
Abstract
Heart failure is a progressive disease, leading to reduced quality of life and premature death. Adverse ventricular remodeling involves changes in the balance between cardiomyocyte protein synthesis and degradation, forcing these myocytes in equilibrium between life and death. In this context, autophagy has been recognized to play a role in the pathophysiology of heart failure. At basal levels, autophagy performs housekeeping functions, maintaining cardiomyocyte function and ventricular mass. Autophagy also occurs in the failing human heart, and upregulation has been reported in animal models of pressure overload–induced heart failure. Although the factors that determine whether autophagy will be protective or detrimental are not well known, the level and duration of autophagy seem important. Autophagy may antagonize ventricular hypertrophy by increasing protein degradation, which decreases tissue mass. However, the rate of protective autophagy declines with age. The inability to remove damaged structures results in the progressive accumulation of ‘garbage’, including abnormal intracellular proteins aggregates and undigested materials such as lipofuscin. Eventually, the progress of these changes results in enhanced oxidative stress, decreased ATP production, collapse of the cellular catabolic machinery, and cell death. By contrast, in load-induced heart failure, the extent of autophagic flux can rise to maladaptive levels. Excessive autophagy induction leads to autophagic cell death and loss of cardiomyocytes and may contribute to the worsening of heart failure. Accordingly, the development of therapies that up-regulate the repair qualities of the autophagic process and down-regulate the cell death aspects would be of great value in the treatment of heart failure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Heart failure and aging
Aging of the population and improved medical care to acute cardiac disease have led to an increased burden of heart failure in the community. According to the latest American Heart Association’s statistics, 9.3% of men and 4.8% of women between 60 and 79 years of age suffers from heart failure, and these incidences increase up to 13.8 and 12.2% respectively, above the age of 80 [1] (Fig. 1). Heart failure is a progressive disease, leading to reduced quality of life and premature death. Twenty percent of patients die the first year after diagnosis. Older patients are more likely to develop heart failure, despite a preserved left ventricular ejection fraction. The heart of these patients is characterized by concentric rather than eccentric left ventricular hypertrophy, and with more pronounced cardiomyocyte stiffening [2]. Within the large spectrum of heart failure phenotypes, the incidence of disease modifiers such as obesity, diabetes, hypertension, and female gender varies, but the overall pathophysiological mechanisms are overlapping [3].

Prevalence of heart failure by age and gender (modified after [1])
Progression of heart failure is the result of several maladaptive processes. These processes initially preserve cardiac output and blood pressure, but also accelerate adverse remodeling of the ventricular tissue. Although several molecular mechanisms of ventricular remodeling have been elucidated, there is still much that remains unknown. Adverse remodeling of the ventricle involves changes in the balance between cellular protein synthesis and degradation, hence, forcing the myocytes in a delicate equilibrium between life and death. In this context, the process of macroautophagy has been recognized to play a direct role in the pathophysiology of heart failure [4–7].
Autophagy
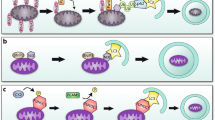
Macroautophagy (further referred to as autophagy) is a housekeeping mechanism cleaning cells from aberrant and dysfunctional molecules and organelles. It is a catabolic trafficking pathway for bulk destruction and turnover of long-lived proteins and organelles via regulated lysosomal degradation. The hallmark of autophagy is the de novo formation of autophagosomes, which are double-membrane vacuoles originating from a largely undefined structure, known as the phagophore or isolation membrane. Autophagy-related genes (ATG) control the process of autophagy. Beclin-1 (Atg6), together with class III phosphoinositide 3-kinase, is needed for vesicle nucleation of autophagosomes. Vesicle elongation requires two conjugation pathways. One of these involves microtubule-associated protein 1 light chain 3 (LC3 or Atg8), which is converted from its soluble form into a vesicle-associated form during the process of elongation. Both Atg6 and Atg8 are important markers for autophagy. The elongation of the isolation membrane is followed by maturation of the autophagosome, which fuses with a lysosome, thereby generating an autophagolysosome or autolysosome. The incorporation of the outer autophagosomal membrane into the lysosomal membrane allows the degradation of the remaining inner single-membrane and the cytoplasmic content of the autophagosome by lysosomal hydrolases (Fig. 2).

