
Abstract
An epidemic shift in Hepatitis A virus (HAV) infection has been observed in recent years in rapidly developing countries, with increasing numbers of severe adult cases which has led to renewed interest in vaccination. Our approach in vaccine development uses recombinant expression of the highly immunogenic HAV antigen VP1-P2a in food-grade lactic acid bacterium Lactococcus lactis and in Escherichia coli. We used genetic constructs that enable nisin-controlled expression of the antigen in L. lactis in three different forms: (a) intracellularly, (b) on the bacterial surface and (c) on the bacterial surface fused with the fragment of the E. coli flagellin molecule that can act as a molecular adjuvant. Expression of the two surface forms of the antigen was achieved in L. lactis, and the resulting antigen-displaying bacteria were administered orally to mice. Half the animals in each of the two groups developed specific IgGs, with titers increasing over time and reaching 1:422 without flagellin and 1:320 with flagellin. A much higher titer 1:25,803 was observed with the parenterally administered antigen, which was purified from E. coli. With the latter, a significant mucosal IgA response was also observed. Despite significant titers, the IgGs elicited with oral or parenteral administration could not prevent HAV from infecting cells in a virus neutralization assay, suggesting that the antibodies cannot recognize viral surface epitopes. Nevertheless, orally administered HAV antigen expressed in L. lactis elicited significant systemic humoral immune response showing the feasibility for development of effective HAV vaccine for mucosal delivery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatitis A virus (HAV) is a positive-strand RNA virus without a lipid envelope and belongs to Picornaviridae family, and genus Hepatovirus (Martin and Lemon 2006). Its genome consists of ca. 7,500 nucleotides, which encompass a single open-reading frame coding for the viral polyprotein. Its icosahedral capsid is composed of VP1, VP2 and VP3 structural proteins. Non-structural proteins 2B, 2C, 3A, 3B, 3C and 3D are involved in RNA replication and viral polyprotein processing (Martin and Lemon 2006).
HAV was first observed and described in 1970 as a causative agent of type A viral hepatitis (Feinstone et al. 1973), which is transmitted through the fecal–oral route. It causes acute hepatitis with clinical symptoms that are indistinguishable from those of other types of viral hepatitis. There were 212 million cases estimated for year 2005 (WHO 2012). The disease is usually asymptomatic in young children, but the severity of symptoms increases with age.
The HAV vaccination policy is based on cost–benefit evaluation and universal immunization has usually not been favoured. Traditionally, vaccination was recommended for people at increased risk of acquiring HAV, such as travellers to regions with high prevalence. Today, several countries (e.g. Israel, Italy, Argentina) include universal childhood immunization in their national immunization programme. This is related to the epidemiological shift that occurs during the transition of countries from developing to developed (Hendrickx et al. 2008). Better sanitation and overall living standards increase the likelihood that people will avoid HAV infection in childhood and acquire it in adulthood. This is accompanied by more severe symptoms and higher health costs (Hendrickx et al. 2008).
Vaccines against HAV have the ability to diminish significantly the disease burden and are gaining importance, especially when taking into account the recent epidemic shift. Inactivated vaccines are highly immunogenic and offer good protection (Innis et al. 1994; Ott et al. 2012). Attenuated vaccines have also been developed and are comparably efficacious (Ott et al. 2012). However, since the vaccination policy is based on cost-benefit evaluation, decreasing the vaccine cost by alternative recombinant antigen production is of great importance. Recombinant surface proteins of hepatitis B virus are already in clinical use (Rustgi et al. 1995).
Another goal in vaccine development is mucosal immunization, which is more patient friendly, mimics the natural infection route and does not require trained personnel for administration. Inactivated HAV, co-administered with adjuvants, was shown to induce strong local immune response in mice through mucosal immunization (Mitchell et al. 2006).
Lactic acid bacteria (LAB) were suggested as vectors for recombinant antigen delivery to mucosal surfaces (Wells et al. 1996). They have little intrinsic immunogenicity, significant adjuvant properties and mucus adherence ability (Berlec et al. 2012). Additionally, they pose no safety concerns and several have “generally recognized as safe” (GRAS) status. Among LAB, Lactococcus lactis has been most frequently used and its applications as an antigen delivery vehicle have recently been reviewed (Bahey-El-Din 2012; Berlec et al. 2012) and include hepatitis B vaccine (Zhang et al. 2010). Cellular localization of the antigen (cytoplasmic, secreted, surface-bound) can influence the immune response, depending on the antigen (Bahey-El-Din et al. 2008; Bermudez-Humaran et al. 2004). The response can be stimulated by concomitant administration of adjuvants. Antigen-producing lactococci have been combined with cholera toxin (Lei et al. 2011), with a strain producing the peptide leptin (Cauchard et al. 2011), or were concomitantly producing cytokine IL-12 (Bermudez-Humaran et al. 2003). Harsh gastrointestinal conditions usually diminish the efficacy of vaccines delivered orally due to the low survival of lactococci (Drouault et al. 1999); however, oral administration is the most patient-friendly.
