
Abstract
Papillary glioneuronal tumors are newly recognized seizure producing tumors. We report two such cases with immunohistochemical characterization of glial and neuronal components and briefly review literature. Co-localization of glial and neuronal markers was demonstrable on confocal microscopy with expression of stem cell markers (Nestin and CD133) suggesting possible origin from neuroepithelial stem cell with biphenotypic differentiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the past decade, several morphologically distinctive neoplasms have been added to the category of mixed glioneuronal tumors. The papillary glioneuronal tumour (PGNT), earlier listed as a variant of ganglioglioma in the WHO 2000 classification, was included as a distinct neurooncological entity, in the 2007 classification of Tumors of Nervous System [1]. These distinctive low-grade tumors share with other mixed lineage tumors, features like seizure causation, relative circumscription, and favorable prognosis following simple resection. Affecting mostly the young and middle aged, it has an anatomical predilection to temporal lobes in cerebral hemispheres [2–4]. Till date, 42 cases have been recorded in literature (Table 1). We report two cases of this relatively rare tumor, one involving left fronto-parietal area in a 47-year-old male and the other occurring in right temporal region in a 25-year-old male. Pathomorphology and immunohistochemical characterization of tumors suggests origin from progenitor stem cells with divergent differentiation towards glial and neuronal lineages.
Case reports
Case 1
A 47-year-old man presented to the neurosurgical services with long standing generalized seizures of 5 years duration with recent onset of headache and right-sided weakness since 2 months. On neurological examination, he was conscious, well oriented. There was right-sided hemiparesis and fundus examination showed bilateral papilledema. Cranial CT scan revealed a well-circumscribed hypodense lesion in the right fronto-parietal region (4.2 × 3.8 × 3.2 cm) with a small enhancing component posteriorly. There was no mass effect, midline shift or calcification. A clinical diagnosis of a high-grade cystic glioma was considered. The patient was subjected to a left frontoparietal craniotomy, and gross total resection of the tumor was achieved. Intraoperatively, the lesion was cystic and contained xanthochromic fluid with a solid component that was soft, suckable, yellowish and moderately vascular. Postoperative period was uneventful and he was symptom free at 3 months follow up.
Case 2
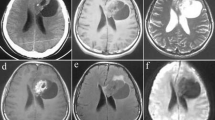
A 25-year-old male presented with a 10-year history of complex partial seizures. On clinical examination, no neurological deficits were detected. Video EEG revealed slow waves and spikes from right frontal and inferior temporal regions. Cranial MR imaging showed a right medial temporal lesion, which was hypointense on T1W and hyperintense on T2W images without perilesional edema or mass effect (Fig. 1a–c). There was a small area of contrast enhancement laterally (Fig. 1d). Clinical and radioimaging impression was that of a glioma and the patient underwent right temporal craniotomy and total excision of the tumor. At surgery the tumor was soft, suckable and mildly vascular with a poor plane of cleavage. Postoperative period was uneventful.

Cranial MR imaging shows a right medial temporal nodular lesion hypointense on T1W (a) and hyperintense on T2W (b) and FLAIR sequences (c) without perilesional edema or mass effect. A small area of ring like contrast enhancement is present laterally while medial portion is non enhancing (d) (Case 2)
Materials and methods
Tissue resected was fixed in 10% buffered formalin, and paraffin embedded sections were stained with routine hematoxylin and eosin stain. Immunohistochemistry was performed using indirect immunoperoxidase method with antibodies to Glial fibrillary acid protein (GFAP, monoclonal, 1:50, Biogenex USA), Synaptophysin (polyclonal, 1:50, Dako USA), Neu-N (monoclonal, 1:50, Biogenex USA), Neurofilament (monoclonal, 1:100, Biogenex USA), Chromogranin (monoclonal, prediluted, Biogenex USA), MIB-1 (anti Ki-67, monoclonal, 1:30 dilution), Nestin (monoclonal, 1:100, ABCAM, USA) and CD133 (polyclonal, 1:10, ABCAM, USA).
