
Abstract
In this review, we focus on the interconnection of inorganic phosphate (Pi) homeostasis in the network of the bone–kidney, parathyroid–kidney, intestine–kidney, and liver–kidney axes. Such a network of organ communication is important for body Pi homeostasis. Normalization of serum Pi levels is a clinical target in patients with chronic kidney disease (CKD). Particularly, disorders of the fibroblast growth factor 23/klotho system are observed in early CKD. Identification of phosphaturic factors from the intestine and liver may enhance our understanding of body Pi homeostasis and Pi metabolism disturbances in CKD patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Inorganic phosphate (Pi) is essential for several biologic functions, such as intracellular signal transduction, energy exchange, production and function of cell membranes, and the composition of hydroxyapatite in the bones and teeth [1, 2]. Serum Pi levels are maintained within the normal range by a number of regulatory hormones. These modulate the intestinal uptake of Pi, its mobilization from bone, and renal excretion. Maintaining serum Pi levels is critical for proper bone development and for skeletal integrity [3]. Hypophosphatemia leads to bone abnormalities such as rickets/osteomalacia, whereas hyperphosphatemia contributes to vascular calcification in patients with chronic kidney disease (CKD) and hemodialysis and is independently associated with cardiac mortality [1, 2, 4]. The kidney is the major regulator of Pi homeostasis. Physiological and pathophysiological regulation of renal epithelial transport of Pi occurs through alterations in the levels of type II sodium-dependent Pi (Na/Pi) cotransporters (SLC34 family). Recent studies demonstrated that Pi reabsorption is regulated through orchestration of the kidney–organ axis, including the bone, intestine, parathyroid, and liver. The factors that regulate renal Na/Pi transporters are dietary Pi, parathyroid hormone (PTH), calcitriol, or 1,25-dihydroxyvitamin D3 (the active form of vitamin D, 1,25(OH)2D3), fibroblast growth factor 23 (FGF23), and pyridine nucleotides [2, 5–8]. In this review, we focus on the regulation of renal NaPi-II transporters by inter-organ communication.
Renal Pi reabsorption
A major regulator of Pi homeostasis is the kidney, and its Pi re-absorptive capacity can be increased or decreased to accommodate the requirements for Pi. Na+-dependent Pi (Na/Pi) transport systems in the brush-border membrane (BBM) mediate the rate-limiting step in the overall Pi re-absorptive process [1, 8]. Pi ions influx from the tubular lumen across the apical BBM and efflux at the basolateral membrane [1, 8]. Na+-dependent transport maintained by basolateral membrane-associated Na+, K+-ATPase drives the Na+-gradient process. Na/Pi cotransport across the BBM is the target for physiologic/pathophysiologic regulation [8].
Three types of Na/Pi cotransporters (types I–III) are located in the apical membrane of renal proximal tubular cells [1, 8]. Type I Na/Pi (SLC17A1/NPT1/NaPi-I/OATv1) is expressed in the liver and the kidney [1]. NPT1 was identified in an expression cloning study using Xenopus laevis oocytes based on the Na/Pi transport, and it was also localized to the apical side of the proximal tubules [1]. Following in vitro studies indicated that this transporter is involved in the proximal tubule transport of organic anions rather than Pi. Recently, Iharada et al. demonstrated that NPT1 mediates urate export into urine [9]. The role of NPT1 in Pi reabsorption remains unclear [1].
The SLC34 family, SLC34A1 (NaPi-IIa), and SLC34A3 (NaPi-IIc), are major functional transporters in the proximal tubular cells [1, 8]. While the overall molecular structure is predicted to be very similar, there are important differences between the two Na/Pi cotransporters. NaPi-IIa is electrogenic, coupling Pi transport (at physiologic pH mainly HPO42−) with the transport of three Na+ ions. In contrast, NaPi-IIc is electroneutral, only transporting two Na+ ions for every Pi. In addition, type III transporters (the SLC20A20 family, PiT2) are localized at the BBM of the proximal tubule cells [10–12].
Little is known about the molecules involved in Pi translocation across the cell membrane and the efflux of Pi across the basolateral membrane. The Pi exporter, xenotropic and polytropic retrovirus receptor 1 (XPR1), was recently identified [13–16]. XPR1 is a highly conserved multi-pass transmembrane protein originally identified as a receptor for xenotropic and polytropic murine retroviruses [13, 14]. Recent studies indicate that XPR1 has a specific role as a Pi exporter in the differentiation of tissue macrophages [15, 16]. XPR1 mutants show impaired bone remodeling that is indicative of osteoclast defects [14]. We previously reported that osteoclasts have a unique Pi efflux system [17] that is involved in the continuous release of Pi at the basolateral membrane to prevent the accumulation of intracellular Pi [17]. Giovannini et al. demonstrated that XPR1 mediates Pi export in cultured mammalian cells [18].
XPR1 mutations are reported to cause primary familial brain calcification [19]. These mutations alter phosphate export, implicating XPR1 in primary familial brain calcification [19]. Thus, XPR1 might be involved in the export of Pi from mammalian cells, but the role of this exporter in renal epithelial cells remains unknown.
A role for type II Na/Pi transporters (NaPi-IIa and NaPi-IIc or Npt2a and Npt2c) on renal Pi reabsorption is reported in rodents and humans [1]. In rodents, NaPi-IIa is important for renal Pi cotransport activity and Npt2a (NaPi-IIa)-knockout (Npt2a−/−) mice have hypophosphatemia and hyperphosphaturia [20]. Serum 1,25(OH)2D concentrations and serum and urine Ca2+ concentrations are significantly increased in Npt2a−/− mice [1, 20, 21]. Npt2a−/− mice exhibit increased urinary Pi excretion, ~70 % decrease in renal BBM vesicle Na/Pi cotransport, and hypophosphatemia [20]. Npt2a−/− mice also over-express Npt2c, which may support the residual renal Pi reabsorption function. These observations indicate that Npt2a has a major role in Pi reabsorption in mice.
In humans, several NaPi-IIa mutations have been reported. Prie et al. reported two patients with NaPi-IIa mutations (A48F and V147M) exhibiting urolithiasis or bone demineralization and persistent idiopathic hypophosphatemia with lower maximal renal Pi reabsorption [22]. Virkki et al., however, demonstrated that reduced Pi transport activity by the NaPi-IIa mutations cannot fully explain the massive phosphaturia observed [23]. In addition, mutation of human NaPi-IIa causes autosomal recessive Fanconi syndrome with hypophosphatemic rickets [24]. This mutation of NaPi-IIa is a homozygous in-frame duplication. Functional studies indicate a complete loss-of-function of mutant NaPi-IIa [24]. Accumulation of mutant NaPi-IIa protein in the cells may have toxic effects leading to Fanconi syndrome. Genetic analysis detected a homozygous in-frame duplication, leading to the insertion of seven additional amino acids [24, 25]. Heterozygous carriers of the mutation are clinically normal, suggesting that the mutation does not have a dominant negative effect [24, 25]. Fanconi syndrome is detected in the human carriers of the mutation, but not in Npt2a−/− mice. Thus, the disruption of human NaPi-IIa impairs overall renal Pi reabsorption, providing evidence for the critical role of NaPi-IIa in human renal Pi handling. The precise role of the NaPi-IIa transporter in humans, however, remains unknown.
