
Abstract
Various flavonoid glycosides are found in nature, and their biological activities are as variable as their number. In some cases, the sugar moiety attached to the flavonoid modulates its biological activities. Flavonoid glycones are not easily synthesized chemically. Therefore, in this study, we attempted to synthesize quercetin 3-O-glucosyl (1→2) xyloside and quercetin 3-O-glucosyl (1→6) rhamnoside (also called rutin) using two uridine diphosphate-dependent glycosyltransferases (UGTs) in Escherichia coli. To synthesize quercetin 3-O-glucosyl (1→2) xyloside, sequential glycosylation was carried out by regulating the expression time of the two UGTs. AtUGT78D2 was subcloned into a vector controlled by a Tac promoter without a lacI operator, while AtUGT79B1 was subcloned into a vector controlled by a T7 promoter. UDP-xyloside was supplied by concomitantly expressing UDP-glucose dehydrogenase (ugd) and UDP-xyloside synthase (UXS) in the E. coli. Using these strategies, 65.0 mg/L of quercetin 3-O-glucosyl (1→2) xyloside was produced. For the synthesis of rutin, one UGT (BcGT1) was integrated into the E. coli chromosome and the other UGT (Fg2) was expressed in a plasmid along with RHM2 (rhamnose synthase gene 2). After optimization of the initial cell concentration and incubation temperature, 119.8 mg/L of rutin was produced. The strategies used in this study thus show promise for the synthesis of flavonoid diglucosides in E. coli.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Many secondary metabolites exist as glycones, and in several instances, the attached sugar moiety modulates their biological activities. In fact, the attachment of diverse sugar moieties to secondary metabolites has been shown to alter their biological activities [9, 28].
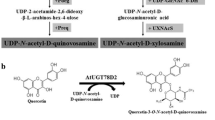
Flavonoids are secondary metabolites, and the availability of several hydroxyl groups and glycosyltransferases (GTs) from multiple sources makes them good candidates for studying the attachment of sugar moieties. GTs use nucleotide sugars, mainly uridine diphosphate sugars (UDP-sugars) or thymine diphosphate sugars (TDP-sugars). Most GTs show a preference for UDP-sugar, and these GTs are called uridine diphosphate-dependent GTs (UGTs) [29, 32]. Diverse sugars, including glucose, galactose, arabinose, glucuronic acid, xylose, and rhamnose, can be attached to flavonoids through an ether linkage [1]. The advent of metabolomics, together with whole genome sequencing of model organisms has led to the identification of various flavonoid glycosides, enabling the characterization of GTs with novel nucleotide sugar selectivities. Plants synthesize flavonoid O-glycosides and flavonoid O-diglycosides. Flavonoid O-diglycosides such as quercetin 3-O-glucose 7-O-rhamnose and kaempferol 3-O-glucose 7-O-rhamnose were detected in Arabidopsis thaliana by a metabolomics approach [30]. AtUGT78D1, which encodes flavonol 3-O-rhamnosyltransferase in A. thaliana, was characterized through these approaches [8]. Flavonol 3-O-arabinosyltransferase (UGT78D3), flavonol 3-O-rhamnosyltransferase (UGT78D1), and anthocyanin 3-O-glucose:2″-O-xylosyltransferase (UGT79B1) were also characterized via metabolomics analysis using mutants [8, 30, 31]. Plants also produce flavonoid O-glycosyl → glycosides. For example, naringenin 7-O-glucose (1→2) rhamnose is responsible for the bitter flavor of citrus [4]. The GTs that generate these flavonoids are a valuable resource for the production of flavonoid glycosides in heterologous systems such as Escherichia coli and Saccharomyces cerevisiae.
Biotransformation of flavonoids using E. coli expressing UGTs has been previously attempted, giving rise to flavonoid O-glucoside as the primary reaction product [16]. Strategies to synthesize flavonoid glycones other than flavonoid glucosides have also been developed (reviewed by Kim et al. [16]). The synthesis of other flavonoid glycosides in E. coli requires the engineering of both nucleotide sugar biosynthesis in E. coli and UGTs, which recognize a novel nucleotide sugar as well as flavonoids. Using these approaches, a range of naturally occurring flavonoid O-glycosides including flavonoid O-galactoside, flavonoid O-glucuronide, flavonoid O-rhamnoside, flavonoid O-xyloside, and flavonoid O-arabinoside have been synthesized [6, 17, 18, 23, 27]. In addition, novel flavonoid glycosides not found in nature have also been synthesized [12, 15, 27, 33]. However, despite their biological activities including anti-inflammatory, antioxidant, and antibacterial activities as well as their established role in the inhibition of platelet aggregation and blood vessel protection [5, 20, 21, 25], the synthesis of flavonoid O-diglycosides in E. coli has not been previously attempted. In the present study, we report the biosynthesis of quercetin 3-O-glucosyl (1→2) xyloside and quercetin 3-O-glucosyl (1→6) rhamnoside in E. coli by engineering the nucleotide pathway and introducing two UGTs either from A. thaliana (AtUGT78D1 and AtUGT79B1) for the synthesis of quercetin 3-O-glucosyl (1→2) xyloside, or from Bacillus subtilis (BcGT1) and from Glycine max (Fg2) for the synthesis of quercetin 3-O-glucosyl (1→6) rhamnoside).