In this work we used recombinant L. lactis and Escherichia coli to express highly immunogenic recombinant HAV antigen. We used fusion of the C-terminal of VP1 to P2a (residues 700–836; AgHAV). AgHAV is a somewhat larger fragment than that previously identified as being highly immunogenic (VP1-P2a residues 767–842; Khudyakov et al. 1999) and includes residues 714–752, previously identified on the virion surface (Robertson et al. 1989). L. lactis was used for oral delivery of the antigen, which was fused to a part of E. coli flagellin molecule, which can act as molecular adjuvant to increase the immune response (Mizel and Bates 2010).
Materials and methods
Bacterial strains, media and culture conditions
Bacterial strains used in this study are shown in Table 1. E. coli strains DH5α and BL21(DE3) were grown at 37 °C with aeration in Luria Bertani (LB) medium supplemented with 100 μg/mL ampicillin or 10 μg/mL kanamycin. L. lactis NZ9000 was grown in M-17 medium (Merck, Darmstadt, Germany) supplemented with 0.5 % glucose (GM-17) and 10 μg/mL of chloramphenicol at 30 °C without aeration.
DNA manipulation and plasmid construction
A fragment of HAV polyprotein, VP1-P2a (amino-acid residues 700–836) (Khudyakov et al. 1999), was used as antigen (AgHAV). Its protein sequence was back-translated and codon-optimized for expression in L. lactis to yield the ag HAV gene (Table 1) which was purchased from ATG:biosynthetics (Merzahusen, Germany).
PCR amplifications were performed with Taq polymerase (Fermentas, St. Leon-Rot, Germany). Plasmid DNA was isolated with Wizard SV Minipreps (Promega, Madison, USA), employing an additional lysozyme treatment step in the case of L. lactis. Electroporation of L. lactis was performed as described (Holo and Nes 1989), using a Gene Pulser II apparatus (Biorad, Hercules, USA). Nucleotide sequencing was performed by Eurofins MWG Operon (Ebersberg, Germany).
The Ag HAV gene was cloned to several plasmids (Table 1, Fig. 1) using the appropriate restriction enzymes (New England Biolabs, Beverly, USA). Recognition sites are highlighted in Fig. 1 and were introduced with appropriate primers (Thermo Fisher Scientific, Ulm, Germany; Table 1). TA cloning was used for pET SUMO. Fragments of flagellin fliC gene (N-terminal, 1–516 bp, and C-terminal, 1,230–1,494 bp) were amplified from E. coli DH5α genome with FliC-1-F-Eco/FliC-1-R-Xma and FliC-2-F-Xma/ FliC-2-R-Eco primer pair, respectively. Fragments were joined via XmaI-site and EcoRI-cloned to pSD HAV, yielding pSD HAV FLIC. Genetic constructs are depicted in Fig. 1.

Schematic representation of the organization of L. lactis and E. coli genetic constructs used in the study. USP sp Usp-LEIS secretion signal, HAV antigen gene ag HAV, FLIC fusion of N- and C-terminal regions of E. coli flagellin gene (fliC-NC), LysM peptidoglycan binding repeats of C-terminal region of AcmA, SUMO Smt3 from Saccharomyces cerevisiae, 6H hexa-histidine tag
Expression of recombinant proteins in L. lactis
Overnight cultures of L. lactis NZ9000 harbouring the appropriate plasmid (pNZ HAV, pSD HAV, pSD HAV FLIC) were diluted (1:100) in 500 mL of fresh GM-17 medium, grown to optical density A 600 = 0.80 and induced with 25 ng/mL nisin (Fluka AG, Buchs, Switzerland). Three hours after induction, the culture was centrifuged at 5,000×g for 20 min and the supernatant decanted. The pellet was resuspended in 10 mL of the growth medium and stored at 4 °C until use (maximum of 3 days). Aliquots (100 μl) were pelleted and resuspended in 0.1 M potassium phosphate buffer (pH 7.0) for SDS PAGE and Western blot analysis.
Expression and purification of AgHAV from E. coli
Overnight cultures of E. coli BL21 (DE3) harbouring pET28::HAV or pET SUMO::HAV (Table 1) were diluted (1:100) in 1 L of fresh LB medium, grown to optical density A 600 = 1.0 and induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Three hours after induction the culture was centrifuged at 5,000×g for 20 min and the supernatant decanted. The pellet was resuspended in 30 mL of equilibration/wash (E/W) buffer (50 mM NaH2PO4, 300 mM NaCl, pH 7.0). The cells were lysed with a freeze/thaw cycle and 10 min sonication with UPS200S sonifier (Hielscher, Teltow, Germany). The cell lysate was centrifuged at 14,500×g for 20 min. Supernatant was removed and stored. Inclusion bodies were dissolved in E/W buffers with increasing concentrations of guanidinium HCl (1 M, 3 M and 6 M) for at least 3 h at 4 °C with each buffer. Fractions soluble in E/W buffers with 1 M and 3 M guanidinium HCl were consecutively loaded on 14 mL of BD Talon metal affinity resin (BD Biosciences, Palo Alto, USA) according to manufacturer’s instructions, using batch/gravity-flow column purification and imidazole elution (elution buffer, 45 mM NaH2PO4, 270 mM NaCl, 1 or 3 M guanidinium HCl, 150 mM imidazole, pH 7.0). Fractions containing pure AgHAV were pooled. Solubilisation of AgHAV was achieved by a rapid 100-fold dilution in phosphate buffered saline (PBS). AgHAV was concentrated by ultrafiltration using Amicon membrane (YM10; Pall, Ann Arbor, USA) and further dialyzed against PBS. Naked AgHAV was used for i.p. administration and AgHAV-SUMO fusion for ELISA experiments.
