
Abstract
Pathological transactivation-responsive DNA-binding protein 43 (TDP-43) has been identified as a component of ubiquitinated inclusions in frontotemporal lobar degeneration with motor neuron disease, as well as in sporadic and some forms of familial amyotrophic lateral sclerosis. To clarify whether pathological TDP-43 is present in other neurodegenerative diseases involving the motor neuron system, we immunohistochemically examined the brain and spinal cord affected by two CAG repeat (polyglutamine) diseases, Machado–Joseph disease (MJD) and spinal and bulbar muscular atrophy (SBMA), using polyclonal antibody against TDP-43. In all the MJD cases, TDP-43-immunoreactive (ir) neuronal cytoplasmic inclusions (NCIs), although few in number, were found only in the lower motor neurons in the brainstem and spinal cord. TDP-43-ir NCIs appeared as linear wisp-like, skein-like, or thick, somewhat rod-like bodies. These inclusions were also visualized with antibodies against phosphoserines 409 and 410 of TDP-43, and ubiquitin, but were not recognized by antibody against expanded polyglutamine stretches or ataxin-3. The ultrastructure of the TDP-43-ir NCIs was similar to that of the inclusions seen in sporadic ALS, consisting of bundles of parallel filaments. None of the SBMA cases showed abnormal TDP-43 immunoreactivity in any of the regions examined. Immunoblot analysis failed to recognize hyperphosphorylated TDP-43 at ~23 kDa in two MJD cases examined. However, the immunohistochemical findings strongly suggested that in MJD, in addition to the polyglutamine-dependent disease process, TDP-43-related pathogenesis is associated with degeneration and death of the lower motor neurons.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transactivation-responsive DNA-binding protein 43 (TDP-43) is a transcription-related protein physiologically localized in the cell nucleus, and also known to be a major component of ubiquitin-positive inclusions in frontotemporal lobar degeneration (FTLD) and sporadic amyotrophic lateral sclerosis (ALS) [2, 22]. Recently, pathological localization of TDP-43 has also been demonstrated in ubiquitin-positive inclusions in other neurodegenerative diseases, including Guam amyotrophic lateral sclerosis/parkinsonism–dementia complex [8, 10, 19], familial FTLD with motor neuron disease linked to chromosome 9p [3], and familial ALS without SOD-1 mutation [18, 29]. Moreover, many missense mutations in the TDP-43 gene have also been identified in familial and sporadic ALS cases, strongly suggesting that TDP-43 plays an essential role in the degeneration and death of motor neurons [4, 9, 15, 16, 25, 27, 31, 36].
Machado–Joseph disease (MJD), also known as spinocerebellar ataxia type 3, is one of the so-called CAG repeat diseases, which are hereditary neurodegenerative disorders caused by the expansion of a polyglutamine tract in each respective disease protein [33]. MJD shows multiple involvement of the nervous system, including the lower motor neurons [33, 34]. Although the affected neurons occasionally show formation of intranuclear inclusions (NIIs) [34], it has also been reported that ubiquitin-immunoreactive (ir) skein-like neuronal cytoplasmic inclusions (NCIs) are detectable in the spinal lower motor neurons [28], although it remains to be clarified whether these filamentous NCIs contain expanded polyglutamine stretches [12]. These findings suggest that ALS and MJD may share certain molecular pathomechanisms responsible for the development of motor neuron degeneration and death.
To clarify whether TDP-43 is involved in the disease process in MJD, we performed immunohistochemical and biochemical studies of brains and spinal cords from patients with MJD, in combination with immunohistochemical studies of the central nervous system (CNS) affected by spinal and bulbar muscular atrophy (SBMA), which is also a CAG repeat disease involving the lower motor neurons [33].
Materials and methods
Subjects
We examined the brain and spinal cord from ten patients with MJD (age range 32–71 years, mean 51.4 years), three patients with SBMA (age range 51–82 years, mean 65.7 years) and five control subjects (age range 42–75 years, mean 61.8 years) (Table 1). In each patient, the diagnosis of MJD or SBMA had been confirmed both clinicopathologically and genetically. None of the cases examined were associated with other neurodegenerative diseases including ALS, FTLD, Alzheimer’s disease (AD) and Lewy body disease. The duration of illness ranged from 3 to 52 years (mean 23.1 years) in MJD cases and from 0.8 to 25 years (mean 11.9 years) in SBMA cases.
