
Abstract
The prevention and correction of hyperphosphatemia are major goals of the treatment of chronic kidney disease (CKD)-bone mineral disorders, and thus, Pi balance requires special attention. Pi balance is maintained by intestinal absorption, renal excretion, and bone accretion. The kidney is mainly responsible for the plasma Pi concentration. In CKD, reduced glomerular filtration rate leads to various Pi metabolism abnormalities, and Pi absorption in the small intestine also has an important role in Pi metabolism. Disturbances in Pi metabolism are mediated by a series of complex changes in regulatory hormones originating from the skeleton, intestine, parathyroid gland, and kidney. In this review, we describe the regulation of type II sodium-dependent Pi co-transporters by the kidney and intestine, including the regulation of Pi transport, circadian rhythm, and the vicious circle between salivary Pi secretion and intestinal Pi absorption in animals with and without CKD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inorganic phosphate (Pi) balance refers to the steady-state difference between Pi input and output over a period of time. When Pi intake exceeds Pi loss (positive balance), Pi accumulates and may lead to growth or weight gain, bone mineralization, or repletion of stores. When Pi loss exceeds Pi intake (negative balance), Pi stores are progressively depleted, eventually resulting in clinical symptoms of deficiency. The Pi balance is maintained mainly by the kidney, bone, and small intestine. It is dependent on the absorption of dietary Pi in the intestine, and re-absorption and excretion of Pi in the kidney and skeletal pools. Absorbed Pi enters the extracellular space and is distributed in three compartments: blood, soft tissue, and bone [1]. The kidney is mainly responsible for the plasma Pi concentration [2]. In chronic kidney disease (CKD), reduced glomerular filtration rate (GFR) leads to various Pi metabolism abnormalities, and Pi absorption in the small intestine also has an important role in Pi metabolism [3]. Disturbances in Pi metabolism are mediated by a series of complex changes in regulatory hormones originating from the skeleton, intestine, parathyroid gland, and kidney [4]. In this review, we focus the regulation of sodium-dependent Pi co-transporters in a number of tissue, including kidney, intestine, and salivary glands.
Control of renal Pi balance
Approximately 80% of filtered Pi is reabsorbed in the proximal tubule. Under normal conditions, ~15% of filtered Pi is ultimately excreted. The renal proximal tubules have a regulatory system that functions through sodium-dependent Pi co-transporters (NaPi transporter). At least two types of transporters (NaPi-II and NaPi-III) play important roles in renal Pi re-absorption [5]. The characteristics of NaPi transport function have been reported in detail by Murer’s group [5]. In the present review, we described the regulation of NaPi-II transporters: NaPi-IIa (SLC34A1, Npt2a) and NaPi-IIc (SLC34A3, Npt2c), because earlier gene knockout (KO) studies for renal Na/Pi co-transporters revealed the importance of NaPi-IIa and NaPi-IIc in renal Pi re-absorption [6].
Many factors affect the Npt2a and Npt2c levels in the proximal tubules
-
1.
Parathyroid hormone (PTH)/fibroblast growth factor 23 (FGF23)—recent excellent reviews have been published regarding the regulation of Pi transport by PTH and FGF23 [7, 8]. PTH is secreted by the parathyroid glands and regulates the endocytosis of NaPi-IIa and NaPi-IIc from the apical membrane of the proximal tubular cells [7, 8]. Scaffolding protein NHERF1 plays an important role in the regulation of NaPi-IIa by PTH and FGF23 [7, 8]. Recent studies demonstrated that the control of phosphorylation of sodium-hydrogen exchanger regulatory factor-1 NHERF1 (by cyclic AMP-dependent protein kinase, protein kinase C, extracellular signal-regulated kinase-1/2 and serine/threonine-protein kinase 1) is a trigger for endocytosis of NaPi-IIa from the apical membrane [7–9]. Furthermore, the bone secretes FGF23, which suppresses Pi re-absorption and vitamin D synthesis in the kidney. Klotho is an essential co-receptor for FGF23 signaling and downregulates the levels of NaPi-IIa protein in the apical membrane [10]. According to recent studies of site-specific klotho deleteon in kidney, NaPi-IIa is regulated by FGF23/klotho signal in proximal and distal tubules [11, 12]. The physiologic roles of secreted klotho on phosphatidic activity, however, remain unknown. NaPi-IIc is also regulated by PTH and FGF23 [4, 13]. Our recent studies suggest that recycling endosomes are involved in NaPi-IIc endocytosis. The regulation of NaPi-IIc, however, is largely unclear.
