
Abstract
There are substantial genotypic differences in the levels of flavonol glycosides (FGs) in soybean leaves. The first objective of this study was to identify and locate genes responsible for FG biosynthesis in the soybean genome. The second objective was to clone and verify the function of these candidate genes. Recombinant inbred lines (RILs) were developed by crossing the Kitakomachi and Koganejiro cultivars. The FGs were separated by high performance liquid chromatography (HPLC) and identified. The FGs of Koganejiro had rhamnose at the 6″-position of the glucose or galactose bound to the 3-position of kaempferol, whereas FGs of Kitakomachi were devoid of rhamnose. Among the 94 RILs, 53 RILs had HPLC peaks classified as Koganejiro type, and 41 RILs had peaks classified as Kitakomachi type. The segregation fitted a 1:1 ratio, suggesting that a single gene controls FG composition. SSR analysis, linkage mapping and genome database survey revealed a candidate gene in the molecular linkage group O (chromosome 10). The coding region of the gene from Koganejiro, designated as GmF3G6″Rt-a, is 1,392 bp long and encodes 464 amino acids, whereas the gene of Kitakomachi, GmF3G6″Rt-b, has a two-base deletion resulting in a truncated polypeptide consisting of 314 amino acids. The recombinant GmF3G6″Rt-a protein converted kaempferol 3-O-glucoside to kaempferol 3-O-rutinoside and utilized 3-O-glucosylated/galactosylated flavonols and UDP-rhamnose as substrates. GmF3G6″Rt-b protein had no activity. These results indicate that GmF3G6″Rt encodes a flavonol 3-O-glucoside (1 → 6) rhamnosyltransferase and it probably corresponds to the Fg2 gene. GmF3G6″Rt was designated as UGT79A6 by the UGT Nomenclature Committee.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Flavonoids are deposited in various tissues of soybean (Glycine max (L.) Merr.) in a tissue-specific manner. Flower petals usually contain four anthocyanins, eight flavonol glycosides, and one dihydroflavonol (Iwashina et al. 2007, 2008). Flavones (apigenin or luteolin) are exclusively deposited in pubescence: flavone aglycones outside of the cell surface (so-called ‘surface flavonoids’) and flavone glycosides inside of the cells (Iwashina et al. 2006a). There are substantial varietal differences in the various FGs in soybean leaves (Buttery and Buzzell 1973). Such structurally divergent flavonoid compounds may play important roles in these tissues.
FGs, derivatives of quercetin with 3′,4′-dihydroxyl groups in the B-ring, and derivatives of kaempferol with 4′-hydroxyl group, are predominant in leaves (Buttery and Buzzell 1973). The T gene encodes flavonoid 3′-hydroxylase (F3′H) that catalyzes hydroxylation of the 3′-position of flavonoids (Buttery and Buzzell 1973). F3′H protein produces quercetin derivatives in leaves (Buttery and Buzzell 1973) and luteolin derivatives in pubescence (Iwashina et al. 2006a). The tawny color in pubescence of cultivars with dominant T allele may be due to highly polymerized luteolin derivatives (Iwashina et al. 2006a). In contrast, cultivars with recessive t allele have gray pubescence color due to apigenin derivatives. In leaves, cultivars with gray pubescence have only kaempferol derivatives, whereas cultivars with tawny pubescence contain both kaempferol and quercetin types (Buttery and Buzzell 1973). Thus, pubescence color is indicative of the dominance of the T locus as well as the types of FGs in leaves. Toda et al. (2002) cloned and characterized the F3′H cDNA and revealed that a single-base deletion occurred in the coding region of this gene in cultivars with gray pubescence; this deletion generated a truncated polypeptide lacking F3′H function.
The color of pubescence in soybean is associated with chilling tolerance in terms of yield and quality of seeds. Takahashi and Asanuma (1996) and Takahashi et al. (2005) evaluated chilling tolerance of near-isogenic lines (NILs) for the T locus. Seed yield and nitrogen-fixation ability of the NILs with T allele was higher than the NILs with t allele only in response to chilling treatment of 15 °C for 4 weeks starting at anthesis. Chilling temperatures (about 15 °C) during flowering induce browning and cracking of the seed coats (Sunada and Ito 1982). Chilling treatment of NILs for the T locus revealed that the dominant T allele completely suppressed the development of browning around the hilum region and partly suppressed cracking of seed coats (Takahashi and Asanuma 1996; Takahashi 1997). Thus, the dominant T allele may be responsible for chilling tolerance in terms of both yield and quality of seeds.
Toda et al. (2011) evaluated the antioxidative activity of flavonols in soybean leaves and found that flavonols with the 3′4′-dihydroxylated configuration had higher antioxidative activity than flavonols with only 4′-hydroxyl group in accordance with previous studies (Rice-Evans et al. 1995; Vinson et al. 1995). Toda et al. (2012) further confirmed a similar relationship between alleles of the T locus and DPPH radical scavenging activity in crude extracts from the hilum of the seed coat. The antioxidant activity of flavonols catalyzed by F3′H may partly explain the association between the T gene and chilling tolerance.
In addition to chilling tolerance, there are additional functions that can be ascribed to FG structures in soybean. Magenta flower color is controlled by a recessive allele of the Wm locus. The recessive wm allele is associated with low levels of FGs in the leaves and flowers (Buzzell and Buttery 1977). The amount of FGs was reduced and dihydroflavonol was increased in flower petals of Harosoy-wm (Iwashina et al. 2007). Takahashi et al. (2007) cloned and characterized the cDNA for flavonol synthase and revealed that a single-base deletion occurred in the coding region of the magenta flower mutant. This deletion generated a non-functional polypeptide. Co-pigmentation between anthocyanins and flavonol glycosides may contribute to the purple flower color in soybeans with the Wm allele (Takahashi et al. 2007). The recessive wm allele for magenta flower reduces photosynthetic rate and seed yield, and causes earlier leaf senescence (Buzzell and Buttery 1977). Either low levels of FGs or high levels of their precursors may be responsible for the deleterious effects.
Buzzell and Buttery (1974) proposed four flavonol glycoside alleles, Fg1 (β(1–6)-glucoside present), Fg2 (α(1–6)-rhamnoside present), Fg3 (β(1–2)-glucoside present), and Fg4 (α(1–2)-rhamnoside present). These alleles are defined by the ability to bind glucose or rhamnose at either 2″- or 6″-position of glucose that is bound to the 3-position of flavonols. Later, Buzzell and Buttery (1992) reported a new allele of the Fg2 locus, resulting in a series of alleles, Fg2-a, Fg2-b and fg2. The Fg3 and Fg4 genes are linked with a recombination frequency of 12.0 % in the molecular linkage group C2 (chromosome 6) (Buzzell 1974).
