
Abstract
Recently, 43-kDa TAR DNA-binding protein (TDP-43) was identified as a component of ubiquitinated inclusions (UIs) in sporadic amyotrophic lateral sclerosis (SALS). To clarify whether TDP-43 immunoreactivity is present in neuronal inclusions in familial ALS (FALS), we examined immunohistochemically the brains and spinal cords from four cases of FALS, two with Cu/Zn superoxide dismutase (SOD1) gene mutation and two without, together with three cases of SALS and three control subjects, using two antibodies, one polyclonal and one monoclonal, against TDP-43. Neuropathologically, the SOD1-related FALS cases were characterized by Lewy body-like hyaline inclusions (LBHIs) in the lower motor neurons. On the other hand, the SOD1-unrelated FALS cases showed degeneration restricted to the upper and lower motor neuron systems, with Bunina bodies (BBs) and UIs in the lower motor neurons, being indistinguishable from SALS. No cytoplasmic TDP-43 immunoreactivity was observed in the control subjects or SOD1-related FALS cases; LBHIs were ubiquitinated, but negative for TDP-43. UIs observed in the SALS and SOD1-unrelated FALS cases were clearly positive for TDP-43. BBs were negative for this protein. Interestingly, in these SALS and FALS cases, glial cells were also found to have cytoplasmic TDP-43-positive inclusions. These findings indicate that the histological and molecular pathology of SALS can occur as a phenotype of FALS without SOD1 mutation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive, fatal neurological disorder that affects both the upper and lower motor neuron systems of adults. In principle, it is a sporadic disorder (sporadic ALS: SALS). Rarely, however, clinically similar diseases can occur as an autosomal dominant or recessive trait (familial ALS: FALS) [12]. In FALS, mutations in the gene encoding Cu/Zn superoxide dismutase (SOD1) are known to be the most common causative abnormalities [3, 20]. Neuropathologically, at least two phenotypes have been recognized; one shows degeneration limited to the upper and lower motor neuron systems (classical form), and the other shows degeneration in the posterior column, Clarke’s column and spinocerebellar tracts in addition to the motor neuron systems (multisystem form, or form with posterior column involvement [4]) [6].
Considering the cellular pathology of SALS, it is of great importance that two characteristic cytoplasmic inclusions—Bunina bodies (BBs) and ubiquitinated skein-like or round inclusions (UIs)—have been described in the lower motor neurons [19]. On the other hand, in FALS, Lewy body-like hyaline inclusions (LBHIs) [5], which are also ubiquitinated [11, 13], are a characteristic feature in the lower motor neurons of most patients with SOD1-related FALS, in whom the neuropathology of the multisystem form is often evident [22, 25, 26]. With regard to the classical form of FALS, we recently encountered a patient with SOD1-unrelated FALS, in whom degenerative changes were limited to the upper and lower motor neuron systems, and interestingly, BBs and UIs were observed in the remaining lower motor neurons, being indistinguishable from SALS [23]. Shortly after studying this case, we had an opportunity of examining another patient with SOD1-unrelated FALS. Again, the neuropathology was essentially that of SALS, with BBs and UIs in the lower motor neurons (case 4 described below).
Significantly, 43-kDa TAR DNA-binding protein (TDP-43), a nuclear protein [18, 29], was recently identified as a component of UIs in frontotemporal lobar degeneration and ALS [1, 15]. In the present study, we performed immunohistochemical examination of the brains and spinal cords from autopsy cases of FALS with or without SOD1 gene mutation in order to clarify whether TDP-43-immunoreactive neuronal inclusions can also occur in these two subgroups of FALS.
Materials and methods
Subjects
Brains and spinal cords from four cases of FALS, two with SOD1 gene mutation (Ala4Thr and Asp101Tyr, respectively) and two without, as well as from three cases of SALS and three control subjects without neurological or psychiatric disorders, were examined in the present study (Table 1). With regard to the FALS cases, the clinical and pathological findings in cases 1 [25], 2 [26] and 3 [23] have been reported previously. Here we describe the clinical and pathological findings in case 4, which have not yet been reported.
