
Abstract
Alzheimer’s disease (AD) can be classified based on the relative density of neurofibrillary tangles (NFTs) in the hippocampus and association cortices into three subtypes: typical AD, hippocampal-sparing AD (HpSp AD), and limbic-predominant AD (LP AD). AD subtypes not only have pathologic, but also demographic, clinical, and genetic differences. Neurofibrillary tangle-predominant dementia (NFTD), a disorder with NFTs relatively restricted to limbic structures, shares this feature with LP AD raising the possibility that NFTD is a variant of AD. The objective criteria for pathologic diagnosis of NFTD are not available. A goal of this study was to design a mathematical algorithm that could diagnose NFTD from NFT and senile plaque (SP) counts in hippocampus and association cortices, analogous to that used to subtype AD. Moreover, we aimed to compare pathologic, demographic, clinical, and genetic features of NFTD (n = 18) with LP AD (n = 19), as well as the other AD subtypes, typical AD (n = 52) and HpSp AD (n = 17). Using digital microscopy, we confirmed that burden of phospho-tau (CP13) and of an NFT conformational epitope (Ab39) correlated with NFT densities and showed expected patterns across AD subtypes. HpSp AD had the highest and LP AD had the lowest burden of cortical CP13 and Ab39 immunoreactivity. On the other hand, cortical β-amyloid burden did not significantly differ between AD subtypes. Semi-quantitative assessment of SPs in the basal ganglia did show HpSp AD to have significantly more frequent presence of SPs compared to typical AD, which was more frequent than LP AD. Compared to LP AD, NFTD had an older age at disease onset and shorter disease duration, as well as lower Braak NFT stage. NFTs and SPs on thioflavin-S fluorescent microscopy, as well as CP13, Ab39, and Aβ immunoreactivities were very low in the frontal cortex of NFTD, differentiating NFTD from AD subtypes, including LP AD. MAPT H1H1 genotype frequency was high (~70 %) in NFTD and LP AD, and similar to typical AD, while APOE ε4 carrier state was low in NFTD. While it shares clinical similarities with regard to female sex predominance, onset in advanced age, and a slow cognitive decline, NFTD has significant pathologic differences from LP AD, suggesting that it may not merely be a variant of AD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) pathology is characterized by the presence of phosphorylated tau in neurofibrillary tangles (NFTs) and dystrophic neurites, as well as abundant extracellular β-amyloid in senile plaques (SPs). Previous work has demonstrated a range of focal clinical presentations that are related to underlying atypical AD pathology, including focal cortical degeneration [1, 12, 28]. A neuroimaging study identified temporoparietal atrophy as an imaging correlate of AD pathology in AD patients with non-amnestic clinical presentations [41]. Historically, age has been used to categorize presenile and senile dementia of the Alzheimer’s type, with differential relative cortical and hippocampal involvement according to age of onset [32]. More recently, age of onset in conjunction with apolipoprotein E (APOE) genotype has been shown to associate with medial temporal lobe vulnerability in AD [39]. Our previous work has shown that AD can be neuropathologically classified into three subtypes based on the relative abundance of NFTs in the hippocampus and cortex [hippocampal-sparing AD (HpSp), typical AD, and limbic-predominant AD(LP)] and that AD subtypes so classified had characteristic demographic, pathologic, clinical, and genetic features [28, 30].
Hippocampal-sparing AD has disproportionate cortical NFTs compared to relative sparing of the hippocampus. This contrasts with typical AD where distribution and density of NFTs follow the pattern suggested by the Braak [6] NFT staging scheme, with relatively greater NFTs in the medial temporal lobe structures than in convexity neocortex. LP AD has disproportionately more hippocampal NFTs, with sparse NFTs in association cortices and little or no NFTs in primary cortices. The pattern of neurofibrillary degeneration in LP AD is reminiscent of limbic neurofibrillary tangle dementia [19, 20] or more commonly known as neurofibrillary tangle-predominant dementia (NFTD) [21, 22]. NFTD is pathologically characterized by NFTs relatively confined to the limbic lobe structures and no, or at most sparse, cortical SPs [9, 21, 22]. If SPs are present, they are typically diffuse β-amyloid deposits without neuritic elements similar to those found in the brains of non-demented elderly individuals [5, 21]. There is currently debate as to whether NFTD is a neurodegenerative disease distinct from AD [21, 33], or whether it is part of the AD spectrum [31]. Preliminary pathologic evidence suggests that NFTD cases would not be classified as LP type of AD [30] given the lack of cortical neuritic type SPs, which are required for neuropathologic diagnosis of AD [15]. Clinically, NFTD patients are often diagnosed to have AD, but their disease process is characterized by slower deterioration [18, 21]. The current genetic findings of NFTD are limited to the observation of low APOE ε4 allele frequency [18, 21], especially compared to AD where the APOE ε4 allele has been shown to have a significant impact on AD risk [40] and amyloid plaque pathology [16, 35]. Given recent evidence that implicates microtubule-associated protein tau (MAPT) H1H1 genotype in AD subtypes [30], we compared NFTD to AD subtypes with respect to both APOE and MAPT.
Until now, our studies have not directly compared LP AD to NFTD, or explored similarities and differences between these two limbic-centric disorders [28, 30]. Furthermore, our AD subtype classification was based on thioflavin-S fluorescent microscopy and has not been validated with tau and β-amyloid immunohistochemistry. Therefore, the present study had three aims: (1) to develop an operational classification algorithm to objectively diagnose NFTD; (2) to compare clinical, pathologic, and genetic features between NFTD and LP AD; and (3) to characterize NFTD and AD subtypes with respect to quantitative burden of cortical tau and β-amyloid with digital microscopic image analysis.