SDS PAGE and Western blots
SDS PAGE was performed with a mini-Protean II apparatus (Bio-Rad, Hercules, USA). PageRuler Plus (Fermentas, St. Leon-Rot, Germany) prestained standard was used for molecular weight comparison. Samples were thawed on ice, briefly sonicated and denatured by heating at 100 °C in the presence of dithiothreitol before loading. Proteins were stained with Coomassie Brilliant Blue or transferred to polyvinylidene fluoride (PVDF) membrane (Immobilon-P, Millipore, Bedford, USA). The membrane was blocked in 5 % skimmed milk and incubated overnight at 4 °C with anti VP1 (HAV) rabbit IgG antibody (eEnzyme, Gaithersburg, USA; 1:5,000 dilution). After washing with TBST (50 mM Tris–HCl, 150 mM NaCl, 0.05 % Tween 20, pH 7.5), the membrane was incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Dianova, Hamburg, Germany; 1:20,000) for 1 h at room temperature. Membrane was washed as above and Lumi-LightPLUS Western Blotting Substrate (Roche, Mannheim, Germany) was used for detection on Hyperfilm ECL (Amersham, Buckinghamshire, UK). Coomassie-stained gels were analysed with ImageJ software to quantify the level of overexpression of different forms of AgHAV in L. lactis.
Immunization of mice
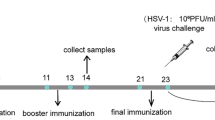
Six week-old female BALB/c mice were obtained from Medical experimental centre of the University of Ljubljana (Ljubljana, Slovenia) and divided into four groups (CONT, SD, FLIC, IP) of twelve animals each. They were housed in pathogen-free conditions and allowed free access to food and water. All experimental procedures were approved by Veterinary administration of the Republic of Slovenia. The vaccination regimen is depicted in Fig. 2 and started at 8 weeks of age. Three immunization events were performed altogether with three weeks intermediate span. Three groups (CONT, SD, FLIC) were administered orally with ca. 2 × 1010 freshly prepared bacteria (containing plasmids pNZ8148, or pSD HAV, or pSD HAV FLIC, respectively) per mouse for three consecutive days in each immunization event. One group (IP) was injected intra-peritoneally with a single dose of ca. 60 μg of naked AgHAV per mouse in each immunization event. Blood samples were obtained by tail vein bleeding before each immunization event. Faecal pellets were collected from the cage of each group. Animals were euthanized by cervical dislocation and fully bled three weeks after the last immunization. Serum and faecal samples were stored at −80 °C.

Scheme of immunization protocol. Oral refers to groups of mice with orally administered bacteria, containing pSD HAV, pSD HAV FLIC or pNZ8148. IP refers to group of mice with intra-peritoneally applied AgHAV. Days of intervention are specified. Square refers to oral administration of bacteria. Star refers to intra-peritoneal administration of the antigen. Arrow refers to bleeding and collection of faecal pellets. Cross refers to euthanization
Analysis of antibody response
Enzyme-linked immunosorbent assay (ELISA) was used to determine serum IgG and IgA antibody responses. Flat transparent 96-well microtiter plates (Corning, Cambridge, USA) were coated overnight with 100 μl of AgHAV-SUMO fusion per well at a concentration of 10 μg/mL at 4 °C. After coating, the wells were blocked with 200 μl of 2 % bovine serum albumine (BSA) in PBS overnight at 4 °C. Wells were then washed three times with PBST (PBS containing 0.05 % Tween 20) and 100 μl of mouse sera diluted in PBS with 0.2 % BSA was added. Serial 2-fold dilutions of sera (ranging from 1:20 to 1:1,310,720) were applied in three parallels and incubated at room temperature for 1 h. This was followed by washing as described previously. Wells were then filled with 100 μl of HRP-conjugated anti-mouse IgG antibodies (γ-chain specific; Sigma-Aldrich, Steinheim, Germany) at 1:2,000 dilution in PBS with 0.2 % BSA and incubated for 1 h at room temperature. The wells were washed again and incubated with 200 μl 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Sigma-Aldrich, Steinheim, Germany) for 15 min at room temperature. Colour development was terminated by the addition of 50 μl 2 M H2SO4 and absorbances were read at 450 nm using Infinite M1000 (Tecan, Salzburg, Austria). The group of mice receiving bacteria with no HAV antigen was used as a negative control. The antibody titer is expressed as the reciprocal of the highest dilution with A450 above the cut off value (i.e. average A 450 of the negative control + 2 × standard deviation of the negative control).