Materials and methods
Constructs
The pTac-pCDFDuet vector was derived from pCDFDuet (Novagen) (Fig. 1). Two primers were designed to eliminate the lacI gene from pCDFDuet and insert a tac promoter [3]. The forward primer contains the tac promoter (shown in uppercase), ribosome binding sequence (underlined), and a pCDFDuet-specific sequence (nucleotides 106 to 125, shown in lowercase) 5′-TGAAATGAGCTGTTGACAATTAATCATCGGCTCGTATAATGTGTGGaggaggattacaaaggatccgaattcgagctcgg-3′. The reverse primer is located from nucleotides 2589 to 2561 in pCDFDuet and its sequence is 5′-GACAGGTTTCCCGACTGGAAAGCGGGCAG-3′. PCR was carried out with the two primers listed above, the pCDFDuet vector, as template and Pyrobest Taq polymerase (Takara, Japan). The PCR products were purified using the Gel Extraction Kit (Bioneer, Korea) and ligated. AtUGT78D2 from A. thaliana, which was cloned previously [15], was cloned into the EcoRI/NotI site of pCDFDuet and pTac-pCDFDuet (Fig. 1) and the resulting plasmids were called pC-UGTD2 and pTac-pC-UGTD2, respectively. AtUGT79B1 (At5g54060) [30] was cloned using reverse-transcription polymerase chain reaction (RT-PCR), as described previously [15]. Two primers containing an EcoRI site and a NotI site (lower case letters in the primer sequence), respectively, 5′-AAgaattcaATGGGTGTTTTTGGATCGAATGA-3′ and 5′-aagcggccgcTCATGACTTCACAAGTTCAATT-3′ were used. The resulting PCR product was digested with EcoRI and NotI and subcloned into the EcoRI/NotI site of the E. coli expression vector pGEX 5X-3, and the resulting plasmid was called pG-UGTB1. UXS from A. thaliana (AtUXS) and ugd (UDP-glucose dehydrogenase) from E. coli (Ecugd) were previously cloned [6] and were subcloned into the EcoRI/NotI site and NdeI/XhoI sites of the pACYCDuet vector, respectively. The resulting construct was called pA-AtUXS-Ecugd. E. coli strains, BarnA and Bpgi, in which arnA (UDP-L-Ara4N formyltransferase/UDP-GlcA C-4″-decarboxylase) and pgi (phosphoglucoisomerase) were deleted, respectively, were generated as described by Kim et al. [12, 15].

Diagram of the pTac-PCDF vector
AtUGT78D2 [5], BcGT1 [19], and RHM [14] were cloned previously.
To synthesize rutin, the BcGT1 gene from B. subtilis was integrated into the tyrR (tyrosine DNA-binding transcriptional repressor) gene of E. coli. BcGT1 was subcloned into the pACYCDuet vector, and the resulting construct was used as a template for PCR. The primers used were 5′-GTGTCATATCATCATATTAATTGTTCTTTTTTCAGGTGAAGGTTCCCATGGCTATCATGCCATACCGCGA-3′ as a forward primer and 5′-TTGCACCATCAGGCATATTCGCGCTTACTCTTCGTTCTTCTTCTGACTCACTGATGTCCGGCGGTGCTTT-3′ as a reverse primer (the E. coli tyrR sequences are underlined and the other sequences are that of the pACYCDuet vector).
UGT from G. max (Fg2, GenBank accession number NM_001288595) [24] was cloned using RT-PCR. RNA was isolated from the leaves of G. max using the Plant Total RNA Isolation Kit (Qiagen), and cDNA was synthesized with Omniscript reverse transcriptase (Qiagen) using oligo dT primer. The primers used to clone Fg2 were 5′-AAGAATTCAATGCCTAGTGAATTAGCTATGAA-3′ (EcoRI site underlined.) as a forward primer and 5′-AAGCGGCCGGCTAAGCCATAGACTTTAACTGGG-3′ (NotI site underlined.) as a reverse primer. The PCR product was subcloned into the EcoRI/NotI site of pGEX 5X-3. Cs1,6RhaT from Citrus sinensis (GenBank accession number DQ119035) was cloned using RT-PCR. Primers 5′-AAGAATTCAATGCACGCCCCTTCGA-3′ (with EcoRI site underlined.) as a forward primer 5′-AAGCGGCCGGC TTAAGCTAAGGCTTTGAGATCC-3′ (NotI site is underlined) as a reverse primer.
Biotransformation of quercetin
To compare the production of quercetin 3-O-glucosyl (1→2) xyloside in the E. coli strains and to optimize the cell concentration, E. coli cells were grown in 2 mL of LB medium containing 50 μg/ml of antibiotics at 37 °C for 18 h. The cultured cells were inoculated into fresh LB medium containing antibiotics, and the cells were grown until the OD600 reached 1.0. The cells were collected by centrifugation, and the cell concentration was adjusted to an OD600 of 1.0 with 2 mL of M9 medium supplemented with 1 % yeast extract, 2 % glucose, and 50 µg/mL antibiotics. Quercetin (200 μM; dissolved in dimethyl sulfoxide [DMSO]) was also added, and the culture was incubated at 30 °C with shaking for 6 h. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to the culture at a final concentration of 1 mM, and the culture was incubated for 20 h at 30 °C. The culture supernatant, which contained most of the product, was boiled for 3 min and centrifuged for 15 min. The supernatant was analyzed by HPLC.