Materials and methods
Mayo Clinic Jacksonville brain bank database was searched for AD and NFTD that had been reviewed and diagnosed by a single neuropathologist (DWD). Classification of AD subtypes using a mathematical algorithm of hippocampal and cortical NFTs has been previously described in detail [30]. The following exclusion criteria were used for AD: Braak NFT stage ≤ IV, absence of paraffin blocks for immunohistochemical studies, and presence of hippocampal sclerosis (initial sample size HpSp = 97, typical = 665, LP = 127). Cases were further required to have antemortem clinical information available. Given the retrospective nature of this study and the absence of standardized clinical assessments, the sum of both HpSp and LP cases was used to set the number of typical AD for clinical history review. AD cases with three or more Mini-Mental State Exam (MMSE) scores [11] were included in the final AD subtype cohort (final sample size HpSp = 17, Typical = 52, LP = 19). Clinical parameters and availability are described later in “Materials and methods”.
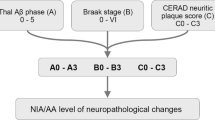
Neurofibrillary tangle-predominant dementia was objectively diagnosed with a mathematical algorithm that used hippocampal and cortical NFT and SP counts in cases with a gestalt diagnosis of NFTD to set standards. NFTD had to have SP counts of ≤30 according to the minimum level described by Khachaturian criteria [24] (see Fig. 1). Of the 62 NFTD cases identified, we excluded cases with concomitant tau pathology (e.g., corticobasal degeneration, progressive supranuclear palsy, Guam parkinsonism–dementia complex, diffuse argyrophilic grain disease) to avoid confounding immunohistochemical differences in tau burden that may not accurately reflect differences among AD subtypes and when compared with NFTD. Other concurrent postmortem diagnoses that could interfere with data interpretation included frontotemporal lobar degeneration, hippocampal sclerosis, amyotrophic lateral sclerosis, and multiple system atrophy. Of the remaining 20 cases, 1 was excluded because of absence of dementia in the clinical history.

Neurofibrillary tangle-predominant dementia (NFTD) was objectively diagnosed with a mathematical algorithm that used hippocampal and cortical NFT and SP counts in cases with a gestalt diagnosis of NFTD to set standards. A flowchart of the algorithm illustrates inclusion and exclusion criteria
There were two primary sources for study cases: the State of Florida Alzheimer’s Disease Initiative (ADI) (n = 53; 50 %) [4] and the Mayo Clinic Jacksonville Memory Disorder Clinic (n = 30; 28 %). Of the remaining cases (n = 22), 2 (2 %) were from the Einstein Aging Study (P01 AG03949), 2 (2 %) from the CurePSP Brain Bank, 2 (2 %) from Mayo Clinic Jacksonville Movement Disorder Clinic, 2 (2 %) from the Florida Alzheimer’s Disease Research Center (P50 AG25711), and 15 (14 %) from various referral sources.
Standardized neuropathologic assessment included gross and microscopic evaluation. Senile plaque (SP, 10× objective) and NFT densities (40× objective), using thioflavin-S fluorescent microscopy, were assessed on an Olympus BH2 fluorescent microscope [36–38]. SP counts were truncated at 50 (i.e., twice the number needed for neuropathologic AD diagnosis [24]) and included primitive, neuritic, and cored type of plaques. NFT counts used in the classification algorithm include intracellular and extracellular tangles from two hippocampal sectors (CA1 and subiculum) and three association cortices (middle frontal, inferior parietal, and superior temporal). The detailed algorithm methods have been previously described, including steps, sample sizes, and median values [30]. To classify NFTD cases objectively, a Microsoft Excel 2003 function was designed. Three algorithm requirements were written to classify NFTD: (1) the hippocampal and average hippocampal NFT densities had to be greater than the minimum NFT found in cases with a gestalt diagnosis of NFTD (minimum of 9 for CA2/3, 5 for CA1, 2 for subiculum, and 10 for hippocampal average); (2) at least three of the cortical and average cortical NFT densities had to be less than the maximum NFT found in cases with a subjective diagnosis of NFTD (maximum of 35 for superior temporal, 4 for inferior parietal, 6 for mid-frontal, and 14 for average cortical); and (3) the SP densities in the association cortex had to be less than or equal to the cutoff according to Khachaturian criteria [24] (≤30 for middle frontal, inferior parietal, and superior temporal cortices).
Five-micrometer-thick sections of formalin-fixed, paraffin-embedded tissue from the middle frontal gyrus were stained with H&E. Additional serial sections were processed using a DAKO Autostainer (Universal Staining System Carpinteria, CA, USA) using the chromogen 3,3′-diaminobenzidine and immunostained for phospho-tau detecting early neuritic and NFT pathology, including pretangles (CP13, mouse IgG1, 1:1,000, generous gift of Peter Davies, Albert Einstein College of Medicine, Bronx, NY, USA), an antibody to a conformational epitope in NFTs detecting late-stage tangles (Ab39, monoclonal, 1:350, generous gift from Shu-Hui Yen, Mayo Clinic, Jacksonville, FL, USA) [42], pan-Aβ (33.1.1, 1:1,000, human Aβ1-16 specific) [25], and Aβ40 (13.1.1, 1:1,000, human Aβ-specific) [25]. Slides were counterstained with hematoxylin after immunostaining. Stained slides were digitally scanned at 20× on the ScanScope XT (Aperio, Vista, CA, USA) and viewed/annotated using ImageScope v10.2 software (Aperio, Vista, CA, USA). The regions of interest (ROI) for each case were initially drawn on the H&E section and the ROI was transferred to the immunostained slides. Slides were edited if there were any differences between the serial sections. The gray–white matter boundary was evaluated at 20× and marked with arrow tools along a parallel gyrus, neither including the gyral ridge nor depth. Two custom algorithms were designed to specifically detect the optical density of the tau and amyloid [8]. The result is a percentage burden that reflects the amount of stained pathology out of the total area that was stained.