Schematic overview of autophagy and its modulation by aging. Autophagy is initiated by formation of nascent autophagosomal structures (isolation membranes), which sequestrate small portions of the cytoplasm and organelles for degradation. Expansion of the isolation membrane and enclosure of the cytoplasmic cargo lead to formation of autophagosomes. Subsequently, autophagosomes dock and fuse with lysosomes to form autolysosomes in which the cargo is degraded. A decreased amount of anti-oxidants in cells enhances chronic oxidative stress. Excessive oxidative damage to proteins, DNA, membrane lipids, and cell organelles plays an important role in aging. In aged cells, autophagy is defective due to: (1) diminished autophagosome formation, (2) impaired ability of lysosomes to fuse with autophagosomes, and (3) decreased proteolytic activity of lysosomes. In particular, presence of undigested materials in lysosomes (lipofuscin) may be responsible for their impaired ability to fuse and/or to degrade the contents of autophagosomes
Autophagy and aging
Aging is characterized by a progressive deterioration of cells and organs. It is to a large extent related to macromolecular damage by mitochondrially produced reactive oxygen species, mostly affecting long-lived postmitotic cells, such as neurons and cardiomyocytes. These cells are rarely or not at all replaced during life. Continuous removal of exhausted components and replacement with newly synthesized ones ensure cellular homeostasis and delay the aging process. However, the rate of autophagosome formation, the maturation, and the efficiency of autophagosome-lysosome fusion, as well as the proteolytic activity of lysosomes decline with age (Fig. 2) [8]. The inability of autophagy and other cellular degradation mechanisms to completely remove damaged structures results in the progressive accumulation of ‘garbage’, including cytosolic protein aggregates and defective mitochondria. In particular, presence of undigested materials in lysosomes (lipofuscin) may be responsible for their impaired ability to fuse and/or to degrade the contents of autophagosomes [8]. Lipofuscin is a non-degradable, yellow-brown pigment composed of lipid and protein residues, which progressively accumulates in cardiomyocytes and other long-lived postmitotic cells. Hydrogen peroxide generated by mitochondria and other organelles permeates in the lumen of secondary lysosomes. These lysosomes contain iron derived from cellular structures undergoing autophagic degradation. The interaction between reactive ferrous iron and hydrogen peroxide results, via fenton reactions, in the generation of hydroxyl radicals inducing lipid peroxidation and eventually intermolecular crosslinking and lipofuscin formation [9]. Crosslinked polymeric lipofuscin cannot be degraded by lysosomal hydrolases and may lead to preferential allocation of lysosomal enzymes to lipofuscin-loaded lysosomes at the expense of active autolysosomes (Fig. 3) [10]. The slow accumulation of lipofuscin within lysosomes depresses autophagy in the aging heart. Impaired autophagy stimulates further accumulation of damaged mitochondria, increases reactive oxygen species generation and enhances lipofuscinogenesis. Moreover, defective and enlarged mitochondria are poorly autophagocytosed. The progress of these changes seems to result in enhanced oxidative stress, decreased ATP production, and collapse of the cellular catabolic machinery, which eventually is incompatible with survival [11]. Finally, these events sensitize cells to undergo apoptosis as released lysosomal enzymes can attack other proteins and mitochondria, triggering cytochrome c release with an amplification of apoptosis [12]. Therefore, integrity of the autophagosomal–lysosomal network appears to be critical in the progression of aging [13].

