
Abstract
Purpose
Papillary glioneuronal tumors (PGNT) have been recently included as a distinct entity in the WHO classification of tumors of the central nervous system. Their molecular pathogenesis is not clear. In the current study, we present the morphological, immunohistochemical, and molecular features of four cases of PGNT reported over the past 11 years.
Methods
Over a period of 11 years (January 2000–February 2010), there were four cases of PGNT, which were reviewed for histomorphological features. TP53 and IDH1 mutations were assessed using antibodies against p53 protein and for mutant IDH1R132H protein, respectively. Immunohistochemistry was also performed for epidermal growth factor receptor (EGFR) protein. Fluorescence in situ hybridization assay was used for analyzing 1p/19q deletion status.
Results
All the tumors showed the characteristic biphasic morphology. Rare findings included minigemistocyte-like cells in one, angiomatous areas in three, focal necrosis in one, and a high MIB-1 labeling index of 12 and 13 %, respectively, in two of the cases. All lacked EGFR, IDH1 expression, and 1p/19q deletions. Interestingly, antibody for p53 labeled the tumor cells, mainly those showing glial differentiation, in two cases. At a mean follow-up of 30 months, there was no evidence of disease progression except in one case which recurred after 24 months.
Conclusion
PGNT are rare CNS neoplasms. Despite showing focal morphological features reminiscent of oligodendroglial tumors and presence of astrocytic component, they usually lack the common genetic alterations involved in the pathogenesis of gliomas. Multi-institutional pooling of cases may aid in elucidating their oncogenetic pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mixed neuronal–glioneuronal neoplasms are a group of slow-growing, indolent tumors, usually involving the temporal lobes and presenting with recurrent seizures. This group includes dysplastic gangliocytoma of the cerebellum, gangliocytoma, ganglioglioma, anaplastic ganglioglioma, desmoplastic infantile astrocytoma/ganglioglioma, dysembryoplastic neuroepithelial tumor, central neurocytoma, extraventricular neurocytoma, cerebellar liponeurocytoma, rosette-forming glioneuronal tumor of the fourth ventricle, paraganglioma, and the recently included papillary glioneuronal tumor (PGNT) [1]. PGNT was initially considered a variant of ganglioglioma as per the 2000 WHO classification [2]. In the world literature, morphologically similar tumors have been described as early as 1996 and 1997 and have been referred to as “pseudopapillary ganglioneurocytoma” and “pseudopapillary neurocytoma with glial differentiation,” respectively [3, 4]. Komori et al. were the first to introduce PGNT as a distinct clinicopathologic entity in 1998, and that was the first and largest series of nine cases to date, describing the clinicopathologic features of this neoplasm [5]. To date, 60 cases of PGNT are reported in the literature mostly as case reports [6–42].
Molecular analysis of these tumors has been performed in a limited number of cases [16, 31, 42]. In a previous study, fluorescent in situ hybridization (FISH) assay performed on six cases of PGNT did not reveal 1p deletion [16]. Faria et al., in their genetic study on a single case of PGNT, demonstrated gains and structural alterations involving chromosome 7, with a high-level amplification region at 7p14-q12. No epidermal growth factor receptor (EGFR) amplification was found [31]. Recently, Myung JK et al. reported on the molecular analysis of four cases of PGNT and found no expression of p53, EGFR, and isocitrate dehydrogenase 1 (IDH1); however, PTEN loss and MGMT methylation were observed in two and three cases, respectively[42].
In the present study, morphological, immunohistochemical, and molecular features of four cases of PGNT diagnosed in our department are presented, along with a brief review of literature.
Material and methods
All the cases of PGNT diagnosed during a period of 11 years (2000 to 2010) in the Department of Pathology, All India Institute of Medical Sciences, one of the largest tertiary-care centers in India, were retrieved from the archives of the department. The tumor tissue had been fixed in 10 % neutral buffered formalin, routinely processed and paraffin embedded. Hematoxylin and eosin (H&E)-stained sections were reviewed by three independent neuropathologists (SA, MCS, and CS), and consensus diagnoses were made as per the 2007 WHO classification [1].
