
Abstract
Cardiac aging is manifested as cardiac remodeling and contractile dysfunction although precise mechanisms remain elusive. This study was designed to examine the role of endothelin-1 (ET-1) in aging-associated myocardial morphological and contractile defects. Echocardiographic and cardiomyocyte contractile properties were evaluated in young (5–6 months) and old (26–28 months) C57BL/6 wild-type and cardiomyocyte-specific ETA receptor knockout (ETAKO) mice. Cardiac ROS production and histology were examined. Our data revealed that ETAKO mice displayed an improved survival. Aging increased plasma levels of ET-1 and Ang II, compromised cardiac function (fractional shortening, cardiomyocyte peak shortening, maximal velocity of shortening/relengthening and prolonged relengthening) and intracellular Ca2+ handling (reduced intracellular Ca2+ release and decay), the effects of which with the exception of ET-1 and Ang II levels was improved by ETAKO. Histological examination displayed cardiomyocyte hypertrophy and interstitial fibrosis associated with cardiac remodeling in aged C57 mice, which were alleviated in ETAKO mice. Aging promoted ROS generation, protein damage, ER stress, upregulated GATA4, ANP, NFATc3 and the autophagosome cargo protein p62, downregulated intracellular Ca2+ regulatory proteins SERCA2a and phospholamban as well as the autophagic markers Beclin-1, Atg7, Atg5 and LC3BII, which were ablated by ETAKO. ET-1 triggered a decrease in autophagy and increased hypertrophic markers in vitro, the effect of which were reversed by the ETA receptor antagonist BQ123 and the autophagy inducer rapamycin. Antagonism of ETA, but not ETB receptor, rescued cardiac aging, which was negated by autophagy inhibition. Taken together, our data suggest that cardiac ETA receptor ablation protects against aging-associated myocardial remodeling and contractile dysfunction possibly through autophagy regulation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cardiac aging, an irreversible biological process, is manifested as a drastic decrease in pump function and contractile reserve [23–25]. Aging-associated cardiac defect en route to heart failure is considered to attribute to the high morbidity and mortality in the elderly [25, 49]. A number of theories have been postulated for senescence-induced changes in myocardial function and remodeling, including diminution of intrinsic cardiac contractile properties resulting from β-myosin heavy chain reexpression, defective intracellular Ca2+ transport by sarcoplasmic reticulum, prolongation of action potential duration, mitochondrial injury, accumulation of free radicals or reactive oxygen species (ROS), deterioration in vascular function, interaction between occult diseases and physical inactivity, as well as loss of ischemic pre- and post-conditioning protection [2–4, 19, 22, 25, 26, 48, 49]. In consequence, aged hearts often display a loss of adaptive capacity in response to mechanical or pathological stresses such as acute ischemia [2, 4]. Recent evidence suggested a rather important role for autophagy, energy metabolism and mitochondrial biogenesis in the onset and progression of cardiac aging [7, 27, 32, 34, 39, 40]. Nonetheless, the precise pathophysiology of cardiac aging has not been carefully defined.
Endothelin-1 (ET-1) is a potent vasoconstrictor secreted by endothelium. ET-1 exerts its biological action through its membrane receptors namely ETA and ETB [47]. ETA is abundant in cardiomyocytes, whereas ETB is mostly found in endothelial cells as well as cardiomyocytes [37]. ET-1 is capable of exerting profound cardiovascular effects including regulation of vascular tone, positive inotropic and chronotropic properties as well as cell hypertrophy in the heart [18, 19, 31]. ET-1 may be produced in the heart in response to a variety of stresses including aging. An aging-related change in circulating ET-1 levels and ET receptors, similar to that of the well-known vasoconstrictor angiotensin II (Ang II), has been reported [1, 8, 43, 50]. Therefore, this study was designed to examine the role of ET-1 cascade in particular ETA receptor in aging-induced cardiac remodeling and contractile dysfunction. We took advantage of a murine model of cardiomyocyte-specific ETA knockout which displays a ~80 % loss in cardiac ETA mRNA level [20]. Myocardial histology, contractile and intracellular Ca2+ properties were examined in wild-type C57/BL6 and ETA knockout mice. To elucidate the underlying mechanism(s) involved in aging and/or ETA knockout-induced cardiac responses, myocardial autophagy and ROS were examined. Autophagy is a cytoprotective mechanism to remove aberrant or dysfunctional molecules and intracellular organelles [7, 9, 13, 14, 35]. Autophagy is initiated with formation of the double-membrane autophagosmes involving elongation of isolated membrane and maturation of autophagosome followed by infusion of autophagosome with lysosome [7, 9, 10, 14]. To this end, the key autophagy-related genes including beclin-1, Atg5, Atg7, microtubule-associated protein 1 light chain 3 (LC3B or Atg8) and the autophagosome cargo protein p62 were scrutinized in young or old C57/BL6 and ETA knockout mice. Given the essential role of autophagosome removal in autophagy regulation [14], LC3B and p62 were evaluated in myocytes exposed to exogenous ET-1 in the absence (steady-state autophagosomes) or the presence (cumulative autophagosomes) of a mixture of lysosomal inhibitors. As ETB receptor becomes the dominant ET-1 receptor in cardiomyocyte-specific ETA knockout mice where ETA receptor may be dispensable for cardiac homeostasis [20], in vivo treatment of the ETA antagonist BQ123 and the ETB receptor antagonist BQ788 was performed in young and old mice prior to mechanical assessment.
Materials and methods
Experimental animals, treatment of ET-1 receptor antagonists and autophagy inhibitor
All animal procedures described here were approved by the Institutional Animal Use and Care Committee at the University of Wyoming (Laramie, WY, USA). The cre/loxP technique was employed to generate mice with the ETA gene deleted specifically in cardiomyocytes. The cre recombinase transgene driven by the α-myosin heavy-chain promoter deleted the floxed ETA allele in the heart, resulting in a 78 % reduction in cardiac ETA mRNA levels [20]. Young (4- to 5-month-old) and old (26- to 28-month-old) male cardiomyocyte-specific ETA receptor knockout (ETAKO) and age-/gender-matched wild-type C57BL/6 J mice were used. To examine the effect of ETA and ETB receptor antagonism on cardiac aging, a cohort of young and old C57BL/6 J mice were treated with the ETA receptor antagonist BQ123 or the ETB receptor antagonist BQ788 (1 mg/kg/d, i.p.) for 28 days [6, 33] in the presence or absence of autophagy inhibitor 3-MA (10 mg/kg, once a week, i.p.) [12]. Mice were maintained on a 12:12-h light–dark illumination cycle with access to food and water ad libitum.
Blood pressure, plasma levels ET-1 and Ang II
Systolic, diastolic and mean blood pressures were examined using a KODA semi-automated, amplified tail cuff device (Kent Scientific Corporation, Torrington, CT, USA). Mice were placed in a temperature-controlled restrainer on a warm pad for 30 min before measurements were taken. A mean of three readings was taken as respective blood pressure value. Upon killing, trunk blood was collected in chilled tubes and microfuged at room temperature before the plasma was transferred to ice-cold methanol (100 %). Plasma ET-1 levels were determined using an ET-1 enzyme immunometric assay kit (Assay Designs, Inc. Ann Arbor, MI, USA) based on a double-antibody sandwich technique [46]. The detection threshold for ET-1 was 0.14 pg/ml. Plasma AngII levels were measured using an ELISA kit (Cayman Chemicals, Ann Arbor, MI, USA) [44]. The detection threshold for Ang II was 1.5 pg/ml.