-
2.
Vitamin D—1,25-dihydroxyvitamin D (1,25(OH)2D), the biologically active form of vitamin D, regulates NaPi-IIa and NaPi-IIc gene expression in the kidney [14, 15]. Administration of 1,25 (OH)2D to vitamin D-deficient rats upregulates the initial rate of renal Pi uptake [14]. Our previous studies demonstrated that the renal Pi uptake as well as the amounts of renal NaPi-IIa and NaPi-IIc protein are downregulated in the global vitamin D receptor (VDR)-KO mice at growth period [16, 17]. Despite the plasma PTH concentration is abnormally high in VDR-KO mice, renal Pi transport activity is not reduced in adult [16, 17]. The mechanisms of the PTH-resistance for Pi transport in VDR-KO mice remain unknown.
-
3.
Vitamin A/thyroid hormone—the vitamin A metabolite ATRA (all-trans-retinoic acid) increases the rate of Pi transport in renal proximal tubular cells [15]. Masuda et al. identified retinoic acid-response elements in both gene promoters [15]. In addition, thyroid hormone increases Pi re-absorption by the elevation of renal NaPi-IIa transcription [18]. Ishiguro et al. identified a thyroid response element in NaPi-IIa gene by 3,5,3-tri-iodothyronine [18].
-
4.
Acute inflammation—Ikeda et al. investigated lipopolysaccharide (LPS)-induced inflammation in mice [19]. LPS administration significantly decreased NaPi-IIa protein expression in the brush border membrane (BBM) after injection. Moreover, tumor necrosis factor-α injection also increased plasma PTH and decreased renal BBM NaPi-IIa expression [19]. The downregulation of NaPi-IIa expression in renal BBM through induction of plasma PTH levels alter Pi homeostasis during LPS-induced acute inflammation [19].
-
5.
NAD/nicotinamide (NAM)—we previously reported that NAM inhibits intestinal and renal Na/Pi transport activity in rodents [20]. Treatment with NAM decreases serum Pi concentration in hemodialysis patients with hyperphosphatemia. Administration of NAM may suppress intestinal Pi absorption [20]. We reported that hepatectomy-induced hypophosphatemia is due to abnormal NAM metabolism, including nicotinamide phosphoribosyltransferase activation in the renal proximal tubular cells [21]. Thus, the liver–kidney axis may be involved in NAD and Pi metabolism [21].
-
6.
Circadian rhythm—the previous studies indicated that the circadian rhythm of serum Pi is remarkably consistent in individuals [22–25]. These studies demonstrated that serum Pi is highest after midnight in normal subjects on normal diets (Fig. 1). In addition, circadian variations in urinary Pi excretion and serum Pi have long been recognized in humans and rodents [24]. The mechanisms involving these diurnal changes remain unclear. The serum Pi peak is observed after midnight between 02:00 and 04:00, and the Pi trough occurs between 08:00 and 10:00 [22, 23, 25]. Because prolonged fasting abolishes the nocturnal peak of Pi, intestinal Pi absorption clearly contributes to this circadian change of serum Pi [22]. Changes in PTH, growth hormone, 1,25(OH)2D, or FGF23, however, cannot fully explain the circadian rhythm of serum Pi and the mechanism of this diurnal variation remains to be clarified [24]. Murer and co-workers reported that the diurnal changes in urinary Pi excretion are mainly related to changes in serum Pi and the tubular threshold, but not to NaPi-IIa expression [26]. The diurnal increase in renal Pi excretion is associated with a mild reduction in the sodium-dependent Pi transport rate in proximal tubular brush border membranes [26]. All of the mineral metabolism-associated hormones have a diurnal rhythm. Recently, Kawai et al. investigated the mechanism of the circadian clock and Pi metabolism in a rodent model [27]. They showed that skeletal FGF23 expression is regulated by the time of food intake, suggesting that the timing of food intake, and not the amount of ingested Pi, is a predominant determinant for the circadian profiles of skeletal FGF23 expression. In addition, a recent study in rodents demonstrated a nearly circadian rhythm for bone mineralization following fluctuations in the activity of the clock gene Per1 [28, 29]. It is possible that variations in the bone mineralization process produce variations in individual serum Pi levels [28, 29]. There is no direct evidence that the bone–kidney axis regulates the circadian rhythm of Pi metabolism. In contrast, considering the importance of the timing of the meal, the intestine–kidney axis, rather than the bone–kidney axis, may provide important factors for the circadian rhythm of Pi metabolism. Further studies are needed to clarify the mechanism of the circadian rhythm of Pi metabolism.
