
Abstract
Background
Papillary glioneuronal tumor (PGNT) is a rare, recently described distinct low-grade brain neoplasm. This study was performed to characterize the clinicopathologic and neuroradiologic features of PGNTs.
Methods
We reviewed the medical records of 16 patients with PGNT who underwent surgery, including 11 males and five females (median age 27 years). The clinical, neuroradiologic, histopathologic, and immunohistochemical findings were documented.
Results
Headache was the principal presentation. Neuroimaging showed contrast-enhancing, cystic-solid or cystic masses with a mural nodule, mostly involved the frontal or parietal lobes. Histologically, the tumors were characterized by glial fibrillary acidic protein (GFAP)-positive small cuboidal cells lining hyalinized vascular pseudopapillae and synaptophysin and/or NeuN-positive interpapillary neuronal elements. Other findings included small angiomatous areas in ten, small islands of neuropil and rosettes in seven, and microvascular proliferation and/or nuclear atypia in six. Mitoses or necrosis were absent. All lacked isocitrate dehydrogenase 1 (IDH1) R132H protein expression. Low expression of p53 was observed in three cases. Ki67 labeling index ranged from less than 1 to 3 %. All but one was totally resected. Median follow-up was 65 months, and one patient had tumor recurrence.
Conclusions
PGNTs display distinct clinicopathologic and imaging characteristics and indicate a favorable prognosis. However, recurrences sometimes occur. Immunohistochemistry facilitates the appropriate diagnosis of these tumors. Complete resection of the tumor is important for a favorable outcome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Papillary glioneuronal tumors (PGNTs) are rare, recently described distinct low-grade glioneuronal neoplasms that usually occur in young adults and generally affect the cerebral hemispheres, predominantly in the deep, paraventricular white matter, with a predilection for the temporal lobe [15, 16, 27]. Tumors are noted on neuroimaging as well-circumscribed, solid to cystic, contrast-enhancing masses with little mass effect, and a cystic mass with mural nodule may be seen [15, 24, 27]. Histologically, PGNTs generally consist of flat to cuboidal, glial fibrillary acidic protein (GFAP)-positive neoplastic glial cells lining hyalinized vascular pseudopapillae and synaptophysin-positive interpapillary collections of sheets of neurocytes, large neurons and intermediate-sized “ganglioid” cells [2, 15, 16, 18]. Histopathologically similar tumor was previously described as a pseudopapillary neurocytoma of temporal lobe with glial differentiation by Kim and Suh in 1997 [14], and was initially considered as a variant of ganglioglioma in the 2000 World Health Organization (WHO) classification of tumors of the central nervous system (CNS). Due to the histological pseudopapillae and biphasic differentiation characters, the term of PGNT first established by Komori et al. [15] in 1998 was currently retained by the updated 2007 WHO classification of tumors of the CNS [16]. To date, only less than one hundred cases have been reported in the literature [6, 23]. Although PGNTs have been identified as WHO grade I based on its indolent clinical behavior following surgical resection, some cases with more pathologically aggressive features, recurrence and malignant transformation have been described [1, 2, 4, 9, 12, 13, 18, 19, 23]. Knowledge of this entity is important to predict the clinical course and make the right therapeutic decisions. Here, we retrospectively review the clinicopathologic, neuroradiologic, and prognostic features of 16 cases of PGNT, to emphasize the processes of diagnosis and treatment and to highlight the prognosis of patients with these lesions.
Materials and methods
Samples and clinical data
The medical records of 16 cases of PGNT during the period 2004 to 2014 at Huashan Hospital of Fudan University and The People’s Hospital of Zhengzhou University were reviewed. PGNTs were identified based on the criteria of the 2007 WHO classification [16]. The clinical features of all patients were documented, including sex, age at diagnosis, initial symptoms, tumor location, tumor size, extent of surgical resection, outcome, and follow-up information. All patients underwent maximal possible surgical resection of the primary lesion consistent with preservation of neurologic function.
Neuroimaging studies
The original magnetic resonance (MR) images and/or computed tomographic (CT) scans where available were reviewed by two neuroradiologists. The preoperative CT analyses were performed on 12 cases, and intravenous administration of a contrast medium was performed on three cases (cases 2, 5, and 12). The preoperative MRI results were available for all cases, including gadolinium injection for 15 cases. The location, size, density, signal and contrast-enhancement features of the lesions, and the presence of hemorrhage, calcification, cystic change, intraventricular expansion, peritumoral edema, or mass effect were evaluated.