Glycosyltransferases (GTs) catalyze the transfer of sugar moieties from activated donor molecules to specific acceptor molecules, forming glycosidic bonds (Lairson et al. 2008). GTs are encoded by large multigene families, and classified into at least 94 families (GT1–GT94, http://www.cazy.org/GlycosylTransferases.html). The family 1 glycosyltransferase, referred to as UDP glycosyltransferases (UGTs), comprise the largest group in plants. UGTs catalyze the transfer of a glycosyl moiety from UDP sugars to a wide range of acceptor molecules including flavonoids (Yonekura-Sakakibara and Hanada 2011).
Soybean plants with the Fg1Fg3 alleles have a lower rate of photosynthesis, lower leaf chlorophyll concentration, lower leaf weight, and lower seed yield (Buttery and Buzzell 1976). Further, Fg1 and Fg3 control waviness of leaf margins in soybean (Buzzell and Buttery 1998). However, wavy leaf is not expressed in the presence of the gene for brown pubescence (TT) (Rode and Bernard 1975). It is unclear how FGs affect a wide variety of characters such as photosynthesis, seed yield, leaf weight, leaf senescence, leaf chlorophyll concentration and leaf waviness.
Preliminary experiments suggested that two Japanese cultivars, Kitakomachi and Koganejiro, had distinct FG composition in their leaves (Iwashina et al. 2010, unpublished results). The first objective of this study was to identify and locate genes for FG biosynthesis in the soybean genome using RILs derived from a cross between Kitakomachi and Koganejiro. The second objective was to clone and verify the function of the candidate genes.
Materials and methods
Plant materials
A Japanese soybean cultivar Koganejiro with yellow hilum, yellow seed coats, gray pubescence, purple flowers and narrow leaflets (IIttW1W1lnln) was crossed with the Japanese cultivar Kitakomachi with yellow hilum, yellow seed coats, gray pubescence, purple flowers and ovate leaflets (IIttW1W1LnLn). Flowers of Koganejiro were emasculated 1 day before opening and fertilized with pollen from Kitakomachi in 2004. Hybridity of F1 plants was ascertained based on ovate leaflets. A total of 120 RILs from the F6 generation were developed without any selection using the single-seed descent method. The pedigree of the parents was reported by Githiri et al. (2007). Seeds were planted at the National Institute of Crop Science, Tsukuba, Japan (36°06′N, 140°05′E) on June 9, 2011. N, P, and K were applied at 3.0, 4.4, and 8.3 g m−2, respectively. Plants were individually planted with spaces between rows and plants at 70 and 10 cm, respectively. On average, nine plants were grown for each parent and RIL.
Extraction of FGs
A total of 94 RILs were randomly selected and used for analysis, because PCR reaction plates and the electrophoresis apparatus were designed for multiples of 96 samples (94 RILs and two parents). Trifoliolate leaves were bulked from four plants from each of the parents and the RILs at the R6 stage (Fehr et al. 1971). Discs (5 mm diameter) were punched out from leaves and a total of 100 mg of leaf discs was prepared in 3 replications. The leaf discs were soaked in 1 ml MeOH in 2 ml plastic tubes, vortexed and kept at 4 °C for 48 h. Vortexing was performed every 24 h. The 1 ml extracts were filtered through disposable filtration units (Maishoridisc H-13-5, Tosoh) and 10 μl from each sample was subjected to high performance liquid chromatography (HPLC) analysis.
HPLC and genetic analysis
Quantitative HPLC analysis of the extracts was performed with the Agilent 1100 HPLC System (Agilent Technologies) using L-column 2 ODS [I.D. 6.0 × 150 mm (Chemicals Evaluation and Research Institute)] at a flow rate of 1.0 ml/min, and a detection wavelength of 190–700 nm. The eluents were solvent A: 0.2 % phosphoric acid and solvent B: phosphoric acid/acetonitrile/H2O (0.2:30:70, v/v/v). The gradient was 0 min, B 20 %; 18 min, B 50 %; 25 min, B 70 %; 31 min, B 50 %; 34 min, B 20 %. The amount of flavonol glycosides was estimated from the pertinent peak area in a HPLC chromatogram generated with a detection wavelength of 350 nm. Seven authentic specimens were selected from soybean cultivars and other plant species (Iwashina et al. 2006b, 2010; Murai et al. 2013). In addition to HPLC comparisons with authentic specimens, eight kaempferol glycosides isolated from Koganejiro and Kitakomachi were identified by UV, LC–MS, acid hydrolysis, and 1H and 13C NMR as described in supplemental data. The genetic model for FG composition was hypothesized based on the HPLC chromatogram of the parents and the RILs, and the chemical structure of the pertinent peaks. Likelihood of the genetic model was estimated by Chi square test.
SSR analysis
Genomic DNA of the parents and the four plants from each of the RILs used for FG analysis were isolated from trifoliolate leaves by the CTAB method (Murray and Thompson 1980). A total of 1,025 SSR markers developed by the USDA (Song et al. 2004) and by the Kazusa DNA Research Institute (Hisano et al. 2007) were used for screening of polymorphisms between the parents. The PCR mixture contained 10 ng of genomic DNA, 2.25 pmol of primer, 625 pmol of nucleotides, and 0.125 unit of ExTaq in 1× ExTaq buffer supplied by the manufacturer (Takara Bio) in a total volume of 5 μl. The initial 2 min denaturation at 94 °C was followed by 35 cycles of 1 min denaturation at 94 °C, 1 min annealing at 49 °C, and 1 min extension at 72 °C. A final extension at 72 °C for 5 min completed the program. PCR was performed in an Applied Biosystems 9700 thermal cycler (Applied Biosystems). The PCR products were separated in 8 % nondenaturing acrylamide gels, and the fragments were visualized by staining with ethidium bromide.
Linkage mapping
The markers were tested by Chi square test for segregation in 1:1 ratio. A linkage map was constructed using the AntMap software version 1.2 (Iwata and Ninomiya 2006). The parameters were as follows: grouping method, all combinations; grouping criterion, LOD; map function, Kosambi; threshold value, 3. Designation of linkage groups followed Cregan et al. (1999).
