shoemaker, B. A., & Landsman, D. (2005). Histone structure and nucleosome stability. Expert Review of Proteomics, 2(5), 719–729. https://doi.org/10.1586/14789450.2.5.719.
Carvalho, D., Taylor, K. R., Olaciregui, N. G., Molinari, V., Clarke, M., Mackay, A., Ruddle, R., Henley, A., Valenti, M., Hayes, A., Brandon, A. D. H., Eccles, S. A., Raynaud, F., Boudhar, A., Monje, M., Popov, S., Moore, A. S., Mora, J., Cruz, O., Vinci, M., Brennan, P. E., Bullock, A. N., Carcaboso, A. M., & Jones, C. (2019). ALK2 inhibitors display beneficial effects in preclinical models of ACVR1 mutant diffuse intrinsic pontine glioma. Communications Biology, 2(1), 156. https://doi.org/10.1038/s42003-019-0420-8.
Castel, D., Philippe, C., Calmon, R., Le Dret, L., Truffaux, N., Boddaert, N., et al. (2015). Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathologica, 130(6), 815–827. https://doi.org/10.1007/s00401-015-1478-0.
Yuen, B. T. K., & Knoepfler, P. S. (2013). Histone H3.3 mutations: a variant path to cancer. Cancer Cell, 24(5), 567–574. https://doi.org/10.1016/j.ccr.2013.09.015.
Kumar, S. S., Sengupta, S., Lee, K., Hura, N., Fuller, C., DeWire, M., et al. (2017). BMI-1 is a potential therapeutic target in diffuse intrinsic pontine glioma. Oncotarget, 8(38), 62962–62975. https://doi.org/10.18632/oncotarget.18002.
Han, H. J., Jain, P., & Resnick, A. C. (2018). Shared ACVR1 mutations in FOP and DIPG: opportunities and challenges in extending biological and clinical implications across rare diseases. Bone, 109, 91–100. https://doi.org/10.1016/j.bone.2017.08.001.
Taylor, K. R., Vinci, M., Bullock, A. N., & Jones, C. (2014). ACVR1 mutations in DIPG: lessons learned from FOP. Cancer Research, 74(17), 4565–4570. https://doi.org/10.1158/0008-5472.CAN-14-1298.
Taylor, K. R., Mackay, A., Truffaux, N., Butterfield, Y. S., Morozova, O., Philippe, C., et al. (2014). Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nature Genetics, 46(5), 457–461. https://doi.org/10.1038/ng.2925.
Viñals, F., & Ventura, F. (2004). Myogenin protein stability is decreased by BMP-2 through a mechanism implicating Id1. Journal of Biological Chemistry, 279(44), 45766–45772. https://doi.org/10.1074/jbc.M408059200.
Hoeman, C. M., Cordero, F. J., Hu, G., Misuraca, K., Romero, M. M., Cardona, H. J., et al. (2019). ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nature Communications, 10(1), 1023. https://doi.org/10.1038/s41467-019-08823-9.
Ségaliny, A. I., Tellez-Gabriel, M., Heymann, M.-F., & Heymann, D. (2015). Receptor tyrosine kinases: characterisation, mechanism of action and therapeutic interests for bone cancers. Journal of bone oncology, 4(1), 1–12. https://doi.org/10.1016/j.jbo.2015.01.001.
Zarghooni, M., Bartels, U., Lee, E., Buczkowicz, P., Morrison, A., Huang, A., et al. (2010). Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor α and poly (ADP-ribose) polymerase as potential therapeutic targets. Journal of Clinical Oncology, 28(8), 1337–1344. https://doi.org/10.1200/JCO.2009.25.5463.
Puget, S., Philippe, C., Bax, D. A., Job, B., Varlet, P., Junier, M.-P., Andreiuolo, F., Carvalho, D., Reis, R., Guerrini-Rousseau, L., Roujeau, T., Dessen, P., Richon, C., Lazar, V., le Teuff, G., Sainte-Rose, C., Geoerger, B., Vassal, G., Jones, C., & Grill, J. (2012). Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One, 7(2), e30313. https://doi.org/10.1371/journal.pone.0030313.