Rutin was produced in E. coli strains BGR-1, BGR-2, and BGR-3. An overnight culture of E. coli was inoculated into 3 mL of a fresh LB medium containing 50 µg/mL antibiotics. The cells were grown until the OD600 reached 0.8, and then IPTG was added to a final concentration of 1 mM. The culture was incubated at 18 °C for 18 h with shaking at 180 rpm. The cells were harvested via centrifugation and resuspended with an OD600 of 3.0 in M9 medium containing 2 % glucose, 1 mM IPTG, 50 µg/mL antibiotics, and 50 µM quercetin. This culture was incubated at 30 °C.
To measure the conversion of quercetin into rutin using strain BGR-3, 50 μM quercetin was added to the culture at 0, 8, 17, and 26 h (for a final concentration of 200 μM). Samples were harvested at 4, 6, 8, 14, 17, 24, 26, 34, and 48 h and were boiled for 5 min. After centrifugation, the supernatant (100 μL) was analyzed by HPLC.
HPLC analysis of reaction products
The reaction products were analyzed using a Thermo Ultimate 3000 HPLC equipped with a photo diode array (PDA) detector and a C18 reversed-phase column (4.60 × 250 mm, 3.5 μm particle size, Varian). For the analysis, the mobile phase consisted of H2O containing 0.1 % formic acid (pH 3.0) with the following gradient: 40 % acetonitrile for 8 min, 90 % acetonitrile for 12 min, 90 % acetonitrile for 15 min, and 10 % acetonitrile for 20 min. The flow rate was 1 mL/min and UV detection was dually performed at 290 and 340 nm. The UV spectrum at 340 nm was used. Authentic rutin was used to calculation of the amount of quercetin 3-O-glucosyl (1→2) xyloside and quercetin 3-O-glucosyl (1→6) rhamnoside because only rutin was commercially available. The means and standard errors were calculated from triplicate experiments. Analysis of variance (ANOVA) was carried out using Tukey’s method with a significance level of 1 % in Excel 2010 (Microsoft).
To determine the structure of the reaction products, the culture supernatant was concentrated using a non-polar copolymer styrene–divinylbenzene adsorbent resin (HP-20, Samyang, Korea). The reaction product was eluted with methanol, and was then evaporated to dryness. The sample was dissolved again in a small amount of methanol and purified by HPLC. The structure of the reaction product was determined using nuclear magnetic resonance (NMR) spectroscopy [33].
The 13C NMR spectrum of the reaction product from the BGX-3 matched that of the known compound, quercetin 3-O-xylosyl (1‴→2″) glucoside [26]: 13C NMR (100 MHz, DMSO-d 6 ) δ ppm 177.4 (C-4), 164.1 (C-7), 161.2 (C-5), 156.2 (C-9), 155.3 (C-2), 148.5 (C-4′), 144.9 (C-3′), 132.9 (C-3), 121.9 (C-6′), 121.2 (C-1′), 115.9 (C-5′), 115.2 (C-2′), 104.5 (C-1‴), 103.8 (C-10), 98.6 (C-6), 97.9 (C-1″), 93.4 (C-8), 81.8 (C-2″), 77.6 (C-5″), 76.8 (C-3″), 76.1 (C-3‴), 73.9 (C-2‴), 69.5 (C-4‴), 69.4 (C-4‴), 65.6 (C-5‴), 60.6 (C-6″); 1H NMR (400 MHz, DMSO-d 6 ) δ ppm 7.66 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 7.56 (d, J = 2.2 Hz, 1H, H-2′), 6.85 (d, J = 8.5 Hz, 1H, H-5′), 6.40 (br s, 1H, H-8), 6.19 (br s, 1H, H-6), 5.72 (d, J = 7.2 Hz, 1H, H-1″), 4.59 (d, J = 7.3 Hz, 1H, H-1‴), 3.68 (dd, J = 11.2, 5.0 Hz, 1H, H-5‴A), 3.55 (d, J = 11.2 Hz, 1H, H-6″A), 3.51 ~ 3.45 (m, 2H, H-2″ and H-4‴), 3.33–3.24 (m, 2H, H-6″B and H-3″), 3.16–3.10 (m, 3H, H-4″, H-5″ and H-3‴), 3.07–3.04 (m, 2H, H-2‴ and H-5‴).
The proton NMR spectrum of the reaction product of strain BGR-2 matched that of rutin [10]. 1H NMR (MeOH, 400 MHz) δ (ppm) 7.69 (1H, d, 2.1 Hz), 7.64 (1H, dd, 2.1, 8.5 Hz), 6.89 (1H, d, 8.5 Hz), 6.41 (1H, d, 2.0 Hz), 6.22(1H, d, 2.0 Hz), 5.11 (1H, d, 7.6 Hz), 4.53 (1H, d, 1.7 Hz), 3.82(1H, dd, 1.2, 11.0 Hz), 3.64(1H, dd, 1.7, 3.4 Hz), 3.55(1H, dd, 3.4, 9.5 Hz), 3.38–3.50(4H, m), 3.24–3.30(2H, m), 1.13(3H, d, 6.2 Hz).