Echocardiographic assessment
Cardiac geometry and function were evaluated in anesthetized (Avertin 2.5 %, 10 μl/g bw, i.p.) mice using a 2-D guided M-mode Sonos 5500 echocardiography (Phillips Medical Systems, Andover, MD, USA) equipped with a 15–6 MHz linear transducer. Hearts were imaged in 2-D mode in the parasternal long-axis view with a depth of 2 cm. A M-mode cursor was then positioned perpendicular to interventricular septum and posterior wall of the left ventricular (LV) at the level of the papillary muscles in the 2-D mode. The sweep speed was 100 mm/s at the M-mode. Diastolic wall thickness, LV end diastolic dimension (EDD) and LV end systolic dimension (ESD) were measured from leading edge to leading edge in accordance with the Guidelines of the American Society of Echocardiography [28]. The percentage of LV fractional shortening was calculated as [(EDD-ESD)/EDD]×100. Heart rates were averaged over 10 cardiac cycles [16].
Isolation of murine cardiomyocytes
Cardiomyocytes were enzymatically isolated as described [16]. Briefly, hearts were removed and perfused (37 °C) with oxygenated (5 % CO2–95 % O2) Krebs–Henseleit bicarbonate (KHB) buffer containing (in mM) 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 10 HEPES and 11.1 glucose. Hearts were subsequently perfused with a Ca2+-free KHB-buffer that containing Liberase Blendzyme (10 mg/ml; Roche, Indianapolis, IN) for 15 min. After perfusion, left ventricles were removed and minced to disperse the individual cardiomyocytes in Ca2+-free KHB-buffer. Extracellular Ca2+ was added incrementally to 1.25 mM. Myocytes with obvious sarcolemmal blebs or spontaneous contractions were not used. Only rod-shaped myocytes with clear edges were selected for mechanical or intracellular Ca2+ transient studies.
Cell shortening/relengthening
Mechanical properties of cardiomyocytes were assessed using a SoftEdge MyoCam system (IonOptix Corporation, Milton, MA, USA) [16]. Cardiomyocytes were placed in a chamber mounted on the stage of an inverted microscope (Olympus, IX-70) and superfused at 25 °C with a buffer containing (in mmol/l): 131 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 Glucose and 10 HEPES, at pH 7.4. Cells were field-stimulated with suprathreshold voltage at the frequency of 0.5 Hz unless otherwise stated, with a 3-ms duration, using a pair of platinum wires placed on opposite sides of the chamber and connected to an electrical stimulator (FHC Inc, Brunswick, NE,USA). The myocyte being studied was displayed on a computer monitor using an IonOptix MyoCam camera. IonOptix SoftEdge software was used to capture changes in cell length during shortening and relengthening. Cell shortening and relengthening were assessed using the following indices: peak shortening (PS), maximal velocities of cell shortening and relengthening (±dL/dt), time-to-PS (TPS), time-to-90 % relengthening (TR90). In the case of altering stimulus frequency, the steady-state contraction of myocyte was achieved (usually after the first six beats) before PS amplitude was recorded at 0.1, 0.5, 1.0, 3.0 and 5.0 Hz.
Intracellular Ca2+ transients
Separate cohorts of myocytes were loaded with fura-2/AM (0.5 μM) for 15 min, and fluorescence intensity was measured with a dual-excitation fluorescence photomultiplier system (IonOptix). Myocytes were placed on an inverted Olympus microscope and imaged through a Fluor 40×-oil objective. Cells were exposed to light emitted by a 75-W mercury lamp and passed through either a 360-nm or a 380-nm filter. The myocytes were stimulated to contract at 0.5 Hz. Fluorescence emissions were detected between 480 and 520 nm by a photomultiplier tube after cells were first illuminated at 360 nm for 0.5 s and then at 380 nm for the duration of the recording protocol (333 Hz sampling rate). The 360-nm excitation scan was repeated at the end of the protocol, and qualitative changes in intracellular Ca2+ concentration were inferred from the ratio of the fluorescence intensity at two wavelengths. Intracellular Ca2+ decay rate was calculated from both single and bi-exponential curve fitting [16].
Histological examination
Hearts were harvested and sliced at mid-ventricular level followed by fixation with normal buffered formalin. Paraffin-embedded transverse sections were cut in 5-μm sections and stained with Masson trichrome [11]. Sections were photographed with a 40× objective of an Olympus BX-51 microscope equipped with an Olympus MaguaFire SP digital camera. Five random fields from each section (3 sections per mouse) were assessed for fibrosis. To determine fibrotic area, pixel counts of blue stained fibers were quantified using Color range and Histogram commands in Photoshop. Fibrotic area was calculated by dividing the pixels of blue stained area to total pixels of non-white area. For cross-sectional measurement, transverse sections of myocardium were stained with the FITC-conjugated wheat-germ agglutinin [41] and were mounted with Vectashield prior to analysis with NIH ImageJ software.
ROS production
Production of intracellular ROS was evaluated by analyzing the fluorescence intensity that resulted from oxidation of the intracellular fluoroprobe 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (DCF). In brief, cardiomyocytes were loaded with 10 μM DCF at 37 °C for 30 min. The fluorescence intensity was measured using a fluorescent microplate reader at an excitation wavelength of 480 nm and an emission wavelength of 530 nm. Untreated cells showed no fluorescence and were used to determine background fluorescence [40].
Protein carbonyl assay
To assess cardiac oxidative damage, the tissue protein carbonyl content was determined as described [11]. Briefly, proteins were extracted and minced to prevent proteolytic degradation. Nucleic acids were eliminated by treating the samples with 1 % streptomycin sulfate for 15 min, followed by a 10-min centrifugation (11,000×g). Protein was precipitated by adding an equal volume of 20 % TCA to protein (0.5 mg) and centrifuged for 1 min. The TCA solution was removed and the sample resuspended in 10 mM 2,4-dinitrophenylhydrazine (2,4-DNPH) solution. Samples were incubated at room temperature for 15–30 min. Following a 500 μl of 20 % TCA addition, samples were centrifuged for 3 min. The supernatant was discarded, the pellet washed in ethanol:ethyl acetate and allowed to incubate at room temperature for 10 min. The samples were centrifuged again for 3 min and the ethanol:ethyl acetate steps repeated two more times. The precipitate was resuspended in 6 M guanidine solution, centrifuged for 3 min and insoluble debris removed. The maximum absorbance (360–390 nm) of the supernatant was read against appropriate blanks (water, 2 M HCl) and the carbonyl content was calculated using the molar absorption coefficient of 22,000 M−1cm−1.