Case report
Case 4
The patient had been healthy until the age of 61 years, when he noticed clumsiness of the left hand. Within the following 2 months, dysarthria and dysphagia became evident. About 4 months after symptom onset, examination demonstrated atrophy, with fasciculation, of the tongue. Muscle weakness, with fasciculation, was also evident in the left upper extremity. Tendon reflexes were exaggerated, and Babinski’s sign was present, in the left extremities. Both electromyography and muscle biopsy showed a neurogenic pattern. The patient’s mental state was apparently normal. A clinical diagnosis of ALS was made. The subsequent clinical course was that of typical ALS, and the patient died of respiratory failure at the age of 62 years, about 20 months after disease onset.
Importantly, there was a family history of a similar neurological disease affecting only the patient’s two younger brothers. Postmortem gene analysis of SOD1 using the patient’s frozen cerebellar tissue showed no mutation (the procedure has been described previously [14]).
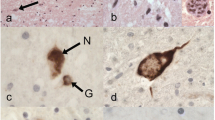
The brain weighed 1,340 g before fixation, showing no apparent abnormalities in external appearance. Histologically, myelin pallor was evident in the anterolateral columns of the spinal cord, especially in the lateral and anterior corticospinal tracts. No degeneration was evident in the posterior column. Moderate to severe neuronal loss and gliosis were evident in the spinal anterior horns and brainstem motor neuron nuclei (XII, VII, and V) (Fig. 1a). Intracytoplasmic eosinophilic inclusions, so-called BBs (Fig. 1b), and UIs (skein-like inclusions) (Fig. 1c) were occasionally found in the remaining lower motor neurons. The BBs observed were immunoreactive for cystatin C [16] and transferrin [10] (data not shown). The ultrastructural presence of these neuronal inclusions was also confirmed in the spinal anterior horn cells by conventional electron microscopy (data not shown). In the precentral cortex, loss of pyramidal neurons, including Betz cells, and gliosis were also evident. In the non-motor neuron systems, there were a few UIs in the putaminal small neurons (Fig. 1d).

Histological findings in case 4. a Severe neuronal loss and gliosis in the hypoglossal nucleus. b A hypoglossal motor neuron, showing a cluster of Bunina bodies (arrow). c A cervical anterior horn cell, showing ubiquitin-positive skeins. d A putaminal small neuron, showing perinuclear ubiquitin-positive structures. a, b Hematoxylin–eosin; c, d Ubiquitin immunostaining. Bars a 100 μm; b, c 10 μm; d 20 μm
Immunohistochemistry
Multiple histological specimens and the original formalin-fixed, paraffin-embedded tissue blocks were available in all cases. In addition, specimens of ubiquitin-immunostained sections covering various brain and spinal cord regions were available in all cases of FALS and SALS. We reviewed these postmortem materials.
For the present immunohistochemical study, we prepared new 4-μm-thick, paraffin-embedded sections from the original blocks of various brain and spinal cord regions. These sections were immunostained by the avidin–biotin–peroxidase complex (ABC) method with a Vectastain ABC kit (Vector, Burlingame, CA, USA), using two antibodies against TDP-43: a rabbit polyclonal antibody (10782-1-AP; ProteinTec Group Inc., Chicago, IL, USA; 1:4,000) and a mouse monoclonal antibody (2E2-D3; Abnova Corpoeation, Taipei, Taiwan; 1:100,000). Pretreatment of the sections by heating for 10 min at 121°C in 10 mM sodium citrate buffer was need for immunostaining with the 2E2-D3 monoclonal antibody [1]. When necessary, selected sections were also immunostained with a rabbit polyclonal antibody against ubiquitin (Dako, Glostrup, Denmark; diluted 1:800). Diaminobenzidine (DAB) was used as the chromogen.
In addition, a double-labeling immunofluorescence study was performed on selected sections, using a rabbit polyclonal antibody against cystatin C (Dako; 1:2,000) and a mouse monoclonal antibody against TDP-43 (2E2-D3; Abnova Corpoeation; 1:40,000), or a rabbit polyclonal antibody against TDP-43 (10782-1-AP; ProteinTec Group Inc.; 1:4,000) and a mouse monoclonal antibody against ubiquitin (Dako; 1:8,000). The second antibodies used were Alexa Fluor 488 goat anti-rabbit IgG (Molecular Probes, Eugene, OR; 1:1,000) and Alexa Fluor 568 goat anti-mouse IgG (Molecular Probes; 1:1,000) in the former, and Alexa Fluor 568 goat anti-rabbit IgG (Molecular Probes; 1:1,000) and Alexa Fluor 488 goat anti-mouse IgG (Molecular Probes; 1:1,000) in the latter.
