
Abstract
Pathological TDP-43 is the major disease protein in frontotemporal lobar degeneration characterized by ubiquitin inclusions (FTLD-U) with/without motor neuron disease (MND) and in amyotrophic lateral sclerosis (ALS). As Guamanian parkinsonism–dementia complex (PDC) or Guamanian ALS (G-PDC or G-ALS) of the Chamorro population may present clinically similar to FTLD-U and ALS, TDP-43 pathology may be present in the G-PDC and G-ALS. Thus, we examined cortical or spinal cord samples from 54 Guamanian subjects for evidence of TDP-43 pathology. In addition to cortical neurofibrillary and glial tau pathology, G-PDC was associated with cortical TDP-43 positive dystrophic neurites and neuronal and glial inclusions in gray and/or white matter. Biochemical analyses showed the presence of FTLD-U-like insoluble TDP-43 in G-PDC, but not in Guam controls (G-C). Spinal cord pathology of G-PDC or G-ALS was characterized by tau positive tangles as well as TDP-43 positive inclusions in lower motor neurons and glial cells. G-C had variable tau and negligible TDP-43 pathology. These results indicate that G-PDC and G-ALS are associated with pathological TDP-43 similar to FTLD-U with/without MND as well as ALS, and that neocortical or hippocampal TDP-43 pathology distinguishes controls from disease subjects better than tau pathology. Finally, we conclude that the spectrum of TDP-43 proteinopathies should be expanded to include neurodegenerative cognitive and motor diseases, affecting the Chamorro population of Guam.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinsonism–dementia complex (PDC) and amyotrophic lateral sclerosis (ALS) of the Chamorro population on Guam are diseases of unknown etiology. Although they have been extensively characterized (for review see [9]), their nosological status is still unclear. The presence of widespread central nervous system Alzheimer’s disease (AD)-like neurofibrillary tangles (NFTs) formed by pathological tau defines these disorders as neurodegenerative tauopathies with clinical and pathological overlap between Guamanian PDC (G-PDC) and Guamanian ALS (G-ALS) [9]. In G-ALS ubiquitinated inclusions in lower motor neurons (LMN) are found, similar to ALS outside of Guam; thus, it is likely that G-PDC and G-ALS represent a continuous spectrum of a single neurodegenerative disease with a common underlying pathogenetic mechanism [6]. However, this notion has been challenged by the presence of NFTs in clinically unaffected Chamorros [1, 4, 26, 28, 30], and it is possible that a different and as yet unidentified pathology or pathogenesis might underlie this group of diseases.
“Ubiquitin only dementia” known as frontotemporal lobar degeneration (FTLD) with ubiquitin positive inclusions (FTLD-U) is mainly characterized by a degeneration of the neocortex and hippocampus with ubiquitin positive, tau and α-synuclein-negative inclusions without motor neuron disease (MND) (for review see [11]); in the presence of a history of MND, the term FTLD-MND is used. Parkinsonism is present in up to 60% of FTLD-U cases [7]. On the basis of ubiquitin and novel monoclonal antibodies (MAbs) generated by using material from FTLD-U brains, at least four subtypes of FTLD-U have been identified [3, 23, 32]. ALS-like LMN inclusions may occur in FTLD-U [20, 24]. Further, ALS cases may show ubiquitinated inclusion pathology in the cortex or hippocampus, especially in association with a history of dementia [19]. Recently, TDP-43, a 43 kDa nuclear TAR DNA-binding protein, was identified as the major disease protein in FTLD-U, FTLD-MND, and ALS, thereby implicating a common pathogenesis of these disorders [24]. As G-PDC or G-ALS may present clinically with symptoms similar or even indistinguishable to FTLD-U, FTLD-MND or ALS, we hypothesized that TPD-43 might also be involved in the pathogenesis of G-PDC and G-ALS. To address this question, we examined 54 Guamanian subjects and found pathological TDP-43 inclusions in both brain and spinal cord of the diseased population, but it was rare in controls. While a report on a small number of cases appeared recently describing similar findings on TDP-43 pathology in atypical Parkinsonism of Guam [13], we independently found TDP-43 proteinopathy in G-PDC and report the first evidence of spinal cord TDP-43 pathology in G-ALS. Thus, TDP-43 may play a major pathogenic role in G-PDC and G-ALS similar to FTLD-U and ALS.
Material and methods
Study subjects
Postmortem brain samples from a cohort of deceased residents of Guam were obtained from the Guam Brain Bank at Mount Sinai School of Medicine or directly from the University of Guam. The patients were evaluated by neurologists from the University of California at San Diego during regular study visits to Guam, and clinical diagnoses were established as described earlier [10]. The study subjects were categorized either as G-PDC, G-ALS, or as Guamanian controls (G-C). Patients with a combination of G-PDC and G-ALS were classified as either G-PDC or G-ALS according to their predominant or earliest clinical features.