Approximately 200 mg of faecal pellets were suspended in PBS with 0.2 % BSA and Halt Protease Inhibitor Cocktail (Thermo Scientific, Rockford, USA) and incubated overnight at 4 °C. Suspension was centrifuged at 16,000×g for 15 min at 4 °C and supernatant stored at −80 °C. ELISA was performed essentially as described for serum samples with minor modifications as follows. Two faecal suspensions were prepared for each group of mice and each was applied in 1:2 dilutions in three parallels. HRP-conjugated anti-mouse IgA antibodies (α-chain specific; Sigma-Aldrich, Steinheim, Germany) at 1:2,000 dilution were used instead of anti-mouse IgG antibodies.
HAV neutralization test
Fetal rhesus monkey kidney (FRhK)-4 cell line was cultivated in a Dulbecco’s minimal essential medium with 10 % foetal bovine serum (Sigma-Aldrich, Steinheim, Germany) in a 96-well plate. The test virus strain was HM175/18f. Sera of three mice with the highest IgG titer from each group (CONT, SD, FLIC, IP) were incubated at 56 °C for 30 min and serially diluted. Two-fold serial dilutions were incubated with 100-fold 50 % tissue culture infective dose (TCID50) of HAV for 2 h at 37 °C and plated on FRhK-4 monolayers in duplicate. Inoculated cells were incubated at 37 °C with 5 % CO2 for 14 days. The cytopathic effect was estimated visually on the monolayer. The cell medium was then decanted and the monolayer washed twice with PBS. Cells were stained with crystal violet (1 %) for contrasting and better visualization of cell monolayer disruption. The cell damage in wells of serum dilutions of tested groups was compared with cell damage obtained in virus control and cell control wells. The reciprocal of the last dilution of serum with which an HAV neutralization effect was observed, is given as the result (Supplemental Table 1).
Nucleotide sequence accession number
The nucleotide sequence of ag HAV gene has been deposited in the GenBank database under accession number KC206063.
Results
Preparation of genetic constructs of HAV antigen
Synthetic HAV antigen gene (ag HAV) codon-optimized for expression in L. lactis was prepared on the basis of a highly-immunogenic fragment of viral polyprotein (Khudyakov et al. 1999), encompassing amino-acid residues 700–836 of VP1_P2a region. Three lactococcal genetic constructs that contain ag HAV were prepared (Fig. 1), enabling intracellular expression of antigen AgHAV (pNZ HAV), surface display of AgHAV (pSD HAV) and surface display of a molecular fusion of AgHAV with a fragment of flagellin as adjuvant (pSD HAV FLIC). Two genetic constructs were prepared for the expression of ag HAV in E. coli (Fig. 1), enabling the expression of AgHAV without fusion (pET28::HAV), and in fusion with SUMO protein to improve folding and solubility (pET SUMO::HAV).
Expression of HAV antigen in L. lactis
Expression of different forms of HAV antigen was achieved by induction with nisin in the exponential phase of the bacterial culture. Fusion proteins were detected in cells containing the plasmids pSD HAV and pSD HAV FLIC with both Coomassie staining and Western blot, using specific antibodies (Fig. 3). Fusion proteins were of the expected molecular weight (41.8 kDa with plasmid pSD HAV and 69.7 kDa with plasmid pSD HAV FLIC). Naked HAV antigen (expressed with plasmid pNZ HAV) could not be detected (Fig. 3), even by testing various expression conditions. Therefore, only pSD HAV and pSD HAV FLIC were used in the animal trial. Single oral dose of bacteria (2 × 1010) was estimated to contain approximately 7 μg of AgHAV with pSD HAV and 4 μg of AgHAV with pSD HAV FLIC.