Neuronal loss
In all the cases of MJD and SBMA, neuronal loss in the cerebral motor cortex as well as the brainstem and spinal cord lower motor neuron nuclei was assessed using hematoxylin–eosin- and Klüver–Barrera-stained sections. Neuronal loss in each region was recorded as absent, mild, moderate, or severe.
Immunohistochemistry
We prepared serial 4-μm thick, formalin-fixed, paraffin-embedded sections from the spinal cord (lower cervical, middle thoracic and lumbar segments), medulla oblongata, pons, midbrain, and cerebral motor cortex. Four sections, 40 μm apart, were pretreated by heating for 10 min at 121°C in 10 mM sodium citrate buffer and immunostained by the avidin–biotin-peroxidase complex (ABC) method with Vectastain ABC kit (Vector, Burlingame, CA, USA) using rabbit polyclonal antibody against TDP-43 (10782-1-AP; ProteinTech Group Inc., Chicago, IL, USA; 1:4,000) [2, 22, 23]. Diaminobenzidine (DAB) was used as the chromogen. Using these four sections, the total number of motor neurons with TDP-43-ir NCIs (described below) was counted in the cerebral motor cortex as well as in the brainstem and spinal cord.
Several selected sections were similarly immunostained with rabbit polyclonal antibody against ubiquitin (Dako, Glostrup, Denmark; 1:800) and mouse monoclonal antibody specific for phosphorylated TDP-43 (pTDP-43), or more strictly, phosphoserines 409 and 410 of TDP-43 (pS409/410; Cosmo Bio Co., Ltd., Tokyo, Japan; 1:3,000) [11, 14]. In addition, double-labeling immunofluorescence studies, with the same pretreatment, were performed using a rabbit polyclonal antibody against TDP-43 (1:2,000) with a combination of either mouse monoclonal antibodies against ubiquitin (Dako, Glostrup, Denmark; 1:8,000), expanded polyglutamine stretches (1C2; Chemicon, Temecula, CA, USA; 1:3,000) or ataxin-3 (Chemicon; 1:1,600). In previous studies [34, 35], we used pretreatment with formic acid for the immunostaining of expanded polyglutamine stretches with 1C2; however, our recent experiences with different types of tissue pretreatment have revealed that autoclaving (heating) is the best one in terms of immunohistochemical sensitivity. The secondary antibodies used were Alexa Fluor 568 goat anti-rabbit IgG (Molecular Probes, Eugene, OR, USA; 1:1,000) and Alexa Fluor 488 goat anti-mouse IgG (Molecular Probes; 1:1,000).
Moreover, to clarify the distribution of TDP-43- and pTDP-43-ir NCIs in MJD, we also examined various other regions of the nervous system in 4 MJD cases (cases 6, 7, 8 and 10) using the anti-TDP-43 rabbit polyclonal antibody (10782-1-AP; ProteinTech Group Inc.; 1:4,000) and anti-pTDP-43 mouse monoclonal antibody (pS409/410; Cosmo Bio Co., Ltd; 1:3,000), respectively, as performed in a previous study of sporadic ALS [23].
Immunoelectron microscopy
The procedures were fundamentally the same as those conducted in a previous study [20]. Briefly, vibratome sections of the spinal anterior horns from one MJD case (case 5) were immunostained using the polyclonal antibody against TDP-43 (1:3,000) for 36 h at 4°C, followed by incubation with a biotinylated secondary antibody (1:200) and ABC. The sections were immersed in 1% glutaraldehyde and developed with DAB. Then, the immunostained sections were post-fixed in 1% osmium tetroxide, dehydrated through a graded ethanol series, and embedded in epoxy resin. Ultrathin sections were cut and lightly counterstained with lead citrate, and examined with a Hitachi H-7100 electron microscope.
Fractionation of frozen tissues and immunoblot analysis
Proteins from the frontal cortex (middle frontal gyrus) and spinal cord from one control subject, two MJD cases (cases 4 and 5) and two sporadic ALS cases were extracted, as described previously [27]. Briefly, frozen tissues were homogenized in buffer A [10 mM Tris–HCl (pH 7.5), 1 mM ethylene glycol-bis[-β-amino-ethylether]-tetra-acetic acid, 1 mM dithiothreitol, 10% sucrose] and centrifuged at 25,000×g for 30 min at 4°C. The resulting pellets were then extracted in buffer A containing 1% Triton X-100 and centrifuged at 100,000×g for 60 min at 4°C. These pellets were subsequently homogenized in buffer A containing 1% sarkosyl, incubated for 1 h, and centrifuged at 100,000×g for 60 min at 22°C. The sarkosyl-insoluble pellets were solubilized in 8 M urea buffer. After centrifugation at 25,000×g for 30 min at 22°C, the supernatants were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analyzed by immunoblotting with the anti-TDP-43 rabbit polyclonal antibody (10782-1-AP; ProteinTech Group Inc.; 1:1,000) [2, 22] and anti-TDP-43 mouse monoclonal antibody (pS409/410; Cosmo Bio Co., Ltd; 1:2,000) [11, 14]. Protein concentration was measured by the Bradfold method, and equal amounts of protein (frontal cortex: 11 μg, spinal cord: 16 μg) were loaded in each lane.