Immunohistochemistry
Fresh or frozen tissues from brain and spinal cord were fixed in 10% neutral buffered formalin or 70% ethanol with 150 mmol NaCl, paraffin-embedded, and cut into 6 μm sections. The latter were stained with hematoxylin and eosin. Immunohistochemistry was performed as previously described using the avidin–biotin complex detection method (Vectastatin ABC kit, Vector Laboratories, Burlingame, CA, USA) with 3,3′-diaminobenzidine as chromogen [19]. The following primary antibodies were used: mouse anti-paired helical filament-1 (PHF-1) MAb (1:1,000) [12], mouse anti-ubiquitin (1510, Chemicon, Temecula, CA, USA; 1:40,000) MAb and rabbit polyclonal anti-TDP-43 (ProteinTech Group, Chicago, IL, USA; 1:3,000). Sections stained for ubiquitin and TDP-43 were microwaved in citrate antigen unmasking solution (Vector Laboratories Burlingame, CA, USA). Double-labeling immunofluorescence using Alexa Fluor 488 and 594 conjugated secondary antibodies (Molecular Probes, Eugene, OR, USA) was performed as previously described [24]. Digital images of immunohistochemistry were obtained by using an Olympus BX 51 (Tokyo, Japan) equipped with bright-field and fluorescence light sources using a ProGres C14 digital camera (Jenoptik AG, Jena, Germany) and Adobe Photoshop, Version 9.0 (Adobe Systems, San Jose, CA, USA) or digital camera-DP71 (Olympus, Orangeburg, NY, USA), and DP manager (Olympus, Orangeburg, NY, USA). Digital images of immunofluorescence were obtained using a Nikon TE2000 microscope and were captured with a CoolSNAP Monochrome camera (Photometrics, Tucson, AZ, USA) and Metamorph (Molecular Devices, Downingtown, PA, USA) software.
Quantitation of pathology
Grading of PHF-1, ubiquitin and TDP-43–immunoreactive pathology was performed on stained tissue sections. The scoring system for LMN neuronal cytoplasmic inclusions (NCIs) is based on total numbers in a single section of spinal cord with maximal pathology. For TDP-43, a total score of NCIs including filamentous, round, or irregular shaped inclusions, small dense granules, and axons with focal positivity/dystrophic neurites was calculated using the following scoring system: none 0; 1–3 inclusions per section, stage 1; 4–10 inclusions per section, stage 2; and >10 inclusions per section, stage 3 [19]. For tau, the presence of NFTs and oligo- or astroglial tau pathology was evaluated, and TDP-43 positive glial inclusions were assessed in the gray and adjacent white matter of the spinal cord on the same grading scale. Neocortical and hippocampal pathology were rated on a similar ordinal grading scale (none 0; mild 1; moderate 2; severe 3), but a 0.5 score was included to account for rare pathology. The specific cortical pathology including neuronal or glial inclusions and dystrophic cellular processes is described in the results section.
Biochemical analyses
Frontal and temporal gray and white matter derived from frozen brain samples were divided and processed separately. To examine the solubility profile of TDP-43, brain tissues were sequentially extracted with buffers of increasing strength as described earlier [38]. Briefly, brain tissue was homogenized in high salt buffer (0.75 M NaCl, 50 mM Tris-HCl, 2 mM EDTA, pH 7.4), high salt Triton-X 100 buffer (high salt buffer + 1% Triton-X 100), radioimmunoprecipitation assay buffer (0.1% sodium dodecyl sulfate (SDS), 1% NP-40, 0.5% deoxycholic acid 5 mM EDTA, 150 mM NaCl, 50 mM Tris-HCl, pH 8.0) and SDS buffer (2% SDS in 50 mM Tris-HCl pH 7.6). Protease inhibitors were added to buffers prior to use. SDS insoluble material was extracted in 70% formic acid (FA). FA was evaporated in an Automatic Environmental SpeedVac System (Savant Instruments, Holbrook, USA). The dried pellets were re-suspended in SDS-polyacrylamide gel electrophoresis sample buffer and the pH was adjusted to neutral with 2 M NaOH.
For immunoblot analysis, proteins were resolved by 5–20% gradient Tris-glycine SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and probed with a mixture of anti-tau MAb T14 (1:3,000) and T46 (1:1,000) [15, 17], or rabbit polyclonal anti-C-terminus TDP-43 antibody (1038). The primary antibodies were detected with horseradish peroxidase conjugated anti-rabbit or anti-mouse IgG (Santa Cruz Biotechnologies, Santa Cruz, CA, USA). Blots were developed with Renaissance Enhanced Luminol Reagents (NEN Life Science Product, Boston, MA), and digital images were acquired using a Fujifilm Intelligent Darkbox II (Fuji Systems USA, Stamford, CT, USA).
