
Abstract
The aim of this study was to determine whether the TAR DNA-binding protein of 43 kDa (TDP-43) has any independent effect on the clinical and neuroimaging features typically ascribed to Alzheimer’s disease (AD) pathology, and whether TDP-43 pathology could help shed light on the phenomenon of resilient cognition in AD. Three-hundred and forty-two subjects pathologically diagnosed with AD were screened for the presence, burden and distribution of TDP-43. All had been classified as cognitively impaired or normal, prior to death. Atlas-based parcellation and voxel-based morphometry were used to assess regional atrophy on MRI. Regression models controlling for age at death, apolipoprotein ε4 and other AD-related pathologies were utilized to explore associations between TDP-43 and cognition or brain atrophy, stratified by Braak stage. In addition, we determined whether the effects of TDP-43 were mediated by hippocampal sclerosis. One-hundred and ninety-five (57 %) cases were TDP-positive. After accounting for age, apolipoprotein ε4 and other pathologies, TDP-43 had a strong effect on cognition, memory loss and medial temporal atrophy in AD. These effects were not mediated by hippocampal sclerosis. TDP-positive subjects were 10× more likely to be cognitively impaired at death compared to TDP-negative subjects. Greater cognitive impairment and medial temporal atrophy were associated with greater TDP-43 burden and more extensive TDP-43 distribution. TDP-43 is an important factor in the manifestation of the clinico-imaging features of AD. TDP-43 also appears to be able to overpower what has been termed resilient brain aging. TDP-43 therefore should be considered a potential therapeutic target for the treatment of AD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Since the time of Alzheimer himself [2], two proteins, beta-amyloid (Aβ) deposited in senile plaques, and tau deposited in neurofibrillary tangles, have become tantamount to Alzheimer’s disease (AD) [9]. Neurofibrillary tangles progress throughout the brain in a stereotypic pattern defined by the Braak staging scheme [9]. This staging scheme correlates with cognitive impairment and brain atrophy in AD [18, 20, 44]. However, a significant proportion of patients with AD pathology remain clinically normal up to the time of death despite the presence of both neurofibrillary tangles and senile plaques [15]. Little is known about this phenomenon and whether other proteins may also be playing a role.
A third protein, the TAR DNA-binding protein of 43 kDa (TDP-43), has, however, recently been found to be present in the brains of subjects with pathologically diagnosed AD [3, 4, 8, 14, 24, 26, 43]. TDP-43 is an RNA-binding protein that functions in exon skipping and is identified in an abnormal phosphorylated state in cellular inclusions [11]. TDP-43 is associated with neurodegeneration and cognitive impairment [34] yet it is unknown whether TDP-43 plays any role in what has been considered “the AD neurodegenerative process” or whether TDP-43 could help explain why some patients remain clinically normal, while others do not, despite both having similar degrees of AD pathology.
The primary aim of this study was to determine whether TDP-43 is independently associated with cognitive impairment and brain atrophy in AD, and hence an important contributor to the AD phenotype. A secondary aim was to determine whether cognitive impairment and atrophy would correlate with TDP-43 burden or the distribution of TDP-43 (number of brain regions showing TDP-43 immunoreactivity).
Materials and methods
Study design and participants
All cases were identified from the Mayo Clinic neuropathological database, Rochester, MN and fulfilled the following inclusion criteria: (1) intermediate–high probability AD diagnosis according to the National Institute on Aging and the Reagan Institute criteria (NIA-Reagan) [47], (2) Braak neurofibrillary tangle stage IV–VI [9] and (3) available formalin-fixed paraffin blocks of brain tissue regions. 346 cases were identified: 4 were excluded for not having available paraffin blocks, resulting in 342 cases being included in this study. All subjects were prospectively recruited and followed in the Alzheimer’s Disease Research Center or Patient Registry between 1992 and 2010.
All subjects had undergone a clinical evaluation by a dementia specialist, completed neuropsychological testing and were determined to be cognitively normal or cognitively impaired before death. The determination of cognitive status was based on consensus of a team of scientists utilizing data generated from detailed clinical and neuropsychological evaluations. For this study, we analyzed the Mini-Mental State Examination (MMSE) [17] as a measure of general cognitive impairment, Clinical Dementia Rating Scale Sum-of-Boxes (CDR-SB) [31] as a measure of functional impairment, Boston Naming Test (BNT) [27] as a measure of confrontational naming, memory subscale of the Dementia Rating Scale (mDRS) [29] as a measure of loss of episodic memory, brief questionnaire version of the Neuropsychiatric Inventory (NPI-Q) [28] as a measure of behavioral impairment, and cognitive status, at the final evaluation before death. Apolipoprotein E (APOE) genotyping was performed, as previously described [13, 23].
