
Abstract
Papillary glioneuronal tumor (PGNT) is a benign (WHO grade I), relatively circumscribed, biphasic cerebral neoplasm occurring predominantly in the temporal lobe; it most commonly affects young adults.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Mixed glioneuronal tumors
- Papillary glioneuronal tumor
- Pseudopapillary ganglioglioneurocytoma
- Pseudopapillary neurocytoma with glial differentiation
1 Overview
-
Papillary glioneuro nal tumor (PGNT) is a benign (WHO grade I), relatively circumscribed, biphasic cerebral neoplasm occurring predominantly in the temporal lobe; it most commonly affects young adults.
-
No population-based epidemiologic data are available to date.
-
PGNT was initially described as “pseudopapillary ganglioglioneurocytoma” or “pseudopapillary neurocytoma with glial differentiation.”
-
◦ Established as a distinct clinicopathologic entity in 1998 by Komori et al.
-
◦ Regarded as a variant of ganglioglioma in the 2000 World Health Organization (WHO) classification of central nervous system tumors.
-
◦ Added as a new entity in the 2007 WHO classification.
-
2 Clinical Features
-
PGNT occurs over a wide age range (4–75 years), with a mean age at presentation of 27 years. It is more prevalent in the second and third decades of life.
-
No gender predilection has been reported.
-
The most common clinical manifestations include headache and seizures.
-
Patients may also present with nausea, vertigo, visual and gait disturbances, focal sensorimotor deficits, syncope, memory loss, and mood changes.
3 Neuroimaging
-
The tumor presents as well-defined, circumscribed lesion centered in subcortical or periventricular cerebral white matter. Small lesions lack peritumoral edema. Mass effect and mild peri tumoral edema are present with large lesions.
-
◦ PGNT occurs in all lobes, with the temporal lobe being the most commonly involved.
-
◦ Rare intraventricular examples have been reported.
-
◦ MRI: The tumors are typically heterogeneous on T2-weighted imaging, although small lesions may be homogeneous, and marked hyperintensity may be present in cystlike regions (Figs. 21.1 and 21.2).
Fig. 21.1 
Papillary glioneuronal tumor (PGNT). Coronal T2-weighted MRI shows a heterogeneous tumor in the left periventricular white matter, with a markedly hyperintense central cystlike com ponent and a less intense peripheral nodular component
Fig. 21.2 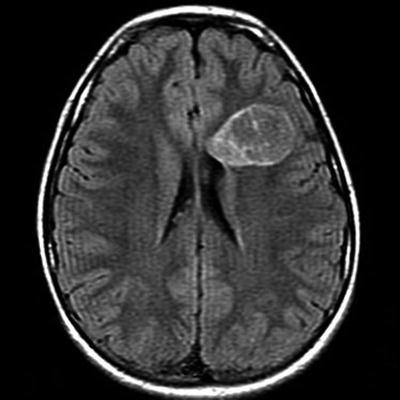
Axial fluid-a ttenuated inversion recovery (FLAIR) MRI showing attenuation of signal in the central, cystlike component. Note the lack of peritumoral edema
-
◦ PGNTs are usually hypointense on T1-weighted imaging (Fig. 21.3) unless intratumoral hemorrhage is present.
Fig. 21.3 
Sagittal T1-weighted MRI shows a well-circumscribed tumor with marked hypointensity in t he central, cystlike component
-
◦ These tumors may show postcontrast enhancement (Fig. 21.4), with the enhancing component isoi ntense or hyperintense to cortex on T1-weighted imaging.
Fig. 21.4 
Axial T1-weighted postcontrast images showing a PGNT with irregular peripheral nodular enhance ment. The central, cystlike component does not enhance
-
◦ Enhancement may be peripheral, solid, or peripheral with mural nodule.
-
◦ Diffusion weighted im aging (DWI) usually shows no restriction of diffusion (Fig. 21.5).
Fig. 21.5 
Axial diffusion weighted imaging (DWI) shows no diffusion restriction within the tumor
-
◦ CT scans: Calcification and intratumoral hemorrhage are uncommon.
-





4 Pathology
-
Smear preparations
-
◦ Smear prepar ations reveal a mixture of cells (Figs. 21.6, 21.7, and 21.8), including ganglion cells with large, eccentric nuclei; prominent nucleoli; delicate nuclear membranes; and Nissl substance in the cytoplasm. Intermediate cells with less prominent nucleoli and relatively scant cytoplasm also are seen. Cells with dense nuclei, scant cytoplasm, and occasional processes represent perivascular astrocytes.