Results
There was no essential difference in immunostaining property between the polyclonal and monoclonal antibodies. In all the examined cases, normal granular nuclear staining for TDP-43 was evident in neurons and glial cells of the brain and spinal cord. In general, nuclei of neurons and glial cells that possessed cytoplasmic TDP-43-positive inclusions (described below) were found to be negative for TDP-43.
In the SALS cases, BBs and UIs were observed in the lower motor neurons. UIs were also observed in the putaminal small neurons (case 6 and 7) and hipocampal dentate granule cells (case 7). TDP-43-positive cytoplasmic inclusions were found in the lower motor neurons (Fig. 2a, b), putaminal small neurons (case 7), and hippocampal granules cells (case 7) (Fig. 2c). No TDP-43-positive structures apparently corresponding to BBs could be detected. Unexpectedly, similar TDP-43-positive neuronal inclusions were also found in the inferior olivary nucleus (case 5 and 7) (Fig. 2d) and reticular formation (Fig. 2e) of the brainstem, the globus pallidus (Fig. 2f) and the thalamus (Fig. 2g). In addition, irregularly shaped inclusions were found in the cytoplasm of glial cells mainly in the affected gray matter of the lower and upper motor neuron systems (Fig. 2h). In the control subjects, no intracytoplasmic inclusions associated with SALS or FALS, or other neurological disorders, were observed. No cytoplasmic TDP-43 immunoreactivity was found in neurons or glial cells of the brain or spinal cord.

a–h Sporadic ALS. TDP-43-positive cytoplasmic inclusions are evident in neurons in the motor nucleus of the facial nerve (a), thoracic anterior horn (b), hippocampal dentate gyrus (c), inferior olivary nucleus (d), midbrain reticular formation (e), globus pallidus (f) and thalamus (g). TDP-43-positive cytoplasmic inclusions are also evident in a glial cell in the motor cortex (h). i–l SOD1-related familial ALS. In the lumbar anterior horn, cytoplasmic Lewy body-like hyaline inclusions (LBHIs) are evident in a motor neuron (i), similar cytoplasmic inclusions observed in another motor neuron are positive for ubiquitin (j), and such LBHIs (k) as well as cord-like, swollen neuronal processes (l) are negative for TDP-43. m–t SOD1-unrelated familial ALS. TDP-43-positive cytoplasmic inclusions are evident in neurons in the motor nucleus of the facial nerve (m), cervical anterior horn (n), putamen (o), inferior olivary nucleus (p), globus pallidus (q) and motor cortex [medium-sized pyramidal neuron (r); Betz cell (s)]. TDP-43-positive cytoplasmic inclusions are also evident in a glial cell in the cervical anterior horn (t). TDP-43-positive neuronal cytoplasmic inclusions are variable in shape, showing a skein-like (a, b, d, m-s), round (c), dense body-like (f, g), or somewhat granular (e) appearance. Arrows indicate nuclei of the neurons (c, e–g, k, o, q, r); note that in a neuron with TDP-43-negative LBHIs, granular nuclear staining of TDP-43 is preserved (k). The shape of TDP-43-positive glial cytoplasmic inclusions can be described as C-like or fibrillary tangle-like (h, t). a, c–g, k–m, o–s TDP-43 immunostaining with the polyclonal antibody; b, h, n, t TDP-43 immunostaining with the monoclonal antibody; i Hematoxylin–eosin; j Ubiquitin immunostaining with the polyclonal antibody. Bar 10 μm for a–t
In the SOD1-related FALS cases, LBHIs observed in the lower motor neurons (Fig. 2i) were positive for ubiquitin (Fig. 2j). Cord-like, swollen neuronal processes, with or without LBHIs, observed in the lower motor neuron nuclei also showed ubiquitin positivity. No cytoplasmic TDP-43 immunoreactivity was found in neurons or glial cells in any of the regions examined; it was evident that LBHIs (Fig. 2k) and cord-like swollen neuronal processes were negative for TDP-43 (Fig. 2l).