Paugh, B. S., Zhu, X., Qu, C., Endersby, R., Diaz, A. K., Zhang, J., Bax, D. A., Carvalho, D., Reis, R. M., Onar-Thomas, A., Broniscer, A., Wetmore, C., Zhang, J., Jones, C., Ellison, D. W., & Baker, S. J. (2013). Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Research, 73(20), 6219–6229. https://doi.org/10.1158/0008-5472.CAN-13-1491.
Broniscer, A., Baker, S. D., Wetmore, C., Pai Panandiker, A. S., Huang, J., Davidoff, A. M., Onar-Thomas, A., Panetta, J. C., Chin, T. K., Merchant, T. E., Baker, J. N., Kaste, S. C., Gajjar, A., & Stewart, C. F. (2013). Phase I Trial, Pharmacokinetics, and pharmacodynamics of vandetanib and dasatinib in children with newly diagnosed diffuse intrinsic pontine glioma. Clinical Cancer Research, 19(11), 3050–3058. https://doi.org/10.1158/1078-0432.CCR-13-0306.
Pollack, I. F., Jakacki, R. I., Blaney, S. M., Hancock, M. L., Kieran, M. W., Phillips, P., Kun, L. E., Friedman, H., Packer, R., Banerjee, A., Geyer, J. R., Goldman, S., Poussaint, T. Y., Krasin, M. J., Wang, Y., Hayes, M., Murgo, A., Weiner, S., & Boyett, J. M. (2007). Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: a Pediatric Brain Tumor Consortium report1. Neuro-Oncology, 9(2), 145–160. https://doi.org/10.1215/15228517-2006-031.
Hoeman, C., Shen, C., & Becher, O. J. (2018). CDK4/6 and PDGFRA signaling as therapeutic targets in diffuse intrinsic pontine glioma. Frontiers in Oncology, 8, 191. https://doi.org/10.3389/fonc.2018.00191.
Paugh, B. S., Broniscer, A., Qu, C., Miller, C. P., Zhang, J., Tatevossian, R. G., Olson, J. M., Geyer, J. R., Chi, S. N., da Silva, N. S., Onar-Thomas, A., Baker, J. N., Gajjar, A., Ellison, D. W., & Baker, S. J. (2011). Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. Journal of Clinical Oncology, 29(30), 3999–4006. https://doi.org/10.1200/JCO.2011.35.5677.
Guglielmi, L., Cinnella, C., Nardella, M., Maresca, G., Valentini, A., Mercanti, D., et al. (2014). MYCN gene expression is required for the onset of the differentiation programme in neuroblastoma cells. Cell Death & Disease, 5(2), e1081–e1081. https://doi.org/10.1038/cddis.2014.42.
Wu, G., Diaz, A. K., Paugh, B. S., Rankin, S. L., Ju, B., Li, Y., Zhu, X., Qu, C., Chen, X., Zhang, J., Easton, J., Edmonson, M., Ma, X., Lu, C., Nagahawatte, P., Hedlund, E., Rusch, M., Pounds, S., Lin, T., Onar-Thomas, A., Huether, R., Kriwacki, R., Parker, M., Gupta, P., Becksfort, J., Wei, L., Mulder, H. L., Boggs, K., Vadodaria, B., Yergeau, D., Russell, J. C., Ochoa, K., Fulton, R. S., Fulton, L. L., Jones, C., Boop, F. A., Broniscer, A., Wetmore, C., Gajjar, A., Ding, L., Mardis, E. R., Wilson, R. K., Taylor, M. R., Downing, J. R., Ellison, D. W., Zhang, J., & Baker, S. J. (2014). The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nature Genetics, 46(5), 444–450. https://doi.org/10.1038/ng.2938.
Plass, C., Pfister, S. M., Lindroth, A. M., Bogatyrova, O., Claus, R., & Lichter, P. (2013). Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nature Reviews Genetics, 14(11), 765–780. https://doi.org/10.1038/nrg3554.
