
Abstract
Introduction
Amongst the mixed glioneuronal tumours, ‘papillary glioneuronal tumour’, a new variant, has been described recently.
Case report
We report a case of papillary glioneuronal tumour in a 4-year-old boy who presented with fever, weakness of left upper and lower limbs and headache for the last 2.5 month. Radiologic examination showed a cystic space-occupying lesion with mural nodule in the right frontal lobe with extension into white matter. Surgical excision of a large cystic mass with small solid nodule was done. Pathological examination revealed a well-circumscribed tumour showing predominantly papillary architecture with focal aggregates of cells in sheets. The papillae were composed of hyalinised blood vessels lined by single to multi-layered cells. The tumour cells showed mild pleomorphism without any necrosis. The individual tumour cells had scant eosinophilic cytoplasm, round to oval hyperchromatic nucleus with occasional mitoses. The tumour cells were immunopositive for glial fibrillary acidic protein, synaptophysin, vimentin, and S-100 protein, but negative for neurofilament, epithelial membrane antigen, cytokeratin and CD34. MIB-1 labelling index was approximately 12% in the highest proliferating areas. In view of subtotal excision of the tumour and high MIB-1 labelling index (LI), the patient was given chemotherapy and he is doing well at 1-year follow-up.
Discussion
This report supports the existence of this rare tumour. Some of its rare clinicopathological features like young age, cyst with mural nodule, presence of mitoses and raised MIB-1 LI need to be documented.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mixed glioneuronal neoplasms of the central nervous system are rare but well-recognised entities. Most common neoplasms of this group include ganglioglioma, desmoplastic infantile astrocytoma and dysembryoplastic neuroepithelial tumour. These neoplasms are known to occur in young age with a history of intractable seizures and are associated with cortical dysplasia. After surgical excision, prognosis is favourable. In recent years, new entities like papillary glioneuronal and rosetted glioneuronal tumour, which are currently not included in the WHO classification [1] have been described. Papillary glioneuronal tumour (PGNT) is a new entity first described by Komori et al. in 1998 [2], and since then only 20 cases of this tumour were reported in English literature [3–14]. Herein we report a rare case of papillary glioneuronal tumour and discuss its clinicopathologic features.
Case report
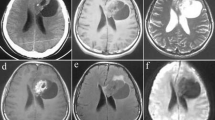
This 4-year-old boy presented with a history of fever, headache and weakness of left upper and lower limbs for the last two and half months. Clinical examination revealed a Glasgow coma score of E4V5M6 and power in left upper and lower limbs of 4+/5. MRI brain (T1-weighted imaging) showed a well-circumscribed mass in the left frontal lobe with a small mural nodule, which was hyperintense on T2-weighted imaging. On gadolinium contrast injection, this nodule was homogenously enhancing with a peripheral ring of enhancement of the cyst wall (Fig. 1). The overlying bone was unremarkable. The radiological differential diagnoses considered were those of a pilocytic astrocytoma, ganglioglioma and pleomorphic xanthoastrocytoma (PXA).

T1-weighted imaging showing a cystic lesion with mural nodule (a) and this lesion is hyperintense on T2WI (b). On contrast injection, nodule and cyst wall are homogenously enhancing (c)
The patient underwent craniotomy and partial excision of the tumour. Macroscopic examination revealed a large cystic mass with solid areas. The operative impression was that of a cystic glioma.
Pathologic examination
Tissue was fixed in 10% neutral buffered formalin, routinely processed and paraffin-embedded. Five-micrometer thick sections were cut for hematoxylin and eosin staining and immunohistochemistry. Immunohistochemical staining was done by streptavidin–biotin peroxidase complex method. The various antibodies used include were glial fibrillary acidic protein (GFAP, dil 1:1,000), synaptophysin (dil 1:50), epithelial membrane antigen (dil 1:50), pancytokeratin (dil 1:100), S-100 protein (dil 1:100), CD34 (dil 1:50), vimentin (dil 1:100), chromogranin (dil 1:50) and MIB-1 (dil 1:50). All antibodies were obtained from M/S Dako Patt Denmark. MIB-1 LI was calculated as percentage of positive nuclei in the highest labelled areas.