SDS-PAGE and Western blot analysis of different AgHAV variants. SDS-PAGE was stained with Coomassie Blue. Anti VP1 (HAV) rabbit IgG antibodies were used in AgHAV detection on Western blot. Ag means AgHAV purified from E. coli. Cont, NZ, SD and FLIC are lysates of L. lactis that contain pNZ8148 (control), pNZ HAV, pSD HAV and pSD HAV FLIC, respectively. Results of Western blot are underlined. Arrows denote the position of over-expressed fusion proteins in L. lactis. MW is molecular weight marker
Expression and isolation of HAV antigen in E. coli
HAV antigen was expressed in the form of inclusion bodies under all conditions tested (growth at 37 °C, 30 °C and 25 °C, induction with 0.1–1 mM IPTG at various optical densities; data not shown) and negligible amount of the protein was in soluble form. Conditions that yielded the highest total amount of protein (growth at 37 °C, 1 mM IPTG at OD600 = 1; Fig. 4) were chosen for large-scale expression. Inclusion bodies were dissolved in incrementally increased concentrations of guanidinium HCl. Most of the denatured protein dissolved in 3 M guanidinium HCl; however the 1 M guanidinium HCl fraction was also used for further purification. A hexa-histidine tag was exploited to purify the protein with immobilized metal affinity chromatography to more than 95 % homogeneity, as judged from Coomassie stained SDS-PAGE gel (Fig. 3). Proteins were refolded, either by slow dialysis against buffers with decreasing guanidinium HCl concentrations, or by rapid dilution in PBS. Only small amounts of naked HAV antigen (ca. 2 mg per litre of bacterial culture) were successfully solubilised in PBS and were used for i.p. administration. HAV antigen-SUMO fusion was readily refolded in PBS with the rapid dilution method (ca. 15 mg per litre of bacterial culture) and was used in all ELISA studies.

SDS-PAGE of cell lysate of E. coli cells expressing AgHAV (denoted with arrows) stained with Coomassie Blue. Pel insoluble fraction of the lysate, Sn soluble fraction of the lysate, Lys total cell lysate
Humoral immune response in mice against HAV antigen
Lactococci with surface expressed AgHAV versions were used for oral administration of the vaccine and the AgHAV purified from E. coli was used in i.p. administration. An increase in the average titer with time was observed in all tested groups (Fig. 5). Animals that received purified antigen intraperitoneally (IP group) reached the geometric mean titer of 1:25,803 and a strong antibody response was observed with all the animals in the group. Animals that were orally immunized with bacteria bearing surface-displayed HAV antigen with flagellin (FLIC) or without flagellin fusion (SD) reached lower titers. Both groups were divided into two equally large sub-groups: one responding to vaccination (mean titer 1:320 for FLIC and 1:422 for SD), and the other with no response. There was no significant difference in the intensity of the immune response between the FLIC and SD groups of animals (Fig. 5).

Titers of antibodies against AgHAV in the sera of mice receiving different immunization at various time intervals. Titers for specific mice that were greater than 1:20 are shown as dots; geometric mean is shown by a horizontal line. SD means orally administered with bacteria containing pSD HAV. FLIC means orally administered with bacteria containing pSD HAV FLIC. IP means intra-peritoneally administered AgHAV
Mucosal IgA response was evaluated in the faeces belonging to SD, FLIC, IP and control group at the end of the vaccination course. A significant increase in IgA response was observed with the IP group (Fig. 6).

Response of IgA against AgHAV in the faeces of mice receiving different immunization routes for 9 weeks as determined with ELISA. CONT means control; orally administered with bacteria containing pNZ8148; IP means intraperitoneally administered AgHAV, SD orally administered with bacteria containing pSD HAV; FLIC means orally administered with bacteria containing pSD HAV FLIC. Error bars represent standard error. Statistically significant difference (t test, P < 0.05) is denoted with an asterisk
In vitro neutralization of HAV with mice sera
The neutralizing ability of anti-HAV antibodies elicited against intraperitoneally (IP) or orally (SD, FLIC) administered AgHAV was evaluated. The ability of antibodies to prevent a viral cytopathic effect on the FRhK-4 cell line was monitored and compared to the sera of mice that did not receive AgHAV (CONT). Anti-AgHAV antibodies, produced by any of the immunization protocols, could not neutralize HAV to a larger extent than the control (Supplemental Table 1).
Discussion
HAV fragment VP1-P2a has been identified as highly immunogenic (Khudyakov et al. 1999) and the importance of VP1/2A genes in HAV virulence has also been established (Emerson et al. 2002). However, to our knowledge this is the first report of the C-terminal of VP1 to P2a fusion being heterologously expressed or applied in immunization. We used E. coli for large-scale expression of purified antigen, and L. lactis for the oral delivery. Since the cellular localization of the lactococcally-delivered antigen has been reported to influence the immune response (Bahey-El-Din et al. 2008; Bermudez-Humaran et al. 2004), we aimed to produce antigen intracellularly as well as displayed on the surface with the use of nisin-controlled expression system (Mierau and Kleerebezem 2005).
Naked antigen could not be detected intracellularly under any condition tested, possibly due to its degradation. However, fusion of the antigen with the Usp45 secretion signal (Berlec et al. 2006; Ravnikar et al. 2010) and C-terminal domain of AcmA peptidoglycan anchor (Moeini et al. 2011; Raha et al. 2005; Ravnikar et al. 2010) was expressed at a considerable level, establishing L. lactis as an expression host for HAV antigens. Fusion partners can assist in proper folding and prevent the degradation that frequently occurs with the naked protein. This has been an established expression strategy in E. coli, but less common in L. lactis, though recently reported there (Douillard et al. 2011). Additionally, protein secretion (which occurs with surface display) has been reported to increase the yield of the recombinant protein (Le Loir et al. 2005). The beneficial effect of the fusion protein was also observed with the expression in E. coli, where both naked antigen and antigen-SUMO fusion were expressed in insoluble form, but the fusion protein was more readily solubilised. Efficient purification was achieved with immobilized metal affinity chromatography. The integrity of all AgHAV variants was established by their recognition by specific anti-VP1 antibodies.