Fig. 21.6 
I ntraoperative cytologic preparation showing a mixed cell population with some of the cells having eosinophilic cytoplasm and eccentric nuclei
Fig. 21.7 
Intraopera tive cytologic preparation showing pseudopapillary architecture
Fig. 21.8 
Intrao perative cytologic preparation showing fibrillary background
-
-
Histology
-
◦ Two distinct histologic components are identified: glial and neuronal.
-
The glial component consists of pseudopapillary structures with hyalinized blood vessels surrou nded by a single or pseudostratified layer of cuboidal glial cells (Figs. 21.9 and 21.10). These cells have rounded nuclei, moderately dense chromatin, and scant eosinophilic cytoplasm and do not elaborate perivascular cytoplasmic processes. Mitotic activity and cellular atypia are absent in this component.
Fig. 21.9 
Charact eristic pseudopapillary pattern in PGNT
Fig. 21.10 
Highe r magnification showing the core of pseudopapillary structures and the mixed glioneuronal population of cells
-
The neuronal component consists of interpapillary sheets or focal collections of neurocytes, gangli on cells, and intermediate “ganglioid cells” in a fibrillary, neuropil-like matrix.
-
-
◦ Neurocytic cells have dark, round, uniform nuclei; scant cytoplasm; and sometimes an oligo-like halo (Fig. 21.11). The ganglion cells feature large, vesicular nuclei; prominent nucleoli; and abundant cytoplasm with well-developed Nissl substance.
Fig. 21.11 
Intervenin g cells with clear cytoplasm may be seen
-
The intermediate-sized “ganglioid cells” feature less-abundant cytoplasm and smaller nucleoli.
-
Occasional pleomorphic and binucleated ganglion cells are identified.
-
-
◦ Minigemistocyt es with eccentrically placed nuclei and eosinophilic hyaline cytoplasm may be found in the interpapillary regions or nestled among neuronal cell types (see Figs. 21.6 and 21.10).
-
◦ A narrow zone of tumor infiltration or a discrete pushing border may be seen with the adjacent neural parenchyma. In this region, prominent gliosis, Rosenthal fibers, eosinophilic granular bodies, hemosiderin deposition, foamy cell infiltration, and dystrophic calcifications may be identified.
-
◦ Mitotic activity is usually inconspicuous.
-
◦Necrosis and microvascular proli feration, when present, are exceptional.
-






5 Immunohistochemistry
-
The small, cuboidal glial cells surrounding the hyalinized vascular pseudopapillae are positive for glial fibrillary acidic protein (GFAP) (Fig. 21.12) and S-100.
Fig. 21.12 
A large subp opulation of tumor cells are positive for glial fibrillary acidic protein (GFAP)
-
Neuronal cells of all sizes are immunopositive for synaptophysin (Fig. 21.13), neuron-specific enolase (NSE), and class III β-tubulin (MAP2) (Fig. 21.14).
Fig. 21.13 
Synapto physin positivity is granular but patchy
Fig. 21.14 
Diffuse positivity for microtubule-associated protein 2 (MAP2) is observed in this tumor
-
Most of the neuronal cells are also positive for NeuN; immunopositivity for neurofilament protein (NFP) is confined to ganglion cells and intermediate “ganglioid cells.”
-
Chromogranin A expression has not been observed.
-
Minigemistocytes show intense GFAP immunoreactivity.
-
Tanaka et al. described a subpopulation of cells that fail to label with GFAP and neuronal markers but express the Olig2 transcription factor. These cells are mainly found in the interpapillary areas surrounding GFAP-positive perivascular elements. They also found that minigemistocytes are in close association with these Olig2-positive cells and may be derived from them.
-
MIB-1 labeling indices are low, in the range of 1–2 %.



6 Electron Microscopy
-
The perivascular glial cells show cytoplasmic bundles of intermediate filaments and form juxtavascular basal lamina.
-
The neuronal cells exhibit cytoplasmic extensions filled with aligned microtubules, dense-core granules, clear vesicles, and synaptic contacts of varying organization.
-
A third cell component of poorly differentiated cells has been described at the ultrastructural level. These cells contain mitochondria, ribosomes, occasional dense bodies, intermediate filaments, and microtubules, but no definitive dense-core granules. They are thought to represent glioneuronal progenitor cells.
-
Hybrid glioneuronal cells have not been described.
7 Molecular Pathology
-
PGNT has been reported in patients with cleft lip and orbital schwannoma, but association with familial syndromes has not been established.
-
Several cases have been studied for 1p status by fluorescence in situ hybridization (FISH) and have been found to be intact.
-
Faria et al. reported a case of PGNT with gains and alterations involving chromosome 7p22. Using comparative genomic hybridization, they also observed high-level amplification at 7p14 ~ q12.
-
SLC44A1-PRKCA fusion has been detected in several cases of PGNT, thus suggesting a potential biomarker for this tumor.