Results
Synthesis of quercetin 3-O-glucosyl (1→2) xyloside in E. coli
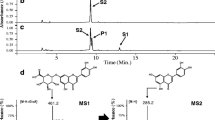
E. coli synthesizes UDP-glucose but does not synthesize UDP-xylose. UDP-xylose is synthesized from UDP-glucuronic acid by the action of UDP-xylose synthase (UXS). UXS has been previously characterized in A. thaliana [7]. To synthesize UDP-xyloside in E. coli, two genes, UDP-glucose dehydrogenase (ugd) from E. coli, which converts UDP-glucose to UDP-glucuronic acid, and UXS were overexpressed. Overexpression of ugd was expected to provide more UDP-glucuronic acid, the direct substrate of UXS. One UGT from A. thaliana, AtUGT78D2, uses quercetin as a sugar acceptor and UDP-glucose as a sugar donor to make quercetin 3-O-glucoside [22]. Another UGT from A. thaliana, AtUGT79B1, uses quercetin 3-O-glucoside as a sugar acceptor and UDP-xyloside as a sugar donor to synthesize quercetin 3-O-glucosyl (1→2) xyloside. To synthesize quercetin 3-O-glucoside (1→2) xyloside from quercetin, quercetin 3-O-glucoside needs to be synthesized before the reaction catalyzed by AtUGT79B1 can occur. Although AtUGT79B1 used quercetin 3-O-glucoside as a sugar acceptor and UDP-xyloside as a sugar donor, it could use quercetin as a sugar acceptor and make quercetin 3-O-xyloside (described below). Therefore, sequential glycosylation of quercetin was necessary to minimize byproduct production and maximize the production of quercetin 3-O-glucoside (1→2) xyloside. As a strategy to promote sequential glycosylation, we used two different promoters to express AtUGT78D2 and AtUGT79B1. AtUGT78D2 was cloned into pTac-pCDFduet and the resulting construct was named pTac-pC-D2. This construct contained a tac promoter instead of a T7 promoter without a lac operator; therefore, the expression of AtUGT78D2 was constitutive. As a control, AtUGT78D2 was also subcloned into pCDFDuet to generate pC-D2, which is controlled by a T7 promoter. AtUGT79B1 was subcloned into pGEX5X-2 to generate pG-B1, which has a lac operator and tac promoter [3]. AtUXS, and Ecugd were subcloned into pACYCDuet vector to generate pA-AtUXS-Ecugd in which expression is controlled by T7 promoter. Their expression was induced by the addition of IPTG. Three constructs, pC-D2, pG-C1, and pA-AtUXS-Ecugd were transformed into an E. coli arnA deletion mutant (BarnA in Table 1). ArnA competes with UXS for UDP-glucuronic acid [2]. Deletion of araA should provide more UDP-glucuronic acid to UXS for the biosynthesis of UDP-xyloside. Our previous study also showed that this BarnA strain produced more UDP-xyloside than the wild-type BL21 (DE3) strain [6]. The resulting transformant was named BGX-1. pTac-pC-D2, pG-C1, and pA-AtUXS-Ecugd were also transformed together into BarnA and the resulting strain was called BGX-2. These two E. coli strains, BGX-1 and BGX-2, were used for the biotransformation of quercetin. The resulting product was analyzed by HPLC (Fig. 2). BGX-1 showed four peaks while BGX-2 showed two peaks. Based on a comparison of the HPLC retention time with the standard molecules, P2, P3, and P4 were determined to be quercetin 3-O-glucoside, quercetin 3-O-xyloside, and quercetin 3-O-N-acetylglucosamine, respectively (data not shown). The structure of reaction product (P1) from BGX-2 was determined to be quercetin 3-O-xylosyl (1‴→2″) glucoside by NMR.

Analysis of the quercetin biotransformation products produced by E. coli strain BGX-1 (A) or BGX-2 (B). P1 quercetin 3-O-glucose (1→2) xylose; P2 quercetin 3-O-glucose; P3 quercetin 3-O-xylose; P4 quercetin 3-N-acetyl glucosamine; substrate, quercetin. Each cell suspension was inoculated into 2 mL of M9 medium supplemented with 1 % yeast extract, 2 % glucose, 50 µg/mL antibiotics and 200 μM quercetin at an OD600 of 1.0. The culture was incubated at 30 °C with shaking for 6 h. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to the culture at a final concentration of 1 mM, and the culture was further incubated for 20 h at 30 °C. The culture was harvested, boiled for 3 min and then centrifuged for 15 min. The supernatant was analyzed at 340 nm using HPLC
BGX-1 accumulated more quercetin 3-O-glucoside (33 mg/L) than quercetin 3-O-glucoside (1→2) xyloside (8 mg/L) while BGX-2 produced more quercetin 3-O-glucoside (1→2) xyloside (35 mg/L) than quercetin 3-O-glucoside (27 mg/L) (Fig. 3). BXG-1 showed the presence of similar amounts of quercetin 3-O-glucoside even after longer incubation without converting into quercetin 3-O-glucoside (1→2) xyloside. These results showed that the sequential expression of UGT is critical for the production of quercetin 3-O-glucoside (1→2) xyloside.