Neuropathological examinations were performed according to recommendations of the Consortium to Establish a Registry for AD (CERAD) [30]. Every specimen was assigned a Braak stage [9] using modified Bielschowsky silver stain, on the basis of the earliest appearance of neurofibrillary tangles. Lewy bodies and infarcts were documented, and Aβ burden measured using CERAD recommendations [30]. Hippocampal sclerosis (HpScl) was diagnosed if neuronal loss in the subiculum and CA1 regions of the hippocampus was out of proportion to the burden of neurofibrillary tangles [16].
This study was approved by the Mayo Clinic IRB. All patients or their proxies provided written informed consent before participating in any research activity.
Pathological analysis
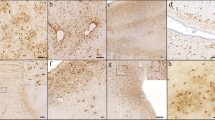
Amygdala blocks were sectioned and immunostained for TDP-43 (polyclonal antibody MC2085 that recognizes a peptide sequence in the 25-kDa C-terminal fragment [48]) with a DAKO-Autostainer (DAKO-Cytomaton, Carpinteria, CA) and 3, 3′-diaminobenzidine as the chromogen. Sections were lightly counterstained with Hematoxylin. Amygdala sections were screened (by DWD) to assess for the presence of TDP-43 immunoreactive neuronal cytoplasmic inclusions, dystrophic neurites, or neuronal intranuclear inclusions (Fig. 1). We screened the amygdala as the amygdala has been shown to be the first region affected in AD by TDP-43 pathology [19, 26]. Any AD case not showing TDP-43 immunoreactivity in the amygdala was considered TDP-negative (Fig. 2a), while any AD case showing any amount of TDP-43 immunoreactivity in the amygdala was considered TDP-positive (Fig. 2b–d). Hence, amygdaloid positivity was all that was necessary to call an individual AD case TDP-43 positive. For TDP-positive cases, we sectioned additional paraffin blocks of the middle frontal, superior temporal, and inferior parietal cortices, nucleus basalis, hippocampus, midbrain and medulla using the same protocol as described above for the amygdala. The following 14 distinct brain regions per case were reviewed simultaneously with a multi-headed microscope (by DWD and KAJ) for TDP-43 immunoreactivity: amygdala, entorhinal cortex, subiculum, hippocampal dentate fascia, occipitotemporal cortex, inferior temporal cortex, basal forebrain, insula, ventral striatum, frontal lobe, basal ganglia, substantia nigra, midbrain tegmentum and inferior olive. A region was considered positive if TDP-43 immunoreactive lesions were observed at 20× magnification screening the entire region, with subsequent confirmation at 40× magnification. The number of cases with TDP-43 immunoreactivity for each of the 14 regions is shown in Fig. 3.

Pathological assessment of TDP-43 immunoreactive inclusions. TDP-43 immunoreactive inclusions identified in the subjects with Alzheimer disease include neuronal cytoplasmic inclusions that were variable in size with some being asterisk-like and small (a), while others were larger, round and more Pick-body like (b). Dystrophic neurites were predominantly thin and thread-like (c) although in some instances there were large thick dystrophic neurites (d). In the CA1 region of the hippocampus, particularly in the cases with widespread TDP-43 immunoreactivity, there were fine neurites (e). Many of these cases with widespread TDP-43 immunoreactivity also had neuronal intranuclear inclusions that were either cat-eye like in appearance (f) or were more rounded (g). TDP-43 immunoreactivity, high-power mag ×40. Dentate fascia of the hippocampus (a–b), frontotemporal neocortices (c–d), CA1 region of the hippocampus (e), entorhinal cortex (f) and occipitotemporal cortex (g)

The range of TDP-43 immunoreactivity observed in the amygdala. Cases without TDP-43 immunoreactivity in the amygdala (a) were classified as TDP-negative, while cases showing any amount of TDP-43 immunoreactivity (b–d) were classified as TDP-positive. Amygdala TDP-43 immunoreactivity varied and included cases with scant (b), moderate (c) and severe (d) immunoreactivity. TDP-43 immunoreactivity at mag ×20

Histograms showing the prevalence of TDP-43 by region (a) and quantitative TDP-43 burden in the dentate gyrus (b). Only TDP-positive subjects are shown for both histograms. Therefore, as would be expected based on our inclusion criteria, 100 % of the subjects had amygdala involvement (a). Involvement in the other 13 regions was variable with the frontal lobe and basal ganglia being involved in the least number of subjects. In the bottom histogram (b), TDP-43 burden was highly skewed to the right. As reported, there was a strong correlation between TDP-43 burden in the dentate gyrus and number of regions affected
To ensure antibody sensitivity, we additionally screened amygdala sections from 10 % of the TDP-negative cases using a different antibody against phosphorylated TDP-43 peptide (1:5,000 rabbit polyclonal anti-human phosphoserine 409/410). None of the cases that had initially screened negative with the polyclonal antibody MC2085 showed TDP-43 immunoreactivity with the phosphorylated antibody, ensuring excellent sensitivity of MC2085.