Immunohistochemistry
Immunohistochemical studies were performed on 5-μ-thick formalin-fixed, paraffin-embedded tumor sections using antibodies directed against synaptophysin (NeoMarker; dilution, 1:100), Neu-N (Chemicon; dil, 1:200), glial fibrillary acidic protein (GFAP) (Dako Denmark; dil, 1:1,000), vimentin (Novocastra; dil, 1:100), S-100 (Dako Denmark; dil, 1:1,500), epithelial membrane antigen(EMA) (Dako Denmark; dil, 1:100), MIB-1 (Dako Denmark; dil, 1:200), p53 (DO-1 monoclonal antibody; dil, 1:100; M/s Santa Cruz, USA), EGFR (Dako Denmark; dil, 1:100), and IDH1 (Dianova; dil, 1:100). A labeled streptavidin biotin kit (Universal) was used as a detection system (Dako, Denmark). Antigen retrieval, when required, was performed in a microwave oven. The MIB-1 labeling index (LI) was calculated in the highest proliferating area as the percentage of labeled nuclei per 1,000 cells.
1p and 19q deletions
Dual-probe FISH assay was performed on paraffin-embedded sections, with locus-specific probes for 1p36 and 19q13 paired, respectively, with the reference probes for 1q25 and 19p13 (Vysis, Downers Grove, IL). The sections were deparaffinized by immersing in xylene thrice for 10 min each followed by two 3-min immersions in 100 % ethanol. Following rinsing in water, target retrieval was achieved using citrate buffer, pH 6.0, and boiling in microwave for 20 min. Sections were digested using 0.04 % pepsin (P-7000; Sigma-Aldrich, St. Louis, MO) for 30 min at 37 °C. The probe mixture (10 μl per slide) was then applied, followed by simultaneous probe/specimen denaturation at 73 °C for 5 min with subsequent overnight incubation at 37 °C, performed in a Thermobrite TM hybridization chamber (Vysis Inc). The sections were washed the next day in 2× SSC (2 min at 73 °C) followed by 0.5× SSC (2 min at room temp) and counterstained with 4,6-diamidino-2-phenylindole (Vysis) and visualized under a fluorescent microscope. Signals were scored in at least 200 non-overlapping, intact nuclei. Sections from non-neoplastic cortical tissue obtained from epilepsy surgery specimens were used as control. At least 50 % or more nuclei had to show one signal to be scored as a deletion [42].
Results
Over a period of 11 years, 21,616 intracranial tumors were diagnosed in the department of pathology, of which four were PGNT comprising 1.85 % of all intracranial tumors. Case 4 was published earlier and was described in details [27].
Clinical and radiological features
The clinical and radiological details of all the four patients are shown in Table 1. The patients' age ranged from 4 to 26 years with a mean age of 13 years. There were three males and one female. All patients presented with symptoms and signs of raised intracranial tension.
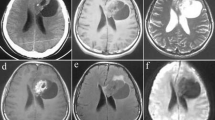
Radiology was available in three cases (Figs. 1 and 2). Cases 1 and 3 showed solid and cystic tumor while case 4 was completely cystic with a mural nodule. Post-gadolinium scans revealed enhancement of the cyst wall and of the solid component.

Photomicrographs showing radiological features. Axial T1-weighted (a) and T2-weighted (b) images of case 1 showing a juxtaventricular mass involving both cerebral hemispheres and crossing the splenium of the corpus callosum. The mass is predominantly cystic with a small solid component. No perilesional edema is seen. Post-gadolinium axial (c) and coronal (d) T1WI show dense enhancement of the solid portion and wall of the cystic portion

Contrast enhanced axial (a) and sagittal (b) T1-weighted images showing a solid-cystic juxtaventricular mass lesion reaching the pial surface with minimal perilesional edema (case 3). Axial T2-weighted image (c) of case 4 showing a predominantly cystic mass in the right frontal lobe deep to white matter with mild edema. On contrast administration, the axial T1-weighted image (d) showed subtle enhancement of cyst wall
The patients were subjected to near total excision in cases 1 and 3, and subtotal in case 4, but the extent of resection was not known in case 2. Follow-up, available in three patients, showed no evidence of disease progression or metastasis in two patients at a mean follow-up period of 30 months (ranging from 12 to 48 months), however in one patient tumor recurrence was seen after 24 months. Even in case 4, the patient had residual tumor and was stable at 48 months postoperatively.