In the SOD1-unrelated FALS cases, BBs and UIs were observed in the lower motor neurons. A few UIs were also evident in the putaminal small neurons. The results of TDP-43 immunohistochemistry were essentially the same as those obtained in the SALS cases. TDP-43-positive cytoplasmic inclusions were found in neurons in the lower motor neuron nuclei (Fig. 2m, n), putamen (Fig. 2o), inferior olivary nucleus (case 3) (Fig. 2p) and reticular formation of the brainstem, globus pallidus (case 4) (Fig. 2q) and thalamus. Moreover, similar TDP-43-positive neuronal inclusions were also found in the motor cortex (Fig. 2r, s). There were no TDP-43-positive structures reminiscent of BBs. Again, irregularly shaped inclusions were found in the cytoplasm of glial cells mainly in the affected gray matter of the lower and upper motor neuron systems (Fig. 2t); such glial inclusions were more frequent and more widespread than in the SALS cases, and also in case 4 than in case 3 (Fig. 3).

TDP-43-positive glial cytoplasmic inclusions observed in the motor cortex (a), internal capsule (b) and cervical anterior horn (c) in case 4. The inclusion-bearing glial cells show relatively uniform, round, pale swollen nuclei, suggesting that most of them are oligodendrocytes. a–c TDP-43 immunostaining with the polyclonal antibody. Bar 30 μm for a–c
In the SOD1-related FALS cases, double-labeling immunofluorescence confirmed that LBHIs observed in the lower motor neurons were ubiquitinated, but negative for TDP-43 (Fig. 4a–c). On the other hand, in the SALS and SOD1-unrelated FALS cases, double-labeling immunofluorescence clearly demonstrated co-localization of TDP-43 and ubiquitin in UIs (Fig. 4d–f), and a lack of TDP-43 immunoreactivity in cystatin C-positive BBs in the lower motor neurons (Fig. 4g–i).

a–c SOD1-related familial ALS. In a lumbar anterior horn cell, there are no apparent cytoplasmic TDP-43-positive structures; only autofluorescence of lipofuscin granules is evident (red, a). Ubiquitinated cytoplasmic inclusions (LBHIs) (green, b) are shown to be negative for TDP-43 (no overlap, c). d–f SOD1-unrelated familial ALS. In a facial nucleus motor neuron, TDP-43 (red, d) and ubiquitin (green, e) are shown to be co-localized (yellow, f) in skein-like inclusions. g–i Sporadic ALS. In the motor nucleus of the facial nerve, cytoplasmic cystatin C-positive Bunina bodies (BBs, arrows) are evident in a motor neuron (green, g). TDP-43-positive skeins (arrows) are evident in another motor neuron (red, h). The cystatin C-positive BBs are negative for TDP-43 (no overlap, i). a–f Double-labeling immunofluorescence with the polyclonal anti-TDP-43 and monoclonal anti-ubiquitin antibodies. g–i Double-labeling immunofluorescence with the polyclonal anti-cystatin C and monoclonal anti-TDP-43 antibodies. Bars 10 μm
Discussion
We have reported an additional autopsy case of FALS showing no mutation in the SOD1 gene (case 4). The disease appeared to be of autosomal recessive inheritance. The neuropathology, including the occurrence of BBs [17, 19, 24, 27] and UIs [7–9, 19, 21] in the remaining lower motor neurons, was indistinguishable from that in patients with SALS [19]. A few UIs were also found in the putaminal small neurons. Such UIs have also been demonstrated in a significant fraction of SALS patients with or without dementia [19, 28]. This family had no relation to the ALS cluster in the Kii peninsula, or the family reported previously by us, in which one autopsy case demonstrated clinical and pathological features very similar, if not identical, to those in the present case [23].
The present study was carried out using two anti-TDP-43 antibodies that were the same as those used in two studies reported previously [1, 15]. The results obtained from the SALS cases and control subjects were also essentially the same as those in the two previous studies; in SALS, UIs observed in the lower motor neurons were shown to be positive for TDP-43 [1, 15], and glial cells were also found to contain TDP-43-positive cytoplasmic inclusions, which were undetectable with the antibody against ubiquitin [1]. In the present study, it was also of significance that BBs were negative for this protein.
Arai et al. [1] reported that the polyclonal antibody recognized TDP-43-positive neuronal intranuclear inclusions in the spinal anterior horn from a case of SALS, and that the monoclonal antibody failed to demonstrate glial cytoplasmic inclusions. In the present study, no intranuclear inclusions were evident. More importantly, however, TDP-43-positive glial cytoplasmic inclusions were detectable with both antibodies, strongly suggesting that both neurons and glial cells can be involved in the disease process in SALS (a neuro-glial proteinopathy of TDP-43). Arai et al. also demonstrated that a proportion of neurofibrillary tangles in Alzheimer’s disease were TDP-43-positive with the polyclonal antibody [1]. In this connection, the present study performed on 4-μm-thick, paraffin-embedded sections showed that a small number of concomitant neurofibrillary tangles (NFTs) encountered in the hippocampus and parahippocampal gyrus (case 2–4, 6–8, and 10) were negative for TDP-43 (data not shown), as shown more recently by Davidson et al [2].