Western blot analysis
Expression of the essential autophagic markers, Beclin-1, Atg 7, Atg 5 and LC3B, the intracellular Ca2+ regulatory proteins, sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2a (SERCA2a) and phospholamban, the hypertrophic markers, GATA4, ANP, NFATc3 and phosphorylated NFATc3 as well as the endoplasmic reticulum stress markers, Bip, CHOP and calcineurin, were examined by Western blot analysis. The protein was prepared as described [16]. Samples containing equal amount of proteins were separated on 10 % SDS–polyacrylamide gels in a minigel apparatus (Mini-PROTEAN II, Bio-Rad) and transferred to nitrocellulose membranes. The membranes were blocked with 5 % milk in TBS-T and were incubated overnight at 4 °C with rabbit polyclonal anti-ETAR (1:1,000), rabbit polyclonal anti-ETBR (1:1,000), rabbit monoclonal anti-Beclin-1 (1:1,000), rabbit monoclonal anti-Atg7 (1:1,000), rabbit anti-Atg5 (1:1,000), rabbit anti-LC3B (1:1,000), guinea pig polyclonal anti-p62/SQSTM1 (1:1,000), rabbit monoclonal anti-SERCA2a (1:1,000), rabbit monoclonal anti-phospholamban (1:1,000), rabbit polyclonal anti-Na+–Ca2+ exchanger, rabbit anti-GATA4 monoclonal (1:1,000), rabbit monoclonal anti-ANP (1:1,000), rabbit monoclonal anti-NFATc3 (1:1,000), rabbit monoclonal anti-pNFATC3 (1:1,000), rabbit anti-Bip monoclonal (1:1,000), mouse monoclonal anti-CHOP (1:1,000) and rabbit monoclonal anti-calcineurin (1:1,000) antibodies. Anti-SERCA2a was purchased from Affinity BioReagents (Golden, CO, USA). Anti-anti-ETAR and anti-phospholamban antibodies were purchased from Abcam (Cambridge, MA, USA). Anti-ANP antibody was purchased from Santa Cruz (Santa Cruz, CA, USA). All other antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). After immunoblotting, the film was scanned and the intensity of immunoblot bands was detected with a Bio-Rad Calibrated Densitometer. GAPDH was used as the loading control.
ETA receptor blockage in H9C2 myoblasts
Given the poor viability of murine cardiomyocytes in culture over time, rat embryonic cardiac myoblast cells (H9C2) [15, 17] obtained from ATCC (Manassas, VA, USA) were used for this part of study. Cells were incubated in 6-well plates at 37 °C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum and 1 % penicillin–streptomycin under a humidified atmosphere with 5 % CO2. Cells were treated with endothelin-1 (ET-1, 10 nM) for 24 h in the absence or presence of the ETA receptor blocker BQ123 or the autophagy inducer rapamycin prior assessment of protein expression.
Statistical analysis
Data were mean ± SEM. The log rank test was used for Kaplan–Meier survival comparison. Statistical significance (p < 0.05) was estimated by a one-way analysis of variance (ANOVA) followed by the Newman-Keuls post hoc analysis.
Results
General biometric and echocardiographic properties of C57BL/6 and ETAKO mice
As expected, increased age significantly elevated body and heart weights with lesser effects on liver and kidney weights. The organ sizes of heart, liver and kidney were unaffected by aging. ETA receptor knockout did not affect body or organ weights although it significantly alleviated aging-induced cardiac hypertrophy. Aging elicited a subtle but significant increase in systolic blood pressure without affecting diastolic and mean blood pressures, the effects of which were unaffected by ETA knockout. Plasma levels of ET-1 and Ang II were both elevated in aging groups, the effects of which were unaffected by ETA knockout. Heart rate and LV EDD were comparable among all groups. While ETA knockout did not affect wall thickness, LV mass, normalized LV mass, ESD and fractional shortening at young age, it attenuated or mitigated aging-induced increase in wall thickness, LV ESD, LV mass and normalized LV mass as well as decline in fractional shortening (Table 1). These findings suggest that ETA knockout ameliorates aging-induced cardiac remodeling and contractile dysfunction. The Kaplan–Meier curve depicts that ETA knockout mice display significantly better survival rates than C57BL/6 mice. The median lifespan was 25.2 and 30.3 months for C57BL/6 and ETA knockout groups, respectively. Survival curves of the two mouse lines begin to separate from each other after ~15 months of age with ETA knockout mice exhibiting a reduced mortality rate (Fig. 1a).

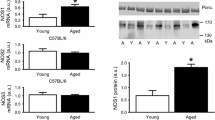
Cumulative survival curve (Kaplan–Meier survival plot), myocardial levels of ETA and ETB receptors in C57BL/6 and ETAKO mice. a The cumulative survival rate was plotted against age in months; b ETA receptor; and c ETB receptor; Insets: Representative gel blots depicting expression of ETA and ETB receptors (GAPDH used as loading control) using specific antibodies; Mean ± SEM, Log rank test was performed for Kaplan–Meier curve; n = 34 and 28 mice for C57BL/6 and ETAKO mice, respectively (a); n = 4–6 mice for b and c, *p < 0.05 vs. C57BL/6 young group; # p < 0.05 vs. C57BL/6 old group
Effects of ETA knockout on aging-induced changes in ETA and ETB receptor expression
As expected, protein levels of ETA receptor were significantly diminished in hearts from cardiomyocyte-specific ETA knockout mice. Aging significantly upregulated expression of ETA receptor, but the effect was blunted in the ETA knockout mice. Consistent with our earlier report [20], ETA knockout triggered a subtle but significant increase in ETB receptor expression. Aging did not affect the level of ETB receptor although it removed ETA knockout-elicited change in ETB receptor seen at young ages (Fig. 1b, c).
Mechanical and intracellular Ca2+ properties of cardiomyocytes
Mechanical properties revealed comparable resting cell length regardless of age or ETA knockout (data not shown). Aging significantly reduced peak shortening and maximal velocity of shortening/relengthening (±dL/dt) and prolonged TR90 associated with similar TPS, the effects of which were attenuated or ablated by ETA knockout. ETA knockout itself did not exert any notable effect at young ages (Fig. 2). Given that rodent hearts normally contract at high frequencies, we evaluated the impact of aging and/or ETA knockout on cardiac contractile function under higher frequencies (up to 5.0 Hz). Cardiomyocytes were initially stimulated to contract at 0.5 Hz for 5 min to ensure steady state before commencing the frequency response. Figure 3 displays a negative staircase of peak shortening amplitude with increased stimulus frequencies in all four groups with a steeper decline in aged C57BL/6 mice. Although ETA knockout did not affect the frequency-peak shortening relationship in young mice, it negated aging-induced steeper decline in peak shortening with increased stimulus frequencies. Resting cell length was comparable among all four groups at the frequencies tested. These data favor a possible role of improved intracellular Ca2+ cycling underneath ETA knockout-elicited cardioprotection against aging.

Cardiomyocyte contractile properties in young or old C57BL/6 and ETAKO mice. a Representative cell shortening traces in young or old C57BL/6 and ETAKO groups; b Peak shortening (PS) amplitude; c Maximal velocity of shortening (+dL/dt); d Maximal velocity of relengthening (−dL/dt); e Time-to-PS (TPS); and f Time-to-90 % relengthening (TR90). Mean ± SEM, n = a total of 104 cells from 4 mice per group (panel B–F), *p < 0.05 vs. corresponding young group; # p < 0.05 vs. C57BL/6 old group

Influence of stimulus frequency (0.1–5.0 Hz) on peak shortening (PS) amplitude of cardiomyocytes from young or old C57BL/6 and ETAKO mice. PS at each stimulus frequency was normalized to that of 0.1 Hz from the same cell. a Resting cell length; b Change in PS from that of 0.1 Hz from the same cell. Mean ± SEM, n = a total of 24–27 cells from 4 mice per group, * p < 0.05 vs. corresponding young group; # p < 0.05 vs. C57BL/6 old group
To better understand the mechanism(s) underneath ETA knockout-offered beneficial effects against cardiac aging, fura-2 fluorescence was monitored to evaluate intracellular Ca2+ handling properties. Cardiomyocytes from aged C57BL/6 mice displayed reduced intracellular Ca2+ release in response to electrical stimuli (ΔFFI) and prolonged intracellular Ca2+ decay (both single or bi-exponential curve fitting) along with unchanged resting intracellular Ca2+ (resting FFI), the effect of which was reconciled by ETA knockout. ETA knockout did not alter intracellular Ca2+ homeostasis at young ages (Fig. 4).