Pathological findings
All the cases showed similar features with subtle differences (Figs. 3, 4, and 5). Two distinct zones of glial and neuronal differentiation were seen which were better highlighted on immunohistochemistry (Figs. 3 and 4). Morphologically, this zonation could be identified on H&E-stained sections in two of the four cases (cases 1 and 2). Case 1 showed a predominant papillary pattern characterized by single to multiple layers of small flattened to cuboidal cells lining fibrovascular cores. The tumor cells had scant amount of cytoplasm and small round to oval nuclei having fine dispersed chromatin. Most of the fibrovascular cores had blood vessels with a narrow lumen and markedly thickened, hyalinized walls. In addition, sheets of small cells with clear cytoplasm, mimicking oligodendroglial cells, were noted in between the pseudopapillary structures. Within the latter area, small islands of neuropil were also identified. Focally, the tumor cells were arranged around a central core of neuropil, forming rosettes. Case 2 showed sheets of non-descript cells having round to oval nuclei with fine granular chromatin, inconspicuous nucleoli, and a moderate amount of cytoplasm with indistinct cell margins. These sheets of cells were interspersed with pale areas, easily discernible even at scanning magnification. In these pale zones, the cells were more loosely dispersed and had clear cytoplasm with indistinct borders. In this case, papillary gliovascular structures were a rare finding and had to be searched for. Cases 3 and 4 showed predominantly pseudopapillary structures lined by multiple layers of cells, and morphologically, no distinction could be made as to the glial or neuronal nature of the tumor cells. Case 3 showed an additional interesting feature in the form of minigemistocyte-like cells lining the hyalinized blood vessels, as well as in the interpapillary areas. These cells had moderate amount of dense eosinophilic cytoplasm with an eccentrically placed nucleus. Ganglioid cells were not identifiable in any of the four cases. Prominent secondary changes including evidence of old hemorrhage in the form of hemosiderin deposition, fibrin exudation, and hyalinization of large blood vessels were present in all four cases. Additionally, there was vascular proliferation without any evidence of endothelial hyperplasia, resulting in formation of small “angioma”-like areas in three of the cases. A rim of foamy macrophages around most of the blood vessels was seen in case 3. Calcification was noted in case 1 only. Only case 4 showed occasional mitosis. There was focal necrosis in one case, but endothelial hyperplasia was absent in all cases.

Photomicrographs showing tumor cells arranged around hyalinized blood vessels forming pseudopapillary structures; at places, there is multilayering of these cells (case 1) (a and b, H&E, ×100 and 400, respectively); some papilla are lined by minigemistocytes (case 3) (c, H&E, ×200); sheets of tumor cells punctuated by islands of loosely arranged cells having clear cytoplasm (case 2) (d and e, H&E, ×200 each); foamy macrophages around blood vessels in an angiomatous area are seen (case 3) (f, H&E, ×200)

Photomicrographs showing tumor cells lining the blood vessels (case 1) showing immunopositivity for GFAP (a, ×200); GFAP staining the sheets of glial cells in case 2 with the neuronal cell islands being immunonegative (b, ×400); synaptophysin and Neu-N positivity (case 2; c and d, ×400) in the GFAP-negative islands

Vimentin immunopositivity in the GFAP-positive areas and negativity in the neuronal areas (case 2, a, ×200); Focal positivity for chromogranin in the neuronal areas (case 2, b, ×400); MIB-1 in case 1 (c, ×400); p53 immunoreactivity more so in the glial cells than in the neuronal cells in case 2 (d, ×400)
Immunohistochemically, GFAP labeled the tumor cells lining the vascular structures in cases 1, 3, and 4. Synaptophysin and Neu-N were positive in the interpapillary zone in case 1. In the latter two cases, the outer layers of the cells lining the vascular cores were synaptophysin positive but negative for Neu-N. Case 2 was characterized by strong GFAP immunoreactivity in the sheets of tumor cells, but the interspersed pale zones were negative, which, in contrast, were immunopositive for synaptophysin and Neu-N. S-100 was focally positive in all the cases. All cases were immunonegative for EMA. The MIB-1 labeling index ranged from 1 to 13 % (Table 2).