Results
Neuronal loss in the lower and upper motor neuron systems
The severity of neuronal loss in each lower motor neuron nucleus is shown in Table 1. In all the MJD cases except one (case 8) with a short disease duration (3 years), various degrees of neuronal loss were observed in the lower motor neuron nuclei. In three SBMA cases, moderate to severe neuronal loss was a feature of the spinal anterior horns, and hypoglossal and facial nuclei. Neuronal loss was not evident in the cerebral motor cortex in none of the MJD or SBMA cases; particularly, Betz cells were well preserved in all the cases.
TDP-43, pTDP-43 and ubiquitin immunohistochemistry
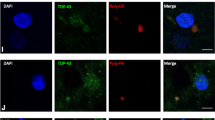
In controls, TDP-43 was clearly visualized as a diffuse pattern in the brain cell nuclei. In ten MJD cases, TDP-43-ir NCIs were detected in the CNS; they were limited to the lower motor neuron nuclei and the number in each case was very small. In the spinal anterior horns, the total number of neurons with TDP-43-ir NCIs in each case ranged between 0 and 4. In the brainstem motor neuron nuclei, only six lower motor neurons were found to have TDP-43-ir NCIs among all the cases. The details are shown in Table 1. The TDP-43-ir NCIs observed varied in shape, appearing as linear wisp-like (Fig. 1a, b), skein-like (Fig. 1c, e, f) or thick, somewhat rod-like bodies (Fig. 1d). TDP-43 staining was also observed in the cytoplasm of a few glial cells and dystrophic neurites in the oculomotor nucleus in cases 4, 7 and 10, as well as in the motor nucleus of the trigeminal nerve in case 4 (Fig. 1g). In general, nuclei of the cells that possessed cytoplasmic TDP-43-ir inclusions were negative (Fig. 1b, c, g), or only weakly positive for TDP-43 (Fig. 1a). In contrast, neurons lacking such inclusions retained endogenous nuclear staining for TDP-43. NCIs indistinguishable from TDP-43-ir NCIs were also detected by pTDP-43 (pS409/410) (Fig. 1h) or ubiquitin (Fig. 1i) immunohistochemistry. No TDP-43-ir lesions, including NCIs, were observed in any of the regions examined in any of the SBMA cases or controls; the brain and spinal cord cells were seen to retain endogenous nuclear staining for TDP-43 in these cases and controls.

TDP-43-immunoreactive (ir) neuronal cytoplasmic inclusions (NCIs) in the lower motor neurons in Machado–Joseph disease (MJD). TDP-43-ir linear wisps (a, b, e), skein-like inclusions (c, f), and thick, somewhat rod-like structures (d) are evident in lower motor neurons in the lumbar anterior horns (a–d), hypoglossal nucleus (e), and motor nucleus of the trigeminal nerve (f). A glial cell with TDP-43-ir cytoplasmic inclusions (arrowhead) and a TDP-43-ir swollen dystrophic neurite (arrow) are also evident in the motor nucleus of the trigeminal nerve (g). Skein-like inclusions are also visualized in the spinal anterior horns (h, lumbar; i, cervical) with antibodies against phosphoserines 409 and 410 of TDP-43 (h) and ubiquitin (i). a, d, h case 5; b case 9; c case 7; e case 8; f, g case 4; i case 10. Bars 20 μm
Immunoelectron microscopy
Ultrastructurally, TDP-43-ir NCIs observed in the spinal anterior horn cells consisted mainly of bundles of filaments (Fig. 2a). The bundles were not membrane-bound and the constituent parallel filaments were apparently thicker than neurofilaments (Fig. 2b).