Double labeling by Confocal microscopy was carried out using GFAP (monoclonal, 1:200) detected by secondary antibody tagged to Cy3 and Synaptophysin (polyclonal, 1:200), detected with secondary antibody conjugated to FITC. For triple labeling, GFAP was detected by FITC, Synaptophysin by Cy5 and stem cell marker CD133 (polyconal, 1:10) by secondary antibody tagged to Cy5. Sections were viewed and images captured using Laser Scanning Confocal microscope (DMIRE-TCS Leica, Germany) with different excitation filters (514 nm for Cy3 fluorochrome, 488 nm laser for FITC and 633 nm for Cy5 fluorochromes, respectively). Emission bandwidth of 495–540 nm for FITC, 550–620 nm for Cy3 and 630–700 nm for Cy5 was maintained to avoid any non specific overlap of emission frequencies. The channel detecting Cy3 and Cy5 were assigned red and blue colour, respectively. All images were captured under 20× magnification at a constant PMT (photo multiplier tube) voltage of 557 V and each image was averaged four times. The image format was kept constant at 1,024 × 1,024 pixel resolution.
Results
Histopathologically in both the cases, the most striking feature was the presence of gliovascular papillae comprised of a central hyalinized core upon which were anchored one or more layers of astrocytic cells (Fig. 2a, inset). These cells lacked the radial tapering processes of ependymal rosettes and more closely resembled neurocytes but strongly expressed glial fibrillary acidic protein (Fig. 2b, inset). Occasional papillae in the first case revealed myxoid stroma. In the second case scattered large cells with abundant bright orange granular cytoplasm with a gemistocytic appearance showed strong cytoplasmic expression of GFAP. Several hypertrophic reactive astrocytes were also highlighted by GFAP stain, the long tapering processes traversing between the neurocytes to reach the vessel walls.

Microphotograph shows characteristic biphasic pattern with hyalinized papillae (P) surrounded by cellular interpapillary zones (I) (a). The papillae are covered by 1–2 layers of glial cells (inset) that express strong GFAP immunoreactivity (b, inset). The interpapillary areas (I) show minimal GFAP labeling whereas synaptophysin labeling is strong and diffuse (c). Focal areas of ganglionic differentiation (d) were seen with synaptophysin localized to cell membrane (e) and neurofilament within the cytoplasm (f). NeuN reactivity (g) was seen in nuclei of neurocytes and cytoplasm of ganglion cells. Focal chromogranin expression was seen in interpapillary zones (h). Strong expression for Nestin (i) was observed in cells lining the papillae with focal expression in interpapillary neuronal areas suggesting common stem cell origin. MIB-1 labeling was low (<1%, j). a HE ×80, (inset: ×500) b GFAP ×200, (inset: ×500), c Synaptophysin ×240, d HE ×320, e E ×320, f ×480, g ×320, h ×480, i ×300, j ×300 (e Synaptophysin, f Neurofilament, g NeuN, h chromogranin, i Nestin, j MIB-1) (a–h: case 1)
The interpapillary areas in both cases were populated by diffuse stretches of cells with neuronal morphology that ranged from small neurocytes dispersed in a finely fibrillar matrix (Fig. 2a), through intermediate sized “ganglioid cells” to mature ganglion cells (Fig. 2d). In focal areas, perinuclear haloes reminiscent of oligodendroglia were seen but these cells expressed neuronal markers. The pattern and localization of neuronal markers varied depending on extent of neuronal differentiation (Fig. 2e–h). All cells expressed diffuse labeling for synaptophysin that was in postsynaptic vesicles in neuropil in mature neurons (Fig. 2c), intracytoplasmic in immature neurons and decorated the presynaptic boutons located on cytoplasmic membrane in ganglioid cells (Fig. 2e). Neurofilament was expressed only by immature and mature ganglion cells in the cytoplasm (Fig. 2f). NeuN expression was seen in nuclei of many cells with cytoplasmic localization in ganglion cells (Fig. 2g). Chromogranin positivity was restricted to mature ganglion cells (Fig. 2h).