Immunohistochemical methods for assessment of TAR DNA binding protein 43 (TDP-43) pathology [2] and Lewy body pathology [38] were performed as previously described. Non-Alzheimer pathologies are reported as frequencies in Table 1. Cerebral amyloid angiopathy (CAA) was graded on a four-point semi-quantitative scale: none, mild, moderate, and severe using thioflavin-S microscopy. The severity of NFTs and SPs in the molecular layer of the dentate fascia and basal ganglia was similarly graded on a four-point semi-quantitative scale by retrospectively reviewing neuropathology reports from each case. Vascular calcification was assessed using a four-point semi-quantitative severity scale in the basal ganglia on a routine hematoxylin and eosin (H&E) stained section by extracting information from neuropathology reports. Ab39 immunohistochemistry in the midbrain and pons was assessed using a four-point scale. Midbrain severity was considered: mild if Ab39-immunopositive neurites and <10 NFTs were confined to the substantia nigra; moderate if significant neuritic and extracellular NFT pathology was found in the substantia nigra, with widespread pathology found in the perinigral and periaqueductal gray; severe if neuritic, extracellular NFT were found in substantia nigra, and perinigral and periaqueductal gray. The locus coeruleus and raphe nuclei were considered as: mild, if NFTs were disproportionate to neurons present and contained no extracellular NFTs; moderate, if of the neurons affected a greater proportion were NFTs; severe, if extracelluar NFTs were greater than the number of NFTs. The severity scores were used to determine the absence (sum of none and mild semi-quantitative severity scores) versus the presence (sum of moderate and severe scores) of pathology. The presence of any NFTs in the basal ganglia, whether mild or moderate, was sufficient to be summed as present given the scarcity of NFT pathology.
Clinic reports were reviewed blind to pathologic diagnosis. This study had Mayo Clinic Institutional Review Board approval. Clinical parameters included: education, age of onset, disease duration, and MMSE scores and dates. Elapsed years between age at death and age of onset were used to calculate disease duration. Three or more MMSE testing dates and scores were required to calculate longitudinal decline as a slope, setting MMSE score as the dependent variable and elapsed years between testing and death as the independent variable. Elapsed time between age of onset or death was not a factor used for exclusion of data in longitudinal decline; data available anywhere along the clinical course were included for evaluation. Antemortem clinical diagnoses compatible with dementia included AD, aphasia, Binswanger’s disease, corticobasal syndrome, Creutzfeldt-Jakob disease, dementia with Lewy bodies, frontotemporal dementia, normal pressure hydrocephalus, Parkinson’s disease dementia, Pick’s disease, progressive supranuclear palsy, and semantic dementia. The available clinical parameters in HpSp, typical, LP, and NFTD, respectively, are: education [11 (65 %), 50 (95 %), 17 (89 %), 11 (61 %)], age of onset and disease duration [16 (94 %), 45 (87 %), 14 (74 %), 16 (89 %)], MMSE initial [10 (59 %), 25 (48 %), 8 (42 %), 7 (39 %)], MMSE final [10 (59 %), 16 (31 %), 11 (58 %), 6(33 %)], and longitudinal MMSE [17 (100 %), 52 (100 %), 19 (100 %), 4 (22 %)]. MAPT and APOE genotyping was available for all cases, except one typical AD which did not have frozen tissue available for genotyping. Genomic DNA was extracted from frozen brain tissue according to previously described methods. Each sample was genotyped for MAPT H1/H2 (SNP rs1052553 A/G, A = H1, G = H2) and APOE alleles (SNP rs429358 C/T and rs7412 C/T) using ABI on-demand Taqman assays (Applied Biosystems, Life Technologies Corporation, Carlsbad, CA, USA) and analyzed with SDS 2.2.2 software (also from Applied Biosystems).
SigmaPlot (Ver. 11, San Jose, CA, USA) was used to analyze all statistical data and create graphs. Group comparisons of continuous variables were performed using Kruskal–Wallis one-way analysis of variance on ranks, and pairwise comparisons were performed with the Mann–Whitney rank sum test. Analyses of categorical data were performed using a χ2 test to determine whether the proportions of observations varied between the groups and between the comparison groups. Each group was evaluated for interrelationships of the hippocampal and cortical NFT using a Spearman correlation. A multiple logistic regression model was constructed to control for APOE ε4 allele status when examining pan-Aβ differences between HpSp and LP.