Statistical analyses
The data were analyzed using SPSS 11.5 or 15.0 for Windows (SPSS, Chicago, IL, USA). The “average” or “spread” of data on patient characteristics or ordinal rating scale was estimated by calculating the mean (and standard deviation) or median (and 25th to 75th percentiles). Further, the medians and 25th to 75th percentiles of the ratings were calculated from “grouped data” taking into account that one stage follows continuous into the other, and therefore, they represent classes rather than clearly distinguishable values on a numerical scale. When a certain percentile was not available from grouped data, a standard percentile was obtained. Non-parametric tests were used for hypothesis testing including the Mann–Whitney U, Friedman and Wilcoxon signed ranks test for continuous or ordinal data. P values of the exact tests were reported for these non-parametric tests. The Fisher’s exact test was applied to compare proportions. Association analyses [31] were performed using Spearman’s rank correlations. The correlation coefficients were interpreted as follows: <0.30 as low, 0.30–0.60 as moderate, 0.60–0.90 as moderately high, and >0.9 as high correlation. The significance level was set at 0.01 rather than the usual 0.05 because multiple statistical tests were done. All statistical tests applied are two sided.
Results
Clinical characteristics
We examined 54 Guamanian subjects including 31 males and 23 females. All the patients (n = 39) had clinical evidence of dementia, parkinsonism, or motor neuron signs in various combinations, and were confirmed at autopsy to have the typical pathology of G-PDC or G-ALS as previously reported [8, 30]. G-C did not show any overt neurological or cognitive dysfunction. The study subjects were categorized as either G-PDC (n = 30, cortical areas available: n = 13, spinal cord available: n = 17), G-ALS (spinal cord available: n = 9), or as G-C (n = 15, cortical areas available: n = 11, spinal cord available: n = 5). The clinical characteristics of the study cohort are summarized in Table 1 (for complete list, see supplementary data Table 1). G-PDC patients had a significantly longer disease duration compared with G-ALS cases and their age at death was significantly higher. There was no significant difference in age at death between G-PDC and G-C. Nearly all study subjects were Chamorros (50/54), one was a mixed Chamorro and Japanese and three were migrants to Guam, two Filipino and one Caucasian (Table 1). All autopsies were performed between the mid 1970s and mid 2000s (see supplementary data Table 1).
Cortical pathology
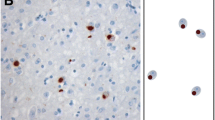
To determine if TDP-43 pathology is present in G-PDC, cortical regions from G-PDC and G-C were examined. All G-PDC cases with cortical tissue available (n = 11) stained positive for tau, ubiquitin, and TDP-43, albeit to varying degrees in the various cortical areas (Table 2). There were widespread tau positive NFTs both in the hippocampal formation and neocortex (Table 2) associated with neuron loss and gliosis. These changes were predominantly located in layers II and III of the neocortex. White matter tau pathology was present as well and included oligodendrogial (or astroglial) inclusions and dystrophic cellular processes. TDP-43 staining revealed a varying degree of, sometimes focal, pathology including neuronal and oligodendroglial inclusions and dystrophic neurites/axonal profiles as well as occasional lentiform shape nuclear inclusions (Table 2, Figs. 1, 2). As with the tau pathology, the degree of TDP-43 pathology was similar in both gray and white matter (Table 2). When comparing PHF-1 with TDP-43 overall average scores (as shown in Table 2), PHF-1 scores in the neocortical gray and white matter and hippocampal formation tended to be higher than the corresponding TDP-43 scores (P values: 0.090 or 0.044 or 0.018). Further, both tau positive and TDP-43 positive pathological structures stained for ubiquitin. Double labeling immunofluorescence experiments demonstrated that pathological TDP-43 co-localized with ubiquitin, but not with tau pathology (Figs. 3a–l, 4). In the hippocampal formation, TDP-43 pathology in the CA1/subiculum was scarce and consisted mainly of dystrophic neurites in sharp contrast to the dentate gyrus and entorhinal cortex with perikaryal inclusions (Fig. 5a, b). Normal nuclear TDP-43 staining was found in unaffected neurons, but not in neurons bearing cytoplasmic inclusion suggesting a shift from TDP-43 from the nucleus into the cytoplasm (“nuclear clearing”) in the diseased neurons. In contrast to the CA1/subiculum of the hippocampus, in the dentate nucleus the tau and TDP-43 burden were similar (Table 2, P = 0.157). A subset of the ubiquitin-positive inclusions in dentate granule cells was TDP-43 positive and another subset was tau and ubiquitin positive, but none was TDP-43 and tau positive (Fig. 5c, d, e).

