
Abstract
Lactococcus lactis is a Gram-positive lactic acid bacterium that, in addition to its traditional use in food fermentations, is increasingly used in modern biotechnological applications. In the last 25 years great progress has been made in the development of genetic engineering tools and the molecular characterization of this species. A new versatile and tightly controlled gene expression system, based on the auto-regulation mechanism of the bacteriocin nisin, was developed 10 years ago—the NIsin Controlled gene Expression system, called NICE. This system has become one of the most successful and widely used tools for regulated gene expression in Gram-positive bacteria. The review describes, after a brief introduction of the host bacterium L. lactis, the fundaments, components and function of the NICE system. Furthermore, an extensive overview is provided of the different applications in lactococci and other Gram-positive bacteria: (1) over-expression of homologous and heterologous genes for functional studies and to obtain large quantities of specific gene products, (2) metabolic engineering, (3) expression of prokaryotic and eukaryotic membrane proteins, (4) protein secretion and anchoring in the cell envelope, (5) expression of genes with toxic products and analysis of essential genes and (6) large-scale applications. Finally, an overview is given of growth and induction conditions for lab-scale and industrial-scale applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Kuipers et al. (1995) published a paper describing the auto-induction of the expression of the lactococcal bacteriocin nisin. In the same paper they described an experiment in which this mechanism was used to drive the induction of the expression of a heterologous protein—a β-glucuronidase from Escherichia coli—by nisin. This marked the birth of one of the most successful and widely used Gram-positive gene expression systems, which was later dubbed as NICE—NIsin-Controlled gene Expression. This review will give an overview of the background and function of the system, on its applications and current and future developments.
The host Lactococcus lactis
Lactococcus lactis is a homofermentative bacterium that is used for the production of fermented milk products such as buttermilk, fermented butter, many varieties of soft and hard cheeses, Scandinavian ropy fermented milk products and as a symbiotic partner in kefir. Its importance cannot be underestimated. Teuber (1995) calculated that annually, approximately 100 million tons of milk are inoculated with Lactococcus species, and that this results at the end of the growth in ca. 500,000 tons of bacteria or the weight of ca. 6 million people, equalling half of the human population of The Netherlands.
The primary function of lactococci is rapid lactic acid production from lactose that leads to the preservation of the otherwise quickly spoiled milk. In secondary processes the bacterial culture contributes to both flavour and texture of the product (Teuber 1995; Leroy and Devuyst 2004). Flavour can be generated by the production of carbon-source-derived aroma compounds, like diacetyl, or by the degradation of milk proteins or fats into specific flavour compounds or their precursors. The texture is influenced by the degradation of milk proteins and the formation of polysaccharides. Lactococci are ubiquitous on plant material and probably came into use with the domestication of farm animals at least 10,000 years ago. Almost 150 years ago Louis Pasteur was the first to recognize that lactic acid fermentation is caused by bacteria. In terms of microbiology history, it is noteworthy that lactococci were the first bacteria ever that were isolated in pure culture by Joseph Lister in 1873 (Teuber 1995). Since this time, especially because of their economic importance, these bacteria have been intensely investigated and characterized. Today, we have a wealth of knowledge of the microbiology, physiology, ecology, technology and genetics of these bacteria (Teuber 1995; Wood and Warner 2003). In the last 25 years impressive progress has been made in the development of genetic engineering tools and the molecular characterization of lactococci (Gasson and de Vos 1994; Wood and Warner 2003). The tools include transformation, the availability of many different vectors, gene integration, gene knockout, conjugation, and constitutive and regulated gene expression systems. Functional characteristics that have extensively been studied in lactococci include the carbon metabolism, the extracellular and intracellular proteolytic system, the production of antibiotic substances, and their interaction with and resistance to bacteriophages (Gasson and de Vos 1994; Wood and Warner 2003). At present the genome information of at least three strains of L. lactis is elucidated and largely publicly available (Bolotin et al. 2001; Klaenhammer et al. 2002). The next stage is the genome-wide characterization of gene transcription, protein composition and metabolic processes and the response of all the aforementioned to changes in the environment or deliberately introduced genetic changes (Kleerebezem et al. 2002; Guillot et al. 2003).