Grill, J., Puget, S., Andreiuolo, F., Philippe, C., MacConaill, L., & Kieran, M. W. (2012). Critical oncogenic mutations in newly diagnosed pediatric diffuse intrinsic pontine glioma. Pediatric Blood & Cancer, 58(4), 489–491. https://doi.org/10.1002/pbc.24060.
Kleiblova, P., Shaltiel, I. A., Benada, J., Ševčík, J., Pecháčková, S., Pohlreich, P., Voest, E. E., Dundr, P., Bartek, J., Kleibl, Z., Medema, R. H., & Macurek, L. (2013). Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. The Journal of Cell Biology, 201(4), 511–521. https://doi.org/10.1083/jcb.201210031.
Yagi, H., Chuman, Y., Kozakai, Y., Imagawa, T., Takahashi, Y., Yoshimura, F., et al. (2012). A small molecule inhibitor of p53-inducible protein phosphatase PPM1D. Bioorganic & Medicinal Chemistry Letters, 22(1), 729–732. https://doi.org/10.1016/j.bmcl.2011.10.084.
Oghabi Bakhshaiesh, T., Majidzadeh-A, K., & Esmaeili, R. (2017). Wip1: a candidate phosphatase for cancer diagnosis and treatment. DNA Repair, 54, 63–66. https://doi.org/10.1016/j.dnarep.2017.03.004.
Giacinti, C., & Giordano, A. (2006). RB and cell cycle progression. Oncogene, 25(38), 5220–5227. https://doi.org/10.1038/sj.onc.1209615.
Paugh, B. S., Qu, C., Jones, C., Liu, Z., Adamowicz-Brice, M., Zhang, J., Bax, D. A., Coyle, B., Barrow, J., Hargrave, D., Lowe, J., Gajjar, A., Zhao, W., Broniscer, A., Ellison, D. W., Grundy, R. G., & Baker, S. J. (2010). Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. Journal of Clinical Oncology, 28(18), 3061–3068. https://doi.org/10.1200/JCO.2009.26.7252.
Warren, K. E., Killian, K., Suuriniemi, M., Wang, Y., Quezado, M., & Meltzer, P. S. (2012). Genomic aberrations in pediatric diffuse intrinsic pontine gliomas. Neuro-Oncology, 14(3), 326–332. https://doi.org/10.1093/neuonc/nor190.
Becher, O. J. (2019). CDK4/6 and diffuse intrinsic pontine glioma - evaluate at diagnosis? EBioMedicine, 44, 16–17. https://doi.org/10.1016/j.ebiom.2019.05.020.
Willems, E., Dedobbeleer, M., Digregorio, M., Lombard, A., Lumapat, P. N., & Rogister, B. (2018). The functional diversity of Aurora kinases: a comprehensive review. Cell Division, 13, 7. https://doi.org/10.1186/s13008-018-0040-6.
Tang, A., Gao, K., Chu, L., Zhang, R., Yang, J., & Zheng, J. (2017). Aurora kinases: novel therapy targets in cancers. Oncotarget, 8(14), 23937–23954. https://doi.org/10.18632/oncotarget.14893.
Buczkowicz, P., Zarghooni, M., Bartels, U., Morrison, A., Misuraca, K. L., Chan, T., Bouffet, E., Huang, A., Becher, O., & Hawkins, C. (2013). Aurora kinase B is a potential therapeutic target in pediatric diffuse intrinsic pontine glioma. Brain Pathology, 23(3), 244–253. https://doi.org/10.1111/j.1750-3639.2012.00633.x.
Matheson, C. J., Backos, D. S., & Reigan, P. (2016). Targeting WEE1 kinase in cancer. Trends in Pharmacological Sciences, 37(10), 872–881. https://doi.org/10.1016/j.tips.2016.06.006.
Geenen, J. J. J., & Schellens, J. H. M. (2017). Molecular pathways: targeting the protein kinase Wee1 in cancer. Clinical Cancer Research, 23(16), 4540–4544. https://doi.org/10.1158/1078-0432.CCR-17-0520.