Results
A summary of the demographic, pathologic, and clinical information of HpSp AD, typical AD, LP AD, and NFTD is found in Table 1. From a large series of brain bank cases with NFT and SP counts, 17 % of all cases met objective algorithmic criteria for NFTD (Fig. 1). Of these cases, a further subset of cases was identified that met both SP criteria and did not have significant coexisting pathology. All cases were required to have a clinical diagnosis consistent with a dementing disorder, causing the exclusion of an additional case giving a final total of 18 NFTD cases. Using quantitative NFT densities to demonstrate that the AD subtypes and NFTD form distinct groups, hippocampal NFTs (i.e., average CA2/3, CA1, and subiculum) and cortical NFTs (i.e., superior temporal, inferior parietal, and middle mid-frontal) were plotted. Figure 2 shows the four scatter plots with minimal overlap observed between diagnostic groups. There were no interrelationships between hippocampal and cortical NFTs for HpSp (R = 0.009, p = 966) or NFTD (R = 0.050, p = 0.837), but there were for both typical AD (R = 0.681, p < 0.001) and LP (R = 0.557, p = 0.013).

Four scatter plots of thioflavin-S hippocampal neurofibrillary tangle (NFT) density are plotted against cortical NFT density. The four plots show minimal overlap between groups, with no interrelationships between hippocampal and cortical NFT for hippocampal-sparing AD (HpSp, red down triangle) or neurofibrillary tangle-predominant dementia (NFTD, yellow square). Significant interrelationships were found for both typical AD (black circle) and limbic-predominant AD (LP, cyan up triangle). The scatter plots were separated based on a MAPT H1H1 carriers and b non-carriers
Neuropathologic and clinical findings in limbic-predominant AD and NFTD
The Braak NFT stage was significantly different between LP AD and NFTD. TDP-43 pathology tended to be more frequent in LP AD compared to NFTD, but was not significantly different. We previously evaluated the frequency of concurrent Lewy body pathology across AD subtypes [30], but did not want to confound Lewy body frequency results by including amygdala-predominant Lewy bodies, a form of Lewy body pathology detected in advanced AD [38], in the previous analysis. Given the predilection of amygdala-predominant Lewy body cases for advanced AD, we elected not to exclude them from our current analysis to detect whether differences existed between LP AD and NFTD. TDP-43 pathology tended to be more frequent in LP AD compared to NFTD, but the difference was not statistically significant. The frequency of Lewy body pathology also approached significance and tended to be higher in LP AD (53 %) than in NFTD (34 %). The majority of LP AD had Lewy bodies restricted to the amygdala (37 %), whereas NFTD cases more frequently had brainstem or limbic Lewy bodies (28 %). NFTD cases were older at symptom onset than LP AD, but had shorter disease duration. There was no difference between the groups in the initial and final MMSE scores. Although the available longitudinal MMSE data were limited in NFTD, both groups lost an average of 1.5 points per year. Sex, education, and fixed brain weight did not differ across the four groups, or between LP AD and NFTD. In order to estimate the frequency of NFTD in the Mayo Clinic Jacksonville brain bank, we identified 3,210 cases with thioflavin-S fluorescent microscopy data, and 2,727 (85 %) who had a clinical diagnosis consistent with a dementing disorder. The frequency of operationally classified LP AD and NFTD in the dementia cohort was 5 and 1 %, respectively.
While it is inherent to the classification algorithms of both LP AD and NFTD that middle frontal gyrus NFT counts are sparse, a comparison between the two revealed significant differences (Table 2). The SP counts reached a maximum of 50 in LP AD, but significantly fewer were found in NFTD. The CAA scores were also different: only 5 cases with CAA compared with 16 in LP AD. Photomicrographs of immunohistochemical stains are shown in Fig. 3. The phospho-tau (CP13) burden was 125-fold lower in NFTD compared to the LP AD, whereas the NFT conformational-epitope (Ab39) burden was threefold lower. Analyses of data from LP AD and NFTD revealed strong correlations between both NFT counts and phospho-tau and NFT conformational epitope (respectively R = 0.74, p < 0.0001; R = 0.78, p < 0.0001); as well as between SP counts and pan-Aβ and Aβ40 burdens (respectively R = 0.54, p < 0.001; R = 0.77, p < 0.0001). Both β-amyloid immunohistochemical burdens were significantly lower in NFTD compared to LP AD, with a ninefold difference for pan-Aβ and threefold difference for Aβ40. TDP-43-positive LP AD did not have higher phospho-tau or conformational-epitope burden, but NFTD TDP-43 cases approached significance with a fourfold higher phospho-tau burden (p = 0.12).