In contrast, NaPi-IIc (Npt2c) may be a major functional Na/Pi cotransporter in the human kidney because the NaPi-IIc mutation causes hereditary hypophosphatemic rickets with hypercalciuria (HHRH) [26–29]. Clinical studies suggest that HHRH is a primary renal Pi wasting disorder, resulting in increased serum 1,25(OH)2D concentrations with associated intestinal Ca2+ hyperabsorption, hypercalciuria, and rickets/osteomalacia [1, 30]. Functional studies suggest that homozygous or compound heterozygous mutations of NaPi-IIc significantly decrease Na/Pi transport activity in Xenopus oocytes and OK cells [31]. In rodents, NaPi-IIc (Npt2c) is important for Pi reabsorption in weanling animals [32], but mediates a very small percentage of Pi reabsorption in adult animals [32]. Npt2c knockout mice (Npt2c−/−) mice exhibit hypercalciuria and higher levels of serum 1,25(OH)2D concentrations, but not hypophosphatemia, rickets, or nephrocalcinosis [33]. Furthermore, only Npt2a/Npt2c double-KO mice exhibit a physiology similar to that of patients with HHRH [34]. NaPi-IIc (Npt2c) has a minor role in Pi reabsorption in mice [1, 21, 33]. More recently, Myakala et al. showed that renal-specific and inducible depletion of NaPi-IIc in mice does not affect Pi or Ca homeostasis [35]. Neither Npt2a−/− nor Npt2c−/− mice exhibit bone abnormalities such as rickets/osteomalacia [20, 33]. Based on these findings, we suggest that human NaPi-IIc is more important than mouse NaPi-IIc for Pi homeostasis.
Finally, the marked differences in the phenotypes of Npt2a−/− and Npt2c−/− mice may be due to differential dominance of the transporters for Pi homeostasis [1, 33]. The role of NaPi-IIa (Npt2a) and NaPi-IIc (Npt2c) in renal Pi reabsorption differs between rodents and human. It remains unclear, however, why regulation of NaPi-IIa does not compensate for the loss-of-function mutation of NaPi-IIc in HHRH as it does in the absence of NaPi-IIc, such as in null mice.
Intestinal Pi absorption
The mechanisms and regulation of intestinal Pi absorption remain poorly defined. Intestinal absorption of Pi is characterized in several mammalian and avian species [36–40]. In intestinal Pi absorption, both saturated and unsaturated systems have been described. Unsaturated systems depend on the paracellular route via tight junctions. Saturated components are Na+-dependent and Na+-independent Pi transport systems in the apical membrane of intestinal epithelial cells [41]. Marks et al. demonstrated that Pi concentrations in the intestinal lumen are typically in the low millimolar range, and under these conditions Na+-independent transport is likely to be the predominant pathway for Pi absorption in vivo [42, 43]. Based on studies using BBM vesicles of the small intestine, both Na+-dependent and Na+-independent components are present, with similar characteristics and pH dependence [44]. Candeal et al. characterized Na+-independent components in the human intestinal cell line Caco2BBE [45]. Na+-independent components are dependent on de novo RNA and protein synthesis [45]. The molecules involved in Na+-independent Pi transport, however, remain unknown.
Na/Pi absorption is mediated primarily via the type IIb sodium-phosphate transporter (NaPi-IIb, Npt2b) [41, 46–52]. Numerous studies revealed that the regional profile for intestinal phosphate absorption differs between rats and mice. In rats, the highest rates of transport occur in the duodenum and jejunum, while in mice maximal absorption occurs in the ileum. During fasting and low dietary phosphate intake, Pi absorption may be mediated by NaPi-IIb, but when Pi levels are elevated post-prandially, transport could also occur via a Na+-independent transcellular or paracellular pathway. The type III Na/Pi cotransporter PiT-1 is expressed in the apical membrane of enterocytes. The role of PiT-1 in Na/Pi cotransport remains unknown.
Mutation of human NaPi-IIb causes pulmonary alveolar microlithiasis [53]. Deposition of Ca/Pi microliths throughout the lungs is observed in patients with pulmonary alveolar microlithiasis. NaPi-IIb is specifically expressed in type II alveolar cells, and NaPi-IIb mutations abolish normal gene function. Homozygous NaPi-IIb KO mice are embryonic lethal. Based on several studies of heterozygous NaPi-IIb KO (Npt2b+/−) mice and conditional KO mice [1, 54–58], NaPi-IIb is the most important transcellular Na/Pi transporter. The roles Pi transporters in the small intestine, however, remain unknown. Particularly, Na+-independent Pi transporters require further characterization. The role of intestinal NaPi-IIb in Pi homeostasis is clearly more important than previously realized in mice.
Regulation of renal NaPi-IIa and NaPi-IIc by inter-organ communication
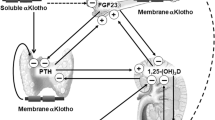
Parathyroid glands secrete PTH into the blood. PTH induces phosphaturia by decreasing Pi reabsorption in the kidney [5, 59]. 1,25(OH)2D3, which is synthesized in the kidney, acts in the gut to increase the absorption of dietary Pi and calcium, and in the bone to promote mobilization of these ions. As a result, blood levels of both Pi and calcium tend to increase. In addition to PTH and 1,25(OH)2D3, recent studies identified a novel regulator of Pi levels: fibroblast growth factor (FGF23) [2, 6]. This bone-derived hormone is currently considered to be the principal regulator of phosphaturia. FGF23 induces Pi excretion and inhibits vitamin D synthesis in the kidney, thus maintaining systemic Pi homeostasis [2, 6]. The parathyroid–kidney and bone–kidney axis are involved in two known phosphaturic factors, PTH and FGF23. In addition, recent studies demonstrated that the intestine and liver also contribute to systemic Pi homeostasis [2, 6]. For example, the control of renal Pi excretion by dietary Pi may involve the intestine–kidney axis. Nicotinamide is controlled by systemic Pi homeostasis. The liver–kidney axis is involved in nicotinamide metabolism. Below we describe the kidney–organ axis in Pi homeostasis (Fig. 1).