Oral and parenteral administration of AgHAV variants resulted in the temporal increase of IgG titer, regardless of the administration regimen. This confirms the specificity of the immune response and justifies multiple doses. I.p. administration of purified HAV antigen elicited a very strong immune response without the addition of adjuvant and mean titer of 1:25,803. The antigen is therefore highly immunogenic, in accordance with previous findings (Khudyakov et al. 1999). Lower, but still significant titers (1:422 without and 1:320 with the molecular adjuvant) were reached with oral lactococcally mediated delivery of HAV antigen. This is to be expected since oral doses of L. lactis bacteria contained lower quantities of AgHAV in comparison to i.p. dose (approximately 9-fold or 15-fold lower dose with or without molecular adjuvant, respectively), and lower quantities still reached the systemic circulation by oral intake. The animals in the oral groups fell into two equally large subgroups—one responding to the orally delivered antigen, and the other showing no detectable antibody response. This may be due to the low amount of the delivered antigen and could be improved by administering larger quantity of bacteria. Differences in digestion or metabolism between individual animals, or postprandial or fasting state of the animal at the time of administration could also be the reason. From the temporal increase of the titer it could be deduced that interference with two or even just one dose can strongly impact on the final titer (no detectable titer was observed in the SD group after one dose or in the FLIC group after two doses).
L. lactis has been reported to possess adjuvant properties, which are beneficial in increasing the immune response against the delivered antigen (Yam et al. 2008). The adjuvant effect was increased by engineering L. lactis to produce molecular adjuvants (Cauchard et al. 2011; Lei et al. 2011). Bacterial flagellin is a ligand for toll-like receptor 5 (TLR5) (Hayashi et al. 2001) and has been used as a molecular adjuvant (Mizel and Bates 2010). The parts of the flagellin molecule that are responsible for TLR5 activation have been mapped to the N- and C-terminal parts, which are highly conserved among Gram-negative bacteria (Murthy et al. 2004). We therefore prepared a fusion of N-terminal (1–172 aa) and C-terminal (410–498 aa) parts of E. coli flagellin FliC. A similar fusion has been described for homologous Salmonella flagellin and enabled activation of TLR5 (Murthy et al. 2004). With the exception of α and ε Proteobacteria, the majority of flagellated Gram-negative bacteria can activate TLR5, whereby E. coli flagellin activates TLR5 even slightly more strongly than that of Salmonella typhimurium (Andersen-Nissen et al. 2005).
In our study, we could not observe any significant differences between the groups that were orally administered with lactococci, which displayed AgHAV with or without flagellin. This implies that the E. coli flagellin in the present form lacked adjuvant or, at least, stimulatory activity on IgG production. This may be due to the positioning of the flagellin fragment in the complex fusion protein, which may not be appropriate for its presentation. Optimization of the antigen/flagellin positioning in the fusion protein has been reported (Song et al. 2009) and should be considered in future work. In particular, positioning of the antigen between the N- and C-terminal flagellin fragments could be beneficial. Additionally, the amount of orally delivered AgHAV was lower when it was in the form of flagellin fusion, which might have contributed to the inability to observe flagellin adjuvant effect.
Mucosal immunization with inactivated HAV in mice elicited significant mucosal IgA immunity, when combined with appropriate adjuvant (Mitchell et al. 2006). In our study, however, significant mucosal humoral immunity was elicited upon i.p. administration, but not upon oral administration. This could be the consequence of technical limitations of IgA determination in faeces, or low total amount of delivered antigen. Additionally, HAV antigens could be intrinsically limited at eliciting mucosal immunity, as HAV infection in human and primates was shown to elicit low mucosal immunity (Stapleton et al. 1991).
The sera elicited against AgHAV antigen upon per os or i.p. administration could not neutralize HAV, indicating that specific IgGs could not bind viral particles and prevent them from infecting target cells. This was unexpected, particularly for the sera of i.p. administered animals, which contained anti-AgHAV antibodies with very high titer. It can, however, be partially explained by the results of previous studies. Immunization with VP1 protein already produced antibodies, which had poor or no neutralizing ability, despite being VP1 specific (Gauss-Muller et al. 1990; Hughes and Stanton 1985). P2a portion of the antigen is essential in virion morphogenesis, but is not present on the virion’s surface (Cohen et al. 2002). Also, the VP1 in a mature virion may differ from recombinant VP1 fragment. In the future, beside the virus neutralization test, HAV challenge should also be used to test the ability of antigen to elicit antibodies that prevent virion formation.
To summarize, AgHAV was successfully expressed in L. lactis and elicited significant systemic humoral immune response upon oral administration, albeit lower than that with i.p. administration of AgHAV purified from E. coli.