Production of quercetin 3-O-glucose (1→2) xylose in different E. coli strains. Authentic rutin was used as a standard to determine the production of quercetin 3-O-glucosyl (1→2) xyloside because rutin is commercially available. The means and standard errors were calculated from triplicate experiments
We optimized the initial cell concentration for the production of quercetin 3-O-glucoside (1→2) xyloside using strain BGX-2. The optimum cell concentration was determined by varying the initial cell density (OD600 = 1–5). The production of quercetin 3-O-glucoside (1→2) xyloside was the highest at an OD600 of 3 (55 mg/L). The conversion of quercetin to quercetin 3-O-glucoside (1→2) xyloside was monitored over time at the optimal initial BXG-2 cell concentration. After incubation of BGX-2 cells with quercetin for 6 h, 85 % of the quercetin was converted to quercetin 3-O-glucoside. The addition of IPTG induced the expression of the enzymes required for the biosynthesis of quercetin 3-O-glucoside (1→2) xyloside from quercetin 3-O-glucoside, resulting in the conversion of approximately 50 % of the synthesized quercetin 3-O-glucoside into quercetin 3-O-glucoside (1→2) xyloside within 2 h. After 2 h, the conversion rate of quercetin 3-O-glucoside or quercetin 3-O-glucoside (1→2) xyloside was reduced (Fig. 4). At 24 h, 65 mg/L quercetin 3-O-glucoside (1→2) xyloside was produced, while approximately 40 mg/L quercetin 3-O-glucoside was still present. Thus, approximately 54.5 % of the quercetin was converted into quercetin 3-O-glucoside (1→2) xyloside.

Production of quercetin 3-O-glucose (1→2) xylose using E. coli strain BGX-2. The cell density was adjusted to OD600 of 5.0 with 10 mL of M9 containing 1 % yeast extract, 2 % glucose, 50 µg/mL antibiotics and 200 μM quercetin. The culture was incubated at 30 °C for 6 h, and then IPTG was added at the final concentration of 1 mM. Samples were harvested at 2, 4, 6, 8, 10, 12, and 24 h, and analyzed by HPLC
Synthesis of quercetin 3-O-glucosyl (1→6) rhamnoside (rutin) in E. coli
Two UGTs, Fg2 from G. max and Cm1,6RhaT from C. sinensis, were implicated in the conversion of quercetin 3-O-glucoside to rutin [4, 24]. We evaluated the ability of Fg2 and Cm1,6RhaT to synthesize rutin from quercetin 3-O-glucoside in E. coli. Each gene, along with RHM2 encoding UDP-rhamnose synthase was transformed into E. coli, and each resulting transformant was tested for its ability to synthesize rutin from quercetin 3-O-glucoside. Analysis of culture filtrates from both transformants showed a peak with the same retention time as that of the rutin standard. In addition, the molecular mass of the product was 609.7 Da, which corresponded with the predicted molecular mass of rutin (data not shown). The structure of the reaction product was determined to be rutin using NMR (See “Materials and methods”). An E. coli harboring Fg2 and RHM2 (25.3 mg/L) produced more rutin than an E. coli harboring Cm1,6RhaT and RHM2 (22.6 mg/L). Therefore, we decided to use Fg2 for further analysis.
We attempted to synthesize rutin from quercetin. This required two reactions: the conversion of quercetin to quercetin 3-O-glucoside catalyzed by AtUGT78D2 or BcGT1 [8, 19] and the conversion of quercetin 3-O-glucoside to rutin by Fg2 and RHM2. For this purpose, we constructed an E. coli strain, BGR-1, harboring three genes, AtUGT78D2, Fg2, and RHM2. BGR-1 cells converted quercetin into quercetin 3-O-glucoside and rutin. However, even after a longer incubation, only the amount of quercetin 3-O-glucoside increased. It is likely that the first reaction (the conversion of quercetin to quercetin 3-O-glucoside) is much faster than the second reaction (the conversion of quercetin 3-O-glucoside to rutin), and the accumulation of quercetin 3-O-glucoside seems to inhibits the second reaction. Usage of pTac-pC-D2 for the biosynthesis of quercetin 3-O-glucoside was unsuccessful. The above results suggested that slow synthesis of quercetin 3-O-glucoside may increase the yield of rutin. Thus, we decided to lower the copy number of the gene encoding the protein responsible for the conversion of quercetin into quercetin 3-O-glucoside. We integrated BcGT1, which also converts quercetin into quercetin 3-O-glucoside, into the chromosome of E. coli. BcGT1 makes quercetin 3-O-glucoside at a slower pace than AtUGT78D2 (see below). Quercetin was added to a reaction with resulting strain BGR-3, and the culture filtrate was analyzed. As shown in Fig. 5, most of quercetin was converted into rutin, and only a small amount of quercetin 3-O-glucoside remained. This indicated that the conversion of quercetin 3-O-glucose to rutin should be faster than the conversion of quercetin to quercetin 3-O-glucose. To confirm that the copy number of BcGT1 was related to the production of rutin, we compared rutin production between strains BGR-2 and BGR-3. The BGR-3 strain contains one copy of BcGT1 because it is integrated into the E. coli chromosome, whereas the BGR-2 strain contains 10–12 copies of BcGT1 because BcGT1 is present on the low copy number plasmid pACYCDuet. The BGR-3 strain produced approximately 25.3 mg/L of rutin, whereas the BGR-1 and BGR-2 strains produced 7.3 and 2.3 mg/L of rutin, respectively, after 7 h. This result supported the observation that the slow production of quercetin 3-O-glucoside is critical to maximize the final product yield.