It was of considerable interest that in the SALS, TDP-43-positive neuronal inclusions were also found in brain regions where no UIs were observed, i.e., the inferior olivary nucleus, brainstem reticular formation, globus pallidus, etc. Only recently, TDP-43-positive neuronal inclusions have been shown to occur in the inferior and superior olivary nuclei in a fraction of SALS cases [2]. It was noteworthy that in the SOD1-unrelated FALS cases, TDP-43-postive cytoplasmic inclusions were also found in pyramidal neurons, including Betz cells, in the motor cortex where no UIs were evident. To discuss the significance of theses findings satisfactorily, further studies including a large number of SALS cases are needed.
In the SOD1-related FALS cases, ubiquitinated LBHIs and cord-like, swollen neuronal processes were shown to be negative for TDP-43. These structures have been reported to contain SOD1 protein [22, 26]. The present finding strongly suggests that in addition to the presence or absence of inheritance, SALS and SOD1-related FALS have essentially different underlying molecular pathomechanisms.
On the other hand, in the SOD1-unrelated FALS cases, the entire pathological picture was that of SALS, and clinically the diseases appeared to show autosomal recessive inheritance (cases 3 [23] and 4). The TDP-43 immunoreactivity in the SOD1-unrelated FALS cases was almost identical to that in the SALS cases, indicating that TDP-43 was also involved in SOD1-unrelated FALS, and strongly suggesting that this protein might play an important role in the pathogenesis as the major disease protein, as discussed previously in the original reports [1, 15].
Finally, although only small amounts were obtained, frozen brain tissue was sampled at autopsy in each of the SOD1-unrelated FALS cases. Using these frozen tissues, it is important and necessary for us to address the next question of whether abnormalities in the gene encoding TDP-43 are present in these cases.
References
Arai A, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP-43 is a component of ubiquitin-positive inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Davidson Y, Kelley T, Mackenzie IRA, Pickering-Brown S, Du Plessis D, Neary D, Snowden JS, Mann DMA (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. doi:10.1007/s00401-006-0189-y
Deng H-X, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, Warner C, Deng G, Soiano E, Smyth C, Parge HE, Ahmed A, Roses AD, Hallewell RA, Pericak-Vance MA, Siddique T (1993) Amyotrophic lateral sclerosis and structural defects in Cu/Zn superoxide dismutase. Science 261:1047–1051
Engel WK, Kurland LT, Klatzo I (1959) An inherited disease similar to amyotrophic lateral sclerosis with a pattern of posterior column involvement. An intermediate form? Brain 82:203–220
Hirano A, Kurland LT, Sayre GP (1967) Familial amyotrophic lateral sclerosis. A subgroup characterized by posterior and spinocerebellar tract involvement and hyaline inclusions in the anterior horn cells. Arch Neurol 16:232–243
Horton WA, Eldridge R, Brody JA (1976) Familial motor neuron disease. Evidence for at least three different types. Neurology 26:460–465
Leigh PN, Anderton BH, Dodson A, Gallo J-M, Swash M, Power DM (1988) Ubiquitin deposits in anterior horn cells in motor neuron disease. Neurosci Lett 93:197–203
Leigh PN, Withwell H, Garofalo O, Buller J, Swash M, Martin JE, Gallo J-M, Weller RO, Anerton BH (1991) Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 114:775–788
Lowe J, Lennox G, Jefferson D, Morrell K, McQuire D, Gray T, Landon M, Doherty FJ, Mayer RI (1988) A filamentous inclusion body within anterior horn neurons in motor neuron disease defined by immunocytochemical localization of ubiquitin. Neurosci Lett 94:203–210
Mizuno Y, Amari M, Takatama M, Aizawa H, Mihara B, Okamoto K (2006) Transferrin localizes in Bunina bodies in amyotrophic lateral sclerosis. Acta Neuropathol 112:597–603
Mizusawa H, Hirano A, Yen S-H (1991) Anterior horn cell inclusions in familial amyotrophic lateral sclerosis contain ubiquitin and phosphorylated neurofilaments epitopes. Neuropathology 11:11–20