This wealth of knowledge and experience has led to the use of lactococci far beyond their original role in food preservation and production. A few examples are the expression of bacterial and viral antigens for safe vaccination via mucosal immunization, the production of human cytokines and other therapeutic agents for in situ treatments, the use of lactococci as a cell factory for the production of specific compounds and the pilot production of pharmaceutical products (Hols et al. 1999; Hugenholtz et al. 2000, 2002; Nouaille et al. 2003; Hanniffy et al. 2004; Leroy and Devuyst 2004; Mierau et al. 2005a). The availability of an easy-to-operate and strictly controlled gene expression system (NICE) has been crucial for the development of many of these applications.
Nisin and the regulation of nisin biosynthesis
Nisin is a 34-amino acid anti-microbial peptide (lantibiotic) with various unusual amino acids and five ring structures (Fig. 1). Nisin first binds to lipidII and then forms, together with this cell-wall synthesis precursor, small pores in the cytoplasmic membrane that lead to leakage of small molecules, including adenosine triphosphate (ATP), and subsequently, to cell death (Hasper et al. 2004). Because of its broad host spectrum it is widely used as a preservative in the food industry (Dodd and Gasson 1994; van Kraaij et al. 1999). Initially, nisin is ribosomally synthesized as a precursor. Subsequent enzymatic modifications introduce the unusual chemical and structural features of the molecule. Finally, the modified molecule is translocated across the cytoplasmic membrane and processed into its mature form (Kleerebezem et al. 1997b). Biosynthesis of nisin is encoded by a cluster of 11 genes (Fig. 2), of which the first gene, nisA, encodes the precursor of nisin. The other genes direct the synthesis of proteins that are involved in the modification, translocation and processing of nisin (nisB, nisC, nisP, and nisT), in the immunity against nisin (nisI, nisF, nisE, and nisG) and in the regulation of the expression of the nisin genes (nisR and nisK) (Kuipers et al. 1995; Kleerebezem et al. 1999). NisR and NisK belong to the family of bacterial two-component signal transduction systems. NisK is a histidine–protein kinase that resides in the cytoplasmic membrane and is proposed to act as a receptor for the mature nisin molecule. Upon binding of nisin to NisK it auto-phosphorylates and transfers the phosphate group to NisR, which is a response regulator that becomes activated upon phosphorylation by NisK. Activated NisR* induces transcription from two of the three promoters in the nisin gene cluster: PnisA and PnisF (Fig. 2). The promoter that drives the expression of nisR and nisK is not affected (Kuipers et al. 1995, 1998; de Ruyter et al. 1996a; Kleerebezem et al. 1999).

Schematic representation of mature nisin. Dhb indicates dehydrobutyrine; Dha, dehydroalanine; Ala–S–Ala, lanthionine; and Abu–S–Ala, β-methyllanthionine

Schematic representation of the nisin gene cluster. Black arrows indicate the three promoters that regulate expression of the nisin genes. P*nisA indicates the nisA promoter that is used for the nisin-controlled gene expression (NICE) systems. A indicates nisin A structural gene; B, T, C and P, genes involved in modification, translocation and processing of nisin; I, F, E and G, genes involved in immunity against nisin; and R and K, genes involved in the regulation of the expression of the nisin gene cluster
The nisin-controlled gene expression system
For exploitation of the auto-induction mechanism of nisin for gene expression, the genes for the signal transduction system nisK and nisR were isolated from the nisin gene cluster and inserted into the chromosome of L. lactis subsp. cremoris MG1363 (nisin-negative), creating the strain NZ9000 (Kuipers et al. 1998) (see Table 1). When a gene of interest is subsequently placed behind the inducible promoter PnisA on a plasmid [e.g. pNZ8048 (Kuipers et al. 1998) (see Table 1)] or on the chromosome (Koebmann et al. 2000; Boels et al. 2004; Simões-Barbosa et al. 2004), expression of that gene can be induced by the addition of sub-inhibitory amounts of nisin (0.1–5 ng/ml) to the culture medium (Fig. 3) (de Ruyter et al. 1996b). Depending on the presence or absence of the corresponding targeting signals, the protein is expressed into the cytoplasm, into the membrane or secreted into the medium.

Nisin-controlled gene expression. NisK indicates histidine–protein kinase; NisR, response regulator; and Gene X, target gene cloned behind the nisA promoter with or without targeting signals
Studies with increasing amounts of nisin, using the β-glucuronidase gene as the reporter, show a linear dose–response curve (Kuipers et al. 1995; de Ruyter et al. 1996a; Kleerebezem et al. 1999). This shows that the NICE system can be used not only for on/off gene expression studies but also to dose the target protein. This property has elegantly been used in a number of metabolic engineering studies to show enzyme concentration-dependant production of specific metabolites (Lopez de Felipe et al. 1998; Hols et al. 1999; Looijesteijn et al. 1999; Hugenholtz et al. 2000).