Histopathology and immunohistochemistry
Surgical specimens were fixed in 10 % neutral buffer formalin, routinely processed and paraffin embedded. 4-μm sections were cut, dewaxed and rehydrated, then stained with hematoxylin and eosin (H&E). Immunohistochemical analysis was performed on selected, representative 4-μm-thick sections using the immunohistochemical EnVision™ detection system (Dakopatts, Glostrup, Denmark) according to kit instructions. An epitope-retrieval method was applied as needed for each specific antibody, and diaminobenzidine was the chromogen. All tumor specimens were studied for the expression of GFAP (catalog M076101, monoclonal, Dakopatts, Glostrup, Denmark), oligodendrocyte transcription factor-2 (Olig-2) (catalog JP18953, polyclonal, IBL International, GmbH), neurofilament protein (NFP) (catalog M076229, monoclonal, Dakopatts, Glostrup, Denmark), neuronal nuclear antigen (NeuN) (catalog MAB377, monoclonal, Chemicon International, Temecula, CA), chromogranin A (CgA) (catalog M086901, monoclonal, Dakopatts, Glostrup, Denmark), synaptophysin (SYN) (catalog M077601, monoclonal, Dakopatts, Glostrup, Denmark), p53 (catalog M700101, monoclonal, Dakopatts, Glostrup, Denmark), IDH1 R132H protein (catalog DIAH09, monoclonal, Dianova, Hamburg, Germany), and Ki67 (catalog M724801, monoclonal, Dakopatts, Glostrup, Denmark). The MIB-1 labeling index (LI) was calculated in the highest proliferating area as the percentage of labeled nuclei per 1,000 cells. The expression of markers was evaluated based on intensity of staining and distribution of positive cells. Both the percentage of positive cells and the staining intensity were evaluated by 2 neuropathologists respectively. The percentage positivity was scored as four classes: “0” (<5 %, negative), “1” (5–25 %, sporadic), “2” (25–50 %, focal), or “3” (>50 %, diffuse). The staining intensity was scored as “0” (no staining), “1” (weakly stained), “2” (moderately stained), or “3” (strongly stained). The immunostaining score of markers was calculated as the percentage positive score × the staining intensity score and ranged as 0, 1, 2, 3, 4, 6, and 9. The sum-indexes (−), (+), (++), and (+++) indicated overall staining score of 0, 1–3, 4–6, and 9, respectively. The histopathologic and immunohistochemical features of all tumors at the 2 medical centers were reviewed by 2 neuropathologists according to the 2007 WHO classification.
Results
Clinical features
The clinical data of all patients are shown in (Table 1). The patients ranged in age from 11 to 63 years (median age, 27 years). The male-to-female ratio was 11:5. In 15 out of 16 patients (94 %) diagnosis was preceded by neurological symptoms, with a median duration of 6 months (range, 0.5 months to 156 months) before surgery. Seven patients (44 %) presented with intermittent headaches, four patients presented with seizures, three patients presented with vertigo (case 2 also had left lower extremity weakness), one patient had fatigability and sluggish reactions (case 16), and one patient (case 14) had an asymptomatic mass discovered incidentally upon neuroimaging secondary to a traumatic injury to the brain. In addition to headaches, one patient (case 8) also had visual disturbance, one patient (case 9) had tinnitus and a focal mass.
A craniotomy and a gross total resection (GTR) of the tumor were performed in 15 cases. One case (case 11) received a subtotal resection (STR). Profuse intraoperative bleeding was the main cause of subtotal removal. The postoperative course was uneventful, and each patient was discharged a few days after surgery. Radiotherapy was administered to three patients (cases 2, 11, and 12). Median follow-up was 65 months (range, 16–105 months). At the latest follow-up, 14 cases were symptom free after surgery, with no evidence of disease progression or recurrence. However, one patient (case 11) developed tumor recurrence at 50 months after the initial excision, and received second surgery and eventually had a good clinical outcome. One patient (case 16) developed seizures at 9 months after surgery and received antiepileptic medication.