None of the cases was immunopositive for IDH1 and EGFR. p53 protein expression was observed in three cases with 25–30 % of cells showing weak to moderate staining. This immunoreactivity was mainly localized to the glial component, and only an occasional cell in the neuronal zone was reactive with the p53 antibody. 1p and 19q deletions were not found in any case (Table 2).
Discussion
Papillary glioneuronal tumor is a distinct entity recently included in the “Neuronal and mixed neuronal-glial” group of CNS tumors under the 2007 WHO classification [1]. These are slow-growing indolent tumors and involve patients over a wide age range varying from 4 to 75 years [41, 42]. A recent review of literature showed no sex predilection, and males and females are affected equally [42]. The mean age of presentation was 23.6 years, and 78.3 % of patients were younger than 30 years of age [42]. In the present series, patients' ages ranged from 4 to 26 years, with a mean age of 13 years. The male predominance and younger mean age of presentation in the present study could be attributed to the small number of cases included. The temporal lobe is the most common site of involvement.
Histologically, PGNT showed stereotypic morphology comprising of a biphasic pattern, showing evidence of both glial and neuronal differentiation. The proportion of these two components varies from case to case. The glial cells usually line vascular structures, forming pseudopapillae, and the neuronal component is present as sheets of cells distributed in the interpapillary zone. Focal oligodendroglial-like areas, ganglioid cells, and immature neurons with accompanying neuropil may also be identified. Though oligodendroglia-like cells were identifiable, ganglioid differentiation was not seen in any of the cases under present study. Case 1 demonstrated the classical histomorphological features consisting of prominent gliovascular structures along with interpapillary sheets of cells showing neuronal differentiation on morphology and immunohistochemistry. Case 2 showed islands of cells with clear cytoplasm interspersed within sheets of glial cells, with the former group of cells being immunopositive for neuronal antigens. Pseudopapillae were only focally present. Cases 3 and 4 showed predominantly papillary structures lined by stratified layers of cells, with scanty solid interpapillary sheets of cells. The inner layer of lining cells was found to be immunopositive for GFAP and the outer layer for synaptophysin and Neu-N.
Degenerative changes are commonly seen in these tumors [5, 13, 14, 20–22]. In the present series, prominent secondary changes including foamy macrophages, vascular hyalinization, fibrin exudation, hemosiderin deposition, and calcification were noted in all the cases. Interestingly, angiomatous areas, which have been described in PGNT only once in the English literature [29], were also found in as many as three of the cases and thus do not appear to be a rare phenomenon. The presence of minigemistocytes in PGNT is a rare finding [16, 22] and found in one case in the present series. Tanaka et al., in their study of six cases of PGNT, demonstrated the presence of a third distinct glial and neuronal marker-negative component, which was immunopositive for Olig2, which is a transcription factor specific for oligodendrocytes, suggesting oligodendroglial participation in these tumors. They also found minigemistocytes in three of their cases, distributed in close proximity to the Olig2-positive cells. However, none of their cases showed 1p deletion by fluorescence in situ hybridization assay [16]. In the current series also, 1p/19q deletions were not found in any of the cases, thereby ruling out the possibility of a role of these molecular alterations in the histogenesis of PGNT.