Host strains
Table 1 gives an overview of commonly used host strains and plasmids and their sources. All strains are derivatives of L. lactis subsp. cremoris MG1363, a plasmid-free progeny of the dairy starter strain NCDO712 (Gasson 1983). NZ9700 (Kuipers et al. 1993, 1998) is a nisin-producing strain that was obtained by conjugation of the nisin–sucrose transposon Tn5276 of the nisin-A-producer NIZO B8 (previously R5) with MG1614, a rifampicin- and streptomycin-resistant derivative of MG1363 (Gasson 1983). This strain is often used as a source of nisin for induction of the NICE system. NZ9800 is a derivative of strain NZ9700, in which four nucleotides of the nisA gene have been deleted (Kuipers et al. 1993). This strain does not produce nisin, and the transcription of the nisin operon is abolished. Transcription of the nisin operon in this strain can be reactivated by the addition of sub-inhibitory amounts of nisin (Kuipers et al. 1995). This was one of the early host strains of the NICE system since it provides the necessary regulatory genes (nisK and nisR) and immunity to nisin. Presently, the most commonly used host strain is NZ9000. To construct this strain the genes for nisK and nisR were integrated into the pepN gene of MG1363 (de Ruyter et al. 1996a; Kuipers et al. 1998). The two genes are transcribed from their own constitutive promoter.
Strain NZ3900 was developed for food-grade applications of the NICE system (de Ruyter et al. 1996a, 1997). It is derived from strain NZ3000, which is a lacF deletion mutant of strain MG5267, a strain with a single chromosomal copy of the lactose operon of the dairy starter strain NCDO712. The lactose operon was transferred to strain MG1363 by transduction, creating strain MG5267. Due to the lacF deletion, strain NZ3000 is unable to grow on lactose. However, growth on lactose can be restored by providing lacF on a plasmid (Platteeuw et al. 1996). Finally, NZ3900 was obtained by inserting nisRK into the pepN gene as described for NZ9000 (de Ruyter et al. 1996a).
In addition to the use of nisK and nisR genes that are integrated into the chromosome, two plasmids have been constructed based on the broad-host-range plasmid pAMβ1 that allow transfer of the NICE system to other species (see below): pNZ9520 (high copy number) and pNZ9530 (low copy number) (Kleerebezem et al. 1997a).
Alongside various alternative food-grade selection systems (de Vos 1999), a new attractive selection strategy has been developed based on the lethal phenotype of an alanine racemase mutation (Bron et al. 2002). In addition to its food-grade nature it allows stringent selection for the presence of the recombinant plasmid, which is not possible with selection on growth of a fermentable sugar, since there are always other fermentable carbon sources in industrial fermentation media.
The elucidation of the genome sequence of L. lactis led to the identification of a hitherto unknown proteinase that is bound to the extracellular surface of the membrane. It functions both in the degradation of abnormally folded secreted proteins and in the maturation of secreted pro-proteins (Poquet et al. 2000). Deletion of this proteinase gene (NZ9000 ΔhtrA) leads to increased stability of heterologous-secreted proteins (Lindholm et al. 2004).
Plasmids
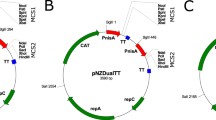
Plasmids have been constructed for translational and transcriptional fusions and for intracellular production or secretion of the gene product. pNZ8048 is the most commonly used plasmid for translational fusions. A gene of interest can be PCR-amplified using primers that introduce the canonical NcoI site around the ATG start codon, allowing direct cloning of the gene fused to the nisA start codon. Recently, two variants of this plasmid were constructed: pNZ8148 and pNZ8150 (Fig. 4a and b). In pNZ8148 a small 60-bp remnant DNA-fragment of Bacillus subtilis, the initial cloning host of the pSH series of plasmids (de Vos 1987), was removed, making the plasmid conform to self-cloning guidelines. In plasmid pNZ8150 (Fig. 4b) the NcoI site was exchanged for an ScaI site, which is situated directly upstream of the ATG start codon. This improved version of pNZ8148 avoids the disadvantage generated by the obligate use of the NcoI site, which in some cases makes it necessary to change the first base of the second codon of the gene of interest. In pNZ8150, the gene of interest can be amplified, starting at the ATG start codon and bluntly fused to the vector, creating an accurate nisA translational fusion. Plasmid pNZ8021 (Fig. 4c) can be used for transcriptional fusions. Prior to the gene of interest a truncated nisin A precursor of 33 amino acids, with a number of additional amino acids (about ten, depending on the restriction site used) from a different reading frame, will be produced. pNZ8110 (Fig. 4d) can be used for protein secretion using the signal sequence of the major secreted protein Usp45 of L. lactis (van Asseldonk et al. 1993). The advent of cheap and fast gene synthesis with codon usage correction makes the availability of a large array of vectors unnecessary, since in most cases, gene synthesis will be faster and cheaper than the preparation of complicated constructs (Fuglsang 2003; Gupta et al. 2004).