Regulation of renal reabsorption by the inter-organ communication. a Bone–kidney axis: FGF23-dependent downregulation of NaPi-IIa is associated with MAPK-dependent NHERF1 phosphorylation and endocytosis into cellular compartments. b Parathyroid–kidney axis: PTH-dependent activation of either PKC or PKA in the renal proximal tubule cells inhibits Na+-dependent Pi transport activity. c Intestine–kidney axis: Intestinal phosphatonin secretion induced by a high Pi diet may be involved in the downregulation of renal NaPi-IIa, NaPi-IIc transporters. d Liver–kidney axis: Liver phosphatonin secretion induced by hepatectomy activates renal Nampt function and downregulates NaPi-IIa, NaPi-IIc transporters
Bone–kidney axis
FGF23 is a hormone that promotes renal Pi excretion by decreasing its reabsorption in the proximal tubules while concurrently reducing serum 1,25(OH)2D3 by decreasing its biosynthesis and increasing its metabolism [60]. FGF23 requires an additional cofactor, klotho, to bind with high affinity and signal efficiently through its cognate FGF receptor. Klotho and FGF receptor 1 (IIIc) form a heterodimeric receptor for FGF23 [61, 62]. We previously reported that administration of FGF23 containing the R179Q mutation (FGF23M) decreased the levels of Na/Pi transport activity and NaPi-IIa and NaPi-IIc expression [63, 64]. FGFR1 is the predominant receptor mediating the hypophosphatemic actions of FGF23 by decreasing BBM levels of NaPi-IIa and NaPi-IIc expression with FGFR4, which has an additional but relatively minor role [65]. FGF23 regulates proximal tubular 1,25(OH)2D3 biosynthesis by a different receptor than that for the inhibition of Pi transport reuptake in the proximal tubule [66–68]. The mechanisms of FGF23 downregulation of NaPi-IIa are well studied include MAPK-dependent NHERF1 phosphorylation and endocytosis into cellular compartments [69–71].
The downregulation of NaPi-IIa by FGF23 is dependent on the presence of klotho, which is mainly expressed in the distal tubules. In the distal tubule-specific depletion of klotho–conditional KO mice, downregulation of NaPi-IIa is not observed after injection of FGF23. These findings suggest that complex FGF23/klotho/FGFR in the distal tubules is essential for the reduction of NaPi-IIa transporters [72].
We previously reported that administration of FGF23 containing the R179Q mutation (FGF23M) decreased the levels of Na/Pi transport activity and NaPi-IIc expression in Npt2a−/− [73]. We used Npt2a−/− mice and analyzed the localization and protein levels of Npt2c [73]. As described previously, the levels of Npt2c protein are increased in the proximal tubules in Npt2a−/− mice [73]. In Npt2a−/− mice, FGF23 treatment led to a reduction of renal BBM Na/Pi cotransport activities at 3 and 12 h after injection (H. Segawa et al., unpublished data). Urinary Pi excretion was significantly increased at 3 and 12 h. After FGF23 treatment, at 3 h, the amounts of NaPi-IIc protein were not changed when compared with non-injected controls. The effect of FGF23 on the rapid reduction of Na/Pi cotransport activity may be mediated by the phosphorylation-dependent inactivation of NaPi-IIc protein, but not NaPi-IIc protein levels. These data suggest that FGF23-dependent phosphaturic actions in Npt2a−/− mice are associated with NaPi-IIc inactivation in the apical membrane. Finally, the difference in the PTH regulation between NaPi-IIa and NaPi-IIc could affect the time scale of the downregulation by PTH. The downregulation of NaPi-IIa by PTH occurs within 1 h, while NaPi-IIc regulation takes up to four times longer (H. Segawa et al., unpublished data).
Parathyroid–kidney axis
PTH decreases the levels of NaPi-IIa and NaPi-IIc protein in the BBM [2, 6, 36, 74]. In response to PTH, NaPi-IIa undergoes clathrin-dependent endocytosis and is targeted to lysosomes [75]. Thus, PTH inhibits Pi transport by promoting Npt2a endocytosis and lysosomal degradation [5, 6, 74–76]. PTH receptors are expressed on both the apical and basolateral surfaces of the renal proximal tubular cells. The apical PTH1 receptor is signaled via a protein kinase C (PKC) pathway while the basolateral PTH1 receptor is signaled via a protein kinase A (PKA) pathway [77]. Previous studies suggest that PTH increases the phosphorylation of Na+/H+ exchanger regulatory factor-1 (NHERF-1) [59, 78–80]. Weinman et al. demonstrated that biochemical modification of the serine77 of NHERF-1 decreases its binding affinity to NaPi-IIa, resulting in dissociation of the NaPi-IIa/NHERF-1 complex, and that this modification is required for PTH-dependent inhibition of Na/Pi transport [59, 79, 80]. They also indicated an important role for threonine 95 of the PDZ1 domain of NHERF-1 [80]. These data suggest that the phosphorylation of threonine 95 and the phosphorylation of serine 77 of NHERF-1 are essential for PTH-mediated inhibition of Pi transport [80]. In addition, NHERF1-KO mice exhibit PTH-resistant urinary Pi wasting, nephrocalcinosis, and osteopenia [81]. Thus, NHERF-1 is involved in the PTH regulation of NaPi-IIa endocytosis.
By contrast, in the regulation of PTH, NaPi-IIc is downregulated through a microtubule-dependent pathway that does not involve lysosomal degradation [74, 82]. After administration of PTH, the intensity of immunoreactive signals in apical and subapical type IIc transporters decrease in the renal proximal tubular cells in thyroparathyroidectomized rats [82]. Colchicine completely blocks the internalization of NaPi-IIc transporters [82]. In addition, leupeptin prevents PTH-mediated degradation of the NaPi-IIa transporter in lysosomes, but has no effect on PTH-mediated degradation of the lysosomal NaPi-IIc transporters [82]. The precise pathway for the decrease in the apical membrane expression of NaPi-IIc has not been determined, but recent evidence suggests that the protein is shifted to the base of the microvillar compartment where it undergoes in situ dissolution. Villa-Bellosta et al. reported that NaPi-IIc interacts with NHERF-3 in the apical membrane in OK cells and proximal tubules [83]. In NHERF-3-KO mice, NaPi-IIc translocation into the apical membrane is impaired after stimulation by a low Pi diet [84]. NaPi-IIc may bind indirectly to ezrin. The PDZ domain-containing protein NHERF-3, as an intermediate linking NaPi-IIc to ezrin, is necessary for many aspects of NaPi-IIc regulation. In addition, internalization of NaPi-IIc might be involved in Rab11-dependent recycling endosomes in the proximal tubular cells (H. Segawa et al., data not shown).