Potential interactions between autophagy in the heart, lipofuscinogenesis, and aging. Hydrogen peroxide (H2O2) generated by mitochondria and other organelles permeates in the lumen of secondary lysosomes. These lysosomes contain iron derived from cellular structures undergoing autophagic degradation. The interaction between reactive ferrous iron and H2O2 results, via fenton-like reactions, in the generation of hydroxyl radicals inducing lipid peroxidation and eventually intermolecular crosslinking and lipofuscin formation. Progressive inhibition of autophagy in the aging heart is at least in part attributed to intralysosomal accumulation of lipofuscin. Crosslinked polymeric lipofuscin cannot be degraded by lysosomal hydrolases and might lead to preferential allocation of lysosomal enzymes to lipofuscin-loaded lysosomes at the expense of active autolysosomes. Impaired autophagy stimulates further accumulation of damaged mitochondria, usually deficient in ATP production and producing increased amounts of reactive oxygen species (ROS), while oxidatively modified cytosolic proteins form large indigestible aggregates. These events enhance lipofuscinogenesis and sensitize cardiomyocytes to undergo apoptosis
Autophagy in the heart: friend or foe?
In the heart, autophagy occurs constitutively at low basal levels to perform housekeeping functions, such as the destruction of dysfunctional organelles, in order to maintain cardiac function and morphology [6, 14]. However, autophagic processes in cardiomyocytes are up-regulated in response to altered internal needs (e.g. removal of protein aggregates) and environmental stress conditions, such as ATP depletion (e.g. during starvation), oxidative damage (e.g. via reactive oxygen species), and mitochondrial permeability transition pore opening [5, 15, 16]. These observations strongly suggest that autophagy is a survival mechanism. Indeed, it has been demonstrated that autophagy plays a critical role in the maintenance of ventricular function during starvation in the adult [17]. Moreover, treatment of mice with the autophagy inhibitor bafilomycin A1 does not affect cardiac function in normally fed animals, but it significantly depresses cardiac function and causes important left ventricular dilatation in mice starved for 3 days [11]. On the other hand, the occurrence of autophagic structures in dying cells of different organisms has led to the hypothesis that autophagy may also play a causative role in stress-induced cell death [18]. It has become clear that simultaneously occurring features of failing hearts include not only apoptosis and necrosis but also autophagy [15, 19]. Autophagy has been observed in the failing human heart [20, 21], and upregulation of this process has been reported in animal models of pressure overload–induced heart failure [5, 22, 23].
Accordingly, the functional role of enhanced autophagy in the diseased heart is still unclear, and studies have reported conflicting results. Is increased autophagic response a sign of failed cardiomyocyte repair or an adaptive suicide pathway for failing cardiomyocytes? Should autophagy be regarded as a protective mechanism or as a process contributing to disease progression [16]? Current evidence indicates that autophagic activity under physiological conditions is important for cellular homeostasis, whereas excessive autophagy is rather detrimental [16]. As already mentioned above, autophagy functions predominantly as a pro-survival pathway during nutrient deprivation and other forms of cellular stress. However, when autophagy is severely triggered, the autophagic machinery may also be used for self-destruction. Hence, when autophagic cell death occurs in cardiac cells, it may contribute to the progress of heart failure. Although it is not well known which factors determine whether autophagy will be protective or detrimental to a cell, the level and duration of autophagy seem important [24].
Autophagy in the failing heart
Autophagy antagonizes cardiac hypertrophy by increasing protein degradation
Inhibition of autophagy in cardiomyocytes by Atg5 deletion induces hypertrophy [22]. Consistently, the mammalian target of rapamycin (mTOR) inhibitor rapamycin, a potent activator of autophagy, prevents ventricular hypertrophy induced by thyroid hormone treatment [25] or aortic banding [26], regresses established ventricular hypertrophy induced by pressure overload and improves ventricular function [27]. Autophagy may antagonize ventricular hypertrophy by increasing protein degradation, which decreases ventricular mass. Without basal levels of autophagic activity in the heart, abnormal aggregates of intracellular proteins develop [22].