RNA extraction and cDNA cloning
Total RNA was extracted from trifoliolate leaves (200 mg) of Kitakomachi and Koganejiro using the TRIZOL Reagent (Invitrogen) according to the manufacturer’s instructions. Reverse transcription of 5 μg of total RNA was done using the Superscript III First-Strand Synthesis System (Invitrogen) and an oligo(dT) primer according to the manufacturer’s instructions. The full-length cDNA was cloned from Kitakomachi and Koganejiro by end-to-end PCR using a pair of PCR primers (Table 1) that were based on the genome sequence of US cultivar Williams 82 deposited in the soybean genome database (Phytozome, http://www.phytozome.net/soybean.php). The PCR mixture contained 0.5 μg of cDNA, 10 pmol of each primer, 5 nmol of nucleotides and 1 unit of ExTaq in 1× ExTaq Buffer in a total volume of 25 μl. A 30 s denaturation at 94 °C was followed by 30 cycles of 30 s denaturation at 94 °C, 1 min annealing at 59 °C and 1 min extension at 72 °C. A final 7 min extension at 72 °C completed the program. The ~ 1.4 kbp PCR product was cloned into pCR 2.1 vector (Invitrogen) and sequenced.
Sequencing analysis
Nucleotide sequences of both strands were determined with the BigDye terminator cycle method using an ABI3100 Genetic Analyzer (Applied Biosystems). Nucleotide sequences and the putative amino acid translations were analyzed with the BLAST program (Altschul et al. 1997). Intron/exon structure of the gene was estimated based on the comparison between the cDNA sequence and the corresponding genome sequences of Williams 82 deposited in the soybean genome database. The nucleotide sequences were aligned using ClustalW and the alignment was used to construct a phylogenetic tree using the neighbor-joining method (http://clustalw.ddbj.nig.ac.jp/top-j.html) and illustrated using TREEVIEW (Page 1996). Bootstrap test (1,000 replications) was performed with MEGA5 version 5.2.2 (http://www.megasoftware.net/) (Tamura et al. 2011).
dCAPS analysis
A pair of PCR primers (Table 1) was designed to detect the two-base deletion unique to Kitakomachi. The first primer contains a nucleotide (G) that is mismatched with its target DNA to artificially create a restriction site for BcgI (CGANNNNNNTGC) in Koganejiro. The deletion within the restriction site of Kitakomachi would abolish of the restriction site in the amplified product to generate a polymorphism. The PCR mixture contained 30 ng of genomic DNA, 10 pmol of each primer, 5 nmol of nucleotides and 1 unit of ExTaq in 1× ExTaq Buffer in a total volume of 25 μl. The initial 30 s denaturation at 94 °C was followed by 30 cycles of 30 s denaturation at 94 °C, 1 min annealing at 59 °C and 1 min extension at 72 °C. A final 7 min extension at 72 °C completed the program. The amplified products were digested with BcgI, and the digests were separated on 8 % nondenaturing polyacrylamide gels. After electrophoresis, the gel was stained with ethidium bromide and the DNA fragments were visualized under UV light.
Expression and purification of recombinant GmF3G6″Rt proteins
The entire coding region of GmF3G6″Rt was amplified from Koganejiro and Kitakomachi by PCR using the high fidelity KOD -Plus- DNA polymerase (Toyobo) and primers containing restriction enzyme sites for SacI and XhoI (Table 1). Since GmF3G6″Rt had no intron, genomic DNA was used as a template. The PCR mixture contained 30 ng of genomic DNA, 10 pmol of each primer, 5 pmol of nucleotides, 50 pmol of MgSO4 and 0.5 unit of KOD -Plus- in 1× KOD -Plus- Buffer supplied by the manufacturer in a total volume of 25 μl. After an initial 2 min denaturation at 94 °C, there were 40 cycles of 15 s denaturation at 94 °C, 30 s annealing at 59 °C and 1 min extension at 68 °C. A final 7 min extension at 68 °C completed the program. The PCR amplicon was digested by SacI and XhoI, and then cloned into the pCold ProS2 vector (Takara Bio) that was previously digested with the same restriction enzymes. Escherichia coli strain BL21star (DE3) was used as a host for expression. Transformed cells were cultured at 37 °C until the optical density at A600 reached 0.5. After the addition of isopropyl-β-d-thiogalactopyranoside to a final concentration of 1 mM, cells were cultured at 15 °C for 24 h. The cells were collected, and the protein was purified as a His fusion using TALON® Metal Affinity Resin (Clontech) according to the manufacturer’s instructions. ProS2 tag was removed using HRV3C protease (Novagen) according to the manufacturer’s instructions. After exchanging the buffer to 50 mM HEPES–KOH, pH 7.5, the proteins were concentrated using an Amicon Ultra filter (10,000 MWCO, Millipore) and were used for enzyme assays.
Enzyme assays
The standard enzyme assay reaction mixture was described as before (Yonekura-Sakakibara et al. 2007). The mixture was pre-incubated at 30 °C for 2 min, and the reaction was started by the addition of the enzyme. Reactions were stopped after 0, 2, 4, 6, 60 or 80 min of incubation at 30 °C by the addition of 50 μl ice-cold 0.5 % (v/v) trifluoroacetic acid/methanol. The supernatant was recovered by centrifugation at 12,000g for 3 min. Flavonoids in the resultant solution were analyzed using a Shimadzu HPLC system with a Unison UK-C18 column (2.0 × 150 mm, 3 μm, Imtakt corporation) at a flow rate of 0.2 ml/min at 35 °C. Compounds were separated with a linear eluting gradient with solvent A (0.1 % trifluoroacetic acid in water) and solvent B (0.1 % trifluoroacetic acid in acetonitrile) set according to the following profile: 0 min, 10 % B; 3 min, 10 % B; 18 min, 75 % B; 18.01 min, 95 % B; 20 min, 95 % B; 30 min, 95 % B. PDA was used for the detection of UV–Visible absorption in the range of 200–600 nm.