TDP-43 burden was assessed in the hippocampal dentate fascia using the Aperio slide scanner and a customized color deconvolution algorithm enabling the detection of any abnormal TDP-43 (Fig. 4). The dentate fascia was selected for the burden analysis since the dentate fascia has been demonstrated to be most strongly associated with memory loss [36]. TDP-43 immunostained sections of the posterior hippocampus at the level of the lateral geniculate were scanned at ultra-resolution on the ScanScope XT (Aperio Technologies, Vista, CA). This instrument permits scanning of the entire slide from which large areas of interest can be annotated using ImageScope version 11.2 (Aperio Technologies, Vista, CA). The method greatly increases the sampling frame compared to some other image analysis systems that are limited to the field of view of the microscope or require image tiling [38]. The entirety of the dentate fascia was assessed to quantitatively determine TDP-43 immunohistochemical burden. To operationalize annotation of the dentate fascia, the ruler tool was used to measure 125 μm across the granule cell layer to molecular layer to avoid quantification variability resulting from tissue sectioning differences. Any dust or dirt particles or tissue folds were excluded using the negative trace tool. Annotated layers were analyzed in Spectrum version 11.2 (Aperio Technologies, Vista, CA) using a custom-designed color deconvolution algorithm, as previously described [22]. After applying the color deconvolution algorithm, each high-resolution image was reviewed independently by two investigators (MM & AL) to ensure that only abnormal TDP-43 was being measured. Cases where the algorithm was unable to separate abnormal TDP-43 from normal nuclear TDP-43, were removed from further analysis of TDP-43 burden (n = 20). TDP-43 burden was then expressed as the area of immunoreactive pixels to the total area of the annotated region.

TDP-43 burden assessment. TDP-43 immunoreactive inclusions in the dentate fascia were assessed quantitatively to determine TDP-43 burden. High-resolution images created with the Aperio slide scanner were utilized to detect abnormal TDP-43 using a custom-designed color deconvolution algorithm. Top panel shows TDP-43 immunoreactive inclusions, while bottom panel shows the results of the color deconvolution algorithm in which abnormal TDP-43 is depicted in red
MRI analysis
Two-hundred and forty-eight subjects had an antemortem volumetric MRI performed with a standardized protocol [21]. The MRI closest to death was selected for each subject to correspond to the clinical data. Two-hundred and twenty subjects were scanned at 1.5 T. A reference group for volumetric analysis included 46 healthy controls that had antemortem MRI and normal pathological diagnosis (Braak 0-III, NIA-Reagan no/low) and were TDP-negative (mean age = 82 ± 6.46 % female). Gradient non-linearity and intensity non-uniformity corrections were applied to all MRI.
Gray matter volumes of specific regions of interest were calculated using atlas-based parcellation in SPM5 and the automated anatomic labeling atlas [42], as previously described [45]. Volumes were calculated for medial temporal regions (hippocampus, amygdala and entorhinal cortex) and association cortices (fusiform gyrus, medial and lateral frontal and parietal lobes and lateral temporal lobe). Left and right volumes were averaged. Regional volumes were scaled by total intracranial volume [46] to correct for head size. Gray matter atrophy was also assessed at the voxel level using voxel-based morphometry (VBM) [6]. Standard preprocessing steps were employed, including normalization to a customized template, unified segmentation [7], modulation and smoothing at 8 mm full width at half-maximum. A VBM “full-factorial” (i.e., ANCOVA) model was used to compare TDP-negative and TDP-positive subjects to controls (p < 0.05 using family-wise error correction), and to each other (uncorrected p < 0.001). Comparisons were performed separately at Braak IV, V and VI. Age, gender and total intracranial volume were included as covariates in all analyses.