PGNT are indolent tumors (WHO grade I). Most patients fare well with the longest recurrence-free survival recorded being 19 years [21]. Morphologically, these tumors usually lack features suggestive of aggressive behavior. Most cases show absence of significant mitotic activity, endothelial proliferation, and necrosis, and have low MIB-1 labeling indices in the range of 1–2 % [5]. Rare cases with high proliferative indices, ranging from 10 to 26 %, have been reported in the literature [22, 26, 27, 30, 34, 37, 42]. In the current series, three cases (cases 1, 2, and 4) harbored a raised MIB-1 labeling index of 5, 13, and 12 %, respectively, in the maximum proliferative area. However, at follow-up, there was evidence of recurrence of disease in case 1 which had low MIB1LI. Interestingly, case 3, which showed prominent minigemistocytes, both lining the pseudopapillae as well as focally in the interpapillary zones, had a low MIB-1 labeling index (<1 %). This is in contrast to the case reported by Ishizawa et al. [22], in which the increased labeling index (10 %) was found in the minigemistocytic component and the aggressive nature of the tumor was attributed to malignant transformation of a coexisting oligodendroglioma having minigemistocytic component. Tanaka et al. also reported three such cases of papillary glioneuronal tumor with a minigemistocytic component, but they did not mention their proliferative indices [16]. Thus, the presence of minigemistocytes is a rare finding in these tumors, and whether it is a determinant of oligodendroglial differentiation and/or aggressive behavior remains to be evaluated.
Histogenesis of this tumor is not exactly known. One hypothesis is that this tumor appears to originate from neuroepithelial stem cells with potential for biphenotypic differentiation. Govindan et al., using confocal microscopy, demonstrated colocalization of glial and neuronal markers, along with expression of the stem cell markers Nestin and CD 133 in these tumors [36]. There are only limited number of cytogenetic studies on mixed glioneuronal neoplasms, with gangliogliomas and desmoplastic gangliogliomas being the most common to be evaluated [43]. Interestingly, most studies have found recurrent chromosome 7 alterations in these tumors. Faria C et al., in a single case of PGNT, had shown gains and structural aberrations involving chromosome 7, with breakpoints at 7p22 by using conventional cytogenetics, FISH, and comparative genomic hybridization. Both glial and neuronal linked genes (RAC1 and NPXH1) are located in this region. Using FISH analysis, they further demonstrated that, unlike primary glioblastomas, EGFR is not amplified in this tumor [31]. Myung JK et al. demonstrated gains of 2q, 3p, 6p, 7q, 10q, 16q, 19p, and 22q and loss of 1q, 6p, 8p, 9p, 9q, 16q in two cases by comparative genomic hybridization, but none of them showed EGFR protein expression by immunohistochemistry or gene amplification by FISH [42]. A similar lack of expression of EGFR protein was observed in all cases under study.
TP53 gene mutations are commonly implicated in the pathogenesis WHO grade II and III astrocytic tumors, as well as with secondary glioblastomas, but the role of this gene in the pathogenesis of PGNT is not known. There are two studies which included a single case and four cases respectively of papillary glioneuronal tumor in which p53 protein expression was analyzed by immunohistochemistry and was found to be negative [7, 42]. TP53 mutations are not found in the other “neuronal and mixed neuronal-glial tumors,” except cerebellar liponeurocytoma [44]. In the present study, two of the PGNT cases showed p53 reactivity in 20–25 % of the cells. Interestingly, the immunoreactivity for p53 protein was seen predominantly in the glial component, and the cells showing neuronal differentiation were largely negative. The significance of this is not known, and mutation analysis is required to establish the exact role of this gene in the pathogenesis of PGNT.
Recently, mutations involving IDH1 and IDH2 genes have been found to occur in various glial tumors, including WHO grade II and III astrocytomas and oligodendrogliomas, as well as in secondary glioblastomas (WHO grade IV), at a rate varying from 60 to 80 % [45–49]. Immunohistochemistry using an antibody against the mutant Arg132His IDH1 protein has been reported to aid in differentiating neoplastic from reactive conditions [50, 51]. These mutations appear to impart an improved outcome [45, 47, 52, 53]. Mixed neuronal–glioneural tumors have not been studied extensively for IDH mutations. Of the 75 gliomas and 57 non-neoplastic mimickers of gliomas studied by Horbinski et al., IDH1 mutations were detected in 2 of 12 gangliogliomas [46]. In a study from our department, none of six cases of extraventricular neurocytoma was found to be positive for mutant IDH1R132H protein [54]. There are no studies on the role of IDH in PGNT. We performed immunohistochemistry using an antibody specifically detecting mutant IDH1R132H protein in four cases of PGNT, and none of them showed immunopositivity for the same, implying absence of a role of IDH1 mutations in the pathogenesis of this group of tumors, unlike in oligodendrogliomas and astrocytomas. Similar findings have been reported recently by Myung LK et al. [42]. However, the antibody used in the study does not identify rarer mutations involving IDH1 (R132C and R132G) and IDH2; thus, their presence cannot be completely ruled out.