Fig. 1 
Copyright permission is obtained from Oxford University Press
Circadian variation of serum Pi levels in normal subjects. This figure is shown as Fig. 1 in Becker et al. [24]. The data from Jubiz et al. describe mean serum phosphate concentration in ten normal individuals (8 males, 2 females) aged 22–32 years (closed circles) [22]. Portale et al. reported similar findings in six healthy men aged 26–44 years, on a normal phosphate diet (open circle) [23]

CKD and Pi excretion
Bricker and co-workers proposed the intact nephron hypothesis that sets the path for kidney research for the next 50 years [30, 31]. The hypothesis states that although the diseased kidney contains fewer nephrons, the remaining nephrons are functionally normal. To maintain the homeostasis of any given solute, renal function of the diseased kidney must undergo adaptive changes, wherein the excretion rate of each functioning nephron must progressively increase to compensate for the damaged nephrons. There are a number of excellent reviews of Pi metabolism abnormalities in CKD [2, 32].
In the early stages of CKD, the plasma Pi concentration is maintained within the normal range by increase in the serum concentrations of PTH and FGF23, which increases urinary excretion of Pi, and decrease in serum active vitamin D concentration, which decreases intestinal Pi absorption [32]. At the late stage of CKD, the decline in GFR induces relative Pi retention with increased plasma Pi. Both FGF23 and PTH plasma levels are elevated in CKD and lead to a compensatory increase in fractional Pi excretion via inhibition of Pi transport to compensate for the decreased Pi filtration [32]. Experimentally, this is confirmed by a reduction in the NaPi-IIa expression in a classic model of advanced CKD [32]. Recent reports also indicate that FGF23 increases in parallel to albuminuria in immunoglobulin A nephropathy [33]. De Seigneux et al. showed that proteinuria indirectly modulates kidney Pi handling via a toxicity that decreases Klotho expression, which thereby alters FGF23 signaling, or via a direct competitive effect for endocytosis in the proximal tubules [33]. They demonstrated that albuminuria correlates with an increased plasma Pi concentration independently of GFR and that induction of glomerular proteinuria modifies kidney tubular Pi handling by altering FGF23 signaling. Thus, it is possible that many factors are involved in resistance to FGF23 [32].
Intestinal Pi handling
In adults, net Pi absorption typically ranges from 55 to 80% of the customary intake. Dietary Pi and 1,25(OH)2D are the most important physiologic regulators of intestinal Pi absorption [34]. There are two pathways for the intestinal Pi absorption, paracellular, and cellular, and at least two mechanisms, passive diffusion and sodium-dependent active transport [3]. The paracellular pathway occurs at tight junctions and utilizes electrochemical gradients. The cellular transport pathway requires sodium-dependent Pi transporters, including NaPi-IIb (SLC34A2, Npt2b), that are expressed in the small intestine [3]. The relative proportion of absorption via each mechanism varies depending on the luminal Pi concentration, with active transport contributing to between 30 and 80%. Intestinal Npt2b levels are regulated by FGF23, 1,25(OH)2D, and dietary Pi levels [35–37].
The effect of FGF23 on Npt2b levels in the small intestine is associated with FGF23-mediated decrease in 1,25(OH)2D levels [38–40]. Indeed, injecting FGF23 into VDR-KO mice does not further decrease the levels of Npt2b in the small intestine [35]. In addition, the FGF23 injection had no effect on Npt2b levels in Npt2a-KO mice, Npt2c-KO mice, or Npt2a/Npt2c double KO mice, although FGF23 administration attenuates the increase of 1,25(OH)2D levels in all these mutant mice [41]. Thus, FGF23-dependent 1,25(OH)2D repression appears to be in important factor for Npt2b expression in the small intestine. Furthermore, additional factors may be involved in FGF23-dependent Npt2b downregulation.