Accumulation of damaged protein aggregates may contribute to activation of autophagy in the failing heart
The regulation of protein degradation is especially important in long-lived postmitotic cells such as cardiomyocytes and neurons. In Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, which are all characterized by toxic aggregations of misfolded proteins, autophagy is activated as a mechanism to clear misfolded proteins [7]. Increased autophagic activity does not directly clear the aggregates themselves but rather clears their precursors, shifting the equilibrium away from aggregate formation [28]. Various forms of cardiovascular stress cause protein aggregation [29]. Stress-induced protein aggregation may be a trigger of cardiomyocyte autophagy through accumulation of ubiquitinated protein aggregates in the left ventricle and the development of aggresome-like structures [20]. Activation of autophagy at early stages of heart disease is considered to be a protective mechanism to scavenge and eliminate misfolded, polyubiquitinated proteins that escape the overwhelmed proteasome pathway. The accumulation of ubiquitinated aggregates is not attributable to ineffective proteasome-mediated degradation but rather results from an increase in protein damage. In human hearts with cardiomyopathies, protein aggregation and accumulation of ubiquitinated proteins have been reported [30]. In load-induced failing hearts, accumulation of ubiquitinated protein aggregates occurs with the same spatial and temporal pattern as increases in autophagic activity [23]. Because in load-induced heart failure, protein aggregation is only one of the potential inducers of autophagy (besides e.g. reactive oxygen species), the extent of autophagic flux can rise to maladaptive levels [5].
Autophagy may transform compensatory cardiac hypertrophy to pump failure
Increased autophagy in hypertrophied hearts may play a role in the transition from ventricular hypertrophy to ventricular failure. Pressure overload induced by aortic banding evokes heart failure and greatly increases ventricular autophagy. Heterozygous disruption of the gene coding for beclin 1 (Atg6) decreases cardiomyocyte autophagy and diminishes pathological remodeling induced by severe pressure stress. Conversely, beclin 1 overexpression increases autophagic activity and accentuates pathological remodeling (hypertrophy upon pressure overload) [23], indicating that load-induced activation of autophagy is maladaptive. These findings are opposite to the effects observed in mice with Atg5 deletion in cardiomyocytes (vide supra) [22]. Several explanations are conceivable for these dissimilar observations [19]. Basal levels of autophagy may be adaptive and stress-related increases in autophagy maladaptive. Moreover, Atg5 inactivation results in almost complete elimination of basal autophagy, which may lead to different findings than in the beclin 1+/− mice, where autophagic flux is reduced but not eliminated. The loss of important functions of basal autophagy in Atg5-deficient hearts, but not in beclin 1+/− hearts, may generate a different response when the hearts are stressed. It has been reported that the number of apoptotic cells is significantly increased in Atg5-deficient hearts [22], but not in beclin 1+/− mice [23]. Moreover, in wild-type mice subjected to pressure overload, beclin 1 expression increases, suggesting maladaptive autophagic activity, which may be enhanced by beclin 1 overexpression. Furthermore, the loss of Atg5 was cardiac-specific [22], whereas the beclin 1+/− mice [23] had reduced autophagic capacity in all organs, which possibly affects cardiac function. In line with the findings of Zhu et al. [23], myocardial biopsies of patients in the transition stage from hypertrophic cardiomyopathy to heart failure display hypertrophy, myocyte atrophy, and an increased amount of fibrosis. Numerous cardiomyocytes contain ubiquitin positive inclusions and show autophagic vacuoles and myelin structures. These findings indicate that myocardial cell degeneration by autophagy leads to cardiomyocyte death, cell loss, and heart failure [31].