Intestine–kidney axis
Dietary Pi is the most important factor in body Pi homeostasis. Dietary Pi levels control active vitamin D synthesis, PTH secretion, and renal Pi reabsorption [36–40, 85]. Renal Pi reabsorption is a key determinant of serum Pi levels in the body [36]. A low-Pi diet can lead to almost 100 % renal reabsorption of filtered Pi, whereas a high-Pi diet leads to decreased proximal tubular Pi reabsorption. The factors controlling the dietary adaptive system are not known, but do not include PTH, vitamin D, growth hormone, thyroid hormone, calcitonin, or FGF23 [36, 85]. Several reports indicate that intestinal mucosal cells sense the dietary Pi content and secrete intestinal phosphatonin (a putative phosphaturic factor) [36]. Intestinal phosphaturic factors might be involved in the rapid regulation of renal type II transporters by the intestinal–kidney axis [86]. Burnts et al. showed that intraduodenal infusion of 1.2 M sodium phosphate, but not sodium chloride, increases Pi excretion within 20 min [86]. The response is not due to altered serum levels of Pi, PTH, FGF23, secreted frizzled-related protein 4; an increase in glomerular filtration rate; or the result of a neural reflex [86]. Burnts et al. concluded that an intestinal Pi sensor triggers the release of a phosphaturic factor from the duodenal mucosa [86]. Marks et al. demonstrated that matrix extracellular phosphoglycoprotein might be the intestinal phosphatonin proposed by Berndt et al. [87]. To date, however, there is no further published information on the mechanisms underlying this proposed entero-renal reflex or the identity of the putative phosphatonin [86, 88]. An intestinal Pi sensor may be involved in the secretion of FGF23 from the bone and also the regulation of the 1,25(OH)2D3 levels.
We also characterized Pi homeostasis in Npt2b+/− mice. Npt2b+/− mice have significantly reduced intestinal Na/Pi cotransport activity in their BBMVs [56]. At 4 weeks of age, the Npt2b+/− mice showed hypophosphatemia and low urinary Pi excretion [56]. Serum FGF23 levels were significantly reduced and 1,25(OH)2D3 levels were significantly increased in the Npt2b+/− mice compared with those in the Npt2b+/+ mice, and their Npt2b mRNA levels were reduced to 50 % that in the Npt2b+/+ mice [56]. In contrast, renal Npt2a and Npt2c transporter protein levels were significantly increased in the Npt2b+/− mice [56]. At 20 weeks of age, the Npt2b+/− mice showed hypophosphaturia and reduced Na/Pi cotransport activity in the distal intestine [56]. Recent reports revealed the importance of Npt2b in both acute and chronic adaptation of intestinal Pi transport, and FGF23 secretion [89]. It is possible that Npt2b is involved in the secretion of an intestinal phosphatonin mediated by dietary Pi. Further studies are needed to clarify the regulation of renal Pi excretion by the intestinal–kidney axis.
Liver–kidney axis
The number of patients receiving liver transplantation has steadily increased, and thus the incidence of partial hepatectomy (PH) has also increased [90]. Hypophosphatemia frequently occurs after liver resection [91–93]. Acute hypophosphatemia causes septicemia and is associated with a poor prognosis [93, 94]. The phenomenon is highly clinically relevant because numerous patients develop significant hypophosphatemia after hepatectomy requiring large doses of Pi replacement to maintain metabolic homeostasis [91–93]. In many patients, urinary Pi excretion is markedly increased [95]. Post-hepatectomy hypophosphatemia is associated with hyperphosphaturia [96]. For a long time, the increased metabolic demands by the regenerating liver were viewed as the underlying pathologic mechanism of hypophosphatemia [93]. The magnitude of Pi uptake by the recovering liver, however, cannot explain the severity of hypophosphatemia. Post-hepatectomy hypophosphatemia is associated with increased FePi unrelated to intact FGF23, FGF7, or secreted frizzled-related protein 4 as phosphaturic factors [97]. Therefore, other factors must have a role in the pathogenesis of hypophosphatemia. These observations suggest that unknown factors are involved in the control of renal Pi in the liver–kidney axis.
Nicotinamide (an amide derivative of the water-soluble vitamin B3) is a potentially interesting alternative to phosphate binders. In vitro and in vivo data show that nicotinamide reduces hyperphosphatemia by inhibiting Na/Pi cotransport in the renal proximal tubules and the intestine [98–100]. Several recent clinical studies explored the potential value of nicotinamide in Pi control (as well as its effects on lipid) [101, 102]. How nicotinamide regulates systemic Pi homeostasis, however, remains unknown. We investigated the mechanisms for the reduction of NaPi-II (Npt2a, Npt2b, Npt2c) protein caused by abnormal cellular NAD metabolism. Nicotinamide phosphoribosyltransferase (Nampt) is a rate-limiting enzyme for NAD+ synthesis from nicotinamide [103–105]. Elevation of Nampt protein is a candidate cause of hyperphosphaturia in PH animals [106]. Abnormal NAD metabolism is also a candidate cause of hyperphosphaturia with PH [106]. Nicotinamide inhibits intestinal and renal Na/Pi transport activity in normal rats [99]. Injections of pharmacologically relevant doses of nicotinamide increase NAD in inverse proportion to Na/Pi transport [98]. Indeed, we demonstrated that NaPi-IIa, IIb, and IIc protein levels in the kidney and small intestine are markedly decreased in PH animals, suggesting that the reduction of the transporters in PH animals is similar to that in nicotinamide-treated animals [106]. Thus, we demonstrated the possibility that hypophosphatemia and hyperphosphaturia are observed after 70 % hepatectomy due to abnormality of NAD/nicotinamide metabolism in the liver and kidney [106]. In addition, the Nampt inhibitor FK866 influences Pi metabolism in normal mice [106]. The expression of NaPi-IIa (Npt2a) and NaPi-IIc (Npt2c) is increased in the proximal tubules by FK866 treatment. Thus, the mechanism for the increase in Nampt following hepatectomy remains unknown, but nicotinamide may be metabolized in the liver–kidney axis and may be involved in the regulation of Nampt protein in the proximal tubules [106]. A putative phosphatonin in the liver may be involved in regulation of the renal Nampt activity and NAD metabolism in the mitochondria of proximal tubular cells (S. Tatsumi et al., data not shown).