Spectrum of neocortical gray matter pathology in G-PDC detected by anti-TDP-43 immunohistochemistry. a, b, e Temporal cortex; c, d, f, g parietal cortex; large arrows neuronal cytoplasmic inclusion; small arrows dystrophic neurite/axonal profile; arrowhead glial inclusion; asterisks intranuclear inclusion (“cat eye sign”). Note cluster of short dystrophic neurites in b; bar = 20 μm in a, b, c, d; bar = 10 μm in e, f, g and insets (higher magnifications) in c and d

Spectrum of subcortical white matter pathology in temporal cortex of G-PDC detected by anti-TDP-43. In a–d, arrows dystrophic neurite/axonal profile, arrowhead glial inclusion; all bars 20 μm

Co-localization of TDP-43 with ubiqutin immunoreactivity in double-label immunofluorescence of cortical regions of G-PDC. a–c Dystrophic neurite in parietal cortex; a 1510; b TDP-43; c merge; d–f neuronal cytoplasmic neuronal inclusion in temporal cortex; d 1510; e TDP-43; f merge; g–i intranuclear neuronal inclusion (“cat eye sign”) in temporal cortex; g 1510; h TDP-43; i merge; j–l, cytoplasmic oligodendroglial inclusion in temporal white matter; j 1510; k TDP-43; l merge; bar 10 μm

Pattern of tau and TDP-43 pathology in temporal cortex of Guam-PDC. Different patterns of tau (a) and TDP-43 (b) pathology in the same brain area (bar 100 μm). a Note the presence of neuronal cytoplasmic tau immunoreactivity (e.g., large arrow) and astrocytic pathology (e.g., small arrow). b Note the presence of TDP-43 positive cytolasmic inclusions (e.g., large arrow) and dystrophic cellular processes (e.g., small arrows). c–e No co-localization of tau with TDP-43 pathology in double-label immunofluorescence of a NFT in the temporal cortex of G-PDC. c PHF-1; d TDP-43; e merge; bar 10 μm

Hippocampal pathology of G-PDC. a Anti-PHF-1 and b, anti-TDP-43 immunohistochemistry (bar 200 μm). Note CA4-CA1 regions/subiculum with the presence of tau-positive neurofibrillary tangles (e.g., arrow) and no apparent TDP-43 pathology; c, d Anti-TDP-43 immunohistochemistry (bar 20 μm). c CA1/subiculum, note single dystrophic cellular process (arrow); d dentate gyrus, note nuclear TDP-43 clearing in hippocampal granular cells bearing TDP-43 positive cytoplasmic inclusions (arrows) as compared with unaffected neurons (e.g., arrowhead); e–g anti-TDP-43 and anti-tau (PHF-1) immunofluorescence histochemistry. Note that the neuronal cytoplasmic inclusion stained for TDP-43 (arrow) does not co-localize with the tau positivity in another neuron (arrowhead). e PHF-1; f TDP-43; g merge; bar 10 μm
Considering the most affected area or regions of focal TDP-43 pathology as the defining feature for severity grading, five cases were categorized as severe, two as moderate, and four as mild. The morphological and distributional pattern of cortical TDP-43 pathology of the moderate and severe cases was reminiscent to FTLD-U subtype 3 with pathology predominantly located in superficial cortical layers including mainly short dystrophic neurites, NCIs, and variable numbers of neuronal intranuclear inclusions. In the mild cases, scattered dystrophic neurites and, to a lesser degree, neuronal inclusions were present, and in one case the pathology was restricted to the dentate nucleus of the hippocampus. Only statistical trends, but no significant differences between any of the various neocortical or hippocampal brain areas for TDP-43 pathology were found (Table 2).
Five of ten G-C cases with cortical tissue available showed tau pathology. When comparing the total tau burden between G-PDC and G-Cs, the former showed greater tau pathology in the neocortex (gray matter: 1.8 [0.8–2.2] vs. 0.4 [0.0–1.3], P = 0.011 and white matter: 1.4 [0.7–1.7] vs. 0.3 [0.0–0.6], P = 0.001; values are median or 25th to 75th percentile), although the P-value for the gray matter is only borderline significant. Such a difference was not present in the hippocampal formation (2.4 [2.1–2.8] vs. 3.0 [2.3–3.0], P = 0.658) (Table 2, supplementary data Table 2). The G-C subjects were virtually free of TDP-43 pathology with only two individuals showing rare or mild TDP-43 NCIs restricted to the dentate gyrus.
Spinal cord
All G-PDC and G-ALS spinal cords examined exhibited pathological changes, albeit different kinds and to various degrees (Fig. 6a–k). For example, NFTs were present in both G-PDC (17/17 cases) and G-ALS (7/9 cases; P = 0.111) and TDP-43 NCIs were more common in G-ALS (9/9 cases) than in G-PDC (5/17 cases; P = 0.001). TDP-43 positive NCIs were found in various shapes including filamentous/skein-like, or round dense frayed or Lewy body-like formations and smaller granules (Fig. 6a, b, e–g). Twelve of the 26 cases with spinal cord available for examination showed both tau positive NFTs and TDP-43 positive neuronal inclusion pathology (Fig. 6a, b, e–i). The degree of tau pathology tended to be higher in G-PDC as compared with G-ALS (Table 3). This contrasted with the TPD-43 pathology, which was significantly more severe in G-ALS as compared with G-PDC (Table 3). Moreover, in G-PDC, tau positive NFT pathology was significantly more severe than TDP-43 positive NCI pathology (P < 0.0001), whereas in G-ALS no significant difference in neurofibrillary changes and TDP-43 NCI pathology was found (P = 0.094; Fig. 7). Further, TDP-43 positive oligodendroglial pathology was found as comma shaped coiled body-like inclusions (Fig. 6c, d) and correlated positively with NCI pathology (r = 0.840, P < 0.01). Association analysis between tau positive (astro-)glial pathology and NFTs in the spinal cord revealed a moderately high correlation (r = 0.662, P < 0.01). These changes were associated with varying degrees of degeneration of the long descending tracts, and variable neuron loss accompanied by gliosis. There was no co-localization of TDP-43 and tau positive pathology.