CP13 immunohistochemistry detecting early neuritic and tangle pathology in the middle frontal cortex of a hippocampal-sparing Alzheimer’s disease (HpSp AD) reveals both neurofibrillary tangles (NFT, arrow) and extensive neuritic pathology (arrowhead) compared to e typical AD, which shows fewer NFT and neurites, i but not as scarce as that seen in limbic-predominant (LP) AD. m Neurofibrillary tangle-predominant dementia (NFTD) has no NFT and only rare tau-positive neurites. NFT immunohistochemistry using a conformational epitope (Ab39) reveals b frequent mature and extracellular NFT (arrows), as well as neuritic elements (arrowhead) in HpSp and f to a lesser extent in typical AD. j Ab39 staining in LP AD showed rare extracellular NFT and neuritic pathology, n while none were observed in NFTD. c, g, k Amyloid immunohistochemistry using a pan-Aβ marker reveals a range of plaques across groups, but differences in total β-amyloid burden. c Compact plaques can be found in HpSp AD (inset; top and bottom, respectively), but many lightly staining plaques (arrow) are more common. d Aβ40 was found to correspond with some of these plaques, suggesting that the lightly stained plaques were not all “diffuse plaques”, commonly found in aging brains. g Typical AD often had a higher amyloid burden than HpSp and tended to have a wider range of plaque morphology (inset) that could be found to be h Aβ40 positive (g arrow, h arrowhead). k Limbic-predominant cases had the highest plaque burden with stereotypic compact and cored plaques (inset; top and bottom, respectively), but more often had amorphous plaques that were immunopositive for l Aβ40. o, p Neither amyloid stain revealed notable positive structures in NFTD. Scale bars 100 μm (d, h, l, p)
Limbic-predominant AD and NFTD were further discriminated based on the presence of subcortical pathology (Table 3). SPs in the molecular layer of the dentate fascia in the hippocampus and basal ganglia were more frequent in LP AD than NFTD. No significant differences in NFT were found in these regions. NFTs in the midbrain were higher in LP AD, but the difference only approached significance. When the midbrain NFT severity scores were tested using a χ2 test, the differences were found to be significant (p = 0.043). No differences were found in the presence of NFT in either the locus coeruleus or the raphe nucleus. Vascular calcifications found in the basal ganglia were limited and showed no difference in frequency, further distinguishing both LP AD and NFTD from the rare tangle-predominant dementia—diffuse neurofibrillary tangles with calcifications (DNTC) [44].
Immunohistochemical differences across AD subtypes
Table 2 shows the results of data from thioflavin-S fluorescent microscopy and quantitative digital microscopic image analysis in the middle frontal gyrus for the AD subtypes and NFTD. As expected, there were significant differences in NFT, but not SP counts across AD subtypes. Although the median values of CAA severity scores were the same, the range in severity resulted in significant differences across AD subtypes. Burden of phospho-tau and NFT conformational epitope in the middle frontal cortex assessed by image analysis strongly correlated with thioflavin-S NFT counts (respectively R = 0.79, p < 0.001; R = 0.87, p < 0.001). The phospho-tau burden was twofold higher in HpSp AD compared to typical AD and sixfold higher than LP AD (Fig. 4a). The conformational-epitope burden was 4-fold higher in HpSp AD compared to typical AD and 19-fold higher than LP AD (Fig. 4b). The range in data for SP counts from thioflavin-S fluorescent microscopy was too narrow to show correlations with either pan-Aβ or Aβ40 burdens. There were no significant differences between AD subtypes for either Aβ40 or pan-Aβ, although the pan-Aβ burden tended to be lower in HpSp AD than LP AD (2.2 vs. 5.2 %, p = 0.113). When both pan-Aβ and APOE ε4 status were added into a multiple logistic regression model of HpSp AD versus LP AD, the difference remained a trend (p = 0.087).

Box plots of immunohistochemical burden measured using digital microscopy are plotted for hippocampal-sparing Alzheimer’s disease (HpSp AD), typical AD, limbic-predominant (LP) AD, and neurofibrillary tangle-predominant dementia (NFTD). a The phospho-tau burden (CP13) was twofold higher in HpSp AD compared to typical AD and sixfold higher than LP AD. b The conformational-epitope burden (Ab39) was 4-fold higher in HpSp AD compared to typical AD and 19-fold higher than LP AD. Box plots show median (interquartile ratio) and error bars represent the 10–90th percentile
Using thioflavin-S microscopy, the frequency of NFTs in the basal ganglia was found to be higher in HpSp AD (n = 16, 94 %) than typical AD (n = 35, 67 %), LP AD (n = 11, 58 %), and NFTD (n = 6, 33 %) (χ2 = 14.6, p = 0.002). A χ2 test showed this difference to be significant among the AD subtypes (χ2 = 6.26, p = 0.044). The frequency of SPs in the basal ganglia was also higher in HpSp AD (n = 15, 88 %) than typical AD (n = 38, 74 %), LP AD (n = 12, 63 %), and NFTD (n = 1, 6 %) (χ2 = 31.5, p < 0.001). A χ2 test showed this difference not to be significant among the AD subtypes (χ2 = 3.00, p = 0.224).
Genetic findings
MAPT and APOE were analyzed across groups, as well as between the NFTD and LP AD (Table 4). NFTD and LP AD had a high frequency of the H1H1 genotype. Figure 2 illustrates the prevalence of H1H1 in typical AD, LP AD, and NFTD compared to the underrepresentation seen in HpSp AD. APOE ε4ε4 genotype was not found in and NFTD cases, but no difference was found when compared with LP AD. Typical AD had the highest APOE ε4ε4 frequency, which approached significance when a group-wise χ2 analysis was performed. Aβ40 burden in APOE ε4 allele carriers versus non-carriers was significantly higher in HpSp AD (0.97 vs. 0.18 %; p = 0.028) and typical AD (1.1 vs. 0.23 %; p = 0.006), but not LP AD (0.53 vs. 0.057 %; p = 0.38). There were no differences found in pan-Aβ burden with regard to APOE ε4 allele status.