Microscopic examination revealed a well-circumscribed tumour composed predominantly of fibrovascular papillae. These papillae showed hyalinised fibrovascular cores and were lined by single to pseudo-stratified layer of cells (Fig. 2). Focally, sheets of tumour cells were also identified amidst papillae. The tumour cells had round to oval hyperchromatic nuclei and scant eosinophilic cytoplasm and occasional mitoses. No significant pleomorphism/necrosis was noted. Immunohistochemistry revealed diffuse positivity for glial fibrillary acidic protein, synaptophysin, vimentin, and S-100 protein (Fig. 3). MIB1 labelling index (LI) was approximately 12% in the highest proliferating areas (Fig. 3). Tumour was immunonegative for neurofilament, chromogranin, epithelial membrane antigen, cytokeratin and CD34. Based on the histologic features and immunoreactivity for GFAP and synaptophysin, a final diagnosis of a papillary glioneuronal tumour was made.

Photomicrograph showing a well-circumscribed tumour arranged in papillary fashion. The interface between tumour and brain is well-demarcated (a, ×40). Higher magnification showing hyalinised fibrovascular cores lined by cuboidal cells at places in stratification (b, ×100). Tumour cells have indistinct cellular outlines, scant amount of cytoplasm, and hyperchromatic nuclei at places showing mitoses (c, ×200)

Photomicrograph showing tumour cells to be immunopositive for GFAP, synaptophysin, and vimentin, respectively (a–c, ×200)
Follow-up
In view of the presence of mitoses, raised MIB-1 LI and subtotal resection, the patient was given chemotherapy. He was doing well at 12 months follow-up without any increase in size of the lesion or metastasis.
Discussion
The term ‘papillary glioneuronal tumour’ was first coined by Komori et al. in 1998 [2] and, to date, 20 cases have been reported in the literature mostly in the forms of single case reports [3–14], except a single report of nine cases from the Mayo clinic [2]. The clinicopathological features of these 19 published cases are summarised in Table 1. The data is obviously very limited to draw definite conclusions; however, there are certain pertinent features.
Age ranged from 4 to 75 years with a mean age of 23.1 years and consisted of 8 male and 11 female patients. Only five cases are reported in the pediatric population [2, 6, 11, 12], and the youngest patient was 4 years [6].The case under discussion is the second youngest patient of 4 years old age. The most common clinical presentations included headache, as in the present case, visual disturbances, mood changes and seizures. The duration of symptoms ranged from 1 day to 2 years (mean 8 months). Radiologically, most of the tumours presented as well-circumscribed solid and cystic masses, which were contrast-enhancing [2, 3, 11]. Like the present lesion, many of them were cystic with mural nodule [2, 4, 10, 14] and closely mimicked pilocytic astrocytomas, gangliogliomas and PXAs. Sometimes even extension into the ventricle was noted [2]. Perilesional oedema was noted only once [4]. The most common locations are the temporal and parietal lobes (36.9%) followed by frontal lobe (21%).
Papillary glioneuronal tumour is classically characterised by pseudo-papillary appearance with a single layer of pseudo-stratified, small, cuboidal cells around hyalinised vessels, accompanied with sheets or focal collections of neurocytes intermingled with ganglion cells, and cells with morphology intermediate between neurocytes and ganglion cells amidst these papillae. Although these lesions appear to be circumscribed in radiologial examinations, they are microscopically shown to be invading surrounding brain and even Rosenthal fibres formation. Hyalinised vessels are common features of several mixed glioneuronal neoplasms including ganglioglioma, intraventricular neurocytoma, extraventricular neurocytoma and dysembryoplastic neuroepithelial tumour, as well as indolent astrocytic tumours such as pilocytic astrocytoma and pleomorphic xanthoastrocytoma (PXA). On the other hand, the perivascular arrangement of cuboidal GFAP and synaptophysin-positive cells is exclusively seen in papillary glioneuronal tumour only. Thus, papillary architecture with GFAP-positive cells lining these papillae is a helpful feature in the diagnosis of PGNT. Absence of nodular arrangement, perivascular lymphocytic collection, calcification, mucoid look and associated cortical dysplasia are helpful features to exclude other mixed glioneural tumours. Papillary neurocytoma reported in the literature probably belong to this category of tumours [15]. Other differential diagnostic considerations of papillary CNS lesions are choroid plexus tumours, germ cell tumour (embryonal carcinoma and yolk cell tumour), papillary ependymoma, papillary meningioma and metastatic adenocarcinoma. Locations of the lesion along with the absence of staining for cytokeratin, epithelial membrane antigen, alfa-feto protein, transthyritin, and c-kit (CD117) are helpful to rule out some of these entities. Most of the PGNTs reported in the literature had benign histology, except one case [6]. Barnes et al. [6] reported a case of PGNT in a 4-year-old boy, which showed occasional mitoses and endothelial proliferation. However, despite subtotal resection, this patient was well until 30 months of follow-up. MIB-1 LI was done in only 12/19 published cases, but none of them showed LI of > 2.5%. However, MIB-1 LI in this case was 12%, which is higher.