To conclude, though morphologically the features of PGNT are now well defined, the genetic pathways involved in their oncogenesis remain ambiguous. This study, using immunohistochemical techniques, reiterates the lack of the common “glioma-associated” mutations in this tumor. Due to their rarity, a multi-institutional pooling of cases is required for more insight into the pathogenesis of these tumors.
References
Nakazato Y, Figarella-Branger D, Becker AJ, Scheithauer BW, Rosenblum MK (2007) Papillary glioneuronal tumor. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) WHO classification of tumours of the central nervous system, 4th edn. IARC Press, Lyon, pp 113–114
Nelson JS, Bruner JM, Weistler OD, VandenBerg SR (2000) Ganglioglioma and gangliocytoma. In: Kleihues P, Cavenee WK (eds) World Health Organization classification of tumours. Pathology and genetic of tumours of the nervous system, 3rd edn. IARC Press, Lyon, p 97
Komori T, Scheithauer BW, Anthony DC, Scott RM, Okazaki H, Kobayashi M (1996) Pseudopapillary ganglioneurocytoma. J Neuropathol Exp Neurol 55:654
Kim DH, Suh YL (1997) Pseudopapillary neurocytoma of temporal lobe with glial differentiation. Acta Neuropathol 94:187–191
Komori T, Scheithauer BW, Anthony DC, Rosenblum MK, McLendon RE, Scott RM, Okazaki H, Kobayashi M (1998) Papillary glioneuronal tumor: a new variant of mixed neuronal-glial neoplasm. Am J Surg Pathol 22:1171–1183
Bouvier-Labit C, Daniel L, Dufour H, Grisoli F, Figarella-Branger D (2000) Papillary glioneuronal tumour: clinicopathological and biochemical study of one case with 7-year follow up. Acta Neuropathol 99:321–326
Prayson RA (2000) Papillary glioneuronal tumor. Arch Pathol Lab Med 124:1820–1823
Barnes NP, Pollock JR, Harding B, Hayward RD (2002) Papillary glioneuronal tumour in a 4-year-old. Pediatr Neurosurg 36:266–270
Broholm H, Madsen FF, Wagner AA, Laursen H (2002) Papillary glioneuronal tumor—a new tumor entity. Clin Neuropathol 21:1–4
Tsukayama C, Arakawa Y (2002) A papillary glioneuronal tumor arising in an elderly woman: a case report. Brain Tumor Pathol 19:35–39
Ebato M, Tsunoda A, Maruki C, Ikeya F, Okada M (2003) Papillary glioneuronal tumor with highly degenerative pseudopapillary structure accompanied by specific abortive glial cells: a case report. No Shinkei Geka 31:1185–1190
Kordek R, Hennig R, Jacobsen E, Kearney M (2003) Papillary glioneuronal tumor—a new variant of benign mixed brain neoplasm. Pol J Pathol 54:75–78
Lamszus K, Makrigeorgi-Butera M, Laas R, Westphal M, Stavrou D (2003) September 2002: 24-year-old female with a 6-month history of seizures. Brain Pathol 13:115–117
Borges G, Bonilha L, Menezes AS, Queiroz Lde S, Carelli EF, Zanardi V, Menezes JR (2004) Long term follow-up in a patient with papillary glioneuronal tumor. Arq Neuropsiquiatr 62:869–872
Stosic-Opincal T, Peric V, Gavrilovic S, Gavrilov M, Markovic Z, Sener RN (2005) Papillary glioneuronal tumor. AJR Am J Roentgenol 185:265–267
Tanaka Y, Yokoo H, Komori T, Makita Y, Ishizawa T, Hirose T, Ebato M, Shibahara J, Tsukayama C, Shibuya M, Nakazato Y (2005) A distinct pattern of Olig2-positive cellular distribution in papillary glioneuronal tumors: a manifestation of the oligodendroglial phenotype? Acta Neuropathol 110:39–47