Statistical analysis
We used linear, binary logistic and ordinal logistic regression models to estimate associations between outcome variables and TDP-43. In our first modeling analysis, we treated TDP-43 status as a binary predictor.
To better understand the relationship between TDP-43 and cognition and brain atrophy while taking into account the Braak neurofibrillary tangle stage, we stratified by Braak stage and tested for additive associations and interactions between TDP-43 and Braak using linear or logistic regression adjusted for age at death. This analysis allowed us to investigate whether the association between TDP-43 and cognition/brain atrophy differed across Braak stage IV, V and VI. We extended this analysis in two ways to account for any potential genetic or other pathological confounders: (1) to account for confounding effects due to APOE, infarctions, Lewy bodies, neuritic plaques (CERAD) we included these variables as covariates in our models; (2) To address the issue of whether the TDP-43 effects were wholly or partly mediated by HpScl we added this factor to the models and evaluated the resulting HpScl-adjusted TDP-43 associations. We encountered the problem of logistic regression separation in models involving the cognitive impairment outcome. This was because all 104 TDP-positive subjects at Braak stage VI were cognitively impaired and thus in this subgroup there were no “non-events”. To address this problem, we used weakly informative variance stabilizing data augmentation priors for the coefficient corresponding to the Braak VI by TDP-43 interaction [40]. This method is very closely related to the popular approach of adding one success and one failure when estimating a proportion [1]. Our priors specified that on the odds ratio scale 95 % of the prior probability was in the range 1/16–16 which serves to conservatively shrink estimates toward the null.
For our secondary analyses we modeled the outcome variables as a function of TDP-43 burden and as a function of the number of brain regions positive for TDP-43 (i.e., distribution). These analyses were limited only to subjects who were TDP-positive. Quantitative TDP-43 burden was expressed as a percentage of pixels showing immunoreactivity. We rank transformed the numeric value because of extreme right skewness (Fig. 3). So that a 1-unit increase was more interpretable, we divided the rank-transformed value by 40 to obtain a uniformly distributed predictor with a max (and range) of ~4. In these analyses we assessed three models that included: age at death alone; age at death and Braak stage given our interest in Braak stage; and age at death, APOE, Braak stage, CERAD, Lewy bodies, infarcts and HpScl as adjustment covariates to remove the potential confounding effect of all these variables.
Results
One-hundred and ninety-five cases (57 %) were TDP-positive. After accounting for age at death, the TDP-positive subjects had a higher proportion of APOE ε4 carriers, and worse performance on MMSE, CDR-SB, BNT and mDRS than the TDP-negative subjects (Table 1). The TDP-positive group had a lower proportion of subjects who were cognitively normal at death (2 vs 19 %). The TDP-positive group had higher Braak stage, and higher proportions of Lewy bodies and HpScl. Volumes of all medial temporal structures and fusiform gyrus were smaller in the TDP-positive group compared to the TDP-negative group, with no differences observed in other association cortices.
Group differences adjusting for potential confounders
After accounting for age at death, CERAD, APOE ε4, infarcts, Braak stage and Lewy bodies, TDP-43 had an additive effect on cognitive impairment, MMSE, mDRS, CDR-SB and BNT (Fig. 5). When we further account for HpScl in our mediation analysis, the additive effect of TDP-43 on cognitive impairment and BNT remained significant with similar trends persisting for MMSE, mDRS and CDR-SB (Fig. 5). An interaction, in addition to an additive effect beyond Braak, was observed for MMSE and CDR-SB. Specifically, a greater difference was observed between TDP-positive and TDP-negative groups at Braak IV and V. For example, a greater than 4-point difference on MMSE was observed at Braak stages IV and V between TDP-positive and TDP-negative subjects, while less than a 2-point difference was observed for MMSE at Braak stage VI.

Models for clinical variables. Estimates and 95 % CIs for the mean difference for each term in the primary model along with the estimates and 95 % CIs for the hippocampal sclerosis and TDP terms from the mediation model. P values for the primary models summarize a test of an additive effect of TDP after accounting for Braak stage, age, CERAD, APOE ε4, infarctions, and Lewy bodies (P add) and a test of an interaction between TDP and Braak stage versus the additive model (P int). The p values for the mediation model additionally account for HpScl. The cognitive impairment outcome was modeled with logistic regression and the estimates were transformed to the percentage scale with age set to the mean (85). All other outcomes were modeled using linear regression
Similar to the clinical outcomes, after accounting for age at death, CERAD, APOE ε4, infarcts, Braak stage and Lewy bodies, TDP-43 had an additive effect on hippocampal, amygdala and entorhinal cortex volumes (Fig. 6). When we further account for HpScl in our mediation analysis, the additive effect of TDP-43 on hippocampal volume remained significant with similar trends persisting for the amygdala and entorhinal cortex volumes (Fig. 6). No added interactions were observed for these medial temporal volumes. Conversely, interactions, but no additive effects of TDP-43 were observed on the association cortex volumes.