Four commonly used basic cloning vectors for the NICE system. Pnis indicates nisA promoter; nisA′, truncated nisA gene; T, transcription terminator; repA and repC, replication proteins; cm, chloramphenicol resistance; and ssUSP, signal sequence of the L. lactis usp45 gene (van Asseldonk et al. 1993)
Transfer of the NICE system to other bacteria
Because of its simplicity and its powerful induction characteristics, the NICE system has been transferred to other low-GC Gram-positive bacteria. This has, for example, allowed the establishment of regulated gene expression where none existed or the establishment of better-regulated systems than those so far available (Eichenbaum et al. 1998; Neu and Henrich 2003). The establishment of the NICE system in other bacteria has facilitated the study of pathogenic streptococci and enterococci, and allowed dose–response studies for live vaccines (Eichenbaum et al. 1998; Pavan et al. 2000; Hughes et al. 2002; Ribardo and McIver 2003; Waters et al. 2003). Using the dual plasmid system pNZ9520/30 and one of the nisA promoter vectors (typically with β-glucuronidase or β-galactosidase as reporter gene, e.g. pNZ8008) the NICE system was introduced into Leuconostoc lactis, Lactobacillus brevis, Lactobacillus helveticus, Lactobacillus plantarum, Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus pneumoniae, Streptococcus zooepidemicus, Enterococcus faecalis and B. subtilis (Kleerebezem et al. 1997a; Eichenbaum et al. 1998; Pavan et al. 2000; Åvall-Jääskeläinen et al. 2002; Chong and Nielsen 2003). In many cases regulated gene expression can be established; however, growth of several species is retarded by the introduction of the double-plasmid system. This is the case for Leu. lactis, Lb. helveticus and Lb. plantarum (Kleerebezem et al. 1997a; Pavan et al. 2000). However, this seems not to be the case for Streptococcus species and B. subtilis (Eichenbaum et al. 1998). Other strategies have been employed for E. faecalis, Lb. plantarum and Lactobacillus gasseri. For E. faecalis a vector (pMSP3535, Table 1) has been developed that carries both the nisRK genes and the nisA promoter on one plasmid, considerably simplifying the transfer procedure (Bryan et al. 2000). In Lb. plantarum and Lb. gasseri the nisRK genes were integrated as single copies into the chromosome (Pavan et al. 2000; Neu and Henrich 2003). This strategy was very successful to improve growth, to establish tight regulation and to accomplish reliable and stable expression results.
In general these examples demonstrate that the NICE system can successfully be transferred to other Gram-positive bacteria; however, each case is different because of variations in nisin sensitivity, in the primary amino acid sequence of the RNA polymerase (possible interaction with NisR) and in other factors. New systems need to be optimized in relation to the location of the nisRK genes (e.g. on a different plasmid in relation to the nisA promoter with the target gene, or both on the same plasmid, or the nisRK genes on the chromosome), the amount of nisin for induction, cell density for optimum induction, production time, medium composition, temperature etc.
NICE-like systems
Two-component regulatory systems are common in the biosynthesis of bacteriocins of Gram-positive bacteria (Kleerebezem and Quadri 2001; Kotelnikova and Gelfand 2002). Because of the excellent properties of the NICE system (tight regulation and high degree of induction) and its universal applicability, the same regulation mechanism has been used to develop expression systems for other Gram-positive bacteria from their own genetic material. NICE-like systems are now available for B. subtilis [SURE (Subtilin Regulated gene Expression) (Kleerebezem et al. 2004)], Lb. plantarum (Mathiesen et al. 2004), Lactobacillus sakei (Axelsson et al. 2003) and Enterococcus (Hickey et al. 2003). Although the NICE system can be transferred into other bacteria, development of homologous systems will be advantageous for the ease and range of use because it will consist only of genetic elements of the relevant bacterium.
Overview of applications of the NICE system
In the following section we will give an overview of known applications of the NICE system with some pertinent examples.