Confocal microscopy was carried out to determine extent of co-localization, if any, of glial and neuronal markers. In case 1, GFAP expression was localized to cells lining the papillae (Fig. 3a, Cy3 red) while synaptophysin was seen in interpapillary zones (Fig. 3b, FITC green) with variable co-localization of glial and neuronal markers along the papillae on merging (Fig. 3c). A striking co-expression of both GFAP (Fig. 3d, Cy3 red) and synaptophysin (Fig. 3e, FITC green) was found in case 2 in cells lining the papillae suggesting biphenotypic differentiation (Fig. 3f, merged) and probable origin from common progenitor cells. This was confirmed by strong expression of Nestin (Fig. 2i), a stem cell marker in the cells lining papillae as well as in interpapillary zones reflecting stem cell lineage. Nestin expression was also noted in reactive astrocytes and vascular endothelium. Possibility of stem cell origin was further supported by staining with CD133, another stem cell marker that showed strong co-expression of CD133 (Fig. 4c, Cy3 red) with GFAP (Fig. 4a, FITC green) and synaptophysin (Fig. 4b, Cy5 blue) in many cells lining papillae on triple labeling (Fig. 4d, merged) seen as white colour on merging. Interestingly, CD 133 labeling was more diffuse with several tumor cells both in perivascular region as well as scattered cells in the interpapillary zones, expressing CD133, compared to Nestin expression. The MIB-1 labeling index was low (2–3%) in both the cases (Fig. 2j). The bordering brain parenchyma revealed degenerative changes in the form of piloid gliosis with Rosenthal fibres, eosinophilic granular bodies and stromal deposits of hemosiderin.

a–c (case 1) Confocal microscopy shows distinct labeling of papillae by GFAP (a, Cy3) and synaptophysin in interpapillary areas (b, FITC) with minimal co-localization in papillary zones (c). d–f (case 2) In contrast to case 1, extensive co-localization is seen in cells lining the papillary structures strongly expressing GFAP (d, Cy3) and synaptophysin (e, FITC) as evidenced in the merged image (f) (P—papillae, I—interpapillary zones)

a–d (case 2) Strong co-expression of GFAP (a, FITC) and synaptophysin (b, Cy5) with CD133 (c, Cy3) is seen on some of the cells lining papillae seen on triple labeling (d, merged) as white on merging (P—papillae)
Discussion
The papillary glioneuronal tumors in the central nervous system are relatively circumscribed, clinically and biologically indolent, histologically biphasic neoplasms of mixed ontological lineage, with intermingled glial and neuronal components. This entity was first recognized by Komori et al. [3] in a report of the clinicopathologic features of nine cases. To date, a total of 42 cases have been recorded in world literature (Table 1), including two from North India [5, 6]. Tumors of this morphology have been previously described as pseudopapillary ganglio-glioneurocytoma and pseudopapillary neurocytoma with glial differentiation [1, 7]. These tumors are distinct from rosette forming glioneuronal tumors of similar biological behavior, the latter showing rosettes and papillae of neurocytic origin and interpapillary zone populated by glial cells, a cytological reversal.
On review, hitherto reported cases occurred in age range of 4–75 years (mean 25.9 years) with female preponderance (Table 1). The common clinical presentations include headache, declining vision and/or cognition and motor weakness. One of the cases reported had intractable seizures for 16 years similar to one of our cases that had CPS for 10 years [8]. Exclusively intracerebral in location, they involve in decreasing order of frequency, the temporal, parietal, frontal and occipital lobes usually near the lateral ventricles. A single case of a large predominantly solid mass with bilateral extension has been reported [6]. On imaging they are often cystic with a mural nodule, the cyst enhancing on contrast, although rarely solid tumors are on record. Intratumour calcification, especially in the stroma is occasional. The two cytological components of the tumor, the glial and neuronal vary in their distribution, either as nodular masses or diffuse. In the present case, there was coexpression of GFAP and synaptophysin in the cells lining the papillae suggesting origin from a common progenitor cell with bilineage differentiation potential. Coexpression of GFAP and synaptophysin in the cells lining the papillae has been reported previously in occasional cases [3, 7, 9] of papillary glioneuronal tumors.