MS/MS analysis
The extracts (1 μl) were analyzed using LC-QTOF-MS (LC, Waters Acquity UPLC system; MS, Waters Xevo G2 Q-Tof). Analytical conditions were as follows: LC column, Acquity bridged ethyl hybrid (BEH) C18 (1.7 μm, 2.1 mm × 100 mm, Waters); solvent system, solvent A (water including 0.1 % formic acid) and solvent B (acetonitrile including 0.1 % formic acid); gradient program, 99.5 %A/0.5 %B at 0 min, 99.5 %A/0.5 %B at 0.1 min, 20 %A/80 %B at 10 min, 0.5 %A/99.5 %B at 10.1 min, 0.5 %A/99.5 %B at 12.0 min, 99.5 %A/0.5 %B at 12.1 min and 99.5 %A/0.5 %B at 15.0 min; flow rate, 0.3 ml/min at 0 min, 0.3 ml/min at 10 min, 0.4 ml/min at 10.1 min, 0.4 min/min at 14.4 min and 0.3 ml/min at 14.5 min; column temperature, 40 °C; MS detection: capillary voltage, +3.0 keV; cone voltage, 25.0 V; source temperature, 120 °C; desolvation temperature, 450 °C; cone gas flow, 50 l/h; desolvation gas flow, 800 l/h; collision energy, 6 V; mass range, m/z 100–1,500; scan duration, 0.1 s; interscan delay, 0.014 s; data acquisition, centroid mode; polarity, positive; Lockspray (Leucine enkephalin): scan duration, 1.0 s; interscan delay, 0.1 s. MS/MS data was acquired in the ramp mode as the following analytical conditions: (1) MS: mass range, m/z 50–1,500; scan duration, 0.1 s; inter-scan delay, 0.014 s; and (2) MS/MS: mass range, m/z 50–1,500; scan duration, 0.02 s; inter-scan delay, 0.014 s; data acquisition, centroid mode; collision energy, ramped from 10 to 50 V. In this mode, MS/MS spectra of the top 10 ions (>1,000 counts) in an MS scan were automatically obtained. If the ion intensity was less than 1,000, MS/MS data acquisition was not performed and the next top 10 ions were scanned.
Gene expression assays
For real-time quantitative reverse transcription PCR, total RNA (5 μg) from each of three replicate leaf samples at R5 stage from parental lines was reverse-transcribed using the Superscript III First-Strand Synthesis System and an oligo d(T) primer. Primer sequences are exhibited in Table 1. The PCR mixture contained 0.4 μl of cDNA synthesis reaction mixture, 6 pmol of each primer, 1× ROX reference dye, 1× SYBR Premix Dimer Eraser (Takara Bio) and water to a final volume of 20 μl. Analysis was done using the StepOnePlus Real-Time PCR System (Applied Biosystems). The initial 30 s denaturation at 95 °C was followed by 40 cycles of 3 s denaturation at 95 °C, 30 s annealing at 58 °C and 30 s extension at 72 °C. The expression level of the soybean actin gene (GenBank accession number: J01298) (Shah et al. 1983) was used to normalize target gene expression. Expression levels were compared by a t test using Statistica 03J (Stadtsoft). Tissue-specific gene expression data from a diverse set of fourteen tissues were extracted from the RNA-Seq Atlas of the SoyBase webpage (http://soybase.org/soyseq/) (Severin et al. 2010).
Accession numbers
Sequence data from this article have been deposited with the DDBJ Data Libraries under accession numbers AB828192 (Kitakomachi) and AB828193 (Koganejiro).
Results
HPLC analysis
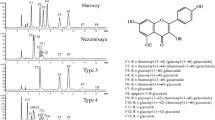
The HPLC chromatograms of Koganejiro and Kitakomachi is exhibited in Fig. 1. Koganejiro had five primary peaks attributed to FGs: 24.7 (F1), 25.1 (F2), 27.7 (F3), 28.8 (F4) and 30.0 min (F5). Kitakomachi had four primary peaks identified as FGs: 26.4 (F6), 26.9 (F7), 28.9 (F8) and 30.0 min (F5). Based on comparison with authentic specimens, these peaks correspond to the followings FGs: F1, kaempferol 3-O-rhamnosyl-(1 → 4)-[rhamnosyl-(1 → 6)-galactoside]; F2, kaempferol 3-O-rhamnosyl-(1 → 2)-[rhamnosyl-(1 → 6)-glucoside]; F3, kaempferol 3-O-rhamnosyl-(1 → 6)-galactoside; F4, kaempferol 3-O-rhamnosyl-(1 → 6)-glucoside; F5, kaempferol 3-O-glucoside; F6, kaempferol 3-O-rhamnosyl-(1 → 2)-galactoside; F7, kaempferol 3-O-rhamnosyl-(1 → 2)-glucoside; F8, kaempferol 3-O-galactoside (Fig. 1). The above results indicate that FGs specific of Koganejiro (F1–F4) have rhamnose at the 6″-position of glucose or galactose that is bound to the 3-position of kaempferol, whereas FGs of Kitakomachi are devoid of rhamnose.

HPLC analysis of flavonol glycosides in leaves of soybean cultivars Koganejiro and Kitakomachi. a HPLC chromatogram. 100 mg of trifoliolate leaves was extracted with 1 ml of MeOH. HPLC conditions: solvent A, 0.2 % phosphoric acid; solvent B, phosphoric acid/acetonitrile/H2O (0.2:30:70, v/v/v); 0 min, B 20 %; 18 min, B 50 %; 25 min, B 70 %; 31 min, B 50 %; 34 min, B 20 %; the flow-rate: 1.0 ml/min; injection volume: 10 μl; temperature, 40 °C; detection: 350 nm. b Chemical structure of flavonol glycosides corresponding to peaks in a. F1 kaempferol 3-O-rhamnosyl-(1 → 4)-[rhamnosyl-(1 → 6)-galactoside], F2 kaempferol 3-O-rhamnosyl-(1 → 2)-[rhamnosyl-(1 → 6)-glucoside], F3 kaempferol 3-O-rhamnosyl-(1 → 6)-galactoside, F4 kaempferol 3-O-rhamnosyl-(1 → 6)-glucoside, F5 kaempferol 3-O-glucoside, F6 kaempferol 3-O-rhamnosyl-(1 → 2)-galactoside, F7, kaempferol 3-O-rhamnosyl-(1 → 2)-glucoside, F8 kaempferol 3-O-galactoside
Genetic analysis
The 94 RILs segregated into 53 RILs that had HPLC peaks of the Koganejiro type and 41 RILs that had peaks of the Kitakomachi type. The segregation fitted a 1:1 ratio (χ2 = 1.53, P = 0.22), suggesting that a single gene, probably encoding flavonol 3-O-glucoside (1 → 6) rhamnosyltransferase, might control the composition of FGs.