Models for neuroimaging variables. Estimates and 95 % CIs for the mean volume difference for each term in the primary model along with the estimates and 95 % CIs for the hippocampal sclerosis and TDP terms from the mediation model. The p values for the primary models summarize the test of an additive effect of TDP after accounting for Braak stage, age, CERAD, APOE ε4, infarctions, and Lewy bodies (P add) and a test of an interaction between TDP and Braak stage versus the additive model (P int). The p values for the mediation model additionally account for HpScl. All outcomes were modeled using linear regression
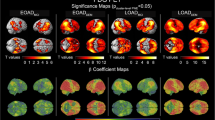
On VBM, the TDP-positive AD subjects showed progressive worsening of volume loss with increasing Braak, when compared to the 46 healthy control subjects (Fig. 7). The TDP-negative AD subjects showed no volume loss at Braak IV and only a tiny area of focal loss at Braak V, when compared to the healthy controls. When the TDP-positive and TDP-negative subjects were directly compared, the TDP-positive subjects showed greater medial temporal loss than the TDP-negative subjects at all Braak stages. Conversely, the TDP-negative subjects showed no regions of greater loss than the TDP-positive subjects, except at Braak VI, where more parieto-occipital loss was observed.

Regional patterns of gray matter volume loss in TDP-negative and TDP-positive AD subjects within Braak stages IV, V and VI. The TDP-negative and TDP-positive subjects are compared to pathologically normal controls (corrected using family-wise error at p < 0.05) and compared to each other (uncorrected p < 0.001). Results are shown on three-dimensional renderings of the brain
TDP-43 burden and distribution
A strong correlation was observed between TDP-43 burden and distribution (Spearman correlation = 0.74, p < 0.001). After accounting for age at death, CERAD, APOE ε4, infarcts, Braak stage, Lewy bodies and HpScl, TDP-43 burden and TDP-43 distribution were associated with many clinical and neuroimaging variables (Tables 2, 3). For all these associations, clinical performance worsened and volume loss increased with higher TDP-43 burden in the dentate gyrus, and as more regions were affected by TDP-43.
Discussion
In this large clinico-imaging pathological study, we demonstrate that TDP-43 is associated with the major features of AD: memory loss and medial temporal atrophy. In fact, when AD pathology was less severe (i.e. Braak Stages IV and V), the absence of TDP-43 was strongly associated with normal cognition. Consequently, TDP-43 appears to play an important role in the cognitive and neuroimaging characteristics that have been linked to AD.
The findings from this study challenge the perception that Aβ and tau are the only important proteins accounting for the clinical features of AD by demonstrating that TDP-43 also has an effect on the core clinical and neuroimaging features considered pathognomonic for AD. Importantly, the effects of TDP-43 on cognition and atrophy occurred after taking into account AD-related pathogenetic potential confounders, including Braak stage, and hence tau, the presence of Lewy bodies, Aβ, infarctions and APOE ε4. The effects of TDP-43 also persisted after taking into account HpScl. Therefore, although HpScl likely influences cognition and brain atrophy when present in AD [10, 33, 35], the effects of TDP-43 on the clinical and imaging variables assessed in our cohort were not mediated by HpScl. This suggests that TDP-43 itself is an important factor. The fact that TDP-43 had an effect on these outcome variables at Braak IV, which represents mild AD, is evidence that TDP-43 is not just a feature of severe pathology. In fact, the differences between TDP-positive and TDP-negative groups were most striking at Braak IV and V. One explanation for this finding is that TDP-43 plays a more important role in the early stages of AD, but once tau deposition is widespread, i.e., Braak VI, the importance of TDP-43 becomes somewhat overshadowed. Alternatively, it is also possible that the effects of TDP-43 at Braak VI are just as strong as they are at Braak IV and V, but are more difficult to detect since the range of cognitive values at Braak VI are likely to be truncated given that most subjects are severely affected at this stage.