Intracellular Ca2+ property of cardiomyocytes from young or old C57BL/6 and ETAKO mice. a–b Representative intracellular Ca2+ transients in young or old C57BL/6 and ETAKO mice; c Resting fura-2 fluorescence intensity (FFI); d Change of intracellular Ca2+ in response to electrical stimuli (ΔFFI); e Intracellular Ca2+ transient decay rate (single exponential); and f Intracellular Ca2+ transient decay (bi- exponential). Mean ± SEM, n = a total of 65 – 66 cells from 4 mice per group, *p < 0.05 vs. corresponding young group; # p < 0.05 vs. C57BL/6 old group
Effect of ETA knockout on myocardial histology in aging
To assess the impact of ETA knockout on myocardial histology in aging, cardiomyocyte cross-sectional area and interstitial fibrosis were examined. Data from the wheat germ agglutinin staining revealed an increased cardiomyocyte cross-sectional area in aging, consistent with the higher LV mass and heart weight in aged C57BL/6 mice. ETA knockout significantly attenuated aging-induced cardiomyocyte hypertrophy. Likewise, the Masson’s trichrome staining revealed overt myocardial interstitial fibrosis in aged C57BL/6 myocardium, the effect of which was significantly dampened by ETA receptor knockout. Last, ETA knockout did not affect cardiomyocyte cross-sectional area and interstitial fibrosis at young ages (Fig. 5a–d).

Histological feature and ROS production from young or old C57BL/6 and ETAKO mice. a and c Representative images of myocardium stained with wheat germ agglutinin (WGA, a, original magnification ×400) and Masson’s trichrome (b, original magnification ×200) depicting cardiomyocyte cross-sectional area (CSA) and interstitial fibrosis, respectively; b and d Quantitative analysis of CSA and interstitial fibrosis using measurements of ~160 myocytes from 5–6 mice per group. e Representative DCF fluorescent images (400×) showing ROS generation in cardiomyocytes from C57BL/6 and ETAKO mice; and f Pooled data of ROS using DCF fluorescence from 6 isolations per group. Mean ± SEM, *p < 0.05 vs. corresponding young group; # p < 0.05 vs. C57BL/6 old group
Effect of ETA knockout on aging-induced ROS production and protein carbonyl formation
To examine the potential mechanism of action behind the ETA receptor knockout-elicited protection against aging-induced cardiac geometric and contractile anomalies, ROS production was examined in cardiomyocytes from C57BL/6 and ETA knockout mice at both young and old ages. Results shown in Fig. 5e–f indicate that ROS production was significantly elevated in aged C57BL/6 mouse cardiomyocytes. Consistent with its mechanical and morphometric responses, ETA knockout attenuated aging-induced increase in ROS production. Furthermore, ETA knockout significantly ameliorated aging-induced protein damage (carbonyl content in n mol/mg protein: C57BL/6-young: 4.09 ± 0.58; C57BL/6-old: 10.75 ± 1.72*; ETAKO-young: 3.94 ± 0.59; and ETAKO-old: 6.44 ± 0.49*#, *p < 0.05 vs. C57BL/6-young; # p < 0.05 vs. C57BL/6-old, n = 5–7 hearts). ETA receptor knockout itself exerted little effects on ROS production and carbonyl formation at young ages, indicating that the genetic manipulation itself is not innately harmful.
Effects of ETA knockout on intracellular Ca2+ regulatory and hypertrophic proteins
Western blot analysis revealed significantly downregulated levels of intracellular Ca2+ regulatory proteins SERCA2a and phospholamban in aged C57BL/6 myocardium. ETA knockout mitigated aging-induced decrease in SERCA2a and phospholamban as well as SERCA2a-to-phospholamban ratio without eliciting any notable effects by itself. Neither ETA knockout nor aging affected the level of Na+–Ca2+ exchanger. Assessment of cardiac hypertrophic markers revealed that elevated expression of GATA4 and ANP along with increased phosphorylation of NFATc3 in aged C57BL/6 hearts. ETA receptor knockout significantly attenuated or nullified aging-induced increase in the levels of GATA4, ANP and phosphorylated NFATc3. Last but not least, ETA knockout did not affect the levels of GATA4, ANP and phosphorylated NFATc3 at young ages (Fig. 6).

Changes of intracellular Ca2+ regulating and hypertrophic proteins in myocardium from young or old C57BL/6 and ETAKO mice. a Representative gel blots depicting expression of SERCA2a, phospholamban, Na+–Ca2+ exchanger, GATA4, phosphorylated NFATc3 (pNFATc3), ANP and GAPDH (loading control) using specific antibodies; b SERCA2a; c Phospholamban; d SERCA2a-to-Phospholamban ratio; e Na+–Ca2+ exchanger; f GATA4; g ANP and h pNFATc3. Mean ± SEM, n = 5–7, *p < 0.05 vs. corresponding young group; # p < 0.05 vs. C57BL/6 old group
Effects of ETA knockout on aging-induced autophagy response
To explore if autophagy contributes to ETA knockout-offered cardioprotection against aging, levels of autophagic markers Beclin-1, Atg7, Atg5, LC3B and the autophagosome cargo adaptor p62 were evaluated in young or old C57BL/6 and ETA knockout mice. Our results depict that aging significantly downregulated the levels of Beclin-1, Atg7, Atg5 and LC3B II/I ratio while upregulated p62 levels, the effects of which were significantly attenuated or mitigated by ETA receptor knockout. ETA knockout failed to alter levels of these autophagic markers at young ages (Fig. 7a–f).

Changes of autophagic and ER stress protein markers in myocardium from young or old C57BL/6 and ETAKO mice. a Representative gel blots depicting expression of Beclin-1, Atg5, Atg7, LC3B, p62, Bip, CHOP, Calcineurin A and GAPDH (loading control) using specific antibodies; b Beclin-1; c Atg5; d Atg7; e LC3B-II-to-LC3B-I ratio; f p62; g Bip; h CHOP; and i Calcineurin A. Mean ± SEM, n = 4–6, *p < 0.05 vs. corresponding young group; # p < 0.05 vs. C57BL/6 old group
Effects of ETA knockout on aging-induced ER stress response
As depicted in Fig. 7g–i, Western blot analysis demonstrated a robust decrease in ER stress proteins including Bip, CHOP and a subtle but significant increase in calcineurin level in aged C57BL/6 hearts. ETA knockout significantly attenuated or obliterated aging-induced changes in these ER stress markers. ETA knockout did not exert any notable effect on the levels of these proteins.