CKD and intestinal Pi absorption
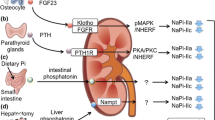
The vicious circle between salivary Pi secretion and intestinal Pi absorption (Fig. 2)—saliva is an important organ for maintaining Pi homeostasis in CKD and dialysis patients [42]. The daily volume of saliva produced by the salivary glands is nearly one-fifth the total plasma volume. Based on the previous data and in accordance with the daily rate of saliva secretion, salivary glands seem to be able to secrete a relevant amount of Pi, possibly ranging from 300 to 600 mg [42]. Savica et al. reported that a high-Pi diet increases saliva Pi levels in CKD and hemodialysis patients [42]. In addition, in dialysis patients, salivary Pi levels correlate with serum Pi and multivariate analysis which confirms that serum Pi is the only independent factor predicting increased salivary Pi secretion [42]. The ingestion of the Pi secreted in saliva and its subsequent absorption in the small intestine starts a vicious circle between salivary Pi secretion and fasting Pi absorption, thereby worsening hyperphosphatemia (Fig. 2). Therefore, salivary Pi binding could be a useful approach for reducing serum Pi levels in dialysis patients. Chitosan is a polymer of glucosamine, similar to sevelamer, which would bind Pi present in high concentration in the saliva of CKD patients [43]. Recent studies, however, have been unable to duplicate these results [44, 45]. A systemic analysis data concluded that the amount of chitosan contained in the chewing gum (20 mg) is too little to account for the originally observed reduction in serum Pi [43]. The development of effective Pi binders and the understanding of mechanisms of Pi handling in the salivary glands is essential. The saliva is primarily produced in the acinar compartment, where 85% of the proteins are ultrafiltrated from the capillary bed adjacent to the glands. In the ductal system, saliva is converted from an isotonic to a hypotonic solution with lower sodium and chloride compared with the plasma [46]. Npt2b is also located in ductal cells and may be involved in the regulation of the salivary Pi concentration [47]. We speculate that cross-talk of Npt2b in the salivary glands and the small intestine is very important for Pi metabolism in the gastrointestinal tract. Understanding the mechanisms of the cross-talk between saliva and intestinal Npt2b with respect to the dietary Pi load may help us to resolve the Pi metabolism disturbances in CKD.

Pi balance in the kidney, small intestine, and salivary glands. Pi balance is dependent on the absorption of dietary Pi in the intestine, and re-absorption and excretion of Pi in the kidney and bone pools. The ingestion of the Pi secreted in saliva and its subsequent absorption in the small intestine starts a vicious circle between salivary Pi secretion and fasting Pi absorption, thereby worsening hyperphosphatemia
Intestinal Pi absorption—the physiology of the intestinal regulation of Pi absorption in humans is poorly understood and the regulators of intestinal Npt2b expression in CKD are uncertain. Experiments conducted using the 5/6 nephrectomy rat model revealed that intestinal Pi transport in the duodenum and jejunum remains unaltered [48]. Studies in mice with adenine-induced CKD also indicate that Pi uptake and Npt2b expression in the intestinal brush border membranes are not significantly altered [49]. Inhibitors of intestinal Npt2b and its molecular targets are expected to be effective for treating CKD-associated hyperphosphatemia [50]. Based on studies using Npt2b-KO mice, intestinal Npt2b is an important target for the treatment of CKD-associated hyperphosphatemia [50]. In a renal failure model of adenine administration in intestinal Npt2b-KO mice, the serum Pi concentration was significantly suppressed [50]. The effectiveness of Npt2b inhibitors in humans, however, is not known. Recent reports indicate that treatment with an NHE3 inhibitor is effective for altering intestinal Pi absorption and corrects hyperphosphatemia [51]. The mechanisms of Pi transport inhibition are not well understood [51].
On the other hand, NAM administration suppresses the expression of intestinal Npt2b and corrects hyperphosphatemia in renal failure models [20]. As described earlier, marked hypophosphatemia is common after major hepatic resection, but the pathophysiologic mechanism remains unknown. We investigated a partial hepatectomy-related hypophosphatemia (PH) model and demonstrated that PH is due to abnormal NAM metabolism, including nicotinamide phosphoribosyltransferase activation in the renal proximal tubular cells [21]. In addition, PH results in marked reduction of intestinal Npt2b expression and possibly suppressed intestinal Pi absorption. We speculate that PH increases intestinal nicotinamide phosphoribosyltransferase activity and suppresses intestinal Npt2b levels. Studies of the NAM metabolism pathway in the liver–intestine axis may contribute to a better understanding of intestinal Pi handling.