One of the most important findings of this study was that the absence of TDP-43 was strongly associated with normal cognition despite subjects with and without TDP-43 having similar degrees of AD pathology. In our sample, approximately one-third of the TDP-negative Braak IV and V subjects were cognitively normal, compared to only 4 % of the TDP-positive Braak IV and V subjects. Given that the TDP-positive Braak IV and V groups showed greater medial temporal atrophy than the TDP-negative Braak IV and V subjects, it appears that the presence of TDP-43 creates a synergistic effect with the AD pathology. A similar effect has also been previously reported to occur with vascular pathology [39] and with argyrophilic grain disease [25, 41]. It therefore appears that the resilience of some subjects to AD pathology can be overpowered by the presence of TDP-43. These findings help further shed light on those subjects that remain cognitively unimpaired despite having AD pathology [5, 15, 37]. Previous studies have suggested that cognitively resilient subjects may have lower burdens of Aβ fibrillar plaques, oligomeric Aβ deposits or hyperphosphorylated tau in synapses [37], or preserved densities of presynaptic terminals and dentritic spines [5], although none of these studies assessed TDP-43. We now know that such subjects are also less likely to have TDP-43.
An association was also identified between APOE ε4, a major risk factor for AD [12], and TDP-43. The proportion of subjects with the APOE ε4 allele was higher in the presence of TDP-43 across all Braak stages. At Braak VI, for example, approximately three-quarters of the TDP-positive subjects had the APOE ε4 allele, a higher proportion than typically observed in AD [32]. It is therefore probable that APOE ε4 increases the risk of TDP-43 pathology, an association not previously recognized. However, APOE ε4, like Lewy body disease, Aβ deposition, infarctions and HpScl, was not a confounder of the observed associations with TDP-43.
An intriguing observation was the atypical characteristics of the Braak VI TDP-negative group. This group showed widespread atrophy, yet the atrophy pattern was unusual when compared to the Braak VI TDP-positive group, with more atrophy in cortical association regions and less atrophy in medial temporal regions. This group of subjects was approximately 10 years younger than all other groups and is reminiscent of the hippocampal sparing variant of AD, which typically is associated with younger age, relatively greater cortical atrophy, an atypical distribution of neurofibrillary tangles [32, 46] and absence of TDP-43 [8]. It is unlikely that TDP-43 is protective against involvement of the cortex and more likely that the greater cortical atrophy in the TDP-negative subjects is being driven by a greater burden of cortical tau in these younger subjects [32, 46]. Conversely, the greater hippocampal atrophy observed in the older TDP-positive subjects is likely associated with the presence of TDP-43. It therefore appears that tau and TDP-43 may have distinct effects in AD as shown in these models from our cohort that presumably generalizes to the general population (Fig. 8).

Summaries illustrating the relationship between normal cognition, MMSE and hippocampal volume loss, and Braak stage, and TDP-43 status in AD. In the absence of TDP-43, there is a progressively higher proportion of subjects with cognitive impairment across Braak stages with the steepest changes in proportion occurring between Braak stages V and VI. On the contrary, TDP-43 is consistently associated with a high proportion of impaired subjects regardless of Braak stage. Similar trends are seen with MMSE and hippocampal volume. While we illustrate this phenomenon using cognition, MMSE and hippocampus, similar trends were observed for other clinical and imaging outcome variables
The co-existence of the hallmark AD pathologies of Aβ and tau, along with TDP-43, could be interpreted in two ways. First, TDP-43 is simply a pathological feature of AD. Arguments supporting this hypothesis are (1) the fact that the TDP-negative subjects were more likely to be cognitively normal and show atypical patterns of atrophy and (2) that important factors that are associated with Braak stage, and hence tau pathology, were also associated with TDP-43, including APOE ε4, memory loss and medial temporal atrophy. One strong argument against this interpretation, however, is the fact that there were many AD subjects without TDP-43 that were cognitively impaired. The second more likely possibility is that the presence of TDP-43 represents a secondary or independent pathology that shares overlapping features with AD by targeting the medial temporal lobe. If this latter interpretation is correct then TDP-43 may have obscured our view of the true AD clinico-imaging phenotype, given that such a high proportion of AD cases have TDP-43.