Excessive autophagy induction leads to autophagic cell death and loss of cardiomyocytes
The above-mentioned observations indicate that the level of autophagy in the heart is a crucial factor in determining whether autophagy will be protective or detrimental [24]. High levels of autophagy observed in failing hearts support the view that excessive induction of autophagy underlies autophagic cell death and loss of cardiomyocytes. The incidence of autophagic cardiomyocytes in failing hearts is greater than the incidence of apoptotic cells (0.03–0.3% versus ≤ 0.002%, based on stainings for granular ubiquitin inclusions or TUNEL, respectively) [20, 32, 33]. In a transgenic mouse model in which human diphtheria toxin receptor is expressed in the heart, intramuscular injections of diphtheria toxin evokes heart failure, and the degenerated cardiomyocytes show characteristics indicative of autophagic cell death [34]. Also in humans with heart failure, both dying and dead cardiomyocytes showing characteristics of autophagy have been reported [20, 32, 35]. Therefore, autophagy is suggested to be an important mechanism underlying the cardiomyocyte dropout responsible for the worsening of heart failure [36].
Modulation of autophagy in heart failure
The development of therapies that up-regulate the repair qualities of the autophagic process and down-regulate the cell death aspects would be of great value in the treatment of heart failure [37, 38].
Targeting Sirt1
Efficient maintenance of autophagic degradation has been associated with the extension of the cellular life span. The formation of autophagic vacuoles is regulated by the enzyme Sirt1, a mammalian homolog of Sir2 longevity factor in yeast. The interaction of Sirt1 with the transcription factors FoxO and p53 regulates both autophagic degradation and life span extension [39]. Transgenic mice with cardiac-specific overexpression of Sirt1 demonstrate delayed aging and protection against oxidative stress in the heart [40]. Moreover, the agonistic effect of resveratrol, a stilbene found in red wine, on Sirt1 diminishes cell death in myocytes from failing hearts [41]. Nicotinamide adenine dinucleotide (NAD+) is a substrate for Sirt1. Nicotinamide phosphoribosyltransferase critically regulates NAD+ and ATP contents and plays an essential role in mediating cell survival by inhibiting apoptosis and stimulating autophagic flux in cardiomyocytes. Cardiac expression of nicotinamide phosphoribosyltransferase is significantly decreased by pressure overload, whereas preventing downregulation of this enzyme inhibits myocardial injury [42]. Therefore, nicotinamide phosphoribosyltransferase seems an important protector of the energy status and survival of cardiomyocytes.
Targeting class I or class III phosphatidylinositol 3-kinase
Class I phosphatidylinositol 3-kinase (PI3 k) suppresses autophagy in which mTOR is an effector kinase. As a result, rapamycin is often used to increase autophagy in experimental settings by suppressing the autophagy-inhibitory effect of mTOR, though the mechanism by which mTOR suppresses autophagy is still not completely understood [43, 44]. On the other hand, class III PI3 k activates autophagy. Beclin 1, a mammalian homolog of Atg6, plays an important role in engaging class III PI3 k to positively regulate autophagy in the experimental setting [45]. Beclin 1 also plays a critical role in the upregulation of autophagy in acute myocardial infarction [46].
Mechanical unloading with a left ventricular assist device
Mechanical unloading with a left ventricular assist device decreases the energy demand of the failing human heart and increases survival compared to medical treatment in patients with advanced heart failure. Left ventricular assist device implantation leads to lower cardiac pressure and a reduction in volume overload in the myocardium. These changes are followed by decreased ventricular wall tension and reduced cardiomyocyte hypertrophy [47]. At the cellular level, unloading decreases energy demand and simultaneously activates cardiomyocyte protein synthesis and degradation [48]. By decreasing the energy requirement of the heart, the available supply of substrate is better matched to demand. A recent study showed that in the left ventricular myocardium of patients with idiopathic dilated cardiomyopathy, mechanical unloading significantly decreases mRNA transcript levels of beclin-1, Atg5, and LC3. Protein levels of beclin-1, Atg5-Atg12 conjugate, and LC3-II are also reduced after left ventricular assist device support. In parallel to the decrease in these autophagy markers, a significant increase in 20S proteasome activity was observed with unloading. Thus, mechanical unloading of the failing human heart decreases markers of autophagy, further indicating that autophagy may be a maladaptive mechanism in the failing heart [49].