Discussion
This study takes advantage of recent advances in our understanding of AD classification to investigate the relationship between AD subtypes and NFTD. We describe demographic, pathologic, cognitive, and genetic features of AD subtypes characterized by their pattern of cortical and limbic neurofibrillary pathology and NFTD, which is characterized by limbic-restricted neurofibrillary degeneration. Using digital microscopy, we present quantitative pathologic evidence confirming that there are distinct AD subtypes, and that NFTD is different from AD subtypes. We particularly focused on similarities and differences between limbic-centric disorders, NFTD and LP AD. Age at death was comparable between NFTD and LP AD, but there was a trend for more females, and significant differences in older age at disease onset and shorter disease duration for NFTD. NFTD also had lower Braak NFT stage and a tendency to have more frequent Lewy body pathology beyond the amygdala. NFTD had almost no NFTs or SPs with thioflavin-S fluorescent microscopy and minimal tau and amyloid burden with image analysis in the middle frontal gyrus, all of which were significantly different from LP AD. As expected, the APOE ε4ε4 genotype was low in NFTD (no ε4ε4 carrier was detected), but surprisingly the frequency of MAPT H1H1 genotype (~ 70 %) was similar in NFTD, typical AD, and LP AD in this series. On the other hand, HpSp AD had a lower frequency of H1H1 genotype.
The AD classification system, originally based on thioflavin-S fluorescent microscopy [30], was examined using immunohistochemistry to demonstrate early- and late-stage NFT pathology in the middle frontal gyrus in this study. Our early-stage phospho-tau marker (CP13) showed neuritic and pre-tangle pathology, but does not stain extracellular tangles [10]. However, our late-stage conformation epitope in NFT (Ab39) stains mature and extracelluar NFT [42], thus providing a spectrum of new and long-standing tau pathology. We report a strong correlation between the thioflavin-S NFT counts and burden of phospho-tau, as well as burden with a monoclonal antibody that recognizes a conformational epitope in NFT, providing immunohistochemical evidence that supports differences detected previously with fluorescence microscopy. We found twofold more cortical phospho-tau in HpSp AD compared to typical AD and a fourfold increase in the NFT conformational epitope. Moreover, LP AD had a 6-fold lower cortical phospho-tau and a 19-fold lower NFT conformational-epitope burden. While it is difficult to know the temporal relationship between disease duration and tau accumulation, the immunohistochemistry results of the NFT conformational epitope, which detects mature NFTs and extracellular NFT, but not pretangles [42], suggest that tau pathology begins in the cortex in HpSp AD. Conversely, we speculate that LP AD has a relatively restricted phospho-tau accumulation that may spread to the neocortex, but does not form mature NFTs. Typical AD had a twofold lower accumulation of phospho-tau and a fourfold lower accumulation of NFT conformational epitope. These findings, along with higher accumulation of thioflavin-S-positive NFTs in the hippocampus and amygdala of typical AD [30], suggest a progressive accumulation from limbic regions to the cortices and support previously described Braak NFT staging scheme [6]. Of interest were the higher NFTs and SPs found in the basal ganglia of HpSp AD compared to LP AD.
Although to our knowledge, HpSp AD does not share a common genetic basis leading to an early age of onset, the higher frequency of moderate-to-severe plaques is reminiscent of higher [11C] Pittsburgh compound B uptake in the striatum of early-onset AD [27]. The plaques found in the basal ganglia in our series were either diffuse or cored, and not ‘cotton wool’ plaques that have been described in familial AD. The ceiling effect for SP counts based on thioflavin-S fluorescent microscopy, which were truncated at 50, limited detection of differences in comparisons of SP density between AD subtypes in previous studies [30]. This problem was alleviated by pan-Aβ and Aβ40 immunostaining, which provided continuous variables and a wide range to determine whether differences existed across AD subtypes. We found a weak difference that approached significance in middle frontal cortex for pan-Aβ burden between HpSp AD and LP AD, which remained a trend when APOE ε4 allele frequency was included as a covariate in a regression model. Further examination of β-amyloid burdens in a larger number of cases may provide evidence of a genetic contribution to this pathologic feature.
Distinct cortical differences were not observed for β-amyloid burden between AD subtypes, although NFTD had significantly lower burden compared to all AD subtypes. Minimal cortical β-amyloid pathology is consistent with previous descriptions of NFTD [3, 5, 17, 18, 20, 21, 31], where neuritic type SPs were absent and SPs that were present had characteristics of diffuse β-amyloid deposits. APOE ε4 status has been shown to influence plaque density [16], but this alone does not explain the lack of SPs in NFTD. For example, over half of HpSp AD are APOE ε4 negative, yet still have abundant cortical β-amyloid pathology. In HpSp AD and typical AD, APOE ε4 allele carriers were found to have significantly higher Aβ40 burden, similar to previous studies in AD without regard to AD subtypes [16, 35].
The MAPT H1H1 genotype was overrepresented in typical AD, LP AD, and NFTD (~70 % frequency). To our knowledge, this finding has not been previously reported in NFTD, but given our small sample size (n = 18) we are cautious not to overinterpret. Recently, another group has independently identified the association of the MAPT H1H1 haplotype with NFTD [34]. It is likely that other genetic factors influence the distribution of NFT, but the MAPT H1 haplotype may play a role in the medial temporal lobe predilection for NFT in these disorders.
The phospho-tau burden was very low in the cortex of NFTD, consistent with previous reports [21, 33]. NFTD is a 3R/4R tauopathy [21] and would not typically be considered for TDP-43 screening [7]. Previous studies have demonstrated TDP-43 positivity in other tauopathies, including AD [2], corticobasal degeneration [43], Guam parkinsonism–dementia complex [13], and NFTD [14, 31]. Clinical and pathologic severity has been reported to be greater in TDP-43-positive corticobasal degeneration and AD compared to TDP-43-negative cases [23, 43]. We examined AD subtypes to determine if frequency of TDP-43 positivity was associated with phospho-tau burden. We did not find increased tau burden in TDP-43-positive AD cases; however, a fourfold increase was found in the frontal cortex between TDP-43-negative and TDP-43-positive NFTD.