Synthesis of rutin from quercetin using strain BGR-3. a Standard rutin; b standard quercetin; c standard quercetin 3-O-glucoside; d reaction products of BGR-2 (P1 and P2 are reaction products). An overnight culture of E. coli was inoculated in 3 mL of a fresh LB medium containing 50 µg/mL antibiotics. The cells were grown until the OD600 reached 0.8 and IPTG was added to the culture at a final concentration of 1 mM. The culture was incubated at 18 °C for 18 h with shaking at 180 rpm. The cells were harvested via centrifugation and resuspended to an OD600 of 3.0 with M9 medium containing 2 % glucose, 1 mM IPTG, 50 µg/mL antibiotics, and 50 µM quercetin. The reaction mixture was incubated at 30 °C
The production of rutin was then further optimized using strain BGR-3. First, the effect of initial cell density was tested. The cell concentration was adjusted to OD600 values of 0.5, 1.0, 2.0, 3.0, and 4.0 after induction, and, the conversion of quercetin to rutin was assessed. The production of rutin continued to increase from an OD600 of 0.5 (14.0 mg/L) to an OD600 of 3.0 (25.3 mg/L), but decreased at an OD600 of 4.0 (19.6 mg/L). Therefore, the optimal initial cell concentration was OD600 of 3.0. Using the optimized cell concentration of BGR-3, rutin was produced. Quercetin (50 μM) was added at 0, 8, 17, and 26 h (a total concentration of 200 μM quercetin was added). The reaction product was periodically sampled and analyzed by HPLC. Approximately 119.8 mg of rutin was obtained after 48 h (Fig. 6) and approximately 98 % of quercetin was converted to rutin.

Production of rutin using E. coli strain BGR-2. To measure the conversion of quercetin into rutin using strain BGR-3, 50 μM quercetin was added to the culture at 0, 8, 17, and 26 h for a final concentration of 200 μM. Samples were harvested at 4, 6, 8, 14, 17, 24, 26, 34, and 48 h and analyzed by HPLC
Discussion
In the present study, we synthesized two quercetin diglycosides: quercetin 3-O-glucose (1→2) xyloside, and rutin, in E. coli. Each quercetin diglycoside was synthesized via a sequential glycosylation reaction using two UGTs. Although the first glycosylation reaction product for both quercetin diglycosides was quercetin 3-O-glucoside, two different strategies were employed for its synthesis. For the synthesis quercetin 3-O-glucoside (1→2) xyloside, a constitutive promoter was used; whereas, for the synthesis of rutin, the copy number of the UGT was varied. The strategy for the first reaction was varied based on the enzyme that catalyzed the second reaction. In cases where the conversion of the substrate using the E. coli expressing the second UGT is faster, a constitutive promoter could be used; whereas, in cases of low conversion rate of the substrate using the E. coli expressing the second UGT, a lower copy number of the first gene proved to be a better choice.
Although rutin is commercially available, most of it is derived from plants, as most natural compounds are generally obtained via extraction and purification from plants sources. However, these methods have a disadvantage owing to the complexity of the refining process. To overcome this, the production of molecules such as rutin using E. coli might be a useful alternative. Various quercetin derivatives have been synthesized using E. coli expressing different genes [13, 16]; however, the low solubility and antibacterial activity of quercetin have limited the final yield. Here, we showed that sequential additional of quercetin to the culture medium could circumvent this problem. In addition, determination of the initial concentration of quercetin and the feeding time as well as controlling the expression of the biosynthetic genes are important for increasing the final product yield without causing cell lysis and a high metabolic load.
Many bioactive flavonoids are modified by O-methylation, glycosylation, and hydroxylation. For example, isorhamnetin 3-O-glucoside, which is known to have an effect on diabetic complications [21], is synthesized by sequential 3′-O-methylation and 3-O-glucosylation of quercetin. The 3′-O-methylation of quercetin should occur before the 3-O-glucosylation, as quercetin 3-O-glucoside does not fit into the substrate binding site of quercetin 3′-O-methyltransferase [11]. The strategies used in this study provide a method for the synthesis of bioactive flavonoids using a single strain of E. coli.