SSR analysis and linkage mapping
Among the 1,025 SSR markers tested, 146 markers exhibited polymorphism between the parents and distinctly segregated in the RILs. These markers were used for linkage mapping. A total of 99 markers fell into 20 linkage groups spanning 748 cM. The low map coverage of this population may be partly due to the shared pedigree of the parents as previously discussed (Githiri et al. 2007). The gene responsible for the attachment of rhamnose to the 6″-position of flavonol 3-O-glucoside was mapped in linkage group O (chromosome 10), between GMES3560 and Satt331, at the position identical with GMES0295 (Fig. 2).

Linkage mapping of a gene (F3G6″Rt) responsible for attachment of rhamnose to the 6″-position of glucose or galactose that is bound to the 3-position of kaempferol using recombinant inbred lines derived from a cross of soybean cultivars, Koganejiro and Kitakomachi. The name of the linkage group is indicated at the top followed by the chromosome number in parenthesis. Distances (cM) of markers from the top of the linkage group are shown on the left
cDNA cloning
A survey of the genome sequence of a US cultivar Williams 82 suggested that two candidate genes similar to GT genes, Glyma10g33790 and Glyma10g33800 were aligned in tandem at a distance of about 6.2 kb in the vicinity of GMES0295. The entire coding region of Glyma10g33790 was amplified by PCR and subsequently cloned. Sequence analysis revealed that coding region of Glyma10g33790 is 1,392 bp long encoding 464 amino acids in Koganejiro. We designated the gene as GmF3G6″Rt. GmF3G6″Rt belongs to the family 1 glycosyltransferase, and it was designated as UGT79A6 by the UGT Nomenclature Committee (Mackenzie et al. 1997). The flavonoid glycosyltransferase phylogenetic tree suggested that GmF3G6″Rt belongs to the 1,2/1,6 branch-forming glucosyltransferase gene cluster (Fig. 3). Flavonoid glucoside (1 → 6) rhamnosyltransferase comprised a separate branch, and it apparently underwent duplication prior to speciation. GmF3G6″Rt had 56 % amino acid similarity with the Rt gene of petunia encoding anthocyanin 3-O-glucoside (1 → 6) rhamnosyltransferase (Kroon et al. 1994) and 58 % similarity with flavanone 7-O-glucoside (1 → 6) rhamnosyltransferase of Citrus sinensis (Frydman et al. 2013) (Fig. 4). Compared with Koganejiro, the cDNA sequence of Kitakomachi had a two-base deletion at the nucleotide position 930 (Fig. 4) and two single-base substitutions in the downstream of the deletion. The cDNAs of Koganejiro and Kitakomachi were designated as GmF3G6″R-a and GmF3G6″R-b, respectively. The deletion generated a truncated polypeptide that consisted of only 314 amino acids in Kitakomachi. GmF3G6″R-b lacked the PSPG (plant secondary product glycosyltransferase)-box around the C-terminal region (Masada et al. 2007) (Fig. 4). Comparison with the genome sequence of Williams 82 suggested that GmF3G6″Rt has no intron.

Unrooted molecular phylogenetic tree of the some flavonoid glycosyltransferases. Bar represents 0.1 amino acid substitutions/site. The GenBank accession numbers for the sequences are shown in parentheses: At3RhaT (NM_102790); At3GlcT (NM_121711); Vv3GlcT (AF000371); Ph3GlcT (AB027454); Pf3GlcT (AB002818); Hv3GlcT (X15694); Zm3GlcT (X13501); At5GlcT (NM_117485); Pf5GlcT (AB013596); Vh5GlcT (BAA36423); Ph5GlcT (AB027455); Db7GlcT 4′GlcT (CAB56231); Nt7GlcT 3GlcT (AAB36653); Sb7GlcT (BAA83484); At7RhaT (AY093133); CmF7G2″RhaT (AAL06646); CsF7G6″RhaT (ABA18631); IpA3G2″GlcT (AB192315); PhA3G6″RhaT (X71059); BpA3G2GlcAT (AB190262); AcA3Ga2″XylT (FG404013); At3G2″XylT (Q9FN26). GlcT glucosyltransferase, RhaT rhamnosyltransferase, XylT xylosyltransferase, GlcAT glucuronosyltransferase. Ac, Actinidia chinensis; At, Arabidopsis thaliana; Bp, Bellis perennis; Cm, Citrus maxima; Cs, Citrus sinensis; Db, Dorotheanthus bellidiformis; Gm, Glycine max; Hv, Hordeum vulgare; Ip, Ipomoea purpurea; Nt, Nicotiana tabacum; Pf, Perilla frutescens; Ph, Petunia hybrida; Sb, Scutellaria baicalensis; Vh, Verbena hybrida; Vv, Vitis vinifera; Zm, Zea mays

Amino acid alignment of soybean GmF3G6″Rt (a cultivar Koganejiro), petunia Rt encoding anthocyanin 3-O-glucoside (1 → 6) rhamnosyltransferase (accession number: X71059) and citrus Cs1,6RhaT encoding flavanone 7-O-glucoside (1 → 6) rhamnosyltransferase (accession number: ABA18631). Identical amino acids are indicated in black, similar amino acids in gray. Dashes represent gaps introduced to improve the alignment. Position of two-nucleotide deletion found in a cultivar Kitakomachi is shown by an asterisk. The plant secondary product glycosyltransferase (PSPG) box is underlined
The database survey suggested that Glyma10g33800 lacks an upstream region and so it may be a pseudogene. Sequencing analysis of the coding region indicated that the transcript of Glyma10g33800 had premature stop codons in all reading frames. In addition, Koganejiro and Kitakomachi had identical sequence for this gene, suggesting that Glyma10g33800 may not be the responsible gene.
dCAPS analysis
Bands with molecular size of about 190 bp were amplified with the dCAPS primers in Koganejiro and Kitakomachi (Fig. 5). BcgI digestion generated a band of about 140 bp in Koganejiro, whereas the PCR amplicon of Kitakomachi was unaffected. Banding pattern of the RILs was classified into Koganejiro type, Kitakomachi type and heterozygous type having both bands (Fig. 5). In total, 50 RILs had bands of Koganejiro type, 41 RILs had bands of Kitakomachi type and 3 RILs had both bands. Banding patterns co-segregated with FG patterns; RILs with FGs of Kitakomachi type had bands classified as Kitakomachi type, whereas RILs with FGs of Koganejiro type had bands classified as Koganejiro type or both types of bands (Fig. 5). The 3 RILs with both bands had the FG pattern classified as Koganejiro type.