Future perspectives
Pi disturbances begin at an early stage in CKD and are associated with cardiovascular disease. In the course of CKD, FGF23, and PTH levels progressively increase, while Klotho and 1,25(OH)2D levels decrease. The following issues remain to be resolved: (1) The molecular mechanisms of the paracellular pathway for intestinal Pi absorption; (2) The factors that determine the circadian rhythm of the serum Pi concentration; (3) The vicious circle between salivary phosphate secretion and intestinal Pi absorption; (4) NAD and Pi metabolism in the liver–kidney axis; and (5) The mechanisms of cross-talk between renal NaPi-IIa (Npt2a) and NaPi-IIc (Npt2c) and intestinal NaPi-IIb (Npt2b).
Finally, the prevention and correction of hyperphosphatemia are major goals of the treatment of CKD-bone mineral disorders, and thus, Pi balance requires special attention. Further studies are necessary to clarify the mechanisms of disturbance of intestinal and renal Pi balance in CKD-bone mineral disorders.
References
Wesseling-Perry K, Juppner H. The osteocyte in CKD: new concepts regarding the role of FGF23 in mineral metabolism and systemic complications. Bone. 2013;54(2):222–9.
Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol. 2015;10(7):1257–72.
Marks J, Debnam ES, Unwin RJ. The role of the gastrointestinal tract in phosphate homeostasis in health and chronic kidney disease. Curr Opin Nephrol Hypertens. 2013;22(4):481–7.
Tatsumi S, Miyagawa A, Kaneko I, Shiozaki Y, Segawa H, Miyamoto K. Regulation of renal phosphate handling: inter-organ communication in health and disease. J Bone Miner Metab. 2016;34(1):1–10.
Forster IC, Hernando N, Biber J, Murer H. Phosphate transporters of the SLC20 and SLC34 families. Mol Aspects Med. 2013;34(2–3):386–95.
Lederer E, Miyamoto K. Clinical consequences of mutations in sodium phosphate cotransporters. Clin J Am Soc Nephrol. 2012;7(7):1179–87.
Weinman EJ, Lederer ED. NHERF-1 and the regulation of renal phosphate reabsorption: a tale of three hormones. Am J Physiol Renal Physiol. 2012;303(3):F321–7.
Andrukhova O, Streicher C, Zeitz U, Erben RG. Fgf23 and parathyroid hormone signaling interact in kidney and bone. Mol Cell Endocrinol. 2016;436:224–39.
Sneddon WB, Ruiz GW, Gallo LI, Xiao K, Zhang Q, Rbaibi Y, et al. Convergent signaling pathways regulate parathyroid hormone and fibroblast growth factor-23 action on NPT2A-mediated phosphate transport. J Biol Chem. 2016;291(36):18632–42.
Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. Faseb J. 2010;24(9):3438–50.
Ide N, Olauson H, Sato T, Densmore MJ, Wang H, Hanai J, et al. In vivo evidence for a limited role of proximal tubular Klotho in renal phosphate handling. Kidney Int. 2016;90(2):348–62.
Olauson H, Lindberg K, Amin R, Jia T, Wernerson A, Andersson G, et al. Targeted deletion of Klotho in kidney distal tubule disrupts mineral metabolism. J Am Soc Nephrol. 2012;23(10):1641–51.
Miyamoto K, Ito M, Tatsumi S, Kuwahata M, Segawa H. New aspect of renal phosphate reabsorption: the type IIc sodium-dependent phosphate transporter. Am J Nephrol. 2007;27(5):503–15.
Taketani Y, Segawa H, Chikamori M, Morita K, Tanaka K, Kido S, et al. Regulation of type II renal Na+-dependent inorganic phosphate transporters by 1,25-dihydroxyvitamin D3. Identification of a vitamin D-responsive element in the human NAPi-3 gene. J Biol Chem. 1998;273(23):14575–81.
Masuda M, Yamamoto H, Kozai M, Tanaka S, Ishiguro M, Takei Y, et al. Regulation of renal sodium-dependent phosphate co-transporter genes (Npt2a and Npt2c) by all-trans-retinoic acid and its receptors. Biochem J. 2010;429(3):583–92.