Immunoelectron microscopy of TDP-43-ir NCIs showing a skein-like appearance in the lumbar anterior horn cells in MJD (case 5). a TDP-43-ir longitudinally and transversely cut bundles of filaments, 19–24 nm in diameter, are evident. b High-magnification view of the bundle indicated by the asterisk in a, showing areas consisting of tightly and loosely arranged parallel filaments, the latter producing rather translucent spaces. Bars a 1 μm, b 250 nm
Association of TDP-43 with other proteins
In MJD, double-labeling immunofluorescence revealed occasional co-localization of TDP-43 with ubiquitin in cytoplasmic inclusions in the spinal lower motor neurons (Fig. 3a–f). Many lower motor neurons were found to show granular polyglutamine cytoplasmic pathology [34]. Occasional lower motor neurons possessing both TDP-43-ir and 1C2-ir (Fig. 3g–i) cytoplasmic inclusions were observed. However, in these lower motor neurons, TDP-43 was never co-localized with polyglutamine stretches (Fig. 3g-i) or ataxin-3. On the other hand, either 1C2-ir or ataxin-3-ir NIIs was rare in the lower motor neurons [34]. No co-occurrence of such NIIs and TDP-43-ir NCIs in the same lower motor neurons was found. In this connection, neuronal nuclei with 1C2-ir or ataxin-3-ir NIIs were well immunostained with the polyclonal TDP-43 antibody (data not shown).

Double-labeling immunofluorescence of NCIs in the lumbar anterior horn cells in MJD. a–c Linear wisps are visualized with the monoclonal anti-ubiquitin (a) and polyclonal anti-TDP-43 antibodies (b); a merged image shows that occasional TDP-43-ir NCIs are ubiquitinated (c). d–f Thick, somewhat rod-like structures are also visualized with the monoclonal anti-ubiquitin (d) and polyclonal anti-TDP-43 antibodies (e); interestingly, a merged image shows absence of TDP-43 in part of the ubiquitinated NCIs (f). g–i In this anterior horn cell, 1C2-ir granular structures (polyglutamine stretches) (g) and TDP-43-ir filamentous inclusions (h) are evident, but both are found to be localized separately in the cytoplasm (i). a–c, d–f case 9; g–h case 7. Bars 20 μm
TDP-43 expression analysis
To characterize TDP-43 protein biochemically, we extracted sarkosyl-insoluble proteins from the frontal cortex and spinal cord of the autopsied patients, and analyzed them by immunoblotting. Although full-length TDP-43 was present in all insoluble fractions, the degradation product was not observed upon immunoblotting with the phosphorylation-independent TDP-43 antibody (10782-1-AP; ProteinTech) (Fig. 4a, b; left panels). Using the phosphorylation-dependent TDP-43 antibody (pS409/410; Cosmo Bio), we failed to find any distinct immunoreactive bands in the MJD cases, whereas a ~23-kDa band and a ~50-kDa band were found in the ALS cases (Fig. 4a, b; right panels).

Immunoblot analyses of sarkosyl-insoluble, urea-soluble fractions of the frontal lobe cortex (a) and spinal cord (b) obtained from control (lane 1), MJD (lanes 2, 3) and ALS (lanes 4, 5) cases. Analyses with the polyclonal anti-TDP-43 antibody yield same bands in all cases (a, b left panels). Analyses with the monoclonal pS409/410 antibody specific for phosphoserines 409 and 410 of TDP-43 reveal no distinct immunoreactive band in control or MJD cases, whereas ~23-kDa (asterisk) and ~50-kDa (double asterisks) bands are visible in ALS cases (a, b right panels)
Discussion
The present study clearly indicates that TDP-43-ir as well as pTDP-43-ir NCIs can occur in the CNS of patients with MJD. However, their occurrence was limited to the lower motor neurons, and mostly confined to the spinal lower motor neurons. These findings suggest that in MJD, the lower motor neuron system is involved in the disease process of polyglutamine-dependent neurodegeneration, as well as in that associated with TDP-43-related pathogenesis. The light microscopic, immunohistochemical and ultrastructural profiles of these NCIs observed in MJD closely resembled those of such inclusions in sporadic ALS [5, 11, 14, 17, 20, 23, 24]. On the other hand, immunoblot analysis revealed no band of pTDP-43 in MJD, unlike the result obtained in ALS [11, 14]. However, it is possible that this failure to detect pTDP-43 might have been due to the very mild pathology in MJD. In other words, there might be no particular difference between MJD and sporadic ALS in the molecular pathomechanisms involving TDP-43 in the process of degeneration of lower motor neurons.