Outline and results of dCAPS analysis of GmF3G6″Rt gene in soybean. a Schematic presentation of dCAPS analysis. Partial nucleotide and amino acid sequences around the region polymorphic between Kitakomachi and Koganejiro are exhibited. The PCR primer has a mismatched base (G) that is double underlined. PCR generated an amplification product having BcgI site (CGANNNNNNTGC) that is single underlined in Koganejiro. The two-base deletion in Kitakomachi abolished the restriction site. b Results of dCAPS analysis of the parents and the recombinant inbred lines derived from a cross between Koganejiro and Kitakomachi. PCR products amplified with dCAPS primers were digested by BcgI and the digests were separated on an 8 % polyacrylamide gel. ϕ, molecular marker ϕ × 174/HaeIII; Ki Kitakomachi, Ko Koganejiro. FG pattern of the recombinant inbred lines is exhibited below the gel. The migration of size markers (bp) is shown to the left of the gel
In vitro characterization of recombinant GmF3G6″Rt-a
The recombinant GmF3G6″Rt protein was expressed in E. coli as a ProS2 protein fusion and was semi-purified as described in Materials and methods. The GmF3G6″Rt-a protein catalyzed the conversion of kaempferol 3-O-glucoside to a single product kaempferol 3-O-rutinoside as confirmed by retention time, MS and MS/MS spectra and UV spectra (Figs. 6, 7). Neither GmF3G6″Rt-b protein nor ProS2 protein alone as a negative control, catalyzed the conversion to 6″-O-rhamnoside. Thus, GmF3G6″Rt-a can be defined as a flavonol 3-O-glucoside (1 → 6) rhamnosyltransferase. The specificity of GmF3G6″Rt-a as a sugar acceptor was also examined. GmF3G6″Rt-a was similarly active on both 3-O-glucoside/galactoside of kaempferol and quercetin (Table 2). GmF3G6″Rt-a prefers kaempferol to quercetin as an aglycon, and 3-O-galactosides to 3-O-glucosides as a glycosylation pattern. The sugar donor specificity of GmF3G6″Rt-a was examined with UDP-rhamnose, UDP-glucose, UDP-galactose, UDP-glucuronic acid and UDP-arabinose as donors and kaempferol 3-O-glucoside as an acceptor. No UGT activity was detected for UDP-glucose, UDP-galactose and UDP-glucuronic acid, indicating that GmF3G6″Rt-a are highly specific to UDP-rhamnose. Only slight activity with UDP-arabinose was found, but the structure of the resultant product was not identified.

HPLC analyses of the reaction products of GmF3G6″Rt-a recombinant protein. Elution profiles of reaction products of ProS2 protein (empty vector) (a), GmF3G6″Rt-b protein (b), GmF3G6″Rt-a protein (c) with kaempferol 3-O-glucoside, and the standards (kaempferol 3-O-rutinoside, EXTRASYNTHESE, France) (d) are shown. Kae3Glc kaempferol 3-O-glucoside, Kae3Rut kaempferol 3-O-rutinoside

Identification of the reaction product of GmF3G6″Rt-a. a Base peak intensity chromatograms of the standard, kaempferol 3-O-rutinoside (EXTRASYNTHESE, France) and the reaction products of GmF3G6″Rt-a recombinant protein. b Mass and MS/MS spectra of the reaction product were identical to those of the standard, kaempferol 3-O-rutinoside (EXTRASYNTHESE, France), respectively. c The MS/MS fragmentation for kaempferol 3-O-rutinoside. d UV spectra of the reaction product and kaempferol 3-O-rutinoside (EXTRASYNTHESE, France)
Gene expression
At the R5 stage, the transcript level of GmF3G6″Rt in leaves of Kitakomachi was 41.1 % of that in Koganejiro (t = 3.43*). According to the RNA-Seq Atlas of soybean, GmF3G6″Rt was expressed predominantly in young leaves, and less so in flowers, pods and pod shells (Table 3). Almost no expression was observed in seeds, roots and root nodules.
Discussion
Soybean cultivars, Koganejiro and Kitakomachi, have gray pubescence and their leaves contain predominantly kaempferol derivatives. However, FG components in leaves were distinct; Koganejiro had five primary HPLC peaks, F1–F5, whereas Kitakomachi had four peaks of F5–F8. Both cultivars had the F5 peak, but its amount was slight in Koganejiro, whereas it was most abundant in Kitakomachi. Comparison with authentic specimens suggested that FGs of Koganejiro had rhamnose at the 6″-position of glucose or galactose that is bound to the 3-position of kaempferol, whereas FGs of Kitakomachi were devoid of rhamnose.
Genetic analysis suggested that a single gene controls FG composition. SSR analysis, linkage mapping and genome database survey revealed the existence of a gene responsible for FG composition at a position similar to GMES0295 in the molecular linkage group O (chromosome 10). The genome sequence of Williams 82 suggested two candidate genes, Glyma10g33790 and Glyma10g33800, that were aligned in tandem in the vicinity of GMES0295. The genome sequence of Williams 82 and results of sequencing analysis suggested that Glyma10g33800 might be a pseudogene. We cloned a cDNA clone corresponding to Glyma10g33790 and designated it as GmF3G6″Rt. The coding region of GmF3G6″Rt is 1,392 bp long, encoding 464 amino acids in Koganejiro (GmF3G6″Rt-a). GmF3G6″Rt belongs to the 1,2/1,6 branch-forming glucosyltransferase gene cluster. It has an amino acid similarity of 56 % with the Rt gene of petunia encoding anthocyanin 3-O-glucoside (1 → 6) rhamnosyltransferase and 58 % similarity with flavanone 7-O-glucoside (1 → 6) rhamnosyltransferase of citrus (Frydman et al. 2013). Multiple alignment of flavonoid glycoside glycosyltransferases (GGTs, three G6″GTs and five G2″GTs) suggested 43 amino acids specific to G6″GTs and 4 amino acids specific to G2″GTs (Supplemental Fig. 1). There are characteristic amino acids in position 42 (V in G6″GTs and I in G2″GTs) and amino acid position 354 in the PSPG box (Y in G6″GTs and F in G2″GTs). Site-directed mutagenesis may reveal amino acids responsible for glycosylation of specific positions of the sugar moiety in flavonoids.