Effect of ETA receptor blockade on ET-1-induced autophagic and hypertrophic responses in H9C2 myoblasts
To further consolidate aging and/or ET-1-elicited responses in cardiac autophagy and hypertrophy, Western blot analysis was performed on the essential autophagic and hypertrophic protein markers including Beclin-1, Atg7, Atg5, LC3B and ANP in H9C2 myoblasts incubated with ET-1 (10 nM) for 24 h in the absence or presence of the ETA receptor blocker BQ123 (1 μM) or the autophagy inducer rapamycin (5 μM). Our data shown in Fig. 8 depicted that ET-1 exposure significantly downregulated levels of Beclin-1, Atg7, Atg5 and LC3B II/I ratio, indicative of suppressed autophagic responses. Interestingly, both BQ123 and rapamycin rescued against ET-1-induced loss of autophagy. Both BQ123 and rapamycin triggered a subtle although significant increase in autophagy (as evidenced by Atg7, Atg5 and the steady-state LC3B ratio but not Beclin-1). Meanwhile, ET-1 significantly elevated expression of the hypertrophic markers ANP, GATA4 and phosphorylated NFATc3, the effect of which was attenuated by both BQ123 and rapamycin (Fig. 8 and supplemental Fig. 1). Neither BQ123 nor rapamycin exerted any notable effect on ANP by themselves. Assessment of autophagic flux revealed that lysosomal inhibition partially reversed ET-1-induced decrease in LC3B II/I ratio (cumulative autophagosome formation) without eliciting any effect by itself. Meanwhile, ET-1 and mixed lysosomal inhibitors both significantly upregulated levels of the autophagosome cargo protein p62 without any additive effect between the two.

Changes of autophagic and hypertrophic markers in H9C2 myoblasts. H9C2 cells were exposed to endothelin-1 (ET-1, 10 nM) for 24 h in the absence or presence of the ETA receptor blocker BQ123 (1 μM), the autophagy inducer rapamycin (RAP, 5 μM) or a mixed lysosomal inhibitors [bafilomycin A1 (50 nM), E64D (2.5 μg/ml) and pepstatin A methyl ester (5 μg/ml). a Representative gel blots depicting Beclin-1, Atg7, Atg5, LC3B, ANP and GAPDH (loading control) using specific antibodies; b Beclin-1; c: Atg 7; d Atg5; e LC3B II/I ratio (steady-state autophagosomes); f ANP; g LC3B II/I ratio (cumulative autophagosomes) in response to ET-1 in the presence or absence of lysosomal inhibitors; and h p62 in response to ET-1 in the presence or absence of lysosomal inhibitors. Mean ± SEM, n = 4–6, *p < 0.05 vs. control (Cont); # p < 0.05 vs. ET-1 group
Effect of ETA and ETB antagonism as well as autophagy inhibition on mechanical and intracellular Ca2+ properties in cardiomyocytes
To further delineate the potential contribution of ET-1 receptor subtypes and autophagy in cardiac aging, young and old mice were treated with the ETA receptor antagonist BQ123 or the ETB receptor antagonist BQ788 for 4 weeks in the absence or presence of autophagy inhibition using 3-methyladenine (3-MA). Resting cell length was comparable among all groups regardless of aging or drug treatment. Aging decreased peak shortening and ±dL/dt, prolonged TR90 associated with similar TPS, the effects of which obliterated by BQ123 but not BQ788 or 3-MA. Interestingly, 3-MA nullified ETA receptor antagonism-induced beneficial effect against cardiac aging. None of the pharmacological inhibitors elicited any notable mechanical effect themselves (Fig. 9). To better understand the mechanism(s) of action behind ET-1 receptor antagonism or autophagy inhibition-induced cardiac mechanical responses, fura-2 fluorescence was monitored to evaluate intracellular Ca2+ handling. Consistent with cardiac mechanical responses, the aging-induced intracellular Ca2+ mishandling shown as dampened ΔFFI and prolonged intracellular Ca2+ decay (single or bi-exponential curve fitting) along with unchanged resting intracellular Ca2+ were significantly improved by BQ123, but not BQ788 or 3-MA. Intriguingly, 3-MA treatment nullified ETA receptor antagonism-induced beneficial effect against aging-induced intracellular Ca2+ derangement. Last but not least, none of the pharmacological inhibitors employed altered intracellular Ca2+ homeostasis themselves (Fig. 10).

Cardiomyocyte mechanical properties in young (4 months) or old (26 months) C57BL/6 mice treated with or without the ETA receptor antagonist BQ123 or the ETB receptor antagonist BQ788 (1 mg/kg/d, i.p.) for 28 days in the presence or absence of autophagy inhibitor 3-MA (10 mg/kg/week, i.p. for 4 weeks). a Resting cell length; b Peak shortening (PS) amplitude; c Maximal velocity of shortening (+dL/dt); d Maximal velocity of relengthening (−dL/dt); e Time-to-PS (TPS); and f Time-to-90 % relengthening (TR90). Mean ± SEM, n = 65–66 cells per group, *p < 0.05 vs. young group; # p < 0.05 vs. old group, † p < 0.05 vs. old-BQ123 group

Intracellular Ca2+ handling properties in young (4 months) or old (26 months) C57BL/6 mice treated with or without the ETA receptor antagonist BQ123 or the ETB receptor antagonist BQ788 (1 mg/kg/d, i.p.) for 28 days in the presence or absence of autophagy inhibitor 3-MA (10 mg/kg/week, i.p. for 4 weeks). a Resting fura-2 fluorescence intensity (FFI); b Change of intracellular Ca2+ in response to electrical stimuli (ΔFFI); c Intracellular Ca2+ transient decay rate (single exponential); and d Intracellular Ca2+ transient decay (bi- exponential). Mean ± SEM, n = 61 cells per group, *p < 0.05 vs. young group; # p < 0.05 vs. old group, † p < 0.05 vs. old-BQ123 group
Discussion
Myocardial morphometric and functional findings from the present study demonstrated that ETA receptor knockout significantly attenuated or ablated aging-induced cardiac geometric and contractile dysfunction. Our data revealed that ETA receptor knockout significantly ameliorated aging-induced ROS generation, protein damage, decrease in autophagosome formation and interruption in autophagy flux. The inhibitory of ET-1 on autophagy flux received further support from lysosomal inhibition findings. Our in vitro data also depicted that ET-1-induced autophagy and increase in hypertrophic markers GATA4, ANP and phosphorylated NFATc3 may be attenuated or reconciled by the ETA receptor antagonist BQ123 and the autophagy inducer rapamycin. Short-term treatment of the ETA, but not ETB receptor antagonist, improved aging-associated cardiomyocyte mechanical properties, the effect of which was offset by autophagy inhibition. Given that plasma ET-1 levels and ETA receptor expression are elevated in aging, our findings supported a pivotal role of ET-1 system in particular ETA receptor in aging-associated cardiac geometric and contractile defect. This is supported by a better survival rate of the ETA knockout mice compared with the C57BL/6 wild-type mice. More importantly, our findings favor a role of autophagy in aging and ETA receptor knockout-induced cardiac geometric and functional changes. These findings suggest the potential of ETA receptor as a target in the treatment of cardiac aging-related remodeling and contractile defects.