Caretti, V., Hiddingh, L., Lagerweij, T., Schellen, P., Koken, P. W., Hulleman, E., van Vuurden, D., Vandertop, W. P., Kaspers, G. J., Noske, D. P., & Wurdinger, T. (2013). WEE1 kinase inhibition enhances the radiation response of diffuse intrinsic pontine gliomas. Molecular Cancer Therapeutics, 12(2), 141–150. https://doi.org/10.1158/1535-7163.MCT-12-0735.
Mueller, S., Hashizume, R., Yang, X., Kolkowitz, I., Olow, A. K., Phillips, J., Smirnov, I., Tom, M. W., Prados, M. D., James, C. D., Berger, M. S., Gupta, N., & Haas-Kogan, D. A. (2014). Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro-Oncology, 16(3), 352–360. https://doi.org/10.1093/neuonc/not220.
Punt, J., & Cartmill, M. (1999). Diffuse brain stem glioma. Child's Nervous System, 15(5), 235–237. https://doi.org/10.1007/s003810050379.
Ratnam, K., & Low, J. A. (2007). Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clinical Cancer Research, 13(5), 1383–1388. https://doi.org/10.1158/1078-0432.CCR-06-2260.
Baxter, P., Su, J., Li, X., Thomas, A. O., Billups, C., Thompson, P., et al. (2016). EPT-15A phase1/2 clinical trial of veliparib (ABT-888) and radiation followed by maintenance therapy with veliparib and temozolomide (TMZ) in patients with newly diagnosed diffuse intrinsic pontine glioma (DIPG): a Pediatric Brain Tumor Consortium interim report of phase II study. Neuro-Oncology, 18(suppl 3), 1–iii27. https://doi.org/10.1093/neuonc/now069.14.
Su, J. M., Thompson, P., Adesina, A., Li, X.-N., Kilburn, L., Onar-Thomas, A., Kocak, M., Chyla, B., McKeegan, E., Warren, K. E., Goldman, S., Pollack, I. F., Fouladi, M., Chen, A., Giranda, V., Boyett, J., Kun, L., & Blaney, S. M. (2014). A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: a pediatric brain tumor consortium report. Neuro-oncology, 16(12), 1661–1668. https://doi.org/10.1093/neuonc/nou103.
Zhou, Z., Luther, N., Ibrahim, G. M., Hawkins, C., Vibhakar, R., Handler, M. H., & Souweidane, M. M. (2013). B7-H3, a potential therapeutic target, is expressed in diffuse intrinsic pontine glioma. Journal of Neuro-Oncology, 111(3), 257–264. https://doi.org/10.1007/s11060-012-1021-2.
Kaye, E. C., Baker, J. N., & Broniscer, A. (2014). Management of diffuse intrinsic pontine glioma in children: current and future strategies for improving prognosis. CNS oncology, 3(6), 421–431. https://doi.org/10.2217/cns.14.47.
Tang, X., Zhao, S., Zhang, Y., Wang, Y., Zhang, Z., Yang, M., Zhu, Y., Zhang, G., Guo, G., Tong, A., & Zhou, L. (2019). B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Molecular therapy oncolytics, 14, 279–287. https://doi.org/10.1016/j.omto.2019.07.002.
Majzner, R. G., Theruvath, J. L., Nellan, A., Heitzeneder, S., Cui, Y., Mount, C. W., et al. (2019). CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 25(8), 2560–2574. https://doi.org/10.1158/1078-0432.CCR-18-0432.
Greenberger, J. S., Cassady, J. R., & Levene, M. B. (1977). Radiation therapy of thalamic, midbrain and brain stem gliomas. Radiology, 122(2), 463–468. https://doi.org/10.1148/122.2.463.
Halperin, E. C. (1985). Pediatric brain stem tumors: patterns of treatment failure and their implications for radiotherapy. International Journal of Radiation Oncology, Biology, and Physics, 11(7), 1293–1298. https://doi.org/10.1016/0360-3016(85)90244-5.