The histogenesis of papillary glioneuronal tumor is uncertain, and origin from multipotent precursor cells capable of divergent glioneuronal differentiation has been suggested, by its common periventricular location suggesting origin from subependymal stem cells. The more superficially located ones probably originate from the secondary germinal layer [3]. Expression of PDGF Rα, Olig2 and Nestin by the tumor cells indicate an origin from subependymal progenitor cells [10]. Immunohistochemical expression of HNK-1 and non-polysialylated NCAM-L species by both components suggests a common origin from a bipotential neuroglial progenitor. Ultrastructural examination of the tumor cells revealed overlapping cell lineage characteristics. Stacks of intracytoplasmic glial fibrils were seen in cells lining papillae and distinct synaptic junctions, dense core vesicles and parallel arrays of microtubules in cytoplasmic processes indicating neuronal origin in interpapillary cells. A third component of non-specific cells lacking cytoplasmic specializations was noted that could represent oligodendroglial lineage. However fluorescent in situ hybridization studies did not detect 1p deletion [11] or 19q on LOH analysis [12]. In the present report, strong co-expression of GFAP and synaptophysin in cells lining the papillae was demonstrable by confocal microscopy. This finding along with expression of Nestin supports origin from neuroepithelial stem cells with biphenotypic differentiation. Nestin expression in reactive astrocytes and endothelial cells of vessels as seen in this case has been noted in other studies also. For instance, Nestin expression has been well documented in endothelial cells of newly formed blood vessels and reactive astrocytes. In the latter it is considered to represent reversal to the embryonic stage while marking endothelial cells indicates neovascularization [13–15]. Transient expression of Nestin is also documented as reparative response following injury/ischemia.
A single genetic study published recently [16] revealed gains and structural alterations in chromosome 7 with breakpoints at 7p22 and amplifications at 7p14–q12. EGFR amplification was not found which is in accordance with other low-grade gliomas and gangliogliomas. Recurrent rearrangements of chromosome 7 have been noted in other mixed glioneuronal tumors suggesting that the involvement of chromosome 7p22, where glial and neuronal linked genes (RAC1 and NXPH1) are known to be located, is involved in the pathogenesis of PGNT and other glioneuronal tumors.
Low MIB labeling index in the cells, absence of necrosis and vascular hyperplasia reflect the benign nature (WHO—Grade I) of these tumors, though rare cases with labeling indices of 10–15% have been recorded [5, 17–19]. A single case has been recently reported with very high MIB-1 LI of 26% suggesting the existence of atypical or aggressive forms [20]. The treatment modality for this tumour is gross total resection. Adjuvant chemotherapy, radiotherapy or a combination has been used in few cases with high MIB-1 labeling index (4–26%) and/or subtotal resection [3, 5, 17–21]. One case each received radiotherapy because of initial pathologic diagnosis of ependymoma or oligodendroglioma [3, 21]. The prognosis of PGNT is favorable and recurrence free follow up period of 6 months to 19 years [21, Table 1] has been recorded. In most cases that received adjuvant chemo/radiotherapy, no tumor recurrence was noted after 1–5 years following initial resection [3, 5, 17, 20] except in one case that recurred within 6 months following surgery and postoperative chemotherapy [18]. In this case, the authors noted a minigemistocytic component in the tumor which showed high proliferative activity (MIB-1 LI 10%) A possible participation of oligodendroglioma with minigemistocytic component coexisting with the glioneuronal element was suggested, expanding the phenotype of this tumor. The aggressive nature of this particular case was attributed to neoplastic transformation in the oligodendroglial component although no immunochemical confirmation (Olig 2) or 1p deletion was demonstrated by FISH. In two children with an aggressive course [22], one had multifocal recurrence 4 years following gross total excision. All lesions however resolved following fractionated radiation and chemotherapy (Temador).
Overall, PGNT is a low grade tumor with low proliferative potential. Gross total resection is the treatment of choice. Adjuvant chemo/radiotherapy has been used with success in the few cases with aggressive behaviour/atypical histology. Role of adjuvant chemo/radiotherapy for these relatively low grade tumors can only be evaluated as more number of cases with long term follow up are reported.