Our data revealed that aging elicited a subtle although significant rise in systolic blood pressure, consistent with the elevated plasma Ang II levels with aging and the reported positive correlation between age and blood pressure [30, 43]. Inhibition of the Ang II cascade has been demonstrated to prolong life and retard aging-associated complications [43]. Our observation that cardiomyocyte-specific ETA knockout failed to affect age-associated changes in systolic blood pressure and plasma Ang II levels did not favor a major role of blood pressure and Ang II in ETA knockout-elicited beneficial effects. Changes in myocardial morphology and contractile function have been reported in aging hearts characterized by cardiac hypertrophy, intracellular Ca2+ dysregulation, compromised contractility and prolonged diastolic duration [21, 26, 48, 49]. This is in line with the findings of our study in that echocardiographic and cardiomyocyte contractile parameters are compromised in aged C57BL/6 mice in conjunction with intracellular Ca2+ mishandling. Interestingly, ETA knockout significantly attenuated or nullified aging-associated cardiac remodeling (increase in wall thickness, LV end systolic diameter, cardiac hypertrophy manifested as increased LV mass, normalized LV mass and cardiomyocyte cross-sectional area), interstitial fibrosis and decreased myocardial contractility (fractional shortening, peak shortening, maximal velocity of shortening/relengthening, prolonged TR90). In addition, ETA knockout ablated intracellular Ca2+ derangement associated with aging including decreased ΔFFI and prolonged intracellular Ca2+ decay rate. Treatment of ETA, but not ETB, receptor antagonist mimicked ETA receptor knockout-elicited beneficial effects on cardiomyocyte mechanical properties. These observations favor a prominent role of ET-1 signaling cascade, in particular ETA receptor, in cardiac geometric and functional alterations associated with aging. ETA knockout attenuated aging-induced increase in ESD along with the preserved EDD, likely to be responsible for the improved fractional shortening parameter in aged ETA knockout mice. Myocardial hypertrophy and fibrosis are common manifestations of aging heart and may lead to heart failure [5, 29, 36, 42]. Our data revealed that ETA knockout attenuated aging-induced changes in LV mass, heart weight, cardiomyocyte size, interstitial fibrosis and hypertrophic markers (ANP, GATA4 and NFATc3 phosphorylation), suggesting a beneficial role of antagonism against ETA receptor against cardiac aging. In addition, ETA knockout itself did not alter cardiac geometric and mechanical properties, indicating the genetic manipulation is not innately harmful to the heart.
Several scenarios may be considered with regard to the possible mechanisms behind the beneficial effects of ETA receptor knockout or antagonism. First, ETA knockout attenuated aging-induced ROS production, suggesting a role of ROS scavenging in ETA knockout-elicited protection against cardiac aging. Generation of ROS in particular free radicals is known to play a pivotal role in cardiac remodeling and contractile dysfunction in aging [19, 23, 25, 40, 49]. Second, aged C57BL/6 cardiomyocytes displayed unchanged resting intracellular Ca2+ levels and decreased intracellular Ca2+ release in response to electrical stimulus and delayed intracellular Ca2+ clearance, somewhat in line with our previous reports [26, 48]. Although ETA knockout or receptor antagonism with BQ123 did not affect intracellular Ca2+ homeostasis in young mice, it attenuated or ablated aging-induced changes in intracellular Ca2+. These findings depict that ETA receptor knockout or inhibition may rescue aging-induced disruption of intracellular Ca2+ handling. This notion is supported by the restored stress tolerance shown as lessened negative peak shortening-frequency in aging. Loss of SERCA2a and the SERCA2a-to-phospholamban ratio may account for, at least in part, intracellular Ca2+ mishandling, prolonged intracellular Ca2+ clearance and cardiac relaxation, as well as reduced stress tolerance in aging hearts although further study is warranted. Third, data from our study also revealed that levels of ER stress markers (Bip and CHOP) were reduced or increased (calcineurin A) in aged C57BL/6 mice, the effect of which was attenuated or ablated by ETA knockout. Up-to-date, little is known for the correlation between aging and ER stress in the heart although findings from our study favor a beneficial role of ER stress in ETA knockout-induced protection against aging. The increase in calcineurin A in aging hearts may be due to a compensatory response. Further study is warranted to explore the role of ER stress in cardiac aging and ETA knockout-exerted beneficial effects.
Perhaps the most interesting findings from our study were that ETA knockout attenuated or reversed aging-induced downregulation of autophagy markers as well as interruption of autophagic flux. ETA knockout attenuated or reversed aging-induced reduction in the levels of Beclin-1, Atg7, Atg5 and LC3B while partially increasing aging-induced rise in the autophagosome cargo protein p62. The potential role of autophagy in ETA knockout-offered cardioprotection in aging was consolidated by the observation that autophagy inhibition using 3-MA ablated ETA receptor antagonist BQ123-induced cardioprotective effect against aging. A number of reports have depicted reduced autophagosome formation or autophagy flux with aging, which may be speculated to contribute to aging-associated accumulation of damaged intracellular components in almost all model organisms thus resulting in altered cellular homeostasis and loss of function in aging [7, 16, 38, 51]. It is likely that the reduced autophagic activity may contribute to the aging-associated cardiac hypertrophy and contractile dysfunction [9, 45]. Autophagy is well known to antagonize cardiac hypertrophy by increasing protein degradation, which decreases tissue mass. However, the rate of protective autophagy declines with aging, leading to a state where the heart is unable to remove damaged structures and thus resulting in garbage accumulation (abnormal intracellular protein aggregates and undigested materials). Reduced autophagy in aging eventually results in enhanced oxidative stress, decreased ATP production, collapse of cellular catabolic machinery and cell death [9, 45]. In our hands, aging overtly inhibited initial autophagosome formation (shown as reduced LC3B) despite interrupted lysosomal degradation (evidenced as elevated p62), the effects of which were partially or completely reversed by ETA knockout. In vitro finding revealed that ET-1 exposure mimicked lysosomal inhibitors in promoting p62 accumulation (Fig. 8h), consolidating the inhibitory effect of ET-1 on autophagic flux. The subtle, although significant, rise of LC3B elicited by lysosomal inhibitors following ET-1 challenge (Fig. 8g) may be due to rather limited initial autophagosome formation. Our finding suggested that ETA knockout attenuated or reversed aging-induced decline in autophagy in conjunction with hypertrophy (gross, histological findings or protein makers GATA4, ANP and pNFATC3). This is in concert with the in vitro findings that autophagy inducer rapamycin rescued against ET-1-induced cardiomyocyte hypertrophy in H9C2 myoblasts. Although it is beyond the scope of our current study, several theories have been postulated to contribute to aging-associated decline in autophagy. First, the slow build-up of the undigested materials such as lipofuscin within lysosomes with age depresses autophagic activity in aged cardiomyocytes and other postmitotic cells [9]. Second, aging-associated change in the integrity of the autophagosomal-lysosomal network as evidenced by our autophagosome cargo protein p62 data plays an essential role in the regulation of cardiac autophagy in aging [9, 16]. Last but not least, an aging-associated decline in AMPK activity in the hearts may also contribute to the reduced autophagy in aging hearts [40]. Data from our present study indicated that ET-1 suppressed autophagy, the effect of which may be reconciled by the ETA blocker BQ123. Although ET-1 has not been previously shown to directly regulate autophagy, one recent report indicated that ET-1-associated vascular injury may be alleviated by autophagy induction [52], favoring an inverse relationship between ET-1 levels and autophagic activity, which is also supported by our current findings. Future study is warranted to better elucidate the precise mechanism of action behind ET-1 and ETA knockout-induced regulation of autophagy and stress in the heart.