Over-expression of homologous and heterologous genes
The NICE system has been used to express genes of various different backgrounds (Gram-positive, Gram-negative and eukaryotic) to study metabolic and enzyme function and to produce larger amounts of an enzyme for food, medical or technical applications. With the homologous pepN gene of L. lactis it has been shown that protein production up to 50% of the total cellular protein is possible without the formation of inclusion bodies (de Ruyter et al. 1996b). The β-glucuronidase gene of the Gram-negative E. coli has been expressed up to 20% of the total cellular protein (de Ruyter et al. 1996a). Furthermore, the NICE system has been used to study the genetics and biology of pathogenic bacteria (McCormick et al. 2001; Antiporta and Dunny 2002; Francia and Clewell 2002; Ribardo and McIver 2003; Waters et al. 2003, 2004; Rigoulay et al. 2004) and to study genetic entities such as chromosomes and bacteriophage genomes (Madsen et al. 1999; Zhou et al. 2000; Campo et al. 2002; McGrath et al. 2002; Zúñiga et al. 2002). In general the experience is that genes of these various backgrounds can be expressed; however, the yield is very much case-dependant. The greatest obstacle is probably codon usage. Genes of closely related Gram-positive organisms (the genera Streptococcus, Enterococcus, Staphylococcus and low-GC Lactobacillus) are almost always expressed effectively and hardly present any problems. The expression of genes of other organisms depends on the codon usage and the distribution of rarely used codons.
There are many examples in which the NICE system has been used to express enzymes that can be used in food applications, although at present, there is, to our knowledge, no commercially used application. Important examples are the expression of phage lysins, various peptidases and esterases to influence, for instance, flavour formation in dairy fermentations (de Ruyter et al. 1997; Wegmann et al. 1999; Fernández et al. 2000; Luoma et al. 2001; Christensson et al. 2002; Hickey et al. 2004). Another important feature of the NICE system is that it is possible to control not only the expression of one gene but of a whole operon, as shown for the eight-gene (F1F0) H+-ATPase operon of L. lactis (Koebmann et al. 2000) or the rfb operon of L. lactis (Boels et al. 2004).
Metabolic engineering
Metabolic engineering is based on the application of genetic engineering methods to manipulate cellular processes and structures with the aim to study, improve or redirect cellular functions (Nielsen 2001). L. lactis has widely been used as a model system for metabolic engineering studies because it has a rather simple carbon and energy metabolism in which the carbon source is mainly transformed into lactic acid via the central metabolite pyruvate (Kleerebezem and Hugenholtz 2003). The possibility to dose the expression of a gene of interest by varying the amount of nisin that is added for induction is unique and makes the NICE system an ideal instrument to study gradual changes in a metabolic route. Therefore, nisin-dosed expression has been used extensively to study, engineer and model sugar catabolism in L. lactis and the conversion of pyruvate into various alternative end products, like diacetyl and l-alanine (Lopez de Felipe et al. 1998; Hols et al. 1999; Lopez de Felipe and Hugenholtz 1999; Hugenholtz et al. 2000; Wouters et al. 2000; Hoefnagel et al. 2002; Neves et al. 2002; Ramos et al. 2004; Smid et al. 2005). As mentioned above, the NICE system cannot only be used to drive expression of single genes, but also of whole operons. This feature has also been used to study and manipulate complex metabolic pathways, like the production of exopolysaccharides (Looijesteijn et al. 1999; Boels et al. 2001, 2003, 2004) and of the vitamins folate and riboflavin, leading to higher product yields (Hugenholtz et al. 2002; Sybesma et al. 2003a,b; Burgess et al. 2004; de Vos and Hugenholtz 2004).