CKD and inter-organ communication
Pi retention is a major harmful complication of CKD [107–109], leading to secondary hyperparathyroidism, uremic bone disease, and progression to end-stage renal disease. Hyperphosphatemia remains a risk factor for arterial calcification contributing to high cardiovascular mortality in patients with CKD [110, 111]. Tubular reabsorption of Pi decreases in proportion to the severity of CKD. As glomerular filtration rate diminishes, the degree of Pi excretion per nephron increases, and at a very low filtration rate levels tubular reabsorption of Pi is 10 to 20 %, or 80 to 90 % of the filtered load of Pi is excreted into the urine. In the early stages of CKD, the serum Pi concentration is maintained within the normal range by increases in the serum concentration of PTH and FGF23 [112–114], which serve to increase the urinary excretion of Pi, and decrease serum 1,25(OH)2D3 concentrations, leading to decreased Pi absorption from the gastrointestinal tract. In more advanced stages of kidney disease, changes in the PTH, FGF23, and 1,25(OH)2D3 levels are no longer sufficient to prevent hyperphosphatemia [115]. In CKD, increased PTH and FGF23 production enhances the excretion of Pi per nephron, thereby restoring normophosphatemia [116]. In CKD patients, however, FGF23 reduces 1,25(OH)2D3 levels, contributing to an increase in PTH secretion, which occurs after FGF23 levels increase [116]. This process disrupts the bone–kidney–parathyroid endocrine axis and eventually fails to prevent the development of hyperphosphatemia as CKD progresses.
In addition, Nampt is a proinflammatory cytokine that has gained considerable attention in recent years with respect to induction of cardiovascular disease [117]. The Nampt pathway (liver–kidney axis) is also activated in CKD and diabetic nephropathy animals [96, 97]. These observations suggest that the bone–parathyroid–kidney and the liver–kidney axes may be activated to maintain Pi homeostasis in CKD. A new pathway of Pi metabolism in the liver–kidney axis may elucidate the abnormal Pi metabolism in CKD.
References
Miyamoto K, Haito-Sugino S, Kuwahara S, Ohi A, Nomura K, Ito M, Kuwahata M, Kido S, Tatsumi S, Kaneko I, Segawa H (2011) Sodium-dependent phosphate cotransporters: lessons from gene knockout and mutation studies. J Pharm Sci 100:3719–3730
Berndt T, Kumar R (2009) Novel mechanisms in the regulation of phosphorus homeostasis. Physiology (Bethesda) 24:17–25
Farrow EG, White KE (2010) Recent advances in renal phosphate handling. Nat Rev Nephrol 6:207–217
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM (2004) Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 15:2208–2218
Weinman EJ, Lederer ED (2012) PTH-mediated inhibition of the renal transport of phosphate. Exp Cell Res 318:1027–1032
Tenenhouse HS (2007) Phosphate transport: molecular basis, regulation and pathophysiology. J Steroid Biochem Mol Biol 103:572–577
Donate-Correa J, Muros-de-Fuentes M, Mora-Fernandez C, Navarro-Gonzalez JF (2012) FGF23/Klotho axis: phosphorus, mineral metabolism and beyond. Cytokine Growth Factor Rev 23:37–46
Murer H, Hernando N, Forster I, Biber J (2000) Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev 80:1373–1409
Iharada M, Miyaji T, Fujimoto T, Hiasa M, Anzai N, Omote H, Moriyama Y (2010) Type 1 sodium-dependent phosphate transporter (SLC17A1 protein) is a Cl(−)-dependent urate exporter. J Biol Chem 285:26107–26113
Biber J, Hernando N, Forster I (2013) Phosphate transporters and their function. Annu Rev Physiol 75:535–550
Wagner CA, Hernando N, Forster IC, Biber J (2014) The SLC34 family of sodium-dependent phosphate transporters. Pflugers Arch 466:139–153
Breusegem SY, Takahashi H, Giral-Arnal H, Wang X, Jiang T, Verlander JW, Wilson P, Miyazaki-Anzai S, Sutherland E, Caldas Y, Blaine JT, Segawa H, Miyamoto K, Barry NP, Levi M (2009) Differential regulation of the renal sodium-phosphate cotransporters NaPi-IIa, NaPi-IIc, and PiT-2 in dietary potassium deficiency. Am J Physiol Ren Physiol 297:F350–F361
Battini JL, Rasko JE, Miller AD (1999) A human cell-surface receptor for xenotropic and polytropic murine leukemia viruses: possible role in G protein-coupled signal transduction. Proc Natl Acad Sci USA 96:1385–1390
Tailor CS, Nouri A, Lee CG, Kozak C, Kabat D (1999) Cloning and characterization of a cell surface receptor for xenotropic and polytropic murine leukemia viruses. Proc Natl Acad Sci USA 96:927–932
Sharma P, Patntirapong S, Hann S, Hauschka PV (2010) RANKL–RANK signaling regulates expression of xenotropic and polytropic virus receptor (XPR1) in osteoclasts. Biochem Biophys Res Commun 399:129–132
Meireles AM, Shiau CE, Guenther CA, Sidik H, Kingsley DM, Talbot WS (2014) The phosphate exporter xpr1b is required for differentiation of tissue-resident macrophages. Cell Rep 8:1659–1667
Ito M, Haito S, Furumoto M, Uehata Y, Sakurai A, Segawa H, Tatsumi S, Kuwahata M, Miyamoto K (2007) Unique uptake and efflux systems of inorganic phosphate in osteoclast-like cells. Am J Physiol Cell Physiol 292:C526–C534
Giovannini D, Touhami J, Charnet P, Sitbon M, Battini JL (2013) Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans. Cell Rep 3:1866–1873
Legati A, Giovannini D, Nicolas G, Lopez-Sanchez U, Quintans B et al (2015) Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat Genet 47:579–581
Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS (1998) Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci USA 95:5372–5377
Lederer E, Miyamoto K (2012) Clinical consequences of mutations in sodium phosphate cotransporters. Clin J Am Soc Nephrol 7:1179–1187
Prie D, Huart V, Bakouh N, Planelles G, Dellis O, Gerard B, Hulin P, Benque-Blanchet F, Silve C, Grandchamp B, Friedlander G (2002) Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med 347:983–991
Virkki LV, Forster IC, Hernando N, Biber J, Murer H (2003) Functional characterization of two naturally occurring mutations in the human sodium-phosphate cotransporter type IIa. J Bone Miner Res 18:2135–2141