Panandiker, A. S. P., Wong, J. K., Nedelka, M. A., Gajjar, A., & Broniscer, A. (2014). Effect of time from diagnosis to start of radiotherapy on children with diffuse intrinsic pontine glioma, (September 2013), (pp. 1180–1183). doi: https://doi.org/10.1002/pbc
Cancer, A. C., Phase, G., Packer, R., Boyett, M., Ph, D., Zimmerman, R. A., … Ph, D. (1994). Outcome of children with brain stem gliomas after treatment with 7800 cGy of hyperfractionated radiotherapy.
Sanford, R. A., & Carpio, R. D. E. L. (1993). ?? Clinical original contribution in children: a Pediatric Oncology Group study, 21(March).
Zaghloul, M. S., Akoush, H., Ahmed, S., Tolba, M., Refaat, A., Youssef, A., Khateeb, N. E., & A. A. A. (2018). Hypofractionated radiation for pediatric diffuse intrinsic pontine glioma (DIPG): younger children have better survival. International Journal of Radiation Oncology, Biology, Physics, 101(4), 1008–1009.
Tsang, D. S., & Laperriere, N. J. (2019). Re-irradiation for paediatric tumours. Clinical Oncology, 31(3), 191–198. https://doi.org/10.1016/j.clon.2018.10.003.
Lassaletta, A., Strother, D., Laperriere, N., Hukin, J., Vanan, M. I., Goddard, K., et al. (2018). Reirradiation in patients with diffuse intrinsic pontine gliomas: the Canadian experience. Pediatric Blood & Cancer, 65(6), e26988. https://doi.org/10.1002/pbc.26988.
Evans, A., Hitrle, R., Ortega, J., Sposto, R., Wara, W., Wilson, C., et al. (1987). Brain-stem tumors in childhood: a prospective randomized trial of irradiation with and without adjuvant CCNU, VCR, and prednisone. Journal of Neurosurgery, 66, 227–233.
Jennings, B. M. T., Sposto, R., Boyett, J. M., Vezina, L. G., Holmes, E., Berger, M. S., et al. (2016). Preradiation chemotherapy in primary high-risk brainstem tumors: phase II study CCG-9941 of the Children’s Cancer Group. Journal of Clinical Oncology, 20(16), 3431–3437. https://doi.org/10.1200/JCO.2002.04.109.
Allen, J., Siffert, J., Donahue, B., Nirenberg, A., Jakacki, R., Robertson, P., et al. (1999). A phase I/II study of carboplatin combined with hyperfractionated radiotherapy for brainstem gliomas. Cancer, 86, 1064–1069.
Broniscer, A., da Leite, C. C., Lanchote, V. L., Machado, T. M. S., & Cristófani, L. M. (2000). Radiation therapy and high-dose tamoxifen in the treatment of patients with diffuse brainstem gliomas: results of a Brazilian cooperative study. Journal of Clinical Oncology, 18(6), 1246–1253. https://doi.org/10.1200/JCO.2000.18.6.1246.
Cohen, K. J., Heideman, R. L., Zhou, T., Holmes, E. J., Lavey, R. S., Bouffet, E., & Pollack, I. F. (2011). with newly diagnosed diffuse intrinsic. Oncology Group, 13(4), 410–416.
Bailey, S., Howman, A., Wheatley, K., Wherton, D., Boota, N., & Pizer, B. (2013). Diffuse intrinsic pontine glioma treated with prolonged temozolomide and radiotherapy – results of a United Kingdom phase II trial CNS 2007 04. European Journal of Cancer, 49(18), 3856–3862. https://doi.org/10.1016/j.ejca.2013.08.006.
Ho, S. L., Singh, R., Zhou, Z., Lavi, E., & Souweidane, M. M. (2015). Toxicity evaluation of prolonged convection-enhanced delivery of small-molecule kinase inhibitors in naïve rat brainstem. Child’s nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery, 31(2), 221–226. https://doi.org/10.1007/s00381-014-2568-3.
Luther, N., Cheung, N.-K., Souliopoulos, E. P., Karempelas, I., Bassiri, D., Edgar, M. A., et al. (2010). Interstitial infusion of glioma-targeted recombinant immunotoxin 8H9scFv-PE38. Molecular Cancer Therapeutics, 9(4), 1039–1046. https://doi.org/10.1158/1535-7163.MCT-09-0996.