Pharmacological inhibition of the renin-angiotensin system
Although autophagy has emerged as an important process in the pathogenesis of cardiovascular diseases, the proximal triggers for autophagy are largely unknown. Angiotensin II is an oligopeptide of eight amino acids, formed from its precursor angiotensinogen by a series of two enzymatic cleavages, renin and angiotensin-converting enzyme (ACE), respectively. It is known for many years that angiotensin II plays a central role in the pathogenesis of cardiac hypertrophy and heart failure [50]. There are two subtypes of angiotensin II receptors, designated AT1 and AT2, both of which have a high affinity for angiotensin II [51, 52]. Angiotensin II contributes via the AT1 receptor directly and indirectly (by increasing blood pressure) to cardiomyocyte hypertrophy, interstitial fibrosis, inflammation, oxidative stress, or apoptosis in cardiac pathologies, thereby promoting disease [53]. In contrast, several studies indicate a tissue-protective effect of the AT2 receptor in cardiac disease because of its anti-fibrotic, anti-inflammatory, and anti-apoptotic features. Recently, it was shown that autophagy is promoted by angiotensin via the AT1 receptor but is diminished via the AT2 receptor in neonatal rat cardiomyocytes overexpressing either the AT1 or the AT2 or both receptors after adenoviral transfection [54, 55]. Cardiomyocytes derived from a genetic model of disturbed heart growth in rats (hypertrophic heart rat) were more susceptible to AT1 receptor–induced autophagy, but also showed a strong reduction of autophagic activity via the AT2 receptor. Thus, autophagy in cardiomyocytes is reciprocally regulated by the AT1 receptor (promotes autophagy) and the AT2 receptor (diminishes autophagy). ACE inhibitors block the formation of angiotensin II, thereby decreasing the amount of angiotensin II available to both AT1 and AT2 receptors. However, AT1 receptor antagonists selectively block the binding of angiotensin II to the AT1 receptor but do not affect the AT2 receptor. Therefore, besides their well-known favorable hemodynamical and neurohumoral effects in the treatment of heart failure, AT1 receptor antagonists may also down-regulate excessive autophagic cell death and consequently preserve cardiomyocytes.
Conclusion
Autophagy occurs in the failing human ventricle, and upregulation has also been reported in animal models of pressure overload–induced heart failure. Although the factors that determine whether autophagy will be protective or detrimental are not well known, the level and duration of autophagy seem important. Autophagy may antagonize ventricular hypertrophy by increasing protein degradation, which decreases ventricular mass. However, the rate of autophagy declines with age. The subsequent inability to remove damaged structures results in the progressive accumulation of ‘garbage’, such as lipofuscin. The progress of these changes results in enhanced oxidative stress, decreased ATP production, collapse of the cellular catabolic machinery, and hence, cell death. By contrast, in load-induced heart failure, the extent of autophagic flux can rise to maladaptive levels. Excessive autophagy induction leads to autophagic cell death and loss of cardiomyocytes, responsible for the worsening of heart failure. Accordingly, the development of therapies that up-regulate the repair qualities of the autophagic process and down-regulate the cell death aspects would be of great value in the treatment of heart failure.