We acknowledge that our outcome measures were not all independent. However, since our aim was not to determine which outcome variable is most affected by TDP-43, and the fact that all the variables we assessed for the study are strongly associated with AD, our approach was very reasonable. We also note that our data for TDP-43 deposition in the dentate gyrus was highly right skewed. However, we addressed this potential problem by rank transforming the burden data prior to our modeling analyses. We did not address the issue of the spatial distribution of TDP-43 in AD given that we have previously done so and describe five stages for TDP-43 deposition in AD for this exact cohort [26]. In fact, all five stages were roughly equally represented. Hence our data was not biased by the overrepresentation of early or late stages.
In this study, we demonstrate that TDP-43 is an important player in the AD field, particularly during the early phases of neurodegeneration. Our findings suggest that in order to be cognitively resilient TDP-43 must be absent. That is, the synergistic effect of having TDP-43 results in cognitive impairment. TDP-43 therefore should be considered a potential therapeutic target for the future treatment of AD.
References
Agresti A, Coull BA (1998) Approximate is better than "exact" for interval estimation of binomial proportions. Am Stat 52:119–126
Alzheimer A (1907) Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift für Psychiatrie und Psychisch-Gerichtliche Medizin 64:146–148
Amador-Ortiz C, Lin WL, Ahmed Z et al (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61:435–445. doi:10.1002/ana.21154
Arai T, Mackenzie IR, Hasegawa M et al (2009) Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol 117:125–136. doi:10.1007/s00401-008-0480-1
Arnold SE, Louneva N, Cao K et al (2013) Cellular, synaptic, and biochemical features of resilient cognition in Alzheimer’s disease. Neurobiol Aging 34:157–168. doi:10.1016/j.neurobiolaging.2012.03.004
Ashburner J, Friston KJ (2000) Voxel-based morphometry—the methods. NeuroImage 11:805–821
Ashburner J, Friston KJ (2005) Unified segmentation. NeuroImage 26:839–851
Bigio EH, Mishra M, Hatanpaa KJ et al (2010) TDP-43 pathology in primary progressive aphasia and frontotemporal dementia with pathologic Alzheimer disease. Acta Neuropathol 120:43–54. doi:10.1007/s00401-010-0681-2
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Brenowitz WD, Monsell SE, Schmitt FA, Kukull WA, Nelson PT (2014) Hippocampal sclerosis of aging is a key Alzheimer’s Disease mimic: clinical-pathologic correlations and comparisons with both Alzheimer’s Disease and non-tauopathic frontotemporal lobar degeneration. J Alzheimers Dis 39:691–702. doi:10.3233/JAD-131880
Buratti E, Brindisi A, Pagani F, Baralle FE (2004) Nuclear factor TDP-43 binds to the polymorphic TG repeats in CFTR intron 8 and causes skipping of exon 9: a functional link with disease penetrance. Am J Hum Genet 74:1322–1325. doi:10.1086/420978
Corder EH, Saunders AM, Strittmatter WJ et al (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923
Crook R, Hardy J, Duff K (1994) Single-day apolipoprotein E genotyping. J Neurosci Methods 53:125–127
Davidson YS, Raby S, Foulds PG et al (2011) TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s syndrome: association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol 122:703–713. doi:10.1007/s00401-011-0879-y
Davis DG, Schmitt FA, Wekstein DR, Markesbery WR (1999) Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol 58:376–388
Dickson DW, Davies P, Bevona C et al (1994) Hippocampal sclerosis: a common pathological feature of dementia in very old (≥80 years of age) humans. Acta Neuropathol 88:212–221
Folstein MF, Folstein SE, McHugh PR (1975) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198
Gosche KM, Mortimer JA, Smith CD, Markesbery WR, Snowdon DA (2002) Hippocampal volume as an index of Alzheimer neuropathology: findings from the Nun Study. Neurology 58:1476–1482
Hu WT, Josephs KA, Knopman DS et al (2008) Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol 116:215–220. doi:10.1007/s00401-008-0400-4
Jack CR Jr, Dickson DW, Parisi JE et al (2002) Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology 58:750–757
Jack CR Jr, Lowe VJ, Senjem ML et al (2008) 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain J Neurol 131:665–680. doi:10.1093/brain/awm336
Janocko NJ, Brodersen KA, Soto-Ortolaza AI et al (2012) Neuropathologically defined subtypes of Alzheimer’s disease differ significantly from neurofibrillary tangle-predominant dementia. Acta Neuropathol 124:681–692. doi:10.1007/s00401-012-1044-y