References
Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, Aderem A (2005) Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci USA 102(26):9247–9252
Bahey-El-Din M (2012) Lactococcus lactis-based vaccines from laboratory bench to human use: An overview. Vaccine 30(4):685–690
Bahey-El-Din M, Casey PG, Griffin BT, Gahan CG (2008) Lactococcus lactis-expressing listeriolysin O (LLO) provides protection and specific CD8(+) T cells against Listeria monocytogenes in the murine infection model. Vaccine 26(41):5304–5314
Berlec A, Jevnikar Z, Majhenic AC, Rogelj I, Strukelj B (2006) Expression of the sweet-tasting plant protein brazzein in Escherichia coli and Lactococcus lactis: a path toward sweet lactic acid bacteria. Appl Microbiol Biotechnol 73(1):158–165
Berlec A, Ravnikar M, Strukelj B (2012) Lactic acid bacteria as oral delivery systems for biomolecules. Die Pharmazie - An International Journal of Pharmaceutical Sciences 67(11):891
Bermudez-Humaran LG, Langella P, Cortes-Perez NG, Gruss A, Tamez-Guerra RS, Oliveira SC, Saucedo-Cardenas O, Montes de Oca-Luna R, Le Loir Y (2003) Intranasal immunization with recombinant Lactococcus lactis secreting murine interleukin-12 enhances antigen-specific Th1 cytokine production. Infect Immun 71(4):1887–1896
Bermudez-Humaran LG, Cortes-Perez NG, Le Loir Y, Alcocer-Gonzalez JM, Tamez-Guerra RS, de Oca-Luna RM, Langella P (2004) An inducible surface presentation system improves cellular immunity against human papillomavirus type 16 E7 antigen in mice after nasal administration with recombinant lactococci. J Med Microbiol 53(Pt 5):427–433
Cauchard S, Bermudez-Humaran LG, Blugeon S, Laugier C, Langella P, Cauchard J (2011) Mucosal co-immunization of mice with recombinant lactococci secreting VapA antigen and leptin elicits a protective immune response against Rhodococcus equi infection. Vaccine 30(1):95–102
Cohen L, Benichou D, Martin A (2002) Analysis of deletion mutants indicates that the 2A polypeptide of hepatitis A virus participates in virion morphogenesis. J Virol 76(15):7495–7505
de Ruyter PG, Kuipers OP, de Vos WM (1996) Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Appl Environ Microbiol 62(10):3662–3667
Douillard FP, O’Connell-Motherway M, Cambillau C, van Sinderen D (2011) Expanding the molecular toolbox for Lactococcus lactis: construction of an inducible thioredoxin gene fusion expression system. Microb Cell Fact 10:66
Drouault S, Corthier G, Ehrlich SD, Renault P (1999) Survival, physiology, and lysis of Lactococcus lactis in the digestive tract. Appl Environ Microbiol 65(11):4881–4886
Emerson SU, Huang YK, Nguyen H, Brockington A, Govindarajan S, St Claire M, Shapiro M, Purcell RH (2002) Identification of VP1/2A and 2C as virulence genes of hepatitis A virus and demonstration of genetic instability of 2C. J Virol 76(17):8551–8559
Feinstone SM, Kapikian AZ, Purcell RH (1973) Hepatitis A: detection by immune electron microscopy of a viruslike antigen associated with acute illness. Science 182(4116):1026–1028
Gauss-Muller V, Zhou MQ, von der Helm K, Deinhardt F (1990) Recombinant proteins VP1 and VP3 of hepatitis A virus prime for neutralizing response. J Med Virol 31(4):277–283
Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A (2001) The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410(6832):1099–1103
Hendrickx G, Van Herck K, Vorsters A, Wiersma S, Shapiro C, Andrus JK, Ropero AM, Shouval D, Ward W, Van Damme P (2008) Has the time come to control hepatitis A globally? Matching prevention to the changing epidemiology. J Viral Hepat 15(Suppl 2):1–15
Holo H, Nes IF (1989) High-Frequency Transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl Environ Microbiol 55(12):3119–3123
Hughes JV, Stanton LW (1985) Isolation and immunizations with hepatitis A viral structural proteins: induction of antiprotein, antiviral, and neutralizing responses. J Virol 55(2):395–401
Innis BL, Snitbhan R, Kunasol P, Laorakpongse T, Poopatanakool W, Kozik CA, Suntayakorn S, Suknuntapong T, Safary A, Tang DB, Boslego JW (1994) Protection against hepatitis A by an inactivated vaccine. Jama 271(17):1328–1334
Khudyakov YE, Lopareva EN, Jue DL, Fang S, Spelbring J, Krawczynski K, Margolis HS, Fields HA (1999) Antigenic epitopes of the hepatitis A virus polyprotein. Virology 260(2):260–272