The frequent and constant appearance of TDP-43-ir NCIs in the lower motor neurons in sporadic ALS implies that these structures are closely related to the degeneration and death of these cells [23]. In contrast, the occurrence of TDP-43-ir NCIs in the lower motor neurons in MJD was rare and confined almost entirely to the spinal lower motor neurons, in spite of the presence of obvious neuronal loss in various regions of the nervous system. In MJD, severe neuronal depletion in the affected brain regions might cause difficulty in detection of TDP-43 pathology there. However, this did not appear to be the case; in the present study, either TDP-43-ir or pTDP-43-ir lesions were detectable outside the lower motor neuron nuclei in any of the MJD cases, including four (cases 6, 7, 8 and 10)—one with a significantly shorter disease duration (case 8)—in which the immunohistochemical examination was performed on various regions of the nervous system.
Based on the present results, it is difficult to discuss whether TDP-43 participates directly in the polyglutamine-dependent disease process in MJD. However, it cannot be ruled out completely that even though secondarily, TDP-43 abnormality may be widespread in the nervous system in MJD, but that its manifestation as NCIs in the lower motor neurons may reflect their peculiar morphology characterized by very large cell bodies and very long axons, and associated with very high metabolic demand. It is noteworthy that recently, the occurrence of TDP-43 pathology has also been reported in AD [1, 2, 30], Lewy body-related diseases [21], Pick disease [2, 6], corticobasal degeneration [2, 30] and argyrophilic grain disease [7], suggesting that TDP-43 pathology, or more strictly, its pathological cytoplasmic localization, is part of a process rather common to neurodegenerative disorders, which is induced in combination with accumulation of the causative protein specific for each disease.
In Huntington’s disease (HD), also a CAG repeat disease, it has been reported that TDP-43 is co-localized with mutant huntingtin in dystrophic neurites and intracellular inclusions [26], suggesting that in this disease, there may be a protein–protein interaction between TDP-43 and huntingtin itself, or between TDP-43 and expanded polyglutamine stretches. In the present study, however, no TDP-43 pathology was observed in the nervous system except for the lower motor neuron nuclei in MJD, or in any CNS regions examined in SBMA. Moreover, in MJD, TDP-43-ir NCIs observed in the lower motor neurons were completely unreactive with 1C2 or anti-ataxin-3 antibody. It has also been reported that ALS-like ubiquitinated filamentous inclusions observed in the cerebellar dentate nucleus neurons in dentatorubral pallidoluysian atrophy (DRPLA), which can be recognized with 1C2 and anti-atrophin-1 antibodies [13, 32, 35], are also completely negative for TDP-43 [33]. These findings suggest that there is little interaction between TDP-43 and expanded polyglutamine stretches in MJD, SBMA or DRPLA.
In conclusion, in CAG repeat diseases, polyglutamine-related changes in the neuronal nuclei and cytoplasm, including the formation of NIIs, appear to be essential for disease progression. However, it is of great interest that in MJD, neuronal TDP-43 pathology can also occur only in the affected lower motor neurons. Although immunoblot analysis failed to demonstrate pathological pTDP-43, the findings of immunohistochemistry, including those at the ultrastructural level, strongly suggest that TDP-43 is a component of the filamentous NCIs. Finally, the results obtained in the present study may help to provide a better understanding of the pathomechanisms underlying motor neuron degeneration and death in ALS, which is also a multisystem neurological disease [23].