Magen D, Berger L, Coady MJ, Ilivitzki A, Militianu D, Tieder M, Selig S, Lapointe JY, Zelikovic I, Skorecki K (2010) A loss-of-function mutation in NaPi-IIa and renal Fanconi’s syndrome. N Engl J Med 362:1102–1109
Wagner CA, Rubio-Aliaga I, Biber J, Hernando N (2014) Genetic diseases of renal phosphate handling. Nephrol Dial Transpl 29:iv45–iv54
Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, Frappier D, Burkett K, Carpenter TO, Anderson D, Garabedian M, Sermet I, Fujiwara TM, Morgan K, Tenenhouse HS, Juppner H (2006) SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet 78:179–192
Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ et al (2014) Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J Am Soc Nephrol 25:2366–2375
Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, Gershoni-Baruch R, Albers N, Lichtner P, Schnabel D, Hochberg Z, Strom TM (2006) Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet 78:193–201
Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner JM, Econs MJ (2006) Intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab 91:4022–4027
Tencza AL, Ichikawa S, Dang A, Kenagy D, McCarthy E, Econs MJ, Levine MA (2009) Hypophosphatemic rickets with hypercalciuria due to mutation in SLC34A3/type IIc sodium-phosphate cotransporter: presentation as hypercalciuria and nephrolithiasis. J Clin Endocrinol Metab 94:4433–4438
Haito-Sugino S, Ito M, Ohi A, Shiozaki Y, Kangawa N, Nishiyama T, Aranami F, Sasaki S, Mori A, Kido S, Tatsumi S, Segawa H, Miyamoto K (2012) Processing and stability of type IIc sodium-dependent phosphate cotransporter mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria. Am J Physiol Cell Physiol 302:C1316–C1330
Segawa H, Kaneko I, Takahashi A, Kuwahata M, Ito M, Ohkido I, Tatsumi S, Miyamoto K (2002) Growth-related renal type II Na/Pi cotransporter. J Biol Chem 277:19665–19672
Segawa H, Onitsuka A, Kuwahata M, Hanabusa E, Furutani J, Kaneko I, Tomoe Y, Aranami F, Matsumoto N, Ito M, Matsumoto M, Li M, Amizuka N, Miyamoto K (2009) Type IIc sodium-dependent phosphate transporter regulates calcium metabolism. J Am Soc Nephrol 20:104–113
Segawa H, Onitsuka A, Furutani J, Kaneko I, Aranami F, Matsumoto N, Tomoe Y, Kuwahata M, Ito M, Matsumoto M, Li M, Amizuka N, Miyamoto K (2009) Npt2a and Npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. Am J Physiol Ren Physiol 297:F671–F678
Myakala K, Motta S, Murer H, Wagner CA, Koesters R, Biber J, Hernando N (2014) Renal-specific and inducible depletion of NaPi-IIc/Slc34a3, the cotransporter mutated in HHRH, does not affect phosphate or calcium homeostasis in mice. Am J Physiol Ren Physiol 306:F833–F843
Miyamoto K, Ito M, Tatsumi S, Kuwahata M, Segawa H (2007) New aspect of renal phosphate reabsorption: the type IIc sodium-dependent phosphate transporter. Am J Nephrol 27:503–515
Murer H, Hernando N, Forster L, Biber J (2001) Molecular mechanisms in proximal tubular and small intestinal phosphate reabsorption (plenary lecture). Mol Membr Biol 18:3–11
Borowitz SM, Ghishan FK (1989) Phosphate transport in human jejunal brush-border membrane vesicles. Gastroenterology 96:4–10
Brandis M, Harmeyer J, Kaune R, Mohrmann M, Murer H, Zimolo Z (1987) Phosphate transport in brush-border membranes from control and rachitic pig kidney and small intestine. J Physiol 384:479–490
Caverzasio J, Danisi G, Straub RW, Murer H, Bonjour JP (1987) Adaptation of phosphate transport to low phosphate diet in renal and intestinal brush border membrane vesicles: influence of sodium and pH. Pflugers Arch 409:333–336
Hilfiker H, Hattenhauer O, Traebert M, Forster I, Murer H, Biber J (1998) Characterization of a murine type II sodium-phosphate cotransporter expressed in mammalian small intestine. Proc Natl Acad Sci USA 95:14564–14569
Marks J, Srai SK, Biber J, Murer H, Unwin RJ, Debnam ES (2006) Intestinal phosphate absorption and the effect of vitamin D: a comparison of rats with mice. Exp Physiol 91:531–537
Marks J, Debnam ES, Unwin RJ (2010) Phosphate homeostasis and the renal-gastrointestinal axis. Am J Physiol Ren Physiol 299:F285–F296
Marks J, Lee GJ, Nadaraja SP, Debnam ES, Unwin RJ (2015) Experimental and regional variations in Na+-dependent and Na+-independent phosphate transport along the rat small intestine and colon. Physiol Rep 3:e12281
Candeal E, Caldas YA, Guillen N, Levi M, Sorribas V (2014) Na+-independent phosphate transport in Caco2BBE cells. Am J Physiol Cell Physiol 307:C1113–C1122
Capuano P, Radanovic T, Wagner CA, Bacic D, Kato S, Uchiyama Y, St-Arnoud R, Murer H, Biber J (2005) Intestinal and renal adaptation to a low-Pi diet of type II NaPi cotransporters in vitamin D receptor- and 1alphaOHase-deficient mice. Am J Physiol Cell Physiol 288:C429–C434
Capuano P, Bacic D, Stange G, Hernando N, Kaissling B, Pal R, Kocher O, Biber J, Wagner CA, Murer H (2005) Expression and regulation of the renal Na/phosphate cotransporter NaPi-IIa in a mouse model deficient for the PDZ protein PDZK1. Pflugers Arch 449:392–402
Hattenhauer O, Traebert M, Murer H, Biber J (1999) Regulation of small intestinal Na-P(i) type IIb cotransporter by dietary phosphate intake. Am J Physiol 277:G756–G762
Katai K, Miyamoto K, Kishida S, Segawa H, Nii T, Tanaka H, Tani Y, Arai H, Tatsumi S, Morita K, Taketani Y, Takeda E (1999) Regulation of intestinal Na+-dependent phosphate co-transporters by a low-phosphate diet and 1,25-dihydroxyvitamin D3. Biochem J 343:705–712
Radanovic T, Wagner CA, Murer H, Biber J (2005) Regulation of intestinal phosphate transport. I. Segmental expression and adaptation to low-P(i) diet of the type IIb Na(+)-P(i) cotransporter in mouse small intestine. Am J Physiol Gastrointest Liver Physiol 288:G496–G500
Segawa H, Kaneko I, Yamanaka S, Ito M, Kuwahata M, Inoue Y, Kato S, Miyamoto K (2004) Intestinal Na-P(i) cotransporter adaptation to dietary P(i) content in vitamin D receptor null mice. Am J Physiol Ren Physiol 287:F39–F47
Xu H, Bai L, Collins JF, Ghishan FK (2002) Age-dependent regulation of rat intestinal type IIb sodium-phosphate cotransporter by 1,25-(OH)(2) vitamin D(3). Am J Physiol Cell Physiol 282:C487–C493