Josephs KA, Tsuboi Y, Cookson N, Watt H, Dickson DW (2004) Apolipoprotein E epsilon 4 is a determinant for Alzheimer-type pathologic features in tauopathies, synucleinopathies, and frontotemporal degeneration. Arch Neurol 61:1579–1584. doi:10.1001/archneur.61.10.1579
Josephs KA, Whitwell JL, Knopman DS et al (2008) Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 70:1850–1857. doi:10.1212/01.wnl.0000304041.09418.b1
Josephs KA, Whitwell JL, Parisi JE et al (2008) Argyrophilic grains: a distinct disease or an additive pathology? Neurobiol Aging 29:566–573. doi:10.1016/j.neurobiolaging.2006.10.032
Josephs KA, Murray ME, Whitwell JL et al (2014) Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol 127:441–450. doi:10.1007/s00401-013-1211-9
Kaplan E, Goodglass H, Weintraub S (1983) The Boston Naming Test. Lea & Febiger, Philadelphia
Kaufer DI, Cummings JL, Ketchel P et al (2000) Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci 12:233–239
Mattis S (1988) Dementia Rating Scale. Psychological Assessment Resources, City
Mirra SS, Heyman A, McKeel D et al (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486
Morris JC (1993) The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43:2412–2414
Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW (2011) Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 10:785–796
Nelson PT, Schmitt FA, Lin Y et al (2011) Hippocampal sclerosis in advanced age: clinical and pathological features. Brain J Neurol 134:1506–1518. doi:10.1093/brain/awr053
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. doi:10.1126/science.1134108
Pao WC, Dickson DW, Crook JE, Finch NA, Rademakers R, Graff-Radford NR (2011) Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord 25:364–368. doi:10.1097/WAD.0b013e31820f8f50
Pavlopoulos E, Jones S, Kosmidis S et al (2013) Molecular mechanism for age-related memory loss: the histone-binding protein RbAp48. Sci Transl Med 5:200ra115. doi:10.1126/scitranslmed.3006373
Perez-Nievas BG, Stein TD, Tai HC et al (2013) Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain J Neurol 136:2510–2526. doi:10.1093/brain/awt171
Rojo MG, Garcia GB, Mateos CP, Garcia JG, Vicente MC (2006) Critical comparison of 31 commercially available digital slide systems in pathology. Int J Surg Pathol 14:285–305. doi:10.1177/1066896906292274
Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR (1997) Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277:813–817
Sullivan SG, Greenland S (2013) Bayesian regression in SAS software. Int J Epidemiol 42:308–317. doi:10.1093/ije/dys213
Thal DR, Schultz C, Botez G et al (2005) The impact of argyrophilic grain disease on the development of dementia and its relationship to concurrent Alzheimer’s disease-related pathology. Neuropathol Appl Neurobiol 31:270–279. doi:10.1111/j.1365-2990.2005.00635.x
Tzourio-Mazoyer N, Landeau B, Papathanassiou D et al (2002) Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage 15:273–289
Uryu K, Nakashima-Yasuda H, Forman MS et al (2008) Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol 67:555–564. doi:10.1097/NEN.0b013e31817713b5
Whitwell JL, Josephs KA, Murray ME et al (2008) MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology 71:743–749
Whitwell JL, Jack CR, Jr., Przybelski SA et al. (2011) Temporoparietal atrophy: A marker of AD pathology independent of clinical diagnosis. Neurobiol Aging 32:1531–1541
Whitwell JL, Dickson DW, Murray ME et al (2012) Neuroimaging correlates of pathologically defined subtypes of Alzheimer’s disease: a case-control study. Lancet Neurol 11:868–877. doi:10.1016/S1474-4422(12)70200-4
WorkingGroup (1997) Consensus recommendation for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for Neuropathologic Assessment of Alzheimer’s Disease. Neurobiol Aging 18:S1–S2
Zhang YJ, Xu YF, Cook C et al (2009) Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci USA 106:7607–7612. doi:10.1073/pnas.0900688106
Acknowledgments
This study was funded by the US National Institute of Heath (NIA) Grants R01-AG037491 (to KAJ), R21-AG038736 (to JLW), P50-AG016574 (to RCP) and R01-AG011378 (to CRJ). We wish to thank the families of the patients who donated their brains to science allowing completion of this study. We further wish to thank Kris Johnson, Linda Rousseau, Virginia Phillips and Monica Casey-Castanedes for pathological support.
Conflict of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Josephs, K.A., Whitwell, J.L., Weigand, S.D. et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 127, 811–824 (2014). https://doi.org/10.1007/s00401-014-1269-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-014-1269-z