Mulder DW, Kurland LT, Offord KP, Beard CM (1986) Familial adult motor neuron disease: amyotrophic lateral sclerosis. Neurology 36:511–517
Murayama S, Ookawa Y, Mori H, Nakano I, Ihara Y, Kuzuhara S, Tomonaga M (1989) Immunocytochemical and ultrastructural study of Lewy body-like hyaline inclusions in familial amyotrophic lateral sclerosis. Acta Neuropathol 78:143–152
Nakano R, Sato S, Inuzuka T, Sakimura K, Mishina M, Takahashi H, Ikuta F, Honma Y, Fujii J, Taniguchi N, Tsuji S (1994) A novel mutation in Cu/Zn superoxide dismutase gene in Japanese familial amyotrophic lateral sclerosis. Biochem Biophys Res Commun 200:695–703s
Neumann M, Sampathu DM, Kwong LK, Truax A, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM-Y (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Okamoto K, Hirai S, Amari M, Watanabe M, Sakurai A (1993) Bunina bodies in amyotrophic lateral sclerosis immunostained with rabbit anti-cystatin C serum. Neurosci Lett 162:125–128
Okamoto K, Morimatsu M, Hirai S, Ishida Y (1980) Intracytoplasmic inclusions (Bunina bodies) in amyotrophic lateral sclerosis. Acta Pathol Jpn 30:591–597
Ou SH, Wu F, Harrich D, García-Martínez LF, Gaynor RB (1995) Cloning and characterization of a novel cellular protein, TDP-43, that bind to human immunodeficiency virus type I TAR DNA sequence motifs. J Viol 69:3584–3596
Piao Y-S, Wakabayashi K, Kakita A, Yamada M, Hayashi S, Morita T, Ikuta F, Oyanagi K, Takahashi H (2003) Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol 13:10–22
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, Oregan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasck D,Cayabyab A, Gaston SM, Berger R,Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horritz HR, Brown RH (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Sasaki S, Murayama S (1992) Ultrastructural study of skein-like inclusions in anterior horn neurons of patients with motor neuron disease. Neurosci Lett 147:121–124
Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, Hung WY, Kato T, Asayama K (1996) Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol 55:481–490
Tagawa A, Tan C-F, Kikugawa K, Fukase M, Nakano R, Onodera O, Nishizawa M, Takahashi H (2007) Familial amyotrophic lateral sclerosis: a SOD1-unrelated Japanese family of bulbar type with Bunina bodies and ubiquitin-positive skein-like inclusions in lower motor neurons. Acta Neuropathol 113:205–211
Takahashi H (1992) Bunina body in amyotrophic lateral sclerosis (in Japanese). No To Shinkei 44:525–532
Takahashi H, Makifuchi T, Nakano R, Sato S, Inuzuka T, Sakimura K, Mishina M, Honma Y, Tsuji S, Ikuta F (1994) Familial amyotrophic lateral sclerosis with a mutation in the Cu/Zn superoxide dismutase gene. Acta Neuropathol 88:185–188
Tan C-F, Piao Y-S, Hayashi S, Obata H, Umeda Y, Sato M, Fukushima T, Nakano R, Tsuji S, Takahashi H (2004) Familial amyotrophic lateral sclerosis with bulbar onset and a novel Asp101Tyr Cu/Zn superoxide dismutase gene mutation. Acta Neuropathol 108:332–336
Tomonaga M, Saito M, Yoshimura M, Shimada H, Tohgi H (1978) Ultrastructure of the Bunina bodies in amyotrophic lateral sclerosis. Acta Neuropathol 42:81–86
Wakabayashi K, Piao Y-S, Hayashi S, Kakita A, Yamada M, Takahashi H (2001) Ubiquitinated neuronal inclusions in the neostriatum in patients with amyotrophic lateral sclerosis with and without dementia—a study of 60 patients 31 to 87 years of age. Clin Neuropathol 20:47–52
Wang H-Y, Wang I-F, Bose J, Shen C-KJ (2004) Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics 83:130–139
Acknowledgments
We thank C. Tanda, J. Takasaki, N. Kaneko, Y. Ota and S. Egawa for their technical assistance. This work was supported by a grant from the Research Committee on Neurodegenerative Diseases, Ministry of Health, Labor and Welfare, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tan, CF., Eguchi, H., Tagawa, A. et al. TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol 113, 535–542 (2007). https://doi.org/10.1007/s00401-007-0206-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-007-0206-9