Spinal cord pathology in G-PDC and G-ALS. a–d Anti-TDP-43 immunohistochemistry in G-ALS (bar 10 μm). a, b, c Gray matter of thoracic spinal cord; d white matter (adjacent to gray matter) of thoracic spinal cord; a filamentous motorneuronal inclusion (large arrow); b dense round motoneuronal inclusion (large arrow), c glial inclusion (arrowhead), d glial inclusion (arrowhead); e–g Anti-TDP-43 and anti-ubiquitin (1510) double-label immunofluorescence histochemistry in G-ALS (bar 10 μm). Co-localization of TDP-43 and ubiquitin in dense round motorneuronal inclusion (large arrow); e 1510; f, TDP-43; g merge. h–k Anti-tau (PHF-1) immunohistochemistry, note neurofibrillary pathology in G-PDC (large arrow) (h) and G-ALS (large arrow) (i), or G-C (large arrow) (j), and astrocytic glial inclusion pathology (small arrow) in G-ALS (k), bar 20 mcm

Severity of tau (PHF-1) positive NFTs and TDP-43 positive NCIs in spinal cord of G-PDC and G-ALS. Figures show error bars with mean and 95% confidence interval (95% CI). Note the significantly higher degree of tau positive NFTs as compared with TDP-43 positive NCIs pathology (P < 0.0001) with no overlap of the 95% CI in G-PDC (n = 17). In G-ALS (n = 9), there is no significant difference between TDP-43 NCI pathology and tau NFT pathology (P = 0.094), and there is a substantial overlap of the 95% CIs
Overall, NFTs were present to a very slight degree in the spinal cord of G-C (see supplementary data Table 3). Indeed, two of five G-C subjects showed mild neurofibrillary pathology. The degree of neurofibrillary pathology in G-C [0.4 (0.0–1.0)] did not differ significantly from G-ALS (1.0 [0.4–1.6]) (P = 0.233), but did so as compared to G-PDC [1.6 (1.0–2.4)] (P = 0.002) (values are median or 25th to 75th percentile; Table 3, supplementary data Table 3). None of the G-C showed pathological TDP-43 in the spinal cord. For both the spinal cord and the brain, the non-Chamorro individuals did not show any pathological distinctions from the native Chamorros.
Biochemistry
To examine the biochemical properties of TDP-43 in Guamanian subjects, sequential extractions of both cortical gray and white matters from 5 G-PDC and 4 G-C brains were analyzed. Biochemical analysis of the fractions revealed TDP-43 is in the SDS fractions in both gray and white matter of G-PDC (Fig. 8a) and G-C cases (Fig. 8b). Furthermore, in all G-PDC brains examined, but not in any of the G-C cases, detectable levels of normal and pathological TDP-43 were observed in the FA-soluble fraction (Fig. 8a, b, e, f and data not shown). Thus, TDP-43 pathological signature in G-PDC may include change in solubility in addition to the already reported presence of a high relative molecular mass smears and 20–25 kDa C-terminal fragments [24]. Further, to examine the solubility profile of tau, the immunoblots were stripped and re-probed with the MAbs T14 and T46. Consistent with previously published results [38], insoluble tau was detected in both the gray and white matter of G-PDC and, to a lesser extent, G-C brains (Fig. 8c, d).

Biochemical analyses of sequential extracts of G-PDC and G-C brains. Soluble high salt (HS), Triton-X 100 (Tx) and radioimmunoprecipitation assay (RIPA) buffer fractions and insoluble sodium dodecyl sulfate (SDS) and formic acid (FA) fractions, from both gray and white matter, were separated by SDS-polyacrylamide gel electrophoresis and immunoblotted with a (a, b) polyclonal anti-C-terminus-TDP-43 (1038) antibody, or (c, d) a mixture of the anti-tau MAbs T14 and T46 (T14/T46). Note the presence of a high molecular weight smear (asterisk), a 45 kDa band (double asterisk) and an approximately 20- to 25-kDa band (triple asterisk) immunoreative for TDP-43 and the prominent PHFtau bands in the gray matter SDS and FA fractions of the G-PDC case (a, c) but not in the C-C case (b, d). e, f SDS- and FA-soluble TDP-43 isolated from gray (G) and white (W) matter regions of the temporal cortex (T) from three additional G-PDC cases. e Short exposure and f, long exposure of the immunoblot, demonstrating the presence of a high molecular weight smear (asterisk), a 45 kDa band and an approximately 20- to 25-kDa band (triple asterisk)
Discussion
The clinical similarities in FTLD-U or FTLD-MND and G-PDC or G-ALS suggest a pathogenetic link between these disorders. Thus, it is significant that, on the basis of our studies of the largest cohort of clinically and pathologically confirmed G-PDC or G-ALS cases examined to date for the presence of TDP-43 proteinopathy, we demonstrate TDP-43 pathologies occur in the cortex of G-PDC, but are virtually absent in G-C subjects of similar age. Notably, we show that both cortical and gray matter spinal cord TDP-43 pathology are coupled with white matter pathology. In addition to these findings, we demonstrated widespread tau pathology in G-PDC, G-ALS and G-C consistent with previous reports including those from our group and other centers [26, 28, 30, 33, 34, 37, 38]. On the basis of a case series with a small sample size, patients with atypical parkinsonism and ALS in the Kii peninsula of Japan have been shown to exhibit NFTs in a similar topographic distribution in PDC and ALS [21], and they have been reported to be associated with dentate gyrus and spinal cord pathological TDP-43 findings similar to our study [18].