Neuroradiologic findings
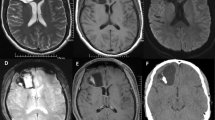
As seen in Table 1, all tumors affected the cerebral hemispheres, including the frontal (n = 7), parietal (n = 5), temporal (n = 3), and occipital lobes (n = 1). Fourteen of the 16 cases were located in the deep, periventricular area, and intraventricular involvement was present in eight cases (50 %). The median tumor size was 5.4 cm (range, 1.8–7.6 cm). In 13 cases, a well-circumscribed, solid-cystic mass was present (Fig. 1). Three cases showed a large cystic mass with an enhancing cyst wall and mural nodules. In CT scans, the solid part of the lesion was generally isodense (n = 9) (Fig. 1a) or slight hypodense (n = 1) in the 12 available cases. Two cases were predominantly cystic hypodense masses. Inhomogeneous enhancement was apparent in two of the three cases given intravenous contrast infusions. Calcific hyperdensities were present in nine of the 12 cases. In MRI, the lesion was heterogeneous isointense and hypointense (n = 10) (Fig. 1b) or hypointense (n = 6) to gray matter in T1-weighted images and hyperintense in T2-weighted images (Fig. 1c), without or with minimal mass effect and peritumoral edema in most of the cases, only three cases showed moderate mass effect and peritumoral edema. Post-gadolinium scans revealed moderate to marked enhancement of the solid component (Fig. 1d) (n = 12) and of the mural nodule (n = 3). In fluid-attenuated inversion-recovery (FLAIR) images the lesion was hyperintense relative to gray matter in seven of ten available cases, two cases showed heterogeneous isointense and hyperintense and one case showed isointense and hypointense (Fig. 1e). Four cases showed hypointensity (Fig. 1f) and three cases showed heterogeneous hypointensity and isointensity in seven available diffusion-weighted images (DWI), indicating no diffusion restriction within these cases.

Axial computerized tomography (CT) scan showed an isodense and hypodense, large cystic and solid mass involving the left frontal lobe and partly extending into the body of left lateral ventricle (a). The mass was heterogeneous isointense and hypointense in axial T1-weighted images (b) and hyperintense in axial T2-weighted images (c), with a prominent heterogeneous contrast enhancement in contrast-enhanced axial T1-weighted image (d). The mass showed isointense and hypointense in fluid-attenuated inversion-recovery images (e), and did not show diffusion restriction in diffusion-weighted images (f)
Pathologic findings
Histopathologically, all tumors showed predominant pseudopapillary structures characterized by a single-to-pseudostratified layer of small, flattened to cuboidal uniform cells with round to oval nuclei and scant cytoplasm lining hyalinized fibrovascular cores (Fig. 2a). The intervening areas were predominantly composed of sheets or focal collections of small round-to-oval cells with coarse granular nuclei and scant eosinophilic or clear cytoplasm, mimicking oligodendrocytes in appearance (Fig. 2b). In seven cases, small islands of neuropil were noted, focally, oligodendroglia-like cells were arranged around a central core of neuropil, similar to rosettes (Fig. 2c). Sporadic or focal medium-sized ganglioid cells and large ganglion cells with abundant cytoplasm and distinct nucleoli were present in the interpapillary areas of eight cases (Fig. 2d). In addition to these pseudopapillary structures, some areas mimicking pilocytic astrocytomas with microcystic changes, microvascular endothelial hyperplasia (Fig. 2e), and/or nuclear atypia (Fig. 2f) were present in six cases; small angioma-like areas were noted in ten cases. Necrosis or mitoses were absent in all cases. Hemosiderin deposition, micro-calcifications, Rosenthal fibers, and eosinophilic granular bodies were common present at the periphery of the lesions.