Our data revealed an elevated ETB receptor abundance in ETA knockout mice at young ages, coinciding with the notion that ETB receptor may become the dominant ET-1 receptor while ETA receptor may be dispensable for the maintenance of cardiac homeostasis [20]. However, the lack of responses from the ETB receptor antagonist BQ788 in cardiac aging as opposed to that elicited by the ETA receptor antagonist BQ123 does not favor a role of ETB receptor in aging-associated cardiac abnormalities. This is also in line with the upregulated ETA receptor express with aging. It is not exactly clear at this point why elevated expression of ETB receptor is lost with aging although caution should be taken with regard to the potential role of ETB receptor in ET-1-associated cardiac aging responses.
Experimental limitation: Measurement of cardiac contractile performance in isolated cardiomyocytes has been established to provide a fundamental assessment of cardiac function in pathological states. However, as in any study of this nature, caution needs to be exercised when correlating cellular findings to whole heart function, as the latter is composed of heterogeneous cell types, including nerve terminals, fibroblasts and connective tissues. For example, the apparent discrepancy in the degree of reduction of contractile capacity between in vivo fractional shortening (~20 %) and in vitro peak shortening (~80 % at 5 Hz) may be attributed to different experimental settings.
In summary, our present study provides evidence, for the first time that ETA receptor knockout rescues against aging-induced cardiac hypertrophy and contractile dysfunction. Our data indicated that ROS production and a decline in autophagy (or autophagy flux) are likely involved in aging- and ETA receptor knockout-elicited changes in cardiac remodeling and function. Given that aging may be associated with reduced rate of formation of autophagosome, poor maturation and efficiency of autophagosome-lysosome fusion, as well as dampened proteolytic activity of lysosomes [7], derangement in autophagy may contribute to cardiac aging through accumulation of cytosolic protein aggregates and defective mitochondria. Although our study sheds some light on the role of autophagy and autophagy flux in aging-induced cardiac geometric, functional and intracellular Ca2+ defects, the mechanism of action behind aging-associated defective autophagy and/or autophagy flux, accumulation of damaged mitochondria and increased ROS generation still deserves further investigation.
Change history
15 November 2022
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1007/s00395-022-00960-5
References
Afiatpour P, Latifpour J, Takahashi W, Yono M, Foster HE Jr, Ikeda K, Pouresmail M, Weiss RM (2003) Developmental changes in the functional, biochemical and molecular properties of rat bladder endothelin receptors. Naunyn Schmiedebergs Arch Pharmacol 367:462–472. doi:10.1007/s00210-003-0715-6
Boengler K, Buechert A, Heinen Y, Roeskes C, Hilfiker-Kleiner D, Heusch G, Schulz R (2008) Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res 102:131–135. doi:10.1161/CIRCRESAHA.107.164699
Boengler K, Konietzka I, Buechert A, Heinen Y, Garcia-Dorado D, Heusch G, Schulz R (2007) Loss of ischemic preconditioning’s cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. Am J Physiol Heart Circ Physiol 292:H1764–H1769. doi:10.1152/ajpheart.01071.2006
Boengler K, Schulz R, Heusch G (2009) Loss of cardioprotection with ageing. Cardiovasc Res 83:247–261. doi:10.1093/cvr/cvp033
Carneiro-Ramos MS, Diniz GP, Nadu AP, Almeida J, Vieira RL, Santos RA, Barreto-Chaves ML (2010) Blockage of angiotensin II type 2 receptor prevents thyroxine-mediated cardiac hypertrophy by blocking Akt activation. Basic Res Cardiol 105:325–335. doi:10.1007/s00395-010-0089-0
Chen Y, Hanaoka M, Droma Y, Chen P, Voelkel NF, Kubo K (2010) Endothelin-1 receptor antagonists prevent the development of pulmonary emphysema in rats. Eur Respir J 35:904–912. doi:10.1183/09031936.00003909
Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A (2005) Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 1:131–140
Dai DF, Chen T, Johnson SC, Szeto H, Rabinovitch PS (2012) Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal 16:1492–1526. doi:10.1089/ars.2011.4179
De Meyer GR, De Keulenaer GW, Martinet W (2010) Role of autophagy in heart failure associated with aging. Heart Fail Rev 15:423–430. doi:10.1007/s10741-010-9166-6
De Meyer GR, Martinet W (2009) Autophagy in the cardiovascular system. Biochim Biophys Acta 1793:1485–1495. doi:10.1016/j.bbamcr.2008.12.011
Doser TA, Turdi S, Thomas DP, Epstein PN, Li SY, Ren J (2009) Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation 119:1941–1949. doi:10.1161/CIRCULATIONAHA.108.823799
Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP (2011) Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J 25:99–110. doi:10.1096/fj.10-164152
Gottlieb RA, Finley KD, Mentzer RM Jr (2009) Cardioprotection requires taking out the trash. Basic Res Cardiol 104:169–180. doi:10.1007/s00395-009-0011-9
Gottlieb RA, Mentzer RM (2010) Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol 72:45–59. doi:10.1146/annurev-physiol-021909-135757
Granata R, Trovato L, Gallo MP, Destefanis S, Settanni F, Scarlatti F, Brero A, Ramella R, Volante M, Isgaard J, Levi R, Papotti M, Alloatti G, Ghigo E (2009) Growth hormone-releasing hormone promotes survival of cardiac myocytes in vitro and protects against ischaemia-reperfusion injury in rat heart. Cardiovasc Res 83:303–312. doi:10.1093/cvr/cvp090
Hua Y, Zhang Y, Ceylan-Isik AF, Wold LE, Nunn JM, Ren J (2011) Chronic Akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res Cardiol 106:1173–1191. doi:10.1007/s00395-011-0222-8
Hwang JT, Kwon DY, Park OJ, Kim MS (2008) Resveratrol protects ROS-induced cell death by activating AMPK in H9c2 cardiac muscle cells. Genes Nutr 2:323–326. doi:10.1007/s12263-007-0069-7
Ito H, Hirata Y, Adachi S, Tanaka M, Tsujino M, Koike A, Nogami A, Murumo F, Hiroe M (1993) Endothelin-1 is an autocrine/paracrine factor in the mechanism of angiotensin II-induced hypertrophy in cultured rat cardiomyocytes. J Clin Invest 92:398–403. doi:10.1172/JCI116579
Kakarla SK, Fannin JC, Keshavarzian S, Katta A, Paturi S, Nalabotu SK, Wu M, Rice KM, Manzoor K, Walker EM Jr, Blough ER (2010) Chronic acetaminophen attenuates age-associated increases in cardiac ROS and apoptosis in the Fischer Brown Norway rat. Basic Res Cardiol 105:535–544. doi:10.1007/s00395-010-0094-3
Kedzierski RM, Grayburn PA, Kisanuki YY, Williams CS, Hammer RE, Richardson JA, Schneider MD, Yanagisawa M (2003) Cardiomyocyte-specific endothelin A receptor knockout mice have normal cardiac function and an unaltered hypertrophic response to angiotensin II and isoproterenol. Mol Cell Biol 23:8226–8232
Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD (1995) High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem 270:17513–17520
Lakatta EG (2003) Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part III: cellular and molecular clues to heart and arterial aging. Circulation 107:490–497
Lakatta EG (2000) Cardiovascular aging in health. Clin Geriatr Med 16:419–444
Lakatta EG, Levy D (2003) Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation 107:139–146
Lakatta EG, Sollott SJ (2002) The “heartbreak” of older age. Mol Interv 2:431–446. doi:10.1124/mi.2.7.431
Li SY, Du M, Dolence EK, Fang CX, Mayer GE, Ceylan-Isik AF, LaCour KH, Yang X, Wilbert CJ, Sreejayan N, Ren J (2005) Aging induces cardiac diastolic dysfunction, oxidative stress, accumulation of advanced glycation endproducts and protein modification. Aging Cell 4:57–64. doi:10.1111/j.1474-9728.2005.00146.x
Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W, Merk M, Zierow S, Bernhagen J, Ren J, Bucala R, Li J (2010) Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation 122:282–292. doi:10.1161/CIRCULATIONAHA.110.953208
Manning WJ, Wei JY, Katz SE, Litwin SE, Douglas PS (1994) In vivo assessment of LV mass in mice using high-frequency cardiac ultrasound: necropsy validation. Am J Physiol 266:H1672–H1675
Mraiche F, Oka T, Gan XT, Karmazyn M, Fliegel L (2011) Activated NHE1 is required to induce early cardiac hypertrophy in mice. Basic Res Cardiol 106:603–616. doi:10.1007/s00395-011-0161-4
Pearson JD, Morrell CH, Brant LJ, Landis PK, Fleg JL (1997) Age-associated changes in blood pressure in a longitudinal study of healthy men and women. J Gerontol A Biol Sci Med Sci 52:M177–M183
Pieske B, Beyermann B, Breu V, Loffler BM, Schlotthauer K, Maier LS, Schmidt-Schweda S, Just H, Hasenfuss G (1999) Functional effects of endothelin and regulation of endothelin receptors in isolated human nonfailing and failing myocardium. Circulation 99:1802–1809
Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, Befroy D, Pypaert M, Hardie DG, Young LH, Shulman GI (2007) Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab 5:151–156. doi:10.1016/j.cmet.2007.01.008
Rivera A (2007) Reduced sickle erythrocyte dehydration in vivo by endothelin-1 receptor antagonists. Am J Physiol Cell Physiol 293:C960–C966. doi:10.1152/ajpcell.00530.2006
Rubinsztein DC, Marino G, Kroemer G (2011) Autophagy and aging. Cell 146:682–695. doi:10.1016/j.cell.2011.07.030
Schlossarek S, Englmann DR, Sultan KR, Sauer M, Eschenhagen T, Carrier L (2012) Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res Cardiol 107:235. doi:10.1007/s00395-011-0235-3
Schneider J, Lother A, Hein L, Gilsbach R (2011) Chronic cardiac pressure overload induces adrenal medulla hypertrophy and increased catecholamine synthesis. Basic Res Cardiol 106:591–602. doi:10.1007/s00395-011-0166-z
Takayanagi R, Kitazumi K, Takasaki C, Ohnaka K, Aimoto S, Tasaka K, Ohashi M, Nawata H (1991) Presence of non-selective type of endothelin receptor on vascular endothelium and its linkage to vasodilation. FEBS Lett 282:103–106
Takemura G, Miyata S, Kawase Y, Okada H, Maruyama R, Fujiwara H (2006) Autophagic degeneration and death of cardiomyocytes in heart failure. Autophagy 2:212–214
Tanno M, Kuno A, Horio Y, Miura T (2012) Emerging beneficial roles of sirtuins in heart failure. Basic Res Cardiol 107:273. doi:10.1007/s00395-012-0273-5
Turdi S, Fan X, Li J, Zhao J, Huff AF, Du M, Ren J (2010) AMP-activated protein kinase deficiency exacerbates aging-induced myocardial contractile dysfunction. Aging Cell 9:592–606. doi:10.1111/j.1474-9726.2010.00586.x
Wang D, Patel VV, Ricciotti E, Zhou R, Levin MD, Gao E, Yu Z, Ferrari VA, Lu MM, Xu J, Zhang H, Hui Y, Cheng Y, Petrenko N, Yu Y, FitzGerald GA (2009) Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc Natl Acad Sci USA 106:7548–7552. doi:10.1073/pnas.0805806106
Wei JY (1992) Age and the cardiovascular system. N Engl J Med 327:1735–1739. doi:10.1056/NEJM199212103272408
Wray DW, Nishiyama SK, Harris RA, Richardson RS (2008) Angiotensin II in the elderly: impact of angiotensin II type 1 receptor sensitivity on peripheral hemodynamics. Hypertension 51:1611–1616. doi:10.1161/HYPERTENSIONAHA.108.111294
Xia H, Suda S, Bindom S, Feng Y, Gurley SB, Seth D, Navar LG, Lazartigues E (2011) ACE2-mediated reduction of oxidative stress in the central nervous system is associated with improvement of autonomic function. PLoS ONE 6:e22682. doi:10.1371/journal.pone.0022682
Xie M, Morales CR, Lavandero S, Hill JA (2011) Tuning flux: autophagy as a target of heart disease therapy. Curr Opin Cardiol 26:216–222. doi:10.1097/HCO.0b013e328345980a
Xu H, Duan J, Dai S, Wu Y, Sun R, Ren J (2008) alpha-Zearalanol attenuates oxLDL-induced ET-1 gene expression, ET-1 secretion and redox-sensitive intracellular signaling activation in human umbilical vein endothelial cells. Toxicol Lett 179:163–168. doi:10.1016/j.toxlet.2008.05.005
Yamamoto S, Matsumoto N, Kanazawa M, Fujita M, Takaoka M, Gariepy CE, Yanagisawa M, Matsumura Y (2005) Different contributions of endothelin-A and endothelin-B receptors in postischemic cardiac dysfunction and norepinephrine overflow in rat hearts. Circulation 111:302–309. doi:10.1161/01.CIR.0000153351.86708.F7
Yang X, Doser TA, Fang CX, Nunn JM, Janardhanan R, Zhu M, Sreejayan N, Quinn MT, Ren J (2006) Metallothionein prolongs survival and antagonizes senescence-associated cardiomyocyte diastolic dysfunction: role of oxidative stress. FASEB J 20:1024–1026. doi:10.1096/fj.05-5288fje
Yang X, Sreejayan N, Ren J (2005) Views from within and beyond: narratives of cardiac contractile dysfunction under senescence. Endocrine 26:127–137. doi:10.1385/ENDO:26:2:127
Yono M, Latifpour J, Takahashi W, Pouresmail M, Afiatpour P, Weiss RM (2004) Age-related changes in the properties of the endothelin receptor system at protein and mRNA levels in the rat vas deferens. J Recept Signal Transduct Res 24:53–66
Zhang C, Cuervo AM (2008) Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 14:959–965. doi:10.1038/nm.1851
Zhang YL, Cao YJ, You SJ, Li RX, Liu HH, Liu CF (2010) Protective effects of autophagy against oxidized LDL-induced injury in endothelial cells. Zhonghua Yi Xue Za Zhi 90:2792–2796
Acknowledgments
The authors wish to thank Miss Haoyu Zhao from University of Wyoming for her assistance in data analysis. This work was supported by NIH/NCRR P20 RR016474 and P20 GM103432.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Asli F. Ceylan-Isik, Maolong Dong and Yingmei Zhang have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article
Cite this article
Ceylan-Isik, A.F., Dong, M., Zhang, Y. et al. RETRACTED ARTICLE: Cardiomyocyte-specific deletion of endothelin receptor A rescues aging-associated cardiac hypertrophy and contractile dysfunction: role of autophagy. Basic Res Cardiol 108, 335 (2013). https://doi.org/10.1007/s00395-013-0335-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-013-0335-3