References
Heart disease & stroke. Statistics. http://www.americanheart.org/downloadable/heart/1240250946756LS-1982+Heart+and+Stroke+Update.042009.pdf. 2009. American Heart Association. Accessed 1 Oct 2009
De Keulenaer GW, Brutsaert DL (2009) The heart failure spectrum: time for a phenotype-oriented approach. Circulation 119:3044–3046
van Heerebeek L, Borbely A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ (2006) Myocardial structure and function differ in systolic and diastolic heart failure. Circulation 113:1966–1973
Martinet W, Knaapen MW, Kockx MM, De Meyer GRY (2007) Autophagy in cardiovascular disease. Trends Mol Med 13:482–491
Rothermel BA, Hill JA (2008) Autophagy in load-induced heart disease. Circ Res 103:1363–1369
De Meyer GRY, Martinet W (2009) Autophagy in the cardiovascular system. Biochim Biophys Acta 1793:1485–1495
Martinet W, Agostinis P, Vanhoecke B, Dewaele M, De Meyer GRY (2009) Autophagy in disease: a double-edged sword with therapeutic potential. Clin Sci (Lond) 116:697–712
Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A (2005) Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 1:131–140
Brunk UT, Jones CB, Sohal RS (1992) A novel hypothesis of lipofuscinogenesis and cellular aging based on interactions between oxidative stress and autophagocytosis. Mutat Res 275:395–403
Terman A, Brunk UT (2005) Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res 68:355–365
Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT (2010) Mitochondrial turnover and aging of long-lived postmitotic cells. Antioxid Redox Signal 12:503–535
Kurz T, Terman A, Brunk UT (2007) Autophagy, ageing and apoptosis: the role of oxidative stress and lysosomal iron. Arch Biochem Biophys 462:220–230
Rajawat YS, Hilioti Z, Bossis I (2009) Aging: central role for autophagy and the lysosomal degradative system. Ageing Res Rev 8:199–213
Gottlieb RA, Finley KD, Mentzer RM Jr (2009) Cardioprotection requires taking out the trash. Basic Res Cardiol 104:169–180
Gustafsson AB, Gottlieb RA (2009) Autophagy in ischemic heart disease. Circ Res 104:150–158
Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K (2008) The role of autophagy in the heart. Cell Death Differ 16:31–38
Kanamori H, Takemura G, Maruyama R, Goto K, Tsujimoto A, Ogino A, Li L, Kawamura I, Takeyama T, Kawaguchi T, Nagashima K, Fujiwara T, Fujiwara H, Seishima M, Minatoguchi S (2009) Functional significance and morphological characterization of starvation-induced autophagy in the adult heart. Am J Pathol 174:1705–1714
Baehrecke EH (2005) Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 6:505–510
Rothermel BA, Hill JA (2007) Myocyte autophagy in heart disease: friend or foe? Autophagy 3:632–634
Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klovekorn WP, Schaper J (2003) Myocytes die by multiple mechanisms in failing human hearts. Circ Res 92:715–724
Hein S, Arnon E, Kostin S, Schonburg M, Elsasser A, Polyakova V, Bauer EP, Klovekorn WP, Schaper J (2003) Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation 107:984–991
Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13:619–624
Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA (2007) Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 117:1782–1793
Gustafsson AB, Gottlieb RA (2008) Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol 44:654–661
Kuzman JA, O’Connell TD, Gerdes AM (2007) Rapamycin prevents thyroid hormone-induced cardiac hypertrophy. Endocrinology 148:3477–3484
Ha T, Li Y, Gao X, McMullen JR, Shioi T, Izumo S, Kelley JL, Zhao A, Haddad GE, Williams DL, Browder IW, Kao RL, Li C (2005) Attenuation of cardiac hypertrophy by inhibiting both mTOR and NFkappaB activation in vivo. Free Radic Biol Med 39:1570–1580
McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S (2004) Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 109:3050–3055
Rubinsztein DC (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780–786
Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, Miller FJ Jr, Rothermel BA, Hill JA (2008) Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation 117:3070–3078
Weekes J, Morrison K, Mullen A, Wait R, Barton P, Dunn MJ (2003) Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 3:208–216
Fidzianska A, Bilinska ZT, Walczak E, Witkowski A, Chojnowska L (2010) Autophagy in transition from hypertrophic cardiomyopathy to heart failure. J Electron Microsc (Tokyo) in press doi:10.1093/jmicro/dfp048