-
Mutations in IDH1 or IDH2 are not detected in PGNT.
8 Differential Diagnosis
-
The pseudopapillae with surrounding astrocytic cells may resemble the perivascular pseudorosettes of ependymoma, but cellular processes radiating to the vessel wall are not seen in PGNT. In addition, PGNT does not show ultrastructural evidence of ependymal differentiation.
-
The presence of neurocytic, ganglionic, and glial components may raise the possibility of ganglioglioneurocytoma as a differential diagnosis, but the presence of pseudopapillary glio-vascular structures and the broad spectrum of neuronal differentiation in PGNT help distinguish between the two entities.
-
PGNT should be distinguished from dysembryoplastic neuroepithelial tumor (DNET), which also presents at a young age with seizures. Histologically, DNET is characterized by the specific glioneuronal element, microcysts with floating neurons, and cortical dysplasia in the adjacent cortex. None of these features are seen in PGNT.
9 Prognosis
-
In general, PGNT s hows a favorable clinical outcome.
-
Gross total resection without adjuvant therapy results in recurrence-free, long-term survival in the majority of cases.
-
One case of atypical papillary glioneuronal tumor has been reported as showing mitotic activity and an MIB-1 labeling index of 15 %. The patient remained symptom-free without tumor recurrence 5 years after radical resection and radiotherapy.
-
Very rare examples of tumor recurrence or progression have also been reported.
Suggested Reading
Bridge JA, Liu XQ, Sumegi J, Nelson M, Reyes C, Bruch LA, et al. Identification of a novel, recurrent SLC44A1-PRKCA fusion in papillary glioneuronal tumor. Brain Pathol. 2013;23:121–8.
Faria C, Miguéns J, Antunes JL, Barroso C, Pimentel J, Martins MC, et al. Genetic alterations in a papillary glioneuronal tumor. J Neurosurg Pediatr. 2008;1:99–102.
Hainfellner JA, Scheithauer BW, Giangaspero F, Rosenblum MK. Rosette-forming glioneuronal tumour of the fourth ventricle. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO Classification of tumours of the central nervous system. 4th ed. Lyon: International Agency for Research on Cancer; 2007. p. 113–4.
Komori T, Scheithauer BW, Anthony DC, Rosenblum MK, McLendon RE, Scott RM, et al. Papillary glioneuronal tumor: a new variant of mixed neuronal-glial neoplasm. Am J Surg Pathol. 1998;22:1171–83.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109.
Matyja E, Grajkowska W, Pucko E, Kunert P, Marchel A. Papillary glioneuronal tumor with an unusual bilateral intraventricular localization. Clin Neuropathol. 2015;34:6–12.
Myung JK, Byeon SJ, Kim B, Suh J, Kim SK, Park CK, et al. Papillary glioneuronal tumors: a review of clinicopathologic and molecular genetic studies. Am J Surg Pathol. 2011;35:1794–805.
Nagaishi M, Nobusawa S, Matsumura N, Kono F, Ishiuchi S, Abe T, et al. SLC44A1-PRKCA fusion in papillary and rosette-forming glioneuronal tumors. J Clin Neurosci. 2016;23:73–5.
Rosenblum MK. The 2007 WHO classification of nervous system tumors: newly recognized members of the mixed glioneuronal group. Brain Pathol. 2007;17:308–13.
Schlamann A, von Bueren AO, Hagel C, Zwiener I, Seidel C, Kortmann RD, Müller K. An individual patient meta-analysis on characteristics and outcome of patients with papillary glioneuronal tumor, rosette glioneuronal tumor with neuropil-like islands and rosette forming glioneuronal tumor of the fourth ventricle. PLoS One. 2014;9:e101211.
Tanaka Y, Yokoo H, Komori T, Makita Y, Ishizawa T, Hirose T, Ebato M, et al. A distinct pattern of Olig2-positive cellular distribution in papillary glioneuronal tumors: a manifestation of the oligodendroglial phenotype? Acta Neuropathol. 2005;110:39–47.
Vaquero J, Coca S. Atypical papillary glioneuronal tumor. J Neurooncol. 2007;83:319–23.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this chapter
Cite this chapter
Rivera-Zengotita, M., Illner, A., Adesina, A.M. (2016). Papillary Glioneuronal Tumor. In: Adesina, A., Tihan, T., Fuller, C., Poussaint, T. (eds) Atlas of Pediatric Brain Tumors. Springer, Cham. https://doi.org/10.1007/978-3-319-33432-5_21
Download citation
DOI: https://doi.org/10.1007/978-3-319-33432-5_21
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-33430-1
Online ISBN: 978-3-319-33432-5
eBook Packages: MedicineMedicine (R0)