References
Nakazato Y, Figarella Branger D, Becker AJ, Scheithauer BW, Rosenblum MK (2007) Papillary glioneuronal tumor. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) 4th edn. International Agency for Research on Cancer: Lyon, pp 113–114
Broholm H, Madsen FF, Wagner AA, Laursen H (2002) Papillary glioneuronal tumor—a new tumor entity. Clin Neuropathol 21:1–4
Komori T, Scheithauer BW, Anthony DC, Rosenblum MK, McLendon RE, Scott RM, Okazaki H, Kobayashi M (1998) Papillary glioneuronal tumor: a new variant of mixed neuronal-glial neoplasm. Am J Surg Pathol 22:1171–1183. doi:10.1097/00000478-199810000-00002
Prayson RA (2000) Papillary glioneuronal tumor. Arch Pathol Lab Med 124:1820–1823
Atri S, Sharma MC, Sarkar C, Garg A, Suri A (2007) Papillary glioneuronal tumour: a report of a rare case and review of literature. Childs Nerv Syst 23:349–353. doi:10.1007/s00381-006-0196-2
Radotra BD, Kumar Y, Bhatia A, Mohindra S (2007) Papillary glioneuronal tumor: a new entity awaiting inclusion in WHO classification. Diagn Pathol 2:6. doi:10.1186/1746-1596-2-6
Kim DH, Suh YL (1997) Pseudopapillary neurocytoma of temporal lobe with glial differentiation. Acta Neuropathol 94:187–191. doi:10.1007/s004010050692
Chen L, Piao YS, Xu OZ, Yang XP, Yang H, Lu DH (2006) Papillary glioneuronal tumor: a clinicopathologic and immunohistochemical study of two cases. Neuropathology 26:243–248. doi:10.1111/j.1440-1789.2006.00687.x
Vajtai I, Kappeler A, Lukes A, Arnold M, Luthy AR, Leibundgut K (2006) Papillary glioneuronal tumor. Pathol Res Pract 202:107–112. doi:10.1016/j.prp.2005.11.008
Gelpi E, Preusser M, Czech T, Slave I, Prayer D, Budka H (2007) Papillary glioneuronal tumor. Neuropathology 27:468–473. doi:10.1111/j.1440-1789.2007.00802.x
Rosenblum Marc K (2007) The 2007 WHO classification of nervous system tumors: newly recognised members of the mixed glioneuronal group. Brain Pathol 17:308–313. doi:10.1111/j.1750-3639.2007.00079.x
Tanaka Y, Yokoo H, Komori T, Makita Y, Ishizawa T, Hirose T, Ebato M, Shibahara J, Tsukayama C, Shibuya M, Nakazato Y (2005) A distinct pattern of Olig2-positive cellular distribution in papillary glioneuronal tumors: a manifestation of oligodendroglial phenotype? Acta Neuropathol 110:39–47. doi:10.1007/s00401-005-1018-4
Tamagno I, Schiffer D (2006) Nestin expression in reactive astrocytes of human pathology. J Neurooncol 80:227–233. doi:10.1007/s11060-006-9181-6
Mokrỳ J, Nemecek S (1999) Cerebral angiogenesis shows Nestin expression in endothelial cells. Gen Physiol Biophys 18(suppl):25–29
Mokrý J, Cízková D, Filip S, Ehrmann J, Osterreicher J, Kolár Z, English D (2004) Nestin expression by newly formed human blood vessels. Stem cells Dev 13:658–664
Faria C, Miguéns J, Antunes JL, Barroso C, Pimentel J, Martins Mdo C, Moura-Nunes V, Roque L (2008) Genetic alterations in a papillary glioneuronal tumor. J Neurosurg Pediatr 1:99–102. doi:10.3171/PED-08/01/099
Vaquero J, Coca S (2007) Atypical papillary glioneuronal tumor. J Neurooncol 83:319–323. doi:10.1007/s11060-007-9333-3
Ishizawa T, Komori T, Shibahara J, Ishizawa K, Adachi J, Nishikawa R, Matsutani M, Hirose T (2006) Papillary glioneuronal tumor with minigemistocytic components and increased proliferative activity. Hum Pathol 37:627–630. doi:10.1016/j.humpath.2005.12.014
Javahery RJ, McComb JG, Davidson L, Gonzalez I (2006) Aggressive variant of papillary glioneuronal tumor. American Association of Neurological Surgeons. http://www.aans.org/library/article.aspx?ArticleId=40451. Article ID 40451. Accessed 13 April 2008
Newton HB, Dalton J, Ray-Chaudhury A, Gahbauer R, McGregor J (2008) Aggressive papillary glioneuronal tumor: case report and literature review. Clin Neuropathol 27:317–324