Knaapen MW, Davies MJ, De BM, Haven AJ, Martinet W, Kockx MM (2001) Apoptotic versus autophagic cell death in heart failure. Cardiovasc Res 51:304–312
Elsasser A, Vogt AM, Nef H, Kostin S, Mollmann H, Skwara W, Bode C, Hamm C, Schaper J (2004) Human hibernating myocardium is jeopardized by apoptotic and autophagic cell death. J Am Coll Cardiol 43:2191–2199
Akazawa H, Komazaki S, Shimomura H, Terasaki F, Zou Y, Takano H, Nagai T, Komuro I (2004) Diphtheria toxin-induced autophagic cardiomyocyte death plays a pathogenic role in mouse model of heart failure. J Biol Chem 279:41095–41103
Shimomura H, Terasaki F, Hayashi T, Kitaura Y, Isomura T, Suma H (2001) Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. Jpn Circ J 65:965–968
Takemura G, Miyata S, Kawase Y, Okada H, Maruyama R, Fujiwara H (2006) Autophagic degeneration and death of cardiomyocytes in heart failure. Autophagy 2:212–214
Dhesi P, Tehrani F, Fuess J, Schwarz ER (2010) How does the heart (not) die? The role of autophagy in cardiomyocyte homeostasis and cell death. Heart Fail Rev 15:15–21
Martinet W, De Meyer GRY (2009) Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res 104:304–317
Salminen A, Kaarniranta K (2009) SIRT1: regulation of longevity via autophagy. Cell Signal 21:1356–1360
Hsu CP, Odewale I, Alcendor RR, Sadoshima J (2008) Sirt1 protects the heart from aging and stress. Biol Chem 389:221–231
Opie LH, Lecour S (2007) The red wine hypothesis: from concepts to protective signalling molecules. Eur Heart J 28:1683–1693
Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J (2009) Nicotinamide phosphoribosyltransferase regulates cell survival through NAD + synthesis in cardiac myocytes. Circ Res 105:481–491
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075
Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306:990–995
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939
Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K (2009) The role of autophagy in the heart. Autophagy 16:31–38
Razeghi P, Myers TJ, Frazier OH, Taegtmeyer H (2002) Reverse remodeling of the failing human heart with mechanical unloading. Emerging concepts and unanswered questions. Cardiology 98:167–174
Razeghi P, Sharma S, Ying J, Li YP, Stepkowski S, Reid MB, Taegtmeyer H (2003) Atrophic remodeling of the heart in vivo simultaneously activates pathways of protein synthesis and degradation. Circulation 108:2536–2541
Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R, Frazier OH, Taegtmeyer H (2009) Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation 120:S191–S197
de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T (2000) International union of pharmacology XXIII. The angiotensin II receptors. Pharmacol Rev 52:415–472
Timmermans PB, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini DJ, Lee RJ, Wexler RR, Saye JA, Smith RD (1993) Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev 45:205–251
Griendling KK, Lassegue B, Alexander RW (1996) Angiotensin receptors and their therapeutic implications. Annu Rev Pharmacol Toxicol 36:281–306
Unger T, Li J (2004) The role of the renin-angiotensin-aldosterone system in heart failure. J Renin Angiotensin Aldosterone Syst 5(Suppl 1):S7–S10
Porrello ER, D’Amore A, Curl CL, Allen AM, Harrap SB, Thomas WG, Delbridge LM (2009) Angiotensin II type 2 receptor antagonizes angiotensin II type 1 receptor-mediated cardiomyocyte autophagy. Hypertension 53:1032–1040
Steckelings UM, Unger T (2009) Angiotensin receptors and autophagy: live and let die. Hypertension 53:898–899
Acknowledgments
The authors thank Liliane Van Den Eynde for secretarial assistance. The authors’ research that is cited in this review was supported by the Fund for Scientific Research (FWO)-Flanders (Belgium) (Projects G.0308.04, G.0113.06 and G.0112.08), the University of Antwerp (NOI-BOF and TOP-BOF), and the Bekales Foundation. W. Martinet is a postdoctoral fellow of the FWO-Flanders.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
De Meyer, G.R.Y., De Keulenaer, G.W. & Martinet, W. Role of autophagy in heart failure associated with aging. Heart Fail Rev 15, 423–430 (2010). https://doi.org/10.1007/s10741-010-9166-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-010-9166-6