In contrast to Koganejiro, GmF3G6″Rt of Kitakomachi (GmF3G6″Rt-b) had a two-base deletion that generated a truncated polypeptide consisting of 314 amino acids. Detection of the two-base deletion by dCAPS analysis in Kitakomachi confirmed the nucleotide polymorphism. Banding patterns of the dCAPS marker co-segregated with FG patterns. RNA-Seq Atlas of soybean suggested that GmF3G6″Rt was primarily expressed in leaves. The transcript level in leaves of Kitakomachi was lower than Koganejiro, probably because of nonsense-mediated mRNA decay, a surveillance mechanism to eliminate aberrant mRNA transcripts that contain premature stop codons (Chang et al. 2007). Recombinant GmF3G6″Rt-a protein had a flavonol 3-O-glucoside (1 → 6) rhamnosyltransferase activity, indicating that GmF3G6″Rt-a encodes a functional flavonol 3-O-glucoside (1 → 6) rhamnosyltransferase; it presumably corresponds to the Fg2 gene. GmF3G6″Rt-a protein can utilize kaempferol 3-O-glucoside/3-O-galactoside as substrates. GmF3G6″Rt-b protein showed no activity probably because of the lack of a C-terminal region including the PSPG-box. These data are consistent with the flavonoid distribution pattern in Koganejiro and Kitakomachi. The GmF3G6″Rt had no intron, consistent with FG genes of other plant species, most of which have either none or one intron (Paquette et al. 2003).
Buzzell and Buttery (1992) reported a new allele for the Fg2 locus, resulting in a series of alleles, Fg2-a, Fg2-b and fg2. Leaves of Kitakomachi and Koganejiro contained FGs with α(1–2)-rhamnoside but lacked FGs with β(1–6)-glucoside or β(1–2)-glucoside suggesting that they might have the allelic combination of fg1 fg3 Fg4. With this genetic background, Fg2-a generates three TLC spots corresponding to rutinoside, 2G-rhamnosylrutinoside and glucoside, whereas Fg2-b generates six spots corresponding to gentiobioside, sophoroside and neohesperidoside in addition to the three spots produced by the Fg2-a allele (Buzzell and Buttery 1992). Leaves of Koganejiro contained rutinoside, 2G-rhamnosylrutinoside and glucoside, and lacked gentiobioside, sophoroside and neohesperidoside, suggesting that the allele of Koganejiro is Fg2-a.
Thus, either glucose or galactose was attached to the 3-position of kaempferol. This contrasts with an earlier report that only glucose was attached to the 3-position (Buttery and Buzzell 1975). Probably, their analytical method could not discriminate between glucose and galactose. Buzzell and Buttery (1974) reported another gene Fg4 encoding flavonol 3-O-glucoside (1 → 2) rhamnosyltransferase. In addition, FGs having rhamnose at the 4″-position of 3-O-galactose have been identified in soybean leaves (Murai et al. 2013), suggesting the existence of flavonol 3-O-glucoside (1 → 4) rhamnosyltransferase. Accordingly, the schematic diagram on the genetic control of FG biosynthesis proposed by Buzzell and Buttery (1974) should be revised. Overall, glucose or galactose is attached to the 3-position of kaempferol or quercetin in the biosynthesis of FGs in soybean leaves. To the glucose or galactose, glucose can be attached to 2″ or 6″-positions and rhamnose can be attached to 2″, 4″ or 6″-positions, resulting in a wide variety of FGs. Currently, there is no evidence that GmF3G6″Rt affects visible phenotypes or agronomic characters. Development of near-isogenic lines may be necessary to obtain information on the effect of GmF3G6″Rt in relation to morphology, productivity and plant fitness in soybean.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Buttery BR, Buzzell RI (1973) Varietal differences in leaf flavonoids of soybeans. Crop Sci 13:103–106
Buttery BR, Buzzell RI (1975) Soybean flavonol glycosides: identification and biochemical genetics. Can J Bot 53:219–224
Buttery BR, Buzzell RI (1976) Flavonol glycoside genes and photosynthesis in soybean. Crop Sci 16:547–550
Buzzell RI (1974) Soybean linkage tests. Soybean Genet Newsl 1:11–14
Buzzell RI, Buttery BR (1974) Flavonol glycoside genes in soybeans. Can J Genet Cytol 16:897–899
Buzzell RI, Buttery BR (1977) Inheritance and linkage of a magenta flower gene in soybeans. Can J Genet Cytol 19:749–751
Buzzell RI, Buttery BR (1992) Inheritance of an anomalous flavonol glycoside gene in soybean. Genome 35:636–638
Buzzell RI, Buttery BR (1998) Genetics of leaf waviness in soybean. Soybean Genet Newsl 25:23–24
Chang YF, Imam JS, Wilkinson MF (2007) The nonsense-mediated decay RNA surveillance pathway. Ann Rev Biochem 76:51–74
Cregan PB, Jarvic T, Bush AL, Shoemaker RC, Lark KG, Kahler AL, Kaya N, VanToai TT, Lohnes DG, Chung J, Specht JE (1999) An integrated genetic map of the soybean genome. Crop Sci 39:1464–1490
Fehr WR, Caviness CE, Burmood DT, Pennington JS (1971) Stage of development descriptions for soybeans, Glucine max (L.) Merrill. Crop Sci 11:929–931
Frydman A, Liberman R, Huhman DV, Carnmeli-Weissberg M, Sapir-Mir M, Ophir R, Sumner LW, Eyal Y (2013) The molecular and enzymatic basis of bitter/non-biter flavor of citrus fruit: evolution of branch-forming rhamnosyltransferases under domestication. Plant J 73:166–178
Githiri SM, Yang D, Khan NA, Xu D, Komatsuda T, Takahashi R (2007) QTL analysis of low temperature-induced browning in soybean seed coats. J Hered 98:360–366
Hisano H, Sato S, Isobe S, Sasamoto S, Wada T, Matsuno A, Fujishiro T, Yamada M, Nakayama S, Nakamura Y, Watanabe S, Harada K, Tabata S (2007) Characterization of the soybean genome using EST-derived microsatellite markers. DNA Res 14:271–281
Iwashina T, Benitez ER, Takahashi R (2006a) Analysis of flavonoids in pubescence of soybean near-isogenic lines for pubescence color loci. J Hered 97:438–443
Iwashina T, Kitajima J, Matsumoto S (2006b) Flavonoids in the species of Cyrtomium (Dryopteridaceae) and related genera. Biochem Syst Ecol 34:14–24
Iwashina T, Githiri SM, Benitez ER, Takemura T, Kitajima J, Takahashi R (2007) Analysis of flavonoids in flower petals of soybean near-isogenic lines for flower and pubescence color genes. J Hered 98:250–257