Surgical excision is the key of management and chemotherapy; radiotherapy is not recommended if completely excised. However, in view of the presence of mitoses, raised MIB-I LI and subtotal excision, this patient was given chemotherapy.
The histogenesis of these tumours is not exactly known. However, based on ultrastructural [4, 6] and double-labelling immunohistochemical studies, it has been proposed that these neoplasms are neuroepithelial tumours arising from the germinal layer in the subependymal zone, which are capable of differentiating into glial and neuronal cells. Recently, some authors have suggested these tumours originated from the oligodendroglial cells in view of their positivity for olig 2 [16].
Thus, based on the analysis of present case and the limited data available from review of literature, we propose that papillary glioneuronal tumour should be included as a distinct entity in the WHO classification of CNS tumours.
References
Genacchi G, Giangaspero F (2004) Emerging tumour entities and variants of CNS neoplasms. J Neuropathol Exp Neurol 63:185–192
Komori T, Scheithauer BW, Anthony DC et al (1998) Papillary glioneuronal tumor: a new variant of mixed neuronal–glial neoplasm. Am J Surg Pathol 22:1171–1183
Prayson RA (2000) Papillary glioneuronal tumor. Arch Pathol Lab Med 124:1820–1823
Bouvier-Labit C, Daniel L, Dufour H, Grisoli F, Figarella-Branger D (2000) Papillary glioneuronal tumor: clinicopathological and biochemical study of one case with 7-year follow. Acta Neuropathol (Berl) 99:321–326
Biemat W, Liberski PP (2001) Papillary glioneuronal tumor (in Polish). Pol J Pathol 52(suppl 4):111–112
Barnes NP, Pollock JR, Harding B, Hayward RD (2002) Papillary glioneuronal tumor in a 4-year-old. Pediatr Neurosurg 36:266–270
Tsukayama C, Arakawa Y (2002) Papillary glioneuronal tumor arising in an elderly woman: a case report. Brain Tumor Pathol 19:35–39
Brohom H, Madsen FF, Wagner AA, Laursen H (2002) Papillary glioneuronal tumor: a new tumor entity. Clin Neuropathol 21:1–4
Lamszus K, Makrigeogri-Butera M, Laas R, Westphel M, Stavrou D (2003) 24-year-old female with a 6-month history of seizures. Brain Pathol 13:115–117
Ebato M, Tsunoda A, Maruki C, Ikeya F, Okada M (2003) Papillary glioneuronal with highly degenerative pseudopapillary structure accompanied by specific abortive glial cells: a case report (in Japanese). No Shinkei Geka 31:1185–1190
Kordek R, Hennig R, Jacobsen E, Kearney M (2003) Papillary glioneuronal tumor: a new variant of benign mixed neoplasms. Pol J Pathol 54:75–78
Borges G, Bonilha L, Menezes AS, Queiroz Lde S, Carelli EF, Zanardi V, Menezes JR (2004) Long term follow-up in a patient with papillary glioneuronal tumor. Arq Neuropsiquiatr 62:869–872
Stosic-Opincal T, Peric V, Gavrilovic S et al (2005) Papillary glioneuronal tumor. AJR Am J Roentgenol 185:265–267
Vajtai I, Kappeler A, Lukes A, Arnold M, Luthy AR, Leibundgut K (2006) Papillary glioneuronal tumor. Pathol Res Pract 202:107–112
Kim DH, Suh YL (1997) Pseudopapillary neurocytoma of temporal lobe with glial differentiation. Acta Neuropathol (Berl) 94:187–191
Tanaka Y, Yokoo H, Komori T, Makita Y, Ishizawa T, Hirose T, Ebato M, Shibahara J, Tsukayama C, Shibuya M, Nakazato Y (2005) A distinct pattern of Olig2-positive cellular distribution in papillary glioneuronal tumors: a manifestation of the oligodendroglial phenotype? Acta Neuropathol (Berl) 110:39–47
Acknowledgements
The authors are thankful to Mr. Pankaj Kumar and Mrs. Kiran Rani for their technical assistance and to Mr. Kamal for secretarial help.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Atri, S., Sharma, M.C., Sarkar, C. et al. Papillary glioneuronal tumour: a report of a rare case and review of literature. Childs Nerv Syst 23, 349–353 (2007). https://doi.org/10.1007/s00381-006-0196-2
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-006-0196-2