Severe atrophy of the frontal and temporal cortex have been previously described in G-PDC [14, 27]. The cortical or hippocampal topographical distribution of both gray and white matter TDP-43 pathology shown here is similar to that in FTLD-U. TDP-43 pathology included NCIs, intranuclear inclusions, dystrophic neurites (or axonal profiles) and oligodendroglial inclusions. In fact, its cortical pattern in the advanced cases was reminiscent of the subtype 3 of FTLD-U, although intranuclear inclusions were present in very variable numbers. Corresponding to the different histological architecture of gray and white matter, we found a different frequency of a similar kind of pathology including NCIs, glial cytoplasmic inclusions and dystrophic cellular processes; however, the overall degree of pathology was similar in both gray and white matter. Whereas overall PHF-1 pathology in hippocampal formation or some phylogenetic younger brain areas trended to be more robust than the TDP-43 pathology, there was clearly no difference in the burden of tau and TDP-43 pathologies in the dentate gyrus. A previous report suggested that the ubiquitinated inclusions in the dentate gyrus of patients with G-PDC or G-ALS seem to be AD-like NFTs and not the typical ubiquitin-positive intracytoplasmic neuronal inclusions seen in ALS with dementia (ALS-D) [16]. Here, we demonstrate that there is a co-occurrence of both tau- and ubiquitin-positive NFT-like inclusions and FTLD-U-like TDP-43 and ubiquitin-positive inclusions in a similar frequency or degree which suggests that TDP-43 pathology, such as tau pathology, contributes to mechanisms of neurodegeneration. Interestingly, TDP-43 positive but not tau positive intranuclear inclusions are present in G-PDC. We did not observe a co-localization of TDP-43 and tau pathology, suggesting that TDP-43 abnormalities are unlikely to be the result of tau-related cellular disruption. Further, tau positive tangle associated neuritic clusters have been reported earlier to be common in early and mild cases of G-PDC and the possibility of a causative interaction between tangle and dystrophic neurite formation has been raised [35]. In our study, some cases showed cortical TDP-43 positive dystrophic neurites predominating over cytoplasmic inclusion, and the CA1/subiculum regions were devoid of TDP-43 pathology except for rare dystrophic neurites. The cerebellum was free of TDP-43 pathology, reminiscent to FTLD-U, and only rare tau pathology was present, which is consistent with the literature [25, 28].
Our spinal cord finding in G-ALS and G-PDC of TDP-43 positive NCIs is novel, and there appeared to be more NCIs in G-ALS as compared to G-PDC. However, the NFT burden in the spinal cord did not differ between PDC and ALS, in contrast to the degree of TDP-43 pathology. Almost half of the cases showed both kinds of pathology, although to a different degree. In G-PDC, but not in G-ALS, a significantly lower degree of NCIs as compared with NFTs were found, suggesting that pre-clinical ALS-like pathology may occur in the former. The presence of ubiquitinated and TDP-43 positive inclusions in the LMN of G-ALS or G-PDC is paralleled by ALS, FTLD-U or FTLD-MND outside Guam [2, 19, 20, 24, 36]. Further, LMN inclusion pathology was associated with glial inclusions in our study. In fact, the correlation of TDP-43 neuronal with TPD-43 glial inclusion pathology appeared to be somewhat higher than the association of neurofibrillary changes with tau-positive glial pathology. Recently, TDP-43 positive white matter pathology, most likely in oligodendroglial cells, was demonstrated in the neocortex and spinal cord of FTLD-U cases [22]. In addition, a large series of ALS cases demonstrated similar glial inclusions in spinal cord gray and white matter [19]. Analogous to α-synucleinopathies, where the concept of “gliodegeneration” is increasingly raised taking into account that glial cells may represent a primary target of an as yet unknown degenerative disease processes [5], glial pathology could contribute to the dysfunction and death of adjacent neurons in TDP-43 proteinopathies [19].