Chen L, Piao YS, Xu QZ, Yang XP, Yang H, Lu DH (2006) Papillary glioneuronal tumor: a clinicopathological and immunohistochemical study of two cases. Neuropathology 26:243–248
Buccoliero AM, Giordano F, Mussa F, Taddei A, Genitori L, Taddei GL (2006) Papillary glioneuronal tumor radiologically mimicking a cavernous hemangioma with hemorrhagic onset. Neuropathology 26:206–211
Celli P, Caroli E, Giangaspero F, Ferrante L (2006) Papillary glioneuronal tumor. Case report and literature review. J Neurooncol 80:185–189
Dim DC, Lingamfelter DC, Taboada EM, Fiorella RM (2006) Papillary glioneuronal tumor: a case report and review of the literature. Hum Pathol 37:914–918
Epelbaum S, Kujas M, Van Effenterre R, Poirier J (2006) Two cases of papillary glioneuronal tumours. Br J Neurosurg 20:90–93
Ishizawa T, Komori T, Shibahara J, Ishizawa K, Adachi J, Nishikawa R, Matsutani M, Hirose T (2006) Papillary glioneuronal tumor with minigemistocytic components and increased proliferative activity. Hum Pathol 37:627–630
Konya D, Peker S, Ozgen S, Kurtkaya O, Necmettin Pamir M (2006) Superficial siderosis due to papillary glioneuronal tumor. J Clin Neurosci 13:950–952
Qi JP, Zhu H, Li DY, Mei HL (2006) Papillary glioneuronal tumor: report of a case. Zhonghua Bing Li Xue Za Zhi 35:764–765, Chinese
Vajtai I, Kappeler A, Lukes A, Arnold M, Lüthy AR, Leibundgut K (2006) Papillary glioneuronal tumor. Pathol Res Pract 202:107–112
Adam C, Polivka M, Carpentier A, George B, Gray F (2007) Papillary glioneuronal tumor: not always a benign tumor? Clin Neuropathol 26:119–124
Atri S, Sharma MC, Sarkar C, Garg A, Suri A (2007) Papillary glioneuronal tumour: a report of a rare case and review of literature. Childs Nerv Syst 23:349–353
Gelpi E, Preusser M, Czech T, Slavc I, Prayer D, Budka H (2007) Papillary glioneuronal tumor. Neuropathology 27:468–473
Radotra BD, Kumar Y, Bhatia A, Mohindra S (2007) Papillary glioneuronal tumor: a new entity awaiting inclusion in WHO classification. Diagn Pathol 2:6–10
Vaquero J, Coca S (2007) Atypical papillary glioneuronal tumor. J Neurooncol 83:319–323
Faria C, Miguéns J, Antunes JL, Barroso C, Pimentel J, Martins Mdo C, Moura-Nunes V, Roque L (2008) Genetic alterations in a papillary glioneuronal tumor. J Neurosurg Pediatr 1:99–102
Guo SP, Zhang F, Li QL, Li Q, Wang WL, Li FF (2008) Papillary glioneuronal tumor—contribution to a new tumor entity and literature review. Clin Neuropathol 27:72–77
Izycka-Swieszewska E, Majewska H, Szurowska E, Mazurkiewicz-Bełdzińska M, Drozyńska E (2008) Papillary glioneuronal tumour of the precentral gyrus. Folia Neuropathol 46:158–163
Newton HB, Dalton J, Ray-Chaudhury A, Gahbauer R, McGregor J (2008) Aggressive papillary glioneuronal tumor: case report and literature review. Clin Neuropathol 27:317–324
Williams SR, Joos BW, Parker JC, Parker JR (2008) Papillary glioneuronal tumor: a case report and review of the literature. Ann Clin Lab Sci 38:287–292
Govindan A, Mahadevan A, Bhat DI, Arivazhagan A, Chakraborti S, Suja MS, Phalguni AA, Sampath S, Chandramouli BA, Shankar SK (2009) Papillary glioneuronal tumor-evidence of stem cell origin with biphenotypic differentiation. J Neurooncol 95:71–80