Expression of integral membrane proteins
Lactococcus lactis is an excellent tool for the expression and study of integral membrane proteins of both prokaryotes and eukaryotes (Kunji et al. 2003). In addition to straightforward cloning and cultivation procedures, (1) many strains of L. lactis are auxotrophic, allowing the incorporation of various labels, (2) the tightly regulated NICE system allows the cloning and induction of membrane proteins that are often toxic for the cell, (3) expressed membrane proteins are only targeted to the cytoplasmic membrane, (4) the cells have a weak proteolytic activity (htrA) that can easily be eliminated (see above), (5) L. lactis has only one membrane, allowing direct functional studies with either intact bacteria or isolated membrane vesicles and (6) the membrane proteins can easily be solubilized with various detergents (Kunji et al. 2003). L. lactis has been used for the expression and functional analysis of various classes of prokaryotic integral membrane proteins such as ATP-binding cassette (ABC) transporters, ABC efflux pumps, major facilitator superfamily proteins, peptide transporters, mechano-sensitive channel, ATP/adenosine diphosphate (ADP) transporters etc. (Hagting et al. 1997; Franke et al. 1999; Heuberger et al. 2001; Sakamoto et al. 2001; Kunji et al. 2003). These transporters have been expressed to sometimes very high levels of up to 30% of all membrane proteins (Margolles et al. 1999) and, in general, to 1–10% of all membrane proteins (Kunji et al. 2003). Recently, it has been shown that L. lactis is also suitable for the expression of eukaryotic membrane proteins, like the KDEL receptor and different mitochondrial and hydrogenosomal carriers (Kunji et al. 2003). These proteins could not only be expressed at between 0.1 and 5% of all membrane proteins, but they were also functionally intact and showed the characteristics that they have in their natural environment.
Protein secretion and surface exposure of proteins
Lactococcus lactis is a Gram-positive bacterium and therefore has only one cellular membrane. This makes it an ideal host for protein secretion with subsequent membrane- or cell-wall-anchoring, or export into the fermentation medium. Another advantage is the low extracellular proteinase activity in lactococci. To date there are only two proteinases known: (1) the cell-wall-anchored proteinase PrtP (200 kD) (Kunji et al. 1996) and (2) the housekeeping membrane-bound proteinase HtrA (Poquet et al. 2000). The first is plasmid-encoded and absent in the plasmid-free host strains (Gasson 1983). For the second a viable mutation can be constructed that helps to stabilize secreted proteins (Miyoshi et al. 2002; Lindholm et al. 2004) (see above).
In comparison to the aerobically growing B. subtilis, which can secrete several grams of protein per litre, protein secretion in Lactococcus is less effective. Nonetheless, Lactococcus is an interesting host for, e.g. surface display of various antigens and the development of live vaccines and other in situ applications (Enouf et al. 2001; Le Loir et al. 2001b; Åvall-Jääskeläinen et al. 2002; Bermúdez-Humarán et al. 2002, 2003; Ribeiro et al. 2002; Chatel et al. 2003; Steen et al. 2003; Lindholm et al. 2004). One of the latest areas of application has been the expression and secretion of a surface-layer protein with a yield of about 100 mg/l for the exploration of nanobiotechnological applications (Novotny et al. 2005).
Research on the improvement of the secretion efficiency of lactococci is still ongoing (Le Loir et al. 2001a; Nouaille et al. 2003, 2004; Ravn et al. 2003) and is now intensified by the elucidation of the genome sequences of many Gram-positive bacteria. To date, two signal peptides are mainly used to effect protein secretion: (1) the signal peptide of the major lactococcal-secreted protein Usp45 (van Asseldonk et al. 1993) and (2) the signal peptide of the cell-wall-associated proteinase PrtP. In general the signal peptide of Usp45 gives better results and is more widely used than that of PrtP (Arnau et al. 1997; Le Loir et al. 2001a; Nouaille et al. 2003).
For the exposure of proteins on the cell wall two principal systems have been developed: (1) the sortase system that uses the LPXTG motive at the N- or C-terminus of the protein (Dieye et al. 2001; Ton-That et al. 2004) and (2) the cell-wall anchor of the major autolysin of L. lactis that also can be attached either N- or C-terminally (Steen et al. 2003).
Expression and analysis of toxic or essential gene products
One of the great strengths of the NICE system is that it is tightly regulated, and genes that would otherwise have a detrimental effect on the cell can be cloned, analysed and expressed. Two examples are the cloning and expression of the lysin and holin genes of a homologous bacteriophage (de Ruyter et al. 1997) and of cell-wall-lytic enzymes of other bacteria (Cibik et al. 2001; Hickey et al. 2004). Other examples are the expression of integral membrane proteins that were mentioned above. In very rare cases the residual leakage of the system can lead to unsuccessful cloning attempts when genes are cloned that encode toxic gene products. This can be amended by placing also the nisA promoter in single copy on the chromosome (Henrich et al. 2002; Simões-Barbosa et al. 2004).