Guamanian subjects without symptoms of PDC or ALS have been shown to exhibit an increased prevalence of neurofibrillary pathology with age, often with a widespread appearance in Chamorros in their 30s and 40s and up to 100% of Chamorros over age of 50 [1, 4, 9, 26, 28, 29]. Thus, in contrast to the neocortex, the degree of tau pathology in the hippocampus was similar in G-PDC and G-C. In the spinal cord, G-PDC and G-ALS showed an overall mild (to moderate) degree of neurofibrillilary pathology, and a few tangles were present in the limited number (i.e., five) of examined G-C subjects. TDP-43 pathology was completely absent in the neocortex and spinal cord in G-C, and only slightly present in the dentate of two subjects with significant tau pathology in hippocampus. These data suggest that TDP-43 pathology in neocortical or hippocampal brain areas discriminates disease from control better than tau pathology.
The finding of a lack of an apparent difference in the pathology between the non-Chamorro individuals including three migrants, i.e., two Filipinos (G-PDC and G-C) and one Caucasian (G-ALS), as well as one mixed Chamorro/Japanese subject (G-C) might have etiological significance. However, as only a few non-Chamorro individuals have been studied, comparative studies with larger sample size are certainly needed in order to establish the nature and degree of pathology in non-native individuals having lived on Guam and eventually draw reasonable pathogenetic conclusions.
Importantly, our studies demonstrated a pathological signature for TDP-43 including a shift in the solubility of TDP-43 toward the FA soluble fraction, a high molecular weight TDP-43 smear plus low molecular weight TDP-43 fragments in G-PDC, which is very similar to that in FTLD-U [24]. Further, consistent with previously published data [38], the immunhistochemically identified tau pathology was confirmed to represent insoluble tau by biochemical analyses.
Taken together, these results demonstrate that G-PDC and G-ALS are associated with pathological TDP-43, suggesting that these disorders represent similar TDP-43 proteinopathy diseases just like FTLD-U, FTLD-MND, ALS, and ALS-D, and we hypothesize that common pathogenetic mechanisms linked to TPD-43 pathology underlie these disorders. Thus, neocortical or hippocampal TDP-43 pathology distinguishes disease from controls better than tau pathology and pathological TDP-43 might therefore reflect a more disease specific process.
References
Anderson FH, Richardson EP Jr, Okazaki H, Brody JA (1979) Neurofibrillary degeneration on Guam: frequency in Chamorros and non Chamorros with no known neurological disease. Brain 102:65–77
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL, III, Schneider JA, Kretzschmar HA, Carter D, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Trojanowski JQ, Lee VM-Y, Mackenzie IRA (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240
Chen L (1981) Neurofibrillary change on Guam. Arch Neurol 38:16–18
Croisier E, Graeber MB (2006) Glial degeneration and reactive gliosis in alpha-synucleinopathies: the emerging concept of primary gliodegeneration. Acta Neuropathol (Berl) 112:517–530
Elizan TS, Hirano A, Abrams BM, Need RL, Van Nuis C, Kurland LT (1966) Amyotrophic lateral sclerosis and parkinsonism–dementia complex of Guam: neurological reevaluation. Arch Neurol 14:356–368
Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, Chatterjee A, Hurtig HI, Karlawish JH, Rosen HJ, Van D, V, Lee VM-Y, Miller BL, Trojanowski JQ, Grossman M (2006) Frontotemporal dementia: clinicopathological correlations. Ann Neurol 59:952–962
Forman MS, Schmidt ML, Kasturi S, Perl DP, Lee VM-Y, Trojanowski JQ (2002) Tau and alpha-synuclein pathology in amygdala of Parkinsonism–dementia complex patients of Guam. Am J Pathol 160:1725–1731
Forman MS, Trojanowski JQ, Lee VM-Y (2004) Hereditary tauopathies and idiopathic frontotemporal dementias. In: Esiri MM, Lee VM-Y, Trojanowski JQ (eds) The neuropathology of dementia. Cambridge University Press, Cambridge, pp 257–288
Galasko D, Salmon D, Craig UK, Wiederholt W (2000) The clinical spectrum of Guam ALS and Parkinson–dementia complex: 1997–1999. Ann N Y Acad Sci 920:120–125
Geser F, Lee VM-Y, Trojanowski JQ (2007) Frontotemporal dementias. In: Rosenberg RN, DiMauro S, Paulson H, Ptacek L, Nestler E (eds) The molecular and genetic basis of neurological and psychiatric disease. Lippincott Williams & Wilkins, Philadelphia (in press)
Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 87:5827–5831
Hasegawa M, Arai T, Akiyama H, Nonaka T, Mori H, Hashimoto T, Yamazaki M, Oyanagi K (2007) TDP-43 is deposited in the Guam parkinsonism–dementia complex brains. Brain 130:1386–1394
Hirano A, Malamud N, Kurland LT (1961) Parkinsonism–dementia complex, an endemic disease on the island of Guam: II. Pathological features. Brain 84:662–679
Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM-Y (1998) Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 282:1914–1917