Morphologic features of papillary glioneuronal tumor (PGNT). Pseudopapillary structures composed of hyalinized vessels surrounded by a single layer of cuboidal cells with round or oval nuclei (a, X 200). Intervening areas with oligodendroglial-like cells (b, X 200), neuropil-like islands (c, X 200), and ganglioid and gangliocytic cells (d, X 400). Vascular endothelial hyperplasia (e, X 200) and nuclear atypia of tumor cells (f, X 400) were noted
The immunohistochemical findings for all 16 cases are summarized in (Table 2). Two distinct zones of glial and neuronal differentiation were better highlighted on immunohistochemistry. The tumor cells lining the fibrovascular core showed strong cytoplasmic immunoreactivity for GFAP (Fig. 3a) and variable focal nuclear immunoreactivity for Olig-2 in 14 cases. However, in two cases, Olig-2-positive, GFAP-negative glial cells surrounded the papillary core. Interpapillary neuronal cells in all cases showed strong cytoplasmic immunoreactivity for synaptophysin (Fig. 3b) and variable nuclear positivity for NeuN (Fig. 3c). NFP expression was mostly confined to larger ganglioid and ganglion cells (Fig. 3d). CgA expression was lacking or scattered. In addition, Olig-2 was variable positive in the interpapillary areas in all cases (Fig. 3e), and focal GFAP-positive tumor cells were also present in the intervening areas in 11 cases. IDH1 R132H protein was negative in all cases. p53 protein expression was observed in three cases with 10–20 % of tumor cells showing weak to moderate staining. The MIB-1-labeling index ranged from less than 1 % to about 3 % (Fig. 3f).

Immunohistochemical features of papillary glioneuronal tumor (PGNT). Tumor cells lining the hyalinized vascular pseudopapillae showing cytoplasmic immunopositivity for GFAP (a, X400). Intervening neuronal cells clearly shown by synaptophysin (b, X400) and NeuN (c, X400) immunostaining. Larger ganglioid and ganglion cells highlighted by NFP immunostaining (d, X400). Olig-2 reactivity cells were seen in the intervening areas or surrounding vessels (e, X400). MIB-1-labeling index was low (f, X400)
Discussion
PGNTs are uncommon, clinically indolent and morphologically biphasic cerebral neoplasms that mostly involve in young adults, with a median age of 23 years, but they occur over a wide age range varying from 4 to 75 years [3, 23, 26]. No gender predilection has been observed [15, 23, 27]. The vast majority of PGNT are located in the supratentorial region, most frequently in the deep, paraventricular white matter, with common involvement of the temporal lobe [15, 16, 27]. Rarely, it may involve the ventricle [6, 22]. PGNTs usually present with single lesion, but it may also involve bilateral cerebral hemispheres [2, 9]. In this study, there was a slight male and frontal lobe predominance, 50 % of patients were 25 years of age or less, which were consistent with the reports of the literature [2, 6, 18].
The clinical manifestations of PGNT were not specific. As seen in this study, headache and seizures were common symptoms [23]. Nausea/vomiting, focal neurological deficit, and disturbances of vision, gait, sensation, cognition and emotional affect may also be encountered [2, 15, 18, 23, 27]. Patients may also be asymptomatic with the mass discovered incidentally upon neuro-imaging for other reasons as seen in case 14 [23].
The neuroimaging characteristics of PGNT reported so far consistently described a supratentorial, periventricular well-defined, contrast-enhancing, solid to cystic mass or a cystic lesion with a mural nodule [2, 15, 16, 18, 24, 27], as detected in this study. Most PGNTs were adjacent to the ventricle, suggesting a possible origin from the germinal zone of the subependymal plate, while more superficially situated PGNTs might arise from the secondary germinal layer [15]. Typically, PGNTs displayed isodense or hypodense in CT scan, isointense and/or hypointense in T1-weighted images, hyperintense in T2-weighted images, and isointense and/or hypointense in DWI. Inhomogeneous marked enhancement may be associated with prominent pathological paraplastic blood vessels within the tumors. As a result, these unique radiological findings can facilitate the diagnosis of PGNT and are helpful in the radiologic differential diagnosis.
Histopathologically, PGNT was characterized by a biphasic pattern of a layer of flat to cuboidal glial cells lining hyalinized vascular pseudopapillae and interpapillary sheets or focal collections of neuronal cells of variable size [15, 16] as seen in our cases. The proportion of these two components varied from case to case [15]. Although the glial elements generally lacked both nuclear atypia and mitotic activity as seen in this study, some aggressiveness criteria, including nuclear atypia, high mitotic rate, high MIB-1 labeling indices, vascular endothelial hyperplasia, and necrosis, may be observed in a few cases of PGNT, it suggested that these patients should need more closely follow-up and may require adjuvant therapy [1, 2, 4, 9, 12, 13, 16, 18, 19]. In this study, areas of microvascular hyperplasia and nuclear atypia may be associated with piloid gliosis, and one case, which recurred after 50 months, did not show any changes suggestive of anaplasia. At the periphery of the lesions, prominent secondary changes including Rosenthal fibers, eosinophilic granular bodies, angiomatous areas, hemosiderin deposition, and calcifications were common noted, reflecting the long-standing course of PGNTs.