References
Amador-Ortiz C, Lin WL, Ahmed Z et al (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61:435–445
Arai T, Hasegawa M, Akiyama H et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Cairns NJ, Neumann M, Bigio EH et al (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240
Daoud H, Valdmanis PN, Kabashi E et al (2009) Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J Med Genet 46:112–114
Dickson DW, Josephs KA, Amador-Ortiz C (2007) TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol 114:71–79
Freeman SH, Spires-Jones T, Hyman BT, Growdon JH, Frosch MP (2008) TAR-DNA binding protein 43 in Pick disease. J Neuropathol Exp Neurol 67:62–67
Fujishiro H, Uchikado H, Arai T (2008) Accumulation of phosphorylated TDP-43 in brains of patients with argyrophilic grain disease. Acta Neuropathol 117:151–158
Geser F, Winton MJ, Kwong LK et al (2008) Pathological TDP-43 in parkinsonism–dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol 115:133–145
Gitcho MA, Baloh RH, Chakraverty S et al (2008) TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol 63:535–538
Hasegawa M, Arai T, Akiyama H et al (2007) TDP-43 is deposited in the Guam parkinsonism–dementia complex brains. Brain 13:1386–1394
Hasegawa M, Arai T, Nonaka T et al (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64:60–70
Hayashi M, Kobayashi K, Furuta H (2003) Immunohistochemical study of neuronal intranuclear and cytoplasmic inclusions in Machado–Joseph disease. Psychiatr Clin Neurosci 57:205–213
Hayashi Y, Kakita A, Yamada M et al (1998) Hereditary dentatorubral-pallidoluysian atrophy: ubiquitinated filamentous inclusions in the cerebellar dentate nucleus neurons. Acta Neuropathol 95:479–482
Inukai Y, Nonaka T, Arai T et al (2008) Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett 582:2899–2904
Kabashi E, Valdmanis PN, Dion P et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Kühnlein P, Sperfeld A-D, Vanmassenhove B et al (2008) Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch Neurol 65:1185–1189
Lin WL, Dickson DW (2008) Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol 116:205–213
Mackenzie IRA, Bigio EH, Ince PG et al (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434
Miklossy J, Steele JC, Yu S et al (2008) Enduring involvement of tau, β-amyloid, α-synuclein, ubiquitin and TDP-43 pathology in the amyotrophic lateral sclerosis/parkinsonism–dementia complex of Guam (ALS/PDC). Acta Neuropathol 116:625–637
Mori F, Tanji K, Zhang HX et al (2008) Maturation process of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol 116:193–203
Nakashima-Yasuda H, Uryu K, Robinson J et al (2007) Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 114:221–229
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Nishihira Y, Tan CF, Onodera O et al (2008) Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol 116:169–182
Piao YS, Wakabayashi K, Kakita A et al (2003) Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol 12:10–22
Rutherford NJ, Zhang YJ, Baker M et al (2008) Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 4:e1000193
Schwab C, Arai T, Hasegawa M, Yu S, McGeer PL (2008) Colocalization of transactivation-responsive DNA-binding protein 43 and Huntingtin in inclusions of Huntington disease. J Neuropathol Exp Neurol 67:1159–1165
Sreedharan J, Blair IP, Tripathi VB et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Suenaga T, Matsushima H, Nakamura S, Akiguchi I, Kimura J (1993) Ubiquitin-immunoreactive inclusions in anterior horn cells and hypoglossal neurons in a case with Joseph’s disease. Acta Neuropathol 85:341–344
Tan CF, Eguchi H, Tagawa A et al (2007) TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol 113:535–542
Uryu K, Nakashima-Yasuda H, Forman M et al (2008) Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol 67:555–564
Van Deelin VM, Leverenz JB, Bekris LM et al (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7:409–416
Yamada M, Piao YS, Toyoshima Y, Tsuji S, Takahashi H (2000) Ubiqutinated filamentous inclusions in cerebellar dentate nucleus neurons in dentatorubral-pallidoluysian atrophy contain expanded polyglutamine stretches. Acta Neuropathol 99:615–618
Yamada M, Sato T, Tsuji S, Takahashi H (2008) CAG repeat disorder models and human neuropathology: similarities and differences. Acta Neuropathol 115:71–86
Yamada M, Tan CF, Inenaga C, Tsuji S, Takahashi H (2004) Sharing of polyglutamine localization by the neuronal nucleus and cytoplasm in CAG-repeat diseases. Neuropathol Appl Neurobiol 30:665–675
Yamada M, Wood JD, Shimohata T et al (2001) Widespread occurrence of intranuclear atrophin-1 accumulation in the central nervous system neurons of patients with dentatorubral-pallidoluysian atrophy. Ann Neurol 49:14–23
Yokoseki A, Shiga A, Tan CF et al (2008) TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol 63:538–542
Acknowledgments
We thank C. Tanda, J. Takasaki, H. Saito and S. Egawa for their technical assistance, and M. Machida for help in preparing the manuscript. This work was supported by Grants-in-Aid for Scientific Research (17300109, 20240037) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and a grant from the Research Committee for Ataxic Diseases, the Ministry of Health, Labour and Welfare, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tan, CF., Yamada, M., Toyoshima, Y. et al. Selective occurrence of TDP-43-immunoreactive inclusions in the lower motor neurons in Machado–Joseph disease. Acta Neuropathol 118, 553–560 (2009). https://doi.org/10.1007/s00401-009-0552-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0552-x