Kaneko I, Segawa H, Furutani J, Kuwahara S, Aranami F, Hanabusa E, et al. Hypophosphatemia in vitamin D receptor null mice: effect of rescue diet on the developmental changes in renal Na+-dependent phosphate cotransporters. Pflugers Arch. 2011;461(1):77–90.
Kido S, Kaneko I, Tatsumi S, Segawa H, Miyamoto K. Vitamin D and type II sodium-dependent phosphate cotransporters. Contrib Nephrol. 2013;180:86–97.
Ishiguro M, Yamamoto H, Masuda M, Kozai M, Takei Y, Tanaka S, et al. Thyroid hormones regulate phosphate homoeostasis through transcriptional control of the renal type IIa sodium-dependent phosphate co-transporter (Npt2a) gene. Biochem J. 2010;427(1):161–9.
Ikeda S, Yamamoto H, Masuda M, Takei Y, Nakahashi O, Kozai M, et al. Downregulation of renal type IIa sodium-dependent phosphate cotransporter during lipopolysaccharide-induced acute inflammation. Am J Physiol Renal Physiol. 2014;306(7):F744–50.
Katai K, Tanaka H, Tatsumi S, Fukunaga Y, Genjida K, Morita K, et al. Nicotinamide inhibits sodium-dependent phosphate cotransport activity in rat small intestine. Nephrol Dial Transplant. 1999;14(5):1195–201.
Nomura K, Tatsumi S, Miyagawa A, Shiozaki Y, Sasaki S, Kaneko I, et al. Hepatectomy-related hypophosphatemia: a novel phosphaturic factor in the liver–kidney axis. J Am Soc Nephrol. 2014;25(4):761–72.
Jubiz W, Canterbury JM, Reiss E, Tyler FH. Circadian rhythm in serum parathyroid hormone concentration in human subjects: correlation with serum calcium, phosphate, albumin, and growth hormone levels. J Clin Invest. 1972;51(8):2040–6.
Portale AA, Halloran BP, Morris RC Jr. Dietary intake of phosphorus modulates the circadian rhythm in serum concentration of phosphorus. Implications for the renal production of 1,25-dihydroxyvitamin D. J Clin Invest. 1987;80(4):1147–54.
Becker GJ, Walker RG, Hewitson TD, Pedagogos E. Phosphate levels—time for a rethink? Nephrol Dial Transplant. 2009;24(8):2321–4.
Ix JH, Anderson CA, Smits G, Persky MS, Block GA. Effect of dietary phosphate intake on the circadian rhythm of serum phosphate concentrations in chronic kidney disease: a crossover study. Am J Clin Nutr. 2014;100(5):1392–7.
Bielesz B, Bacic D, Honegger K, Biber J, Murer H, Wagner CA. Unchanged expression of the sodium-dependent phosphate cotransporter NaPi-IIa despite diurnal changes in renal phosphate excretion. Pflugers Arch. 2006;452(6):683–9.
Kawai M, Kinoshita S, Shimba S, Ozono K, Michigami T. Sympathetic activation induces skeletal Fgf23 expression in a circadian rhythm-dependent manner. J Biol Chem. 2014;289(3):1457–66.
McElderry JD, Zhao G, Khmaladze A, Wilson CG, Franceschi RT, Morris MD. Tracking circadian rhythms of bone mineral deposition in murine calvarial organ cultures. J Bone Miner Res. 2013;28(8):1846–54.
Lederer E. Regulation of serum phosphate. J Physiol. 2014;592(18):3985–95.
Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest. 1968;47(8):1865–74.
Slatopolsky E, Caglar S, Pennell JP, Taggart DD, Canterbury JM, Reiss E, et al. On the pathogenesis of hyperparathyroidism in chronic experimental renal insufficiency in the dog. J Clin Invest. 1971;50(3):492–9.
Ritter CS, Slatopolsky E. Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol. 2016;11(6):1088–100.
de Seigneux S, Courbebaisse M, Rutkowski JM, Wilhelm-Bals A, Metzger M, Khodo SN, et al. Proteinuria increases plasma phosphate by altering its tubular handling. J Am Soc Nephrol. 2015;26(7):1608–18.
Katai K, Miyamoto K, Kishida S, Segawa H, Nii T, Tanaka H, et al. Regulation of intestinal Na+-dependent phosphate co-transporters by a low-phosphate diet and 1,25-dihydroxyvitamin D3. Biochem J. 1999;343(Pt 3):705–12.