The tight control of gene expression displayed by the NICE system also allows the functional study of essential genes in any background in which the NICE system can be implemented. By integration of the nisA promoter upstream of the gene under study, expression of essential genes can be controlled by the addition of nisin to the medium, thereby allowing growth. Subsequent removal of nisin from the growth medium leads to increasing depletion of the essential gene product, allowing the investigation of the mutant phenotype. This strategy has been employed successfully to study the phenotypes of the (F1F0) H+-ATPase (Koebmann et al. 2000), the rfb locus in L. lactis (Boels et al. 2004) and the alr gene in Lb. plantarum (Bron et al. 2002). This application of the NICE system raises possibilities for high-throughput essential gene screening methods that can be of great value for pharmaceutical drug target research.
Industrial-scale applications
To our knowledge, nothing has been published yet on large-scale use of the NICE system, although clearly, there is great potential. Recently, in our own facilities, we conducted experiments to upscale the nisin-induced gene expression, using the anti-microbial protein lysostaphin as a model. This process was straightforward and without complications. Fermentation protocols can be scaled up from the 1- to 300-l scale and to the 3,000-l scale, with almost identical fermentation characteristics and product yields (100 mg/l). Four consecutive fermentations with nisin induction at 3,000 l were virtually identical (Mierau et al. 2005a). In the same process it was also demonstrated that the downstream processing is straightforward and easy. With four unit operations—microfiltration, homogenization, second microfiltration and chromatography—a product with 90% purity was obtained (Mierau et al. 2005a).
Optimization of the NICE system for large-scale applications
As can be seen from the above, the NICE system is widely used in the scientific community for very different purposes. However, so far, we are aware of only one study on the optimization of the fermentation and induction scheme to maximize product formation. In our laboratory, in a series of 58 fermentations, a number of key parameters were tested and optimized using the antibiotic protein lysostaphin as a model: pH of the fermentation, the neutralizing agent, the fermentation temperature, the optical density (OD) at which the culture is induced, the amount of nisin that is used for induction and the medium composition (C, N and minerals). This optimization led to a threefold increase in product formation (300 mg/l) and shows that a substantial increase in yield can be achieved with careful optimization of the complete process (Mierau et al. 2005b).
Next-generation applications
Lactococcus lactis, in combination with the NICE gene expression system, has a number of properties that make it an interesting host for novel applications:
Straightforward and functional expression and placement of membrane proteins
This property may allow the use of L. lactis for the expression of pharmaceutically relevant receptors and the search for new drug targets.
Expression of genes with potentially toxic gene products
With the advent of genome sequencing there is an increased need for the functional analysis of known and unknown genes. L. lactis is an ideal host for low-GC Gram-positive bacteria, especially the Gram-positive pathogens like streptococci, enterococci and staphylococci. High-throughput cloning of silent genes (not induced with nisin) can help to increase the gene cloning success rate and allow the study of gene products that otherwise would be missed.
Furthermore, control of the expression of essential genes allows high-throughput screening for pharmaceutical anti-microbial targets.
Lactococcus lactis can be grown in micro-titre plates without vigorous agitation
This feature allows simple high-throughput applications for enzyme screening and enzyme evolution.
Lactococcus lactis has a basic amino-sugar metabolism that allows the production of various heteropolysaccharides
The ability of L. lactis to produce polysaccharides makes it a possible alternative for the production of capsule polysaccharides of, e.g. pathogenic streptococci and other Gram-positive bacteria.
Lactococcus lactis has a high reduction potential because of its fermentative metabolism and thus allows in situ regeneration of NAD(P)H for redox reactions
This feature can be exploited for the construction of cell factories for the production of specific stereoisomers using various dehydrogenases.
Bottlenecks for gene expression in L. lactis
Fermentation conditions
Aerobic bacteria can be grown to cell densities far above 100 g/l dry biomass concentration (Riesenberg and Guthke 1999). Because of the fermentative metabolism this is not possible with L. lactis. In a simple acidifying buffered culture in, for instance, M17 medium (Terzaghi and Sandine 1975), the maximum cell density is about OD600=3 (1 g/l dry cell mass) (Pedersen et al. 2002). Growth will stop when a pH of about 5.0 is reached. With neutralization using NaOH or NH4OH the cell density can rise to OD600=15 (5 g/l dry cell mass). The main reason for this limitation is the accumulation of lactic acid that eventually will stop the growth. There have been attempts to develop high cell density cultivation methods for lactic acid bacteria (Schiraldi et al. 2003), but so far, none of these have been applied to increase gene expression. Efficient methods to extend logarithmic growth of L. lactis would allow further increase in the product yield.
Recently, it has been rediscovered that lactococci can grow under aerobic conditions when haem is added to the medium (Duwat et al. 2001). Under these conditions the growth period and the long-term survival of the cells is greatly extended. This observation can be employed to considerably increase the cell density of lactococcal cultures and initiate nisin-controlled gene expression at higher cell densities, leading to increased product formation.