References
Bowles D, Lim E-K, Poppenberger B, Vaistij FE (2006) Glycosyltransferases of lipophilic small molecules. Annu Rev Plant Biol 57:567–597. doi:10.1146/annurev.arplant.57.032905.105429
Breazeale SD, Ribeiro AA, McClerren AL, Raetz CRH (2005) A formyltransferase required for polymyxin resistance in Escherichia coli and the modification of lipid A with 4-amino-4-deoxy-L-arabinose. Identification and function of UDP-4-deoxy-4-formamido-L-arabinose. J Biol Chem 280(14):14154–14167. doi:10.1074/jbc.M414265200
De Boer HA, Comstock LJ, Vasser M (1983) The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc Natl Acad Sci 80(1):21–25. doi:10.1073/pnas.80.1.21
Frydman A, Weisshaus O, Bar-Peled M, Huhman DV, Sumner LW, Marin FR, Lewinsohn E, Fluhr R, Gressel J, Eyal Y (2004) Citrus fruit bitter flavors: isolation and functional characterization of the gene Cm1,2RhaT encoding a 1,2 rhamnosyltransferase, a key enzyme in the biosynthesis of the bitter flavonoids of citrus. Plant J 40(1):88–100. doi:10.1111/j.1365-313X.2004.02193.x
Guardia T, Rotelli AE, Juarez AO, Pelzer LE (2001) Anti-inflammatory properties of plant flavonoids. Effects of rutin, quercetin and hesperidin on adjuvant arthritis in rat. Farmaco 56(9):683–687. doi:10.1016/S0014-827X(01)01111-9
Han SH, Kim B-G, Yoon JA, Chong Y, Ahn J-H (2014) Synthesis of flavonoid O-pentosides by Escherichia coli through engineering nucleotide sugar synthesis pathway and glycosyltransferase. Appl Env Microbiol 80(9):2754–2762. doi:10.1128/AEM.03797-13
Harper AD, Bar-Peled M (2002) Biosynthesis of UDP-xylose. Cloning and characterization of a novel Arabidopsis gene family, UXS, encoding soluble and putative membrane-bound UDP-glucuronic acid decarboxylase isoforms. Plant Physiol 130(4):2188–2198. doi:10.1104/pp.009654
Jones P, Messner B, Nakajima J-I, Schäffner AR, Saito K (2003) UGT73C6 and UGT78D1, glycosyltransferase involved in flavonol glycoside biosynthesis in Arabidopsis thaliana. J Biol Chem 278(45):43910–43918. doi:10.1074/jbc.M303523200
Jüergenliemk G, Boje K, Huewel S, Lohmann C, Galla HJ, Nahrstedt A (2003) In vitro studies indicate that miquelianin (quercetin 3-O-beta-d-glucuronopyranoside) is able to reach the CNS from the small intestine. Planta Med 69(11):1013–1017. doi:10.1055/s-2003-45148
Kazuma K, Noda N, Suzuki M (2003) Malonylated flavonol glycosides from the petals of Clitoria ternatea. Phytochemistry 62(2):229–237. doi:10.1016/S0031-9422(02)00486-7
Kim B-G, Sung SH, Jung NR, Chong Y, Ahn J-H (2010) Biological synthesis of isorhamnetin 3-O-glucoside using engineered glucosyltransferase. J Mol Catalysis B: Enzymatic 63(3–4):194–199. doi:10.1016/j.molcatb.2010.01.012
Kim B-G, Jung NR, Joe EJ, Hur H-G, Lim Y, Chong Y, Ahn J-H (2010) Bacterial synthesis of a flavonoid deoxyaminosugar conjugate in Escherichia coli expressing a glycosyltransferase of Arabidopsis thaliana. ChemBioChem 11(17):2389–2392. doi:10.1002/cbic.201000456
Kim B-G, Sung SH, Chong Y, Lim Y, Ahn J-H (2010) Plant flavonoid O-methyltransferases: substrate specificity and application. J Plant Biol 53(5):321–329. doi:10.1007/s12374-010-9126-7
Kim BG, Kim HJ, Ahn J-H (2012) Production of bioactive flavonol rhamnosides by expression of plant genes in Escherichia coli. J Agr Food Chem 60(44):11143–11148. doi:10.1021/jf302123c
Kim B-G, Sung SH, Ahn J-H (2012) Biological synthesis of quercetin 3-O-N-acetylglucosamine conjugate using engineered Escherichia coli expressing UGT78D2. Appl Microbiol Biotechnol 93(6):2447–2453. doi:10.1007/s00253-011-3747-8
Kim BG, Yang SM, Kim SY, Cha MN, Ahn J-H (2015) Biosynthesis and production of glycosylated flavonoids in Escherichia coli: current state and perspectives. Appl Microbiol Biotechnol 99(7):2979–2988. doi:10.1007/s00253-015-6504-6
Kim HS, Kim B-G, Sung S, Kim M, Mok H, Chong Y, Ahn J-H (2013) Engineering flavonoid glycosyltransferases for enhanced catalytic efficiency and extended sugar donor selectivity. Planta 238(4):683–693. doi:10.1007/s00425-013-1922-0
Kim SY, Lee HR, K-s Park, Kim BG, Ahn J-H (2015) Metabolic engineering of Escherichia coli for the biosynthesis of flavonoid O-glucuronides and flavonoid O-galactoside. Appl Microbiol Biotechnol 99(5):2233–2242. doi:10.1007/s00253-014-6282-6