Miyamoto K, Ito M, Kuwahata M, Kato S, Segawa H. Inhibition of intestinal sodium-dependent inorganic phosphate transport by fibroblast growth factor 23. Ther Apher Dial. 2005;9(4):331–5.
Segawa H, Kaneko I, Yamanaka S, Ito M, Kuwahata M, Inoue Y, et al. Intestinal Na–P(i) cotransporter adaptation to dietary P(i) content in vitamin D receptor null mice. Am J Physiol Renal Physiol. 2004;287(1):F39–47.
Miyamoto K, Haito-Sugino S, Kuwahara S, Ohi A, Nomura K, Ito M, et al. Sodium-dependent phosphate cotransporters: lessons from gene knockout and mutation studies. J Pharm Sci. 2011;100(9):3719–30.
Inoue Y, Segawa H, Kaneko I, Yamanaka S, Kusano K, Kawakami E, et al. Role of the vitamin D receptor in FGF23 action on phosphate metabolism. Biochem J. 2005;390(Pt 1):325–31.
Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, et al. Circulating FGF-23 is regulated by 1 alpha, 25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005;280(4):2543–9.
Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, et al. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1 alpha, 25-dihydroxyvitamin D3 production. J Biol Chem. 2003;278(4):2206–11.
Tomoe Y, Segawa H, Shiozawa K, Kaneko I, Tominaga R, Hanabusa E, et al. Phosphaturic action of fibroblast growth factor 23 in Npt2 null mice. Am J Physiol Renal Physiol. 2010;298(6):F1341–50.
Savica V, Calo LA, Santoro D, Monardo P, Santoro G, Muraca U, et al. Salivary glands: a new player in phosphorus metabolism. J Renal Nutr. 2011;21(1):39–42.
Oh MS, Uribarri J. What can we learn from the saga of chitosan gums in hyperphosphatemia therapy? Clin J Am Soc Nephrol. 2014;9(5):967–70.
Savica V, Calo LA, Monardo P, Davis PA, Granata A, Santoro D, et al. Salivary phosphate-binding chewing gum reduces hyperphosphatemia in dialysis patients. J Am Soc Nephrol. 2009;20(3):639–44.
Akizawa T, Tsuruta Y, Okada Y, Miyauchi Y, Suda A, Kasahara H, et al. Effect of chitosan chewing gum on reducing serum phosphorus in hemodialysis patients: a multi-center, randomized, double-blind, placebo-controlled trial. BMC Nephrol. 2014;15:98.
Savica V, Bellinghieri G, Calo LA. Salivary glands: a ‘third kidney’ for phosphate excretion in kidney disease? Blood Purif. 2009;28(4):364.
Homann V, Rosin-Steiner S, Stratmann T, Arnold WH, Gaengler P, Kinne RK. Sodium-phosphate cotransporter in human salivary glands: molecular evidence for the involvement of NPT2b in acinar phosphate secretion and ductal phosphate reabsorption. Arch Oral Biol. 2005;50(9):759–68.
Marks J, Churchill LJ, Srai SK, Biber J, Murer H, Jaeger P, et al. Intestinal phosphate absorption in a model of chronic renal failure. Kidney Int. 2007;72(2):166–73.
Ohi A, Hanabusa E, Ueda O, Segawa H, Horiba N, Kaneko I, et al. Inorganic phosphate homeostasis in sodium-dependent phosphate cotransporter Npt2b(+)/(−) mice. Am J Physiol Renal Physiol. 2011;301(5):F1105–13.
Schiavi SC, Tang W, Bracken C, O’Brien SP, Song W, Boulanger J, et al. Npt2b deletion attenuates hyperphosphatemia associated with CKD. J Am Soc Nephrol. 2012;23(10):1691–700.
Labonte ED, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, et al. Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol. 2015;26(5):1138–49.
Acknowledgements
This supplement is supported by the grants from the Japanese Society for Kidney Bone Disease (JSKBD) and from the Research Meeting on Kidney and Metabolic Bone Disease.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflict of interest exists.
About this article

Cite this article
Kaneko, I., Tatsumi, S., Segawa, H. et al. Control of phosphate balance by the kidney and intestine. Clin Exp Nephrol 21 (Suppl 1), 21–26 (2017). https://doi.org/10.1007/s10157-016-1359-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-016-1359-4