Epelbaum S, Kujas M, Van Effereterre R, Poirier J (2006) Two cases of papillary glioneuronal tumours. Br J Neurosurg 20:90–93. doi:10.1080/02688690600682465
Konya D, Peker S, Ozgen S, Kurtkaya O, Necmettin Pamir M (2006) Superficial siderosis due to papillary glioneuronal tumor. J Clin Neurosci 13:950–952. doi:10.1016/j.jocn.2005.10.014
Bouvier-Labit C, Daniel L, Dufour H, Grisoli F, Figarella-Branger D (2000) Papillary glioneuronal tumour: clinicopathological and biochemical study of one case with 7-year follow up. Acta Neuropathol 99:321–326. doi:10.1007/PL00007445
Tsukayama C, Arakawa Y (2002) A papillary glioneuronal tumor arising in an elderly woman: a case report. Brain Tumor Pathol 19:35–39. doi:10.1007/BF02482454
Barnes NP, Pollock JR, Harding B, Hayward RD (2002) Papillary glioneuronal tumour in a 4 year old. Pediatr Neurosurg 36:266–270. doi:10.1159/000058431
Ebato M, Tsunoda A, Maruki C, Ikeya F, Okada M (2003) Papillary glioneuronal tumor with highly degenerative pseudopapillary structure accompanied by specific abortive glial cells: a case report. No Shinkei Geka 31:1185–1190
Kordek R, Hennig R, Jacobsen E, Kearney M (2003) Papillary glioneuronal tumor—a new variant of benign mixed brain neoplasm. Pol J Pathol 54:75–78
Lamszus K, Makrigeorgi-Butera M, Laas R, Westphal M, Stavrou D (2003) September 2002: 24-year-old female with a 6-month history of seizures. Brain Pathol 13:115–117
Borges G, Bonilha L, As Menezes, Queiroz Lde S, Carelli EF, Zanardi V, Menezes JR (2004) Long term follow-up in a patient with papillary glioneuronal tumor. Arq Neuropsiquiatr 62:869–872
Stosic-Opincal T, Peric V, Gavrilovic S, Gavrilov M, Markovic Z, Sener RN (2005) Papillary glioneuronal tumor. AJR Am J Roentgenol 185:265–267
Qi JP, Zhu H, DY LI, Mei HL (2006) Papillary glioneuronal tumor: report of a case. Zhonghua Bing Li Xue Za Zhi 35:764–765
Dim DC, Lingamfelter DC, Taboada EM, Fiorella RM (2006) Papillary glioneuronal tumor: a case report and review of literature. Hum Pathol 37:914–918. doi:10.1016/j.humpath.2006.01.031
Buccoliero AM, Giordano F, Mussa F, Taddei A, Genitori L, Taddei GL (2006) Papillary glioneuronal tumor radiologically mimicking a cavernous hemangioma with hemorrhagic onset. Neuropathology 26:206–211. doi:10.1111/j.1440-1789.2006.00674.x
Celli P, Caroli E, Giangaspero F, Ferrante L (2006) Papillary glioneuronal tumor. Case report and review of literature. J Neurooncol 80:185–189. doi:10.1007/s11060-006-9170-9
Guo SP, Zhang F, Li QL, Li Q, Wang WL, Li FF (2008) Papillary glioneuronal tumor—contribution to a new tumor entity and literature review. Clin Neuropathol 27:72–77
Williams SR, Joos BW, Parker JC, Parker JR (2008) Papillary glioneuronal tumor: a case report and review of the literature. Ann Clin Lab Sci 38:287–292
Izycka-Swieszewska E, Majewska H, Szurowska E, Mazurkiewicz-Bełdzińska M, Drozyńska E (2008) Papillary glioneuronal tumour of the precentral gyrus. Folia Neuropathol 46:158–163
Acknowledgements
The authors wish to acknowledge the assistance of Dr. Lily Pal, Additional Professor, Department of Pathology, Sanjay Gandhi Institute of Postgraduate Medical Education & Research, Lucknow, India in providing help with immunohistochemistry for Nestin. The authors also are thankful to Department of Neurophysiology, NIMHANS, Bangalore for assistance with double labeling and confocal microscopy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Govindan, A., Mahadevan, A., Bhat, D.I. et al. Papillary glioneuronal tumor—evidence of stem cell origin with biphenotypic differentiation. J Neurooncol 95, 71–80 (2009). https://doi.org/10.1007/s11060-009-9893-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-009-9893-5