Le Loir Y, Azevedo V, Oliveira SC, Freitas DA, Miyoshi A, Bermudez-Humaran LG, Nouaille S, Ribeiro LA, Leclercq S, Gabriel JE, Guimaraes VD, Oliveira MN, Charlier C, Gautier M, Langella P (2005) Protein secretion in Lactococcus lactis: an efficient way to increase the overall heterologous protein production. Microb Cell Fact 4(1):2
Lei H, Sheng Z, Ding Q, Chen J, Wei X, Lam DM, Xu Y (2011) Evaluation of oral immunization with recombinant avian influenza virus HA1 displayed on the Lactococcus lactis surface and combined with the mucosal adjuvant cholera toxin subunit B. Clin Vaccine Immunol 18(7):1046–1051
Martin A, Lemon SM (2006) Hepatitis A virus: from discovery to vaccines. Hepatology 43(2 Suppl 1):S164–S172
Mierau I, Kleerebezem M (2005) Ten years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Appl Microbiol Biotechnol 68(6):705–717
Mitchell LA, Joseph A, Kedar E, Barenholz Y, Galun E (2006) Mucosal immunization against hepatitis A: antibody responses are enhanced by co-administration of synthetic oligodeoxynucleotides and a novel cationic lipid. Vaccine 24(25):5300–5310
Mizel SB, Bates JT (2010) Flagellin as an adjuvant: cellular mechanisms and potential. J Immunol 185(10):5677–5682
Moeini H, Rahim RA, Omar AR, Shafee N, Yusoff K (2011) Lactobacillus acidophilus as a live vehicle for oral immunization against chicken anemia virus. Appl Microbiol Biotechnol 90(1):77–88
Murthy KG, Deb A, Goonesekera S, Szabo C, Salzman AL (2004) Identification of conserved domains in Salmonella muenchen flagellin that are essential for its ability to activate TLR5 and to induce an inflammatory response in vitro. J Biol Chem 279(7):5667–5675
Ott JJ, Irving G, Wiersma ST (2012) Long-term protective effects of hepatitis A vaccines. A systematic review. Vaccine. doi:10.1016/j.vaccine.2012.04.104
Raha AR, Varma NR, Yusoff K, Ross E, Foo HL (2005) Cell surface display system for Lactococcus lactis: a novel development for oral vaccine. Appl Microbiol Biotechnol 68(1):75–81
Ravnikar M, Strukelj B, Obermajer N, Lunder M, Berlec A (2010) Engineered lactic acid bacterium Lactococcus lactis capable of binding antibodies and TNFalpha. Appl Environ Microbiol 76(20):6928–6932
Robertson BH, Brown VK, Holloway BP, Khanna B, Chan E (1989) Structure of the hepatitis A virion: identification of potential surface-exposed regions. Arch Virol 104(1–2):117–128
Rustgi VK, Schleupner CJ, Krause DS (1995) Comparative study of the immunogenicity and safety of Engerix-B administered at 0, 1, 2 and 12 months and Recombivax HB administered at 0, 1, and 6 months in healthy adults. Vaccine 13(17):1665–1668
Song L, Zhang Y, Yun NE, Poussard AL, Smith JN, Smith JK, Borisevich V, Linde JJ, Zacks MA, Li H, Kavita U, Reiserova L, Liu X, Dumuren K, Balasubramanian B, Weaver B, Parent J, Umlauf S, Liu G, Huleatt J, Tussey L, Paessler S (2009) Superior efficacy of a recombinant flagellin:H5N1 HA globular head vaccine is determined by the placement of the globular head within flagellin. Vaccine 27(42):5875–5884
Stapleton JT, Lange DK, LeDuc JW, Binn LN, Jansen RW, Lemon SM (1991) The role of secretory immunity in hepatitis A virus infection. J Infect Dis 163(1):7–11
Wells JM, Robinson K, Chamberlain LM, Schofield KM, Le Page RW (1996) Lactic acid bacteria as vaccine delivery vehicles. Antonie Van Leeuwenhoek 70(2–4):317–330
WHO (2012) WHO position paper on hepatitis A vaccines - June 2012. Weekly epidemiological record 28–29(87):261–276
Yam KK, Pouliot P, N’Diaye MM, Fournier S, Olivier M, Cousineau B (2008) Innate inflammatory responses to the Gram-positive bacterium Lactococcus lactis. Vaccine 26(22):2689–2699
Zhang Q, Zhong J, Huan L (2010) Expression of hepatitis B virus surface antigen determinants in Lactococcus lactis for oral vaccination. Microbiol Res 166(2):111–120
Acknowledgements
This study was supported by the Slovenian Research Agency Grant No. P4-0127. The authors are grateful to Prof. Roger Pain for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM 1
(PDF 68 kb)
Rights and permissions
About this article
Cite this article
Berlec, A., Malovrh, T., Zadravec, P. et al. Expression of a hepatitis A virus antigen in Lactococcus lactis and Escherichia coli and evaluation of its immunogenicity. Appl Microbiol Biotechnol 97, 4333–4342 (2013). https://doi.org/10.1007/s00253-013-4722-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-4722-3