TDP-43 is highly implicated in another limbic-centric disorder, hippocampal sclerosis. Cases where hippocampal sclerosis do not occur in the setting of frontotemporal lobar degeneration or AD are uncommon, but share similar clinical features with NFTD and LP AD. All three are more common in elderly women with a slowly progressive amnestic syndrome. We speculate that both NFTD and hippocampal sclerosis would likely be included in the recently described “SNAP” (suspected non-Alzheimer’s pathophysiology) group [26]. The SNAP group was described as having positive signs for neurodegeneration (e.g., hippocampal volume loss), but no cortical β-amyloid on positron emission tomography amyloid imaging. In a recent study, we found that “pure” hippocampal sclerosis cases lacked NFTs in the middle frontal gyrus (median NFT = 0) and had minimal SPs (median SP = 12) [29]. The focus of the current study, however, was on medial temporal tauopathies. Hippocampal sclerosis cases were excluded, given that the neuronal loss observed in the hippocampus is disproportionate to the neurofibrillary pathology. Future studies should aim at characterizing the pathology of SNAP, to determine whether neuroimaging studies can differentiate hippocampal sclerosis from NFTD. Our current work suggests that antemortem β-amyloid imaging would considerably help in determining whether elderly patients’ amnestic syndrome is a result of underlying NFTD or LP AD.
Although we are limited by the small sample size for NFTD, the clinical features of our NFTD were very similar to those of a larger series of NFTD reviewed by Kurt A. Jellinger, [21] a leader in neuropathologic aspects of this disease. Our study sought to examine potential overlaps of NFTD with recently defined AD subtypes [30]. NFTD is considered to affect the very old (>80 years) and, in reference to LP AD, age and sex are suggested factors in the susceptibility of medial temporal lobe to neurofibrillary pathology in both NFTD and LP AD. Although NFTD cases were significantly older at disease onset than LP AD, they had shorter disease duration. It is interesting to consider that one of the major differences between NFTD and LP AD cases is the paucity of cortical β-amyloid pathology in NFTD. Given this difference, future studies should consider these two disease entities in the investigation of how amyloid influences tau pathology.
References
Alladi S, Xuereb J, Bak T et al (2007) Focal cortical presentations of Alzheimer’s disease. Brain 130:2636–2645
Amador-Ortiz C, Lin WL, Ahmed Z et al (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61:435–445
Bancher C, Egensperger R, Kosel S, Jellinger K, Graeber MB (1997) Low prevalence of apolipoprotein E epsilon 4 allele in the neurofibrillary tangle predominant form of senile dementia. Acta Neuropathol 94:403–409
Barker WW, Luis CA, Kashuba A et al (2002) Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord 16:203–212
Braak H, Braak E (1990) Neurofibrillary changes confined to the entorhinal region and an abundance of cortical amyloid in cases of presenile and senile dementia. Acta Neuropathol 80:479–486
Braak H, Braak E (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 82:239–259
Cairns N, Bigio E, Mackenzie I et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114:5–22
Dugger BN, Tu M, Murray ME, Dickson DW (2011) Disease specificity and pathologic progression of tau pathology in brainstem nuclei of Alzheimer’s disease and progressive supranuclear palsy. Neurosci Lett 491:122–126
Duyckaerts C, Delatour B, Potier M-C (2009) Classification and basic pathology of Alzheimer disease. Acta Neuropathol 118:5–36
Espinoza M, de Silva R, Dickson DW, Davies P (2008) Differential incorporation of tau isoforms in Alzheimer’s disease. J Alzheimers Dis 14:1–16
Folstein MF, Folstein SE, McHugh PR (1975) “Mini-Mental State”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198
Galton CJ, Patterson K, Xuereb JH, Hodges JR (2000) Atypical and typical presentations of Alzheimer’s disease: a clinical, neuropsychological, neuroimaging and pathological study of 13 cases. Brain 123(Pt 3):484–498
Hasegawa M, Arai T, Akiyama H et al (2007) TDP-43 is deposited in the Guam parkinsonism–dementia complex brains. Brain 130:1386–1394
Hatanpaa KJ, Bigio EH, Cairns NJ et al (2008) TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a Midwest-Southwest Consortium for FTLD study. J Neuropathol Exp Neurol 67:271–279
Hyman BT, Trojanowski JQ (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 56:1095–1097
Hyman BT, West HL, Rebeck GW et al (1995) Quantitative analysis of senile plaques in Alzheimer disease: observation of log-normal size distribution and molecular epidemiology of differences associated with apolipoprotein E genotype and trisomy 21 (Down syndrome). Proc Natl Acad Sci USA 92:3586–3590
Ikeda K, Akiyama H, Arai T et al (1999) Clinical aspects of ‘senile dementia of the tangle type’— a subset of dementia in the senium separable from late-onset Alzheimer’s disease. Dement Geriatr Cogn Disord 10:6–11
Ikeda K, Akiyama H, Arai T et al (1997) A subset of senile dementia with high incidence of the apolipoprotein E epsilon2 allele. Ann Neurol 41:693–695
Iseki E, Yamamoto R, Murayama N et al (2006) Immunohistochemical investigation of neurofibrillary tangles and their tau isoforms in brains of limbic neurofibrillary tangle dementia. Neurosci Lett 405:29–33
Jellinger KA (2012) Neuropathological subtypes of Alzheimer’s disease. Acta Neuropathol 123:153–154