Souweidane, M. M., Occhiogrosso, G., Mark, E. B., & Edgar, M. A. (2004). Interstitial infusion of IL13-PE38QQR in the rat brain stem. Journal of Neuro-Oncology, 67(3), 287–293 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15164984.
Grasso, C. S., Tang, Y., Truffaux, N., Berlow, N. E., Liu, L., Debily, M.-A., Quist, M. J., Davis, L. E., Huang, E. C., Woo, P. J., Ponnuswami, A., Chen, S., Johung, T. B., Sun, W., Kogiso, M., du, Y., Qi, L., Huang, Y., Hütt-Cabezas, M., Warren, K. E., le Dret, L., Meltzer, P. S., Mao, H., Quezado, M., van Vuurden, D., Abraham, J., Fouladi, M., Svalina, M. N., Wang, N., Hawkins, C., Nazarian, J., Alonso, M. M., Raabe, E. H., Hulleman, E., Spellman, P. T., Li, X. N., Keller, C., Pal, R., Grill, J., & Monje, M. (2015). Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nature Medicine, 21(6), 555–559. https://doi.org/10.1038/nm.3855.
Hennika, T., Hu, G., Olaciregui, N. G., Barton, K. L., Ehteda, A., Chitranjan, A., et al. (2017). Pre-clinical study of panobinostat in xenograft and genetically engineered murine diffuse intrinsic pontine glioma models. PLoS One, 12(1), e0169485. https://doi.org/10.1371/journal.pone.0169485.
Helder, F., Felix, C., & Leite, O. (2014). Retrospective evaluation of the outcomes of children with diffuse intrinsic pontine glioma treated with radiochemotherapy and valproic acid in a single center, (pp. 261–266). doi: https://doi.org/10.1007/s11060-013-1280-6
Pollack, I. F., Stewart, C. F., Kocak, M., Poussaint, T. Y., Broniscer, A., Banerjee, A., Douglas, J. G., Kun, L. E., Boyett, J. M., & Geyer, J. R. (2011). A phase II study of gefitinib and irradiation in children with newly diagnosed brainstem gliomas: a report from the Pediatric Brain Tumor Consortium. Neuro-Oncology, 13(3), 290–297. https://doi.org/10.1093/neuonc/noq199.
Carceller, F. (2019). Long-term survivors of diffuse intrinsic pontine glioma (DIPG): myth or reality. Translational Cancer Research, 8(20). https://doi.org/10.21037/tcr.2019.03.12.
Veldhuijzen van Zanten, S. E. M., Lane, A., Heymans, M. W., Baugh, J., Chaney, B., Hoffman, L. M., Doughman, R., Jansen, M. H. A., Sanchez, E., Vandertop, W. P., Kaspers, G. J. L., Vuurden, D. G., Fouladi, M., Jones, B. V., & Leach, J. (2017). External validation of the diffuse intrinsic pontine glioma survival prediction model: a collaborative report from the International DIPG Registry and the SIOPE DIPG Registry. Journal of Neuro-Oncology, 134(1), 231–240. https://doi.org/10.1007/s11060-017-2514-9.
Hoffman, L. M., Veldhuijzen van Zanten, S. E. M., Colditz, N., Baugh, J., Chaney, B., Hoffmann, M., et al. (2018). Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): a collaborative report from the International and European Society for Pediatric Oncology DIPG Registries. Journal of Clinical Oncology, 36(19), 1963–1972. https://doi.org/10.1200/JCO.2017.75.9308.
Rechberger, J. S., Lu, V. M., Zhang, L., Power, E. A., & Daniels, D. J. (2019). Clinical trials for diffuse intrinsic pontine glioma: the current state of affairs. Child's Nervous System. https://doi.org/10.1007/s00381-019-04363-1.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rashed, W.M., Maher, E., Adel, M. et al. Pediatric diffuse intrinsic pontine glioma: where do we stand?. Cancer Metastasis Rev 38, 759–770 (2019). https://doi.org/10.1007/s10555-019-09824-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-019-09824-2