Javahery RJ, Davidson L, Fangusaro J, Finlay JL, Gonzalez-Gomez I, McComb JG (2009) Aggressive variant of a papillary glioneuronal tumor. Report of 2 cases. J Neurosurg Pediatr 3:46–52
Pimentel J, Barroso C, Miguéns J, Firmo C, Antunes JL (2009) Papillary glioneuronal tumor—prognostic value of the extension of surgical resection. Clin Neuropathol 28:287–294
Mahajan H, Varikatt W, Dexter M, Boadle R, Ng T (2010) Papillary glioneuronal tumor of the frontal lobe. J Clin Neurosci 17:534–536
Phi JH, Park SH, Chae JH, Wang KC, Cho BK, Kim SK (2010) Papillary glioneuronal tumor present in a patient with encephalocraniocutaneous lipomatosis: case report. Neurosurgery 67:E1165–E1169
Xiao H, Ma L, Lou X, Gui Q (2011) Papillary glioneuronal tumor: radiological evidence of a newly established tumor entity. J Neuroimaging 21:297–302
Myung JK, Byeon SJ, Kim B, Suh J, Kim SK, Park CK, Chung CK, Chang KH, Park SH (2011) Papillary glioneuronal tumors: a review of clinicopathologic and molecular genetic studies. Am J Surg Pathol 35:1794–1805
Squire JA, Arab S, Marrano P, Bayani J, Karaskova J, Taylor M, Becker L, Rutka J, Zielenska M (2001) Molecular cytogenetic analysis of glial tumors using spectral karyotyping and comparative genomic hybridization. Mol Diagn 6:93–108
Horstmann S, Perry A, Reifenberger G, Giangaspero F, Huang H, Hara A, Masuoka J, Rainov NG, Bergmann M, Heppner FL, Brandner S, Chimelli L, Montagna N, Jackson T, Davis DG, Markesbery WR, Ellison DW, Weller RO, Taddei GL, Conti R, Del Bigio MR, González-Cámpora R, Radhakrishnan VV, Söylemezoglu F, Uro-Coste E, Qian J, Kleihues P, Ohgaki H (2004) Genetic and expression profiles of cerebellar liponeurocytomas. Brain Pathol 14:281–289
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN (2009) Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol 68:1319–1325
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, Weller M, Herold-Mende C, Unterberg A, Jeuken JW, Wesseling P, Reifenberger G, von Deimling A (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118:469–474
Ichimura K, Pearson DM, Kocialkowski S, Bäcklund LM, Chan R, Jones DT, Collins VP (2009) IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol 11:341–347
Capper D, Zentgraf H, Balss J, Hartmann C, von Deimling A (2009) Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol 118:599–601
Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL (2010) Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol 119:509–511
Sanson M, Marie Y, Paris S et al (2009) Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27:4150–4154
Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, Tigchelaar W, Troost D, Vandertop WP, Bardelli A, Van Noorden CJ (2010) The prognostic IDH1 (R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol 119:487–494
Agarwal S, Sharma MC, Sarkar C, Suri V, Jain A, Sharma MS, Ailawadhi P, Garg A, Mallick S (2011) Extraventricular neurocytomas: a morphological and histogenetic consideration. A study of six cases. Pathology 43:327–334
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Agarwal, S., Sharma, M.C., Singh, G. et al. Papillary glioneuronal tumor—a rare entity: report of four cases and brief review of literature. Childs Nerv Syst 28, 1897–1904 (2012). https://doi.org/10.1007/s00381-012-1860-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-012-1860-3