Ko JH, Kim BG, Ahn J-H (2006) Glycosylation of flavonoids with a glycosyltransferase from Bacillus cereus. FEMS Microbiol Lett 258(2):263–268. doi:10.1111/j.1574-6968.2006.00226.x
La Casa C, Villegas I, Alarcón de la Lastra C, Motilva V, Martín Calero MJ (2000) Evidence for protective and antioxidant properties of rutin, a natural flavone, against ethanol induced gastric lesions. J Ethnopharmacol 71(1–2):45–53. doi:10.1016/S0378-8741(99)00174-9
Lee YS, Lee S, Lee HS, Kim BK, Ohuchi K, Shin KH (2005) Inhibitory effects of isorhamnetin-3-O-beta-d-glucoside from Salicornia herbacea on rat lens aldose reductase and sorbitol accumulation in streptozotocin-induced diabetic rat tissues. Biol Pharm Bull 28(5):916–918. doi:10.1248/bpb.28.916
Lim E-K, Ashford DA, Hou B, Jackson RG, Bowles DJ (2004) Arabidopsis glycosyltransferases as biocatalysts in fermentation for regioselective synthesis of diverse quercetin glucosides. Biotechnol Bioeng 87(5):623–631. doi:10.1002/bit.20154
Pandey RP, Malla S, Simkhada B, Kim B-G, Sohng JG (2013) Production of 3-O-xylosyl quercetin in Escherichia coli. Appl Microbiol Biotechnol 97(5):1889–1901. doi:10.1007/s00253-012-4438-9
Rodas FR, Rodriguez TO, Mura Y, Iwashina T, Sufawara S, Suzuki M, Nakabayashi R, Yonekura-Sakakibara Saito K, Kitajima J, Toda K, Takahashi R (2014) Linkage mapping, molecular cloning and functional analysis of soybean Fg2 encoding flavonol 3-O-glucose (1→6) rhamnosyltransferase. Plant Mol Biol 84(3):287–300. doi:10.1007/s11103-013-0133-1
Sheu JR, Hsiao G, Chou PH, Shen MY, Chou DS (2004) Mechanisms involved in the antiplatelet activity of rutin, a glycoside of the flavonol quercetin, in human platelets. J Agric Food Chem 52(14):4414–4418. doi:10.1021/jf040059f
Shui G, Peng LL (2004) An improved method for the analysis of major antioxidants of Hibiscus esculentus Linn. J Chromatogr A 1048(1):17–24. doi:10.1016/j.chroma.2004.07.032
Simkhada D, Lee HC, Sohng JK (2010) Genetic engineering approach for the production of rhamnosyl and allosyl flavonoids from Escherichia coli. Biotechnol Bioeng 107(1):154–162. doi:10.1002/bit.22782
Soundararajan R, Wishart AD, Rupasinghe HP, Arcellana-Panlilio M, Nelson CM, Mayne M, Robertson GS (2008) Quercetin 3-glucoside protects neuroblastoma (SH-SY5Y) cells in vitro against oxidative damage by inducing sterol regulatory element-binding protein-2-mediated cholesterol biosynthesis. J Biol Chem 283(4):2231–2245. doi:10.1074/jbc.M703583200
Vogt T, Jones P (2000) Glycosyltransferases in plant natural product synthesis: characterization of a supergene family. Trend Plant Sci 5(9):380–386. doi:10.1016/S1360-1385(00)01720-9
Yonekura-Sakakibara K, Fukushima A, Nakabayashi R, Hanada K, Matsuda F, Sugawara S, Inoue E, Kuromori T, Ito T, Shinozaki K, Wangwattana B, Yamazaki M, Saito K (2012) Two glycosyltransferases involved in anthocyanin modification delineated by transcriptome independent component analysis in Arabidopsis thaliana. Plant J 69(1):154–167. doi:10.1111/j.1365-313X.2011.04779.x
Yonekura-Sakakibara K, Tohge T, Matsuda F, Nakabayashi R, Takayama H, Niida R, Watanabe-Takahashi A, Inoue E, Saito K (2008) Comprehensive flavonol profiling and transcriptome coexpression analysis leading to decoding gene-metabolite correlation in Arabidopsis. Plant Cell 20(8):2160–2176. doi:10.1105/tpc.108.058040
Yonekura-Sakakibara K, Hanada K (2011) An evolutionary view of functional diversity in family 1 glycosyltransferases. Plant J 66(1):182–193. doi:10.1111/j.1365-313X.2011.04493.x
Yoon J-A, Kim B-G, Lee WJ, Lim Y, Chong Y, Ahn J-H (2012) Production of a novel quercetin glycoside through metabolic engineering of Escherichia coli. Appl Env Microbiol 78(12):4256–4262. doi:10.1128/AEM.00275-12
Acknowledgments
This work was supported by a Grant from the Next-Generation BioGreen 21 Program (PJ00948301), the Rural Development Administration, and the Priority Research Centers Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2009-0093824).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
An, D.G., Yang, S.M., Kim, B.G. et al. Biosynthesis of two quercetin O-diglycosides in Escherichia coli . J Ind Microbiol Biotechnol 43, 841–849 (2016). https://doi.org/10.1007/s10295-016-1750-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-016-1750-x