Huqun, Izumi S, Miyazawa H, Ishii K, Uchiyama B et al (2007) Mutations in the SLC34A2 gene are associated with pulmonary alveolar microlithiasis. Am J Respir Crit Care Med 175:263–268
Marks J, Debnam ES, Unwin RJ (2013) The role of the gastrointestinal tract in phosphate homeostasis in health and chronic kidney disease. Curr Opin Nephrol Hypertens 22:481–487
Sabbagh Y, Giral H, Caldas Y, Levi M, Schiavi SC (2011) Intestinal phosphate transport. Adv Chronic Kidney Dis 18:85–90
Ohi A, Hanabusa E, Ueda O, Segawa H, Horiba N et al (2011) Inorganic phosphate homeostasis in sodium-dependent phosphate cotransporter Npt2b(+)/(-) mice. Am J Physiol Ren Physiol 301:F1105–F1113
Sabbagh Y, O’Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, Schiavi SC (2009) Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol 20:2348–2358
Hernando N, Myakala K, Simona F, Knopfel T, Thomas L, Murer H, Wagner CA, Biber J (2015) Intestinal depletion of NaPi-IIb/Slc34a2 in mice: renal and hormonal adaptation. J Bone Miner Res. doi:10.1002/jbmr.2523
Weinman EJ, Biswas RS, Peng G, Shen L, Turner CL, E X, Steplock D, Shenolikar S, Cunningham R (2007) Parathyroid hormone inhibits renal phosphate transport by phosphorylation of serine 77 of sodium-hydrogen exchanger regulatory factor-1. J Clin Invest 117:3412–3420
Martin A, David V, Quarles LD (2012) Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 92:131–155
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444:770–774
Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281:6120–6123
Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, Miyamoto K, Fukushima N (2003) Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production. J Biol Chem 278:2206–2211
Segawa H, Kawakami E, Kaneko I, Kuwahata M, Ito M, Kusano K, Saito H, Fukushima N, Miyamoto K (2003) Effect of hydrolysis-resistant FGF23-R179Q on dietary phosphate regulation of the renal type-II Na/Pi transporter. Pflugers Arch 446:585–592
Gattineni J, Alphonse P, Zhang Q, Mathews N, Bates CM, Baum M (2014) Regulation of renal phosphate transport by FGF23 is mediated by FGFR1 and FGFR4. Am J Physiol Ren Physiol 306:F351–F358
Gattineni J, Twombley K, Goetz R, Mohammadi M, Baum M (2011) Regulation of serum 1,25(OH)2 vitamin D3 levels by fibroblast growth factor 23 is mediated by FGF receptors 3 and 4. Am J Physiol Ren Physiol 301:F371–F377
Inoue Y, Segawa H, Kaneko I, Yamanaka S, Kusano K, Kawakami E, Furutani J, Ito M, Kuwahata M, Saito H, Fukushima N, Kato S, Kanayama HO, Miyamoto K (2005) Role of the vitamin D receptor in FGF23 action on phosphate metabolism. Biochem J 390:325–331
Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, Ghishan FK (2005) 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal–gastrointestinal–skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol 289:G1036–G1042
Yamashita T, Konishi M, Miyake A, Inui K, Itoh N (2002) Fibroblast growth factor (FGF)-23 inhibits renal phosphate reabsorption by activation of the mitogen-activated protein kinase pathway. J Biol Chem 277:28265–28270
Weinman EJ, Steplock D, Shenolikar S, Biswas R (2011) Fibroblast growth factor-23-mediated inhibition of renal phosphate transport in mice requires sodium-hydrogen exchanger regulatory factor-1 (NHERF-1) and synergizes with parathyroid hormone. J Biol Chem 286:37216–37221
Andrukhova O, Zeitz U, Goetz R, Mohammadi M, Lanske B, Erben RG (2012) FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone 51:621–628
Olauson H, Lindberg K, Amin R, Jia T, Wernerson A, Andersson G, Larsson TE (2012) Targeted deletion of Klotho in kidney distal tubule disrupts mineral metabolism. J Am Soc Nephrol 23:1641–1651
Tomoe Y, Segawa H, Shiozawa K, Kaneko I, Tominaga R, Hanabusa E, Aranami F, Furutani J, Kuwahara S, Tatsumi S, Matsumoto M, Ito M, Miyamoto K (2010) Phosphaturic action of fibroblast growth factor 23 in Npt2 null mice. Am J Physiol Ren Physiol 298:F1341–F1350
Picard N, Capuano P, Stange G, Mihailova M, Kaissling B, Murer H, Biber J, Wagner CA (2010) Acute parathyroid hormone differentially regulates renal brush border membrane phosphate cotransporters. Pflugers Arch 460:677–687
Bacic D, Lehir M, Biber J, Kaissling B, Murer H, Wagner CA (2006) The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int 69:495–503
Nashiki K, Taketani Y, Takeichi T, Sawada N, Yamamoto H, Ichikawa M, Arai H, Miyamoto K, Takeda E (2005) Role of membrane microdomains in PTH-mediated down-regulation of NaPi-IIa in opossum kidney cells. Kidney Int 68:1137–1147
Nagai S, Okazaki M, Segawa H, Bergwitz C, Dean T, Potts JT Jr, Mahon MJ, Gardella TJ, Juppner H (2011) Acute down-regulation of sodium-dependent phosphate transporter NPT2a involves predominantly the cAMP/PKA pathway as revealed by signaling-selective parathyroid hormone analogs. J Biol Chem 286:1618–1626
Deliot N, Hernando N, Horst-Liu Z, Gisler SM, Capuano P, Wagner CA, Bacic D, O’Brien S, Biber J, Murer H (2005) Parathyroid hormone treatment induces dissociation of type IIa Na+-P(i) cotransporter-Na+/H+ exchanger regulatory factor-1 complexes. Am J Physiol Cell Physiol 289:C159–C167
Voltz JW, Brush M, Sikes S, Steplock D, Weinman EJ, Shenolikar S (2007) Phosphorylation of PDZ1 domain attenuates NHERF-1 binding to cellular targets. J Biol Chem 282:33879–33887
Weinman EJ, Steplock D, Zhang Y, Biswas R, Bloch RJ, Shenolikar S (2010) Cooperativity between the phosphorylation of Thr95 and Ser77 of NHERF-1 in the hormonal regulation of renal phosphate transport. J Biol Chem 285:25134–25138
Shenolikar S, Voltz JW, Minkoff CM, Wade JB, Weinman EJ (2002) Targeted disruption of the mouse NHERF-1 gene promotes internalization of proximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc Natl Acad Sci USA 99:11470–11475
Segawa H, Yamanaka S, Onitsuka A, Tomoe Y, Kuwahata M, Ito M, Taketani Y, Miyamoto K (2007) Parathyroid hormone-dependent endocytosis of renal type IIc Na-Pi cotransporter. Am J Physiol Ren Physiol 292:F395–F403
Villa-Bellosta R, Barac-Nieto M, Breusegem SY, Barry NP, Levi M, Sorribas V (2008) Interactions of the growth-related, type IIc renal sodium/phosphate cotransporter with PDZ proteins. Kidney Int 73:456–464