Jellinger KA, Attems J (2007) Neurofibrillary tangle-predominant dementia: comparison with classical Alzheimer disease. Acta Neuropathol 113:107–117
Jellinger KA, Bancher C (1998) Senile dementia with tangles (tangle predominant form of senile dementia). Brain Pathol 8:367–376
Josephs KA, Whitwell JL, Knopman DS et al (2008) Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 70:1850–1857
Khachaturian ZS (1985) Diagnosis of Alzheimer’s disease. Arch Neurol 42:1097–1105
Kim J, Miller VM, Levites Y et al (2008) BRI2 (ITM2b) inhibits Abeta deposition in vivo. J Neurosci 28:6030–6036
Knopman DS, Jack CR, Wiste HJ et al (2012) Short-term clinical outcomes for stages of NIA-AA preclinical Alzheimer disease. Neurology 78:1576–1582
Koivunen J, Verkkoniemi A, Aalto S et al (2008) PET amyloid ligand [11C]PIB uptake shows predominantly striatal increase in variant Alzheimer’s disease. Brain 131:1845–1853
Murray ME, Dickson DW (2008) O1-01-02: Alzheimer’s disease with relative hippocampal sparing: a distinct clinicopathologic variant. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association 4:T106–1106
Murray ME, Graff-Radford NR, Ross OA et al (2012) Differentiating clinicopathologic and genetic aspects of hippocampal sclerosis in Alzheimer’s disease from limbic predominant Alzheimer’s disease and “pure” hippocampal sclerosis. Alzheimer’s Dementia J Alzheimer’s Assoc 8:P620–P621
Murray ME, Graff-Radford NR, Ross OA et al (2011) Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 10:785–796
Nelson PT, Abner EL, Schmitt FA et al (2009) Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J Neuropathol Exp Neurol 68:774–784
Nitrini R, Lopes K, Brucki SMD (2011) Previous description of subtypes of Alzheimer’s pathology. Dement Neuropsychol 5:346–348
Noda K, Sasaki K, Fujimi K et al (2006) Quantitative analysis of neurofibrillary pathology in a general population to reappraise neuropathological criteria for senile dementia of the neurofibrillary tangle type (tangle-only dementia): the Hisayama Study. Neuropathology 26:508–518
Santa-Maria I, Haggiagi A, Liu X et al (2012) The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Neuropathol. doi:10.1007/s00401-012-1017-1
Schmechel DE, Saunders AM, Strittmatter WJ et al (1993) Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA 90:9649–9653
Terry RD, Hansen LA, DeTeresa R et al (1987) Senile dementia of the Alzheimer type without neocortical neurofibrillary tangles. J Neuropathol Exp Neurol 46:262–268
Togo T, Sahara N, Yen SH et al (2002) Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol 61:547–556
Uchikado H, Lin WL, DeLucia MW, Dickson DW (2006) Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol 65:685–697
van der Flier WM, Pijnenburg YAL, Fox NC, Scheltens P (2011) Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE [var epsilon]4 allele. Lancet Neurology 10:280–288
van Duijn CM, de Knijff P, Cruts M et al (1994) Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer’s disease. Nat Genet 7:74–78
Whitwell JL, Jack CR, Jr., Przybelski SA et al (2009) Temporoparietal atrophy: a marker of AD pathology independent of clinical diagnosis. Neurobiol Aging 32:1531–1541
Yen SH, Dickson DW, Crowe A, Butler M, Shelanski ML (1987) Alzheimer’s neurofibrillary tangles contain unique epitopes and epitopes in common with the heat-stable microtubule associated proteins tau and MAP2. Am J Pathol 126:81–91
Yokota O, Davidson Y, Bigio EH et al (2010) Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol 120:55–66
Yokota O, Terada S, Ishizu H et al (2002) NACP/alpha-synuclein immunoreactivity in diffuse neurofibrillary tangles with calcification (DNTC). Acta Neuropathol 104:333–341
Acknowledgments
The project was supported by the Mayo ADRC Grant (P50 AG16574), Mayo Clinic Study on Aging (U01 AG06786), Florida ADRC (P50 AG215711), Einstein Aging Study (P01 AG03949), Mangurian Foundation, and the State of Florida Alzheimer’s Disease Initiative. MEM and this project were supported by a fellowship from the Robert and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program and the Mayo ADRC Pilot Grant. OAR and DWD were supported by the Mayo Clinic Udall Center (P50 NS072187). OAR was partially supported by R01 NS078086. DWD was supported by the Robert E Jacoby Professorship for Alzheimer’s Research. We thank the patients and their families who donated brains to help further our knowledge in neurodegeneration. The authors would like to acknowledge the endless hours of commitment and teamwork offered by Linda G. Rousseau, Virginia R. Phillips, John Gonzalez, and Monica Castanedes-Casey. Without the sampling design and procedures for thioflavin-S fluorescent microscopy developed originally by Dr. Robert D. Terry [36], this study would not have been possible.
Ethical approval
All research reported is on postmortem material, which is considered exempt from human subject research. All brains were acquired with appropriate ethical approval and the research described has approval from the Mayo Clinic Institutional Review Board.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Janocko, N.J., Brodersen, K.A., Soto-Ortolaza, A.I. et al. Neuropathologically defined subtypes of Alzheimer’s disease differ significantly from neurofibrillary tangle-predominant dementia. Acta Neuropathol 124, 681–692 (2012). https://doi.org/10.1007/s00401-012-1044-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-012-1044-y