The immunochemical features of the PGNT suggested its possible origin from multipotent precursors capable of divergent glioneuronal differentiation [15]. As seen in this study, immunoreactivity for GFAP in the glial cells adjacent to the papillary core, and neuronal markers synaptophysin, NeuN, neuron-specific enolase (NSE) and class III β -tubulin staining in the interpapillary neuronal component assisted in establishing the diagnosis of PGNT [2, 15, 16, 18]. Oligodendroglia-like cells in the interpapillary spaces were also positive for Olig-2 (a marker of oligodendrocyte and neuroectodermal progenitor cell), indicating the existence of glial and neuronal elements between the pseudopapilla and suggesting possible origin from the pluripotential neural stem cells of PGNTs [10, 17, 25]. All cases showed a low MIB-1-labeling index, in keeping with its indolent nature.
The major differential diagnosis should include extraventricular neurocytoma (EVN), ganglioglioma, dysembryoplastic neuroepithelial tumor (DNT), papillary ependymoma, and astroblastoma etc [6, 15, 16, 18, 27]. Although hyalinized blood vessels and monotonous neurocyte-like cells can be encountered in EVN and GG, the absence of pseudopapillary architecture can differentiate these tumors from PGNT. Moreover, EVN has a more homogeneous histologic appearance and more mature synapses, and GG usually has more numerous and larger dysmorphic ganglion cells mixed with astrocytic glial components and a typically arborizing vascularity. DNT usually shows multinodular growth patterns and specific glioneuronal elements containing floating neurons and does not exhibit solid neuronal component or pseudopapillary patterns. Ependymoma usually contains a high degree of fibrillarity, which PGNTs lack. Astroblastoma has a perivascular cell population that is columnar or has stout processes, and also demonstrates extensive vascular hyaline changes. The location of the lesion, immunostaining patterns of EMA, and the absence of neuronal differentiation are helpful in excluding ependymoma and astroblastoma.
The underlying molecular pathogenesis of PGNTs remains unclear. By using comparative genomic hybridization (CGH), Faria et al. [8] reported that PGNT was characterized by gains and structural alterations involving only chromosome 7 (7p14-q12) with breakpoints at 7p22, including high-level amplification of 7p14 ∼ q12. MGMT methylation was observed in some cases of PGNT using fluorescence in situ hybridization (FISH) [18]. Recently, on the base of FISH and reverse transcription-polymerase chain reaction (RT-PCR) analysis, the t(9;17)(q31;q24) with the resultant novel fusion oncogene SLC44A1-PRKCA had been described as the defining molecular feature of PGNT that may be responsible for its pathogenesis [5] and seems to be a specific characteristic of PGNT with a high diagnostic value [20]. EGFR gene amplification, 1p/19q deletion or BRAF V600E mutation were not found in PGNT, implying absence of a role of these molecular alterations in the pathogenesis of PGNT [2, 18]. However, an FGFR1 N546K mutation detected in a case of PGNT suggested that at least a subgroup of PGNTs may show close molecular relationships with pilocytic astrocytomas (PAs) [11]. In this study, no expression of IDH1 R132H protein was found by immunohistochemistry, which was consistent with the previous reports [2, 18]. However, three cases of PGNT showed p53 immunoreactivity in 10–20 % of the cells. The significance of this is not known, and mutation analysis may be required to establish the exact role of this gene in the pathogenesis of PGNT.
PGNT is generally considered as a benign neoplasm and can be curable with adequate surgery alone [6, 15, 16, 23, 27]. Most patients remain well with the longest recurrence-free survival recorded being 19 years [7] and the extension of surgical removal was the main prognostic factor [21]. But tumor progression or recurrence sometimes occur after initial subtotal or even gross total resection [2, 18]. Rare PGNTs may show aggressive biological behavior [1, 9, 12, 13, 19]. More recently, Bourekas EC et al. [4] reported a single case of anaplastic PGNT with extraneural metastases. Therefore, accurate diagnosis, completeness of resection and follow-up observation are the optimal treatment for PGNT. Postoperative adjuvant radiotherapy and/or chemotherapy is considered if anaplastic [4, 18, 19] or recurrent [2, 13]. In this study, the follow-up period ranged from 16 to 105 months, most of patients had a favorable prognosis after surgery, suggesting that the usual biological behavior of PGNTs is not aggressive. Only one patient (case 11) demonstrated recurrence of tumor after a long symptom-free period (50 months), which may be the result of subtotal resection of tumor, although radiotherapy was administered postoperatively. This patient received second surgery and was reported to be well at 34 months. One patient (case 16) represented 9 months later with seizures after GTR and remained seizure-free on medication at the latest follow-up, no tumor regrowth was observed on MRI. Our findings suggested that regular follow-up may be important for identifying the rare cases of PGNT recurrence, especially in cases following incomplete resection of the tumor.