Giral H, Lanzano L, Caldas Y, Blaine J, Verlander JW, Lei T, Gratton E, Levi M (2011) Role of PDZK1 protein in apical membrane expression of renal sodium-coupled phosphate transporters. J Biol Chem 286:15032–15042
Kido S, Kaneko I, Tatsumi S, Segawa H, Miyamoto K (2013) Vitamin D and type II sodium-dependent phosphate cotransporters. Contrib Nephrol 180:86–97
Berndt T, Thomas LF, Craig TA, Sommer S, Li X, Bergstralh EJ, Kumar R (2007) Evidence for a signaling axis by which intestinal phosphate rapidly modulates renal phosphate reabsorption. Proc Natl Acad Sci USA 104:11085–11090
Marks J, Churchill LJ, Debnam ES, Unwin RJ (2008) Matrix extracellular phosphoglycoprotein inhibits phosphate transport. J Am Soc Nephrol 19:2313–2320
Scanni R, vonRotz M, Jehle S, Hulter HN, Krapf R (2014) The human response to acute enteral and parenteral phosphate loads. J Am Soc Nephrol 25:2730–2739
Schiavi SC, Tang W, Bracken C, O’Brien SP, Song W, Boulanger J, Ryan S, Phillips L, Liu S, Arbeeny C, Ledbetter S, Sabbagh Y (2012) Npt2b deletion attenuates hyperphosphatemia associated with CKD. J Am Soc Nephrol 23:1691–1700
White SA, Al-Mukhtar A, Lodge JP, Pollard SG (2004) Progress in living donor liver transplantation. Transpl Proc 36:2720–2726
Buell JF, Berger AC, Plotkin JS, Kuo PC, Johnson LB (1998) The clinical implications of hypophosphatemia following major hepatic resection or cryosurgery. Arch Surg 133:757–761
Pomposelli JJ, Pomfret EA, Burns DL, Lally A, Sorcini A, Gordon FD, Lewis WD, Jenkins R (2001) Life-threatening hypophosphatemia after right hepatic lobectomy for live donor adult liver transplantation. Liver Transpl 7:637–642
Datta HK, Malik M, Neely RD (2007) Hepatic surgery-related hypophosphatemia. Clin Chim Acta 380:13–23
Giovannini I, Chiarla C, Nuzzo G (2002) Pathophysiologic and clinical correlates of hypophosphatemia and the relationship with sepsis and outcome in postoperative patients after hepatectomy. Shock 18:111–115
Salem RR, Tray K (2005) Hepatic resection-related hypophosphatemia is of renal origin as manifested by isolated hyperphosphaturia. Ann Surg 241:343–348
Nafidi O, Lepage R, Lapointe RW, D’Amour P (2007) Hepatic resection-related hypophosphatemia is of renal origin as manifested by isolated hyperphosphaturia. Ann Surg 245:1000–1002
Nafidi O, Lapointe RW, Lepage R, Kumar R, D’Amour P (2009) Mechanisms of renal phosphate loss in liver resection-associated hypophosphatemia. Ann Surg 249:824–827
Kempson SA, Colon-Otero G, Ou SY, Turner ST, Dousa TP (1981) Possible role of nicotinamide adenine dinucleotide as an intracellular regulator of renal transport of phosphate in the rat. J Clin Invest 67:1347–1360
Katai K, Tanaka H, Tatsumi S, Fukunaga Y, Genjida K, Morita K, Kuboyama N, Suzuki T, Akiba T, Miyamoto K, Takeda E (1999) Nicotinamide inhibits sodium-dependent phosphate cotransport activity in rat small intestine. Nephrol Dial Transpl 14:1195–1201
Eto N, Miyata Y, Ohno H, Yamashita T (2005) Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol Dial Transpl 20:1378–1384
Sampathkumar K, Selvam M, Sooraj YS, Gowthaman S, Ajeshkumar RN (2006) Extended release nicotinic acid—a novel oral agent for phosphate control. Int Urol Nephrol 38:171–174
Takahashi Y, Tanaka A, Nakamura T, Fukuwatari T, Shibata K, Shimada N, Ebihara I, Koide H (2004) Nicotinamide suppresses hyperphosphatemia in hemodialysis patients. Kidney Int 65:1099–1104
Martin PR, Shea RJ, Mulks MH (2001) Identification of a plasmid-encoded gene from Haemophilus ducreyi which confers NAD independence. J Bacteriol 183:1168–1174
Imai S (2009) The NAD World: a new systemic regulatory network for metabolism and aging—Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem Biophys 53:65–74
Garten A, Petzold S, Korner A, Imai S, Kiess W (2009) Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab 20:130–138
Nomura K, Tatsumi S, Miyagawa A, Shiozaki Y, Sasaki S, Kaneko I, Ito M, Kido S, Segawa H, Sano M, Fukuwatari T, Shibata K, Miyamoto K (2014) Hepatectomy-related hypophosphatemia: a novel phosphaturic factor in the liver–kidney axis. J Am Soc Nephrol 25:761–772
Kuro-o M (2012) Klotho in health and disease. Curr Opin Nephrol Hypertens 21:362–368
Dhingra R, Sullivan LM, Fox CS, Wang TJ, D’Agostino RB Sr, Gaziano JM, Vasan RS (2007) Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med 167:879–885
Block GA, Hulbert-Shearon TE, Levin NW, Port FK (1998) Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 31:607–617
Hallan SI, Matsushita K, Sang Y, Mahmoodi BK, Black C, Ishani A, Kleefstra N, Naimark D, Roderick P, Tonelli M, Wetzels JF, Astor BC, Gansevoort RT, Levin A, Wen CP, Coresh J (2012) Age and association of kidney measures with mortality and end-stage renal disease. JAMA 308:2349–2360
Palmer SC, Hayen A, Macaskill P, Pellegrini F, Craig JC, Elder GJ, Strippoli GF (2011) Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA 305:1119–1127
Isakova T, Wahl P, Vargas GS, Gutierrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M (2011) Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 79:1370–1378
Gutierrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, Juppner H, Wolf M (2005) Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol 16:2205–2215
Wolf M (2012) Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int 82:737–747
Isakova T, Xie H, Yang W, Xie D, Anderson AH et al (2011) Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 305:2432–2439
Slatopolsky E (2011) The intact nephron hypothesis: the concept and its implications for phosphate management in CKD-related mineral and bone disorder. Kidney Int Suppl (121):S3–S8. doi:10.1038/ki.2011.23
Peiro C, Romacho T, Carraro R, Sanchez-Ferrer CF (2010) Visfatin/PBEF/Nampt: a new cardiovascular target? Front Pharmacol 1:135
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors have no conflicts of interest.
About this article

Cite this article
Tatsumi, S., Miyagawa, A., Kaneko, I. et al. Regulation of renal phosphate handling: inter-organ communication in health and disease. J Bone Miner Metab 34, 1–10 (2016). https://doi.org/10.1007/s00774-015-0705-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00774-015-0705-z