Iwashina T, Oyoo ME, Khan NA, Matsumura H, Takahashi R (2008) Analysis of flavonoids in flower petals of soybean flower color variants. Crop Sci 48:1918–1924
Iwashina T, Yamaguchi M, Nakayama M, Onozaki T, Yoshida H, Kawanobu S, Ono H, Okamura M (2010) Kaempferol glycosides in the flowers of carnation and their contribution to the creamy white flower color. Nat Prod Commun 5:1903–1906
Iwata H, Ninomiya S (2006) AntMap: constructing genetic linkage maps using an ant colony optimization algorithm. Breed Sci 56:371–377
Kroon J, Souer E, de Graaff A, Xue Y, Mol J, Koes R (1994) Cloning and structural analysis of the anthocyanin pigmentation locus Rt of Petunia hybrida: characterization of the insertion sequences in two mutant alleles. Plant J 5:69–80
Lairson LL, Henrissat B, Davies GJ, Withers SG (2008) Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem 77:521–555
Mackenzie PI, Owens IS, Burchell B, Bock KW, Bairoch A, Belanger A, Fournel-Gigleux S, Green M, Hum DW, Iyanagi T, Lancet D, Louisot P, Magdalou J, Chowdhury JR, Ritter JK, Schachter H, Tephly TR, Tipton KF, Nebert DW (1997) The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence. Pharmacogenetics 7:255–269
Masada S, Terasawa K, Mizukami H (2007) A single amino acid in the PSPG-box plays an important role in the catalytic function of CaUGT2 (Curcumin glucosyltransferase), a Group D Family 1 glucosyltransferase from Catharanthus roseus. FEBS Lett 581:2605–2610
Murai Y, Takahashi R, Rojas Rodas F, Kitajima J, Iwashina T (2013) New flavonol triglycosides from the leaves of soybean cultivars. Nat Prod Commun 8:453–456
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Page RDM (1996) TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358
Paquette S, Møller BL, Bak S (2003) On the origin of family 1 plant glycosyltransferases. Phytochemistry 62:399–413
Rice-Evans CA, Miller NJ, Bolwell PG, Bramley PM, Pridham JB (1995) The relative antioxidant activities of plant-derived polyphenolic flavonoids. Free Radic Res 22:375–383
Rode MW, Bernard RL (1975) Inheritance of wavy leaf. Soybean Genet Newsl 2:42–44
Severin AJ, Woody JL, Bolon YT, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, Graham MA, Cannon SB, May GD, Vance CP, Shoemaker RC (2010) RNA-Seq Atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol 10:160
Shah DM, Hightower RC, Meagher RB (1983) Genes encoding actin in higher plants: intron positions are highly conserved but the coding sequences are not. J Mol Appl Genet 2:111–126
Song QJ, Marek LF, Shoemaker RC, Lark KG, Concibido VC, Delannay X, Specht JE, Cregan PB (2004) A new integrated genetic linkage map of the soybean. Theor Appl Genet 109:122–128
Sunada K, Ito T (1982) Soybean grain quality as affected by low temperature treatments in plants (color of hilum, seed coat cracking) (in Japanese). Rep Hokkaido Branch, Crop Sci Soc Jpn and Hokkaido Branch. Jpn Soc Breed 22:34
Takahashi R (1997) Association of soybean genes I and T with low-temperature induced seed coat deterioration. Crop Sci 37:1755–1759
Takahashi R, Asanuma S (1996) Association of T gene with chilling tolerance in soybean. Crop Sci 36:559–562
Takahashi R, Benitez ER, Funatsuki H, Ohnishi S (2005) Soybean maturity and pubescence color genes improve chilling tolerance. Crop Sci 45:1387–1393
Takahashi R, Githiri SM, Hatayama K, Dubouzet EG, Shimada N, Aoki T, Ayabe S, Toda K, Matsumura H (2007) A single-base deletion in soybean flavonol synthase gene is associated with magenta flower color. Plant Mol Biol 63:125–135
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony method. Mol Biol Evol 28:2731–2739
Toda K, Yang D, Yamanaka N, Watanabe S, Harada K, Takahashi R (2002) A single-base deletion in soybean flavonoid 3′-hydroxylase gene is associated with gray pubescence color. Plant Mol Biol 50:187–196
Toda K, Takahashi R, Iwashina T, Hajika M (2011) Difference in chilling-induced flavonoid profiles, antioxidant activity and chilling tolerance between soybean near-isogenic lines for pubescence color gene. J Plant Res 124:173–182
Toda K, Kuroiwa H, Senthil K, Shimada N, Aoki T, Ayabe S, Shimada S, Sakuta M, Miyazaki Y, Takahashi R (2012) The soybean F3′H protein is localized to the tonoplast in the seed coat hilum. Planta 236:79–89
Vinson JA, Dabbagh YA, Serry MM, Jang JH (1995) Plant flavonoids, especially tea flavones, are powerful antioxidants using an in vitro oxidation model for heart disease. J Agric Food Chem 43:2800–2802
Yonekura-Sakakibara K, Hanada K (2011) An evolutionary view of functional diversity in family 1 glycosyltransferases. Plant J 66:182–193
Yonekura-Sakakibara K, Tohge T, Niida R, Saito K (2007) Identification of a flavonol 7-O-rhamnosyltransferase gene determining flavonoid pattern in Arabidopsis by transcriptome coexpression analysis and reverse genetics. J Biol Chem 282:14932–14941
Acknowledgments
The authors are grateful to Dr. Joseph G. Dubouzet (Scion Research, New Zealand) for critical reading of the manuscript, and to Mr. Tetsuya Mori (RIKEN CSRS, Japan) for his technical support. This study was partially supported by the Japanese Government (MEXT) Scholarship (to F.R.R. and T.O.R) and the JSPS KAKENHI Grant Number 25440148 (to K.Y.-S.).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11103_2013_133_MOESM1_ESM.eps
Multiple alignment of flavonoid glycoside glycosyltransferases (GGTs). The PSPG motif is within the box. Amino acid residues conserved in flavonoid GGTs are highlighted in black. Amino acid residues conserved in flavonoid G6″GTs and G2″GTs are highlighted in red and blue, respectively. (EPS 1059 kb)
Rights and permissions
About this article
Cite this article
Rojas Rodas, F., Rodriguez, T.O., Murai, Y. et al. Linkage mapping, molecular cloning and functional analysis of soybean gene Fg2 encoding flavonol 3-O-glucoside (1 → 6) rhamnosyltransferase. Plant Mol Biol 84, 287–300 (2014). https://doi.org/10.1007/s11103-013-0133-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-013-0133-1