In summary, PGNTs are rare, indolent distinct neoplasms, although some cases of tumor recurrence and aggression have been described. Precise diagnoses, complete resection, and postoperative follow-up regularly using MRI are important for favorable long-term outcome. The underlying molecular pathogenesis or genetic alterations of PGNTs should be better studied in the future.
References
Adam C, Polivka M, Carpentier A, George B, Gray F (2007) Papillary glioneuronal tumor: not always a benign tumor? Clin Neuropathol 26(3):119–124
Agarwal S, Sharma MC, Singh G, Suri V, Sarkar C, Garg A, Kumar R, Chandra PS (2012) Papillary glioneuronal tumor—a rare entity: report of four cases and brief review of literature. Childs Nerv Syst 28(11):1897–1904
Barnes NP, Pollock JR, Harding B, Hayward RD (2002) Papillary glioneuronal tumour in a 4-year-old. Pediatr Neurosurg 36(5):266–270
Bourekas EC, Bell SD, Ladwig NR, Gandhe AR, Shilo K, McGregor JM, Lehman NL, Newton HB (2014) Anaplastic papillary glioneuronal tumor with extraneural metastases. J Neuropathol Exp Neurol 73(5):474–476
Bridge JA, Liu XQ, Sumegi J, Nelson M, Reyes C, Bruch LA, Rosenblum M, Puccioni MJ, Bowdino BS, McComb RD (2013) Identification of a novel, recurrent SLC44A1-PRKCA fusion in papillary glioneuronal tumor. Brain Pathol 23(2):121–128
Demetriades AK, Al Hyassat S, Al-Sarraj S, Bhangoo RS, Ashkan K (2013) Papillary glioneuronal tumour: a review of the literature with two illustrative cases. Br J Neurosurg 27(3):401–404
Epelbaum S, Kujas M, Van Effenterre R, Poirier J (2006) Two cases of papillary glioneuronal tumours. Br J Neurosurg 20(2):90–93
Faria C, Miguéns J, Antunes JL, Barroso C, Pimentel J, Martins Mdo C, Moura-Nunes V, Roque L (2008) Genetic alterations in a papillary glioneuronal tumor. J Neurosurg Pediatr 1(1):99–102
Flannery T, Purce A, Harney J, McKinstry S, Ironside JW, Herron B (2012) Bilateral non-contiguous atypical papillary glioneuronal tumor: case report. Clin Neuropathol 31(2):77–80
Gelpi E, Preusser M, Czech T, Slavc I, Prayer D, Budka H (2007) Papillary glioneuronal tumor. Neuropathology 27(5):468–473
Gessi M, Abdel Moneim Y, Hammes J, Waha A, Pietsch T (2014) FGFR1 N546K mutation in a case of papillary glioneuronal tumor (PGNT). Acta Neuropathol 127(6):935–9361
Ishizawa T, Komori T, Shibahara J, Ishizawa K, Adachi J, Nishikawa R, Matsutani M, Hirose T (2006) Papillary glioneuronal tumor with minigemistocytic components and increased proliferative activity. Hum Pathol 37(5):627–630
Javahery RJ, Davidson L, Fangusaro J, Finlay JL, Gonzalez-Gomez I, McComb JG (2009) Aggressive variant of a papillary glioneuronal tumor. Report of 2 cases. J Neurosurg Pediatr 3(1):46–52
Kim DH, Suh YL (1997) Pseudopapillary neurocytoma of temporal lobe with glial differentiation. Acta Neuropathol 94(2):187–191
Komori T, Scheithauer BW, Anthony DC, Rosenblum MK, McLendon RE, Scott RM, Okazaki H, Kobayashi M (1998) Papillary glioneuronal tumor: a new variant of mixed neuronal-glial neoplasm. Am J Surg Pathol 22(10):1171–1183
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114(2):97–109
Matsumura N, Yokoo H, Mao Y, Yin W, Nakazato Y (2013) Olig2-positive cells in glioneuronal tumors show both glial and neuronal characters: the implication of a common progenitor cell? Neuropathology 33(3):246–255
Myung JK, Byeon SJ, Kim B, Suh J, Kim SK, Park CK, Chung CK, Chang KH, Park SH (2011) Papillary glioneuronal tumors: a review of clinicopathologic and molecular genetic studies. Am J Surg Pathol 35(12):1794–1805