Ikemoto A, Hirano A, Akiguchi I, Kimura J (1997) Comparative study of ubiquitin immunoreactivity of hippocampal granular cells in amyotrophic lateral sclerosis with dementia, Guamanian amyotrophic lateral sclerosis and Guamanian parkinsonism–dementia complex. Acta Neuropathol (Berl) 93:265–270
Kosik KS, Orecchio LD, Binder L, Trojanowski JQ, Lee VM-Y, Lee G (1988) Epitopes that span the tau molecule are shared with paired helical filaments. Neuron 1:817–825
Kuzuhara S (2007) TDP-43 accumulation in ALS/parkinsonism-dementia complex (ALS/PDC) of the Kii peninsula of Japan. Neuropathology 27:161
Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM-Y, Trojanowski JQ (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434
Mackenzie IR, Feldman HH (2005) Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. J Neuropathol Exp Neurol 64:730–739
Mimuro M, Kokubo Y, Kuzuhara S (2007) Similar topographical distribution of neurofibrillary tangles in amyotrophic lateral sclerosis and parkinsonism–dementia complex in people living in the Kii peninsula of Japan suggests a single tauopathy. Acta Neuropathol (Berl) 113:653–658
Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM, Clark CM, Grossman M, Miller BL, Trojanowski JQ, Lee VM-Y (2007) TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol 66:177–183
Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS (2007) TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol 66:152–157
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM-Y (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Oyanagi K (2005) The nature of the parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam and magnesium deficiency. Parkinsonism Relat Disord 11(Suppl 1):S17–S23
Oyanagi K, Makifuchi T, Ohtoh T, Chen KM, van der ST, Gajdusek DC, Chase TN, Ikuta F (1994) Amyotrophic lateral sclerosis of Guam: the nature of the neuropathological findings. Acta Neuropathol (Berl) 88:405–412
Oyanagi K, Makifuchi T, Ohtoh T, Ikuta F, Chen KM, Chase TN, Gajdusek DC (1994) Topographic investigation of brain atrophy in parkinsonism–dementia complex of Guam: a comparison with Alzheimer’s disease and progressive supranuclear palsy. Neurodegeneration 3:301–304
Oyanagi K, Wada M (1999) Neuropathology of parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam: an update. J Neurol 246(Suppl 2):II19–II27
Perl DP, Hof PR, Purohit DP, Loerzel A, Belli D (1995) Changes in the outbreak of ALS/parkinsonism–dementia complex: neuropathologic studies of asympomatic Chamorros. J Neuropathol Exp Neurol 54:416
Perl DP, Hof PR, Purohit DP, Loerzel AJ, Kakulas BA (2003) Hippocampal and entorhinal cortex neurofibrillary tangle formation in Guamanian Chamorros free of overt neurologic dysfunction. J Neuropathol Exp Neurol 62:381–388
Persaud R (1994) Correlation, regression, and repeated data. BMJ 308:1510
Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VM-Y (2006) Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol 169:1343–1352
Schmidt ML, Garruto R, Chen J, Lee VM-Y, Trojanowski JQ (2000) Tau epitopes in spinal cord neurofibrillary lesions in Chamorros of Guam. Neuroreport 11:3427–3430
Schmidt ML, Zhukareva V, Perl DP, Sheridan SK, Schuck T, Lee VM-Y, Trojanowski JQ (2001) Spinal cord neurofibrillary pathology in Alzheimer disease and Guam Parkinsonism–dementia complex. J Neuropathol Exp Neurol 60:1075–1086
Schwab C, Steele JC, McGeer PL (1997) Dystrophic neurites are associated with early stage extracellular neurofibrillary tangles in the parkinsonism–dementia complex of Guam. Acta Neuropathol (Berl) 94:486–492
Tan CF, Eguchi H, Tagawa A, Onodera O, Iwasaki T, Tsujino A, Nishizawa M, Kakita A, Takahashi H (2007) TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol (Berl) 113:535–542
Trojanowski JQ, Ishihara T, Higuchi M, Yoshiyama Y, Hong M, Zhang B, Forman MS, Zhukareva V, Lee VM-Y (2002) Amyotrophic lateral sclerosis/parkinsonism dementia complex: transgenic mice provide insights into mechanisms underlying a common tauopathy in an ethnic minority on Guam. Exp Neurol 176:1–11
Winton MJ, Joyce S, Zhukareva V, Practico D, Perl DP, Galasko D, Craig U, Trojanowski JQ, Lee VM-Y (2006) Characterization of tau pathologies in gray and white matter of Guam parkinsonism-dementia complex. Acta Neuropathol (Berl) 111:401–412
Acknowledgments
The authors would like to thank T. Schuck, A. Truax, M. Getahun, J. Robinson, J. McBride, and N. Camacho for their expert technical assistance. Further, they thank their patients and families and the dedicated efforts of the research team at the University of Guam who made this research possible. This work was funded by the National Institutes of Health (AG10124, AG17586, AG14382).
Author information
Authors and Affiliations
Corresponding author
Additional information
VM-YL is the John H. Ware III Chair of Alzheimer’s Research and JQT is the William Maul Measey-Truman G. Schnabel, Jr., MD, Professor of Geriatric Medicine and Gerontology.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Geser, F., Winton, M.J., Kwong, L.K. et al. Pathological TDP-43 in parkinsonism–dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol 115, 133–145 (2008). https://doi.org/10.1007/s00401-007-0257-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-007-0257-y