Newton HB, Dalton J, Ray-Chaudhury A, Gahbauer R, McGregor J (2008) Aggressive papillary glioneuronal tumor: case report and literature review. Clin Neuropathol 27(5):317–324
Pages M, Lacroix L, Tauziede-Espariat A, Castel D, Daudigeos-Dubus E, Ridola V, Gilles S, Fina F, Andreiuolo F, Polivka M, Lechapt-Zalcman E, Puget S, Boddaert N, Liu XQ, Bridge JA, Grill J, Chretien F, Varlet P (2015) Papillary glioneuronal tumors: histological and molecular characteristics and diagnostic value of SLC44A1-PRKCA fusion. Acta Neuropathol Commun 3(1):85
Pimentel J, Barroso C, Miguéns J, Firmo C, Antunes JL (2009) Papillary glioneuronal tumor—prognostic value of the extension of surgical resection. Clin Neuropathol 28(4):287–294
Portela-Oliveira E, Torres US, Lancellotti CL, Souza AS, Ferraz-Filho JR (2014) Solid intraventricular papillary glioneuronal tumor: magnetic resonance imaging findings with histopathological correlation. Pediatr Neurol 50(2):199–200
Schlamann A, von Bueren AO, Hagel C, Zwiener I, Seidel C, Kortmann RD, Müller K (2014) An individual patient data meta-analysis on characteristics and outcome of patients with papillary glioneuronal tumor, rosette glioneuronal tumor with neuropil-like islands and rosette forming glioneuronal tumor of the fourth ventricle. PLoS One 9(7):e101211
Tan W, Huang W, Xiong J, Pan J, Geng D, Jun Z (2014) Neuroradiological features of papillary glioneuronal tumor: a study of 8 cases. J Comput Assist Tomogr 38(5):634–638
Tanaka Y, Yokoo H, Komori T, Makita Y, Ishizawa T, Hirose T, Ebato M, Shibahara J, Tsukayama C, Shibuya M, Nakazato Y (2005) A distinct pattern of Olig2-positive cellular distribution in papillary glioneuronal tumors: a manifestation of the oligodendroglial phenotype? Acta Neuropathol 110(1):39–47
Tsukayama C, Arakawa Y (2002) A papillary glioneuronal tumor arising in an elderly woman: a case report. Brain Tumor Pathol 19(1):35–39
Xiao H, Ma L, Lou X, Gui Q (2011) Papillary glioneuronal tumor: radiological evidence of a newly established tumor entity. J Neuroimaging 21(3):297–302
Acknowledgments
We gratefully acknowledge Jing-Jing Zhu, Hai-Xia Cheng, and Fang-Fang Guo for data collection and the photographic assistance.
Author contributions
Rui-Jiao Zhao conceived the study, collected the data, and wrote the first draft of this article. Xia-Ling Zhang has contributed equally to this work. Shu-Guang Chu and Ming Zhang assisted with analyzing neuroimaging data. Ling-Fei Kong and Yin Wang contributed to the data interpretation, reviewing immunostaining data, and the subsequent drafting of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Ethical approval
The authors declared that this manuscript represented a descriptive, retrospective cases analysis without any intervention, and, ethical approval was given by the medical ethics committee of the People’s Hospital of Zhengzhou University. Consent was obtained from all participants prior to their inclusion in the study.
Additional information
Rui-Jiao Zhao and Xia-Ling Zhang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zhao, RJ., Zhang, XL., Chu, SG. et al. Clinicopathologic and neuroradiologic studies of papillary glioneuronal tumors. Acta Neurochir 158, 695–702 (2016). https://doi.org/10.1007/s00701-016-2744-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-016-2744-1