Alternatively, NICE-like systems that display highly similar characteristics, but can be employed in respiring bacteria, such as SURE (SUbtilin Regulated gene Expression) in B. subtilis, can potentially overcome the fermentative biomass yield restrictions encountered with L. lactis.
Codon usage
Until very recently codon usage was an important factor in the possibility and efficiency to express heterologous genes in L. lactis (GC content of the DNA of 35–37%) (see above). When a gene donor organism is closely related to L. lactis, or the DNA GC content is similar to that of L. lactis, the probability that a gene can successfully be expressed is high. With the availability of cheap and reliable custom DNA synthesis, there are no longer restrictions as to the origin of a specific target gene, since, from a known amino acid sequence, a gene can be designed that fits the codon usage pattern of the host organism (Fuglsang 2003; Gupta et al. 2004). In addition to a general codon optimization, specific codon tables can be used, such as the codon table for the highly expressed ribosomal genes, to further increase product formation.
Growth and nisin induction for the NICE system in L. lactis
Various media are available for growth of lactococci. The most commonly used laboratory medium is M17 (Terzaghi and Sandine 1975) supplemented with glucose, lactose or other sugars as carbon source and a relevant antibiotic for plasmid selection. For large-scale applications this medium is too expensive (2% of β-glycerophosphate as buffer) and not compatible with pharmaceutical applications because it contains animal-derived material (beef extract), posing a Bovine spongiform encephalopathy (BSE) hazard. The basic ingredients for a large-scale medium are 1–3% peptone, 0.5–2% yeast extract, 1–10% carbon source and small amounts of magnesium and manganese ions. Since large-scale fermentations are often conducted under pH-controlled conditions, a buffer is not needed. Individual processes need specific optimization of the medium components and fermentation conditions (see Mierau et al. 2005b for an example).
For experiments in which specific metabolites are addressed or cell components need to be labelled, a chemically defined medium can be used (Poolman and Konings 1988; Jensen and Hammer 1993). Lactococci are auxotrophic for a number of amino acids that can be added in a labelled form and are then integrated into newly formed proteins (Kunji et al. 2003).
For nisin induction of gene expression, various protocols have been reported. Nisin can either be used as pure nisinA, as crude preparation from Sigma (Catalogue no. N5764) or obtained from the culture supernatant of the strain NZ9700 (see Table 1). Culture supernatant is mostly used in a 1:1,000 dilution and can be stored frozen in aliquots. For induction, the exact concentration of the supernatant needs to be determined by testing dilutions between 1:100 and 1:10,000 (Kunji et al. 2003). In general, it is somewhat easier to use commercially available nisin; however, the exact amount needed for full induction also needs to be determined because of batch differences.
At a laboratory scale, an overnight culture is most commonly inoculated into fresh medium with a dilution of 1:100, grown to an optical density OD600=0.2–0.5 and induced with 0.1–2 ng/ml nisin. After that, the culture is continued for 0.5–3 h and then harvested for further use or testing. In this set-up pH is not controlled, and the culture will stop growing at low cell densities because of lactic acid production and the consequent pH drop. Alternatively, the culture can be grown with pH control to higher cell densities. In this case, induction can be carried out at cell densities as high as OD600=5 or more, leading to a substantial yield increase (Mierau et al. 2005b).
Finally, it has been shown that nisin induction also operates at large scales, as recently demonstrated with the production of lysostaphin in four subsequent 3,000-l fermentations (Mierau et al. 2005a).
References
Antiporta MH, Dunny GM (2002) ccfA, the genetic determinant for the cCF10 peptide pheromone in Enterococcus faecalis OG1RF. J Bacteriol 184:1155–1162
Arnau J, Hjerl-Hansen E, Israelsen H (1997) Heterologous gene expression of bovine plasmin in Lactococcus lactis. Appl Microbiol Biotechnol 48:331–338
Åvall-Jääskeläinen S, Kylä-Nikkilä K, Kahala M, Miikkulainen-Lahti T, Palva A (2002) Surface display of foreign epitopes on the Lactobacillus brevis S-layer. Appl Environ Microbiol 68:5943–5951
Axelsson L, Lindstad G, Naterstad K (2003) Development of an inducible gene expression system for Lactobacillus sakei. Lett Appl Microbiol 37:115–120
Bermúdez-Humarán LG, Langella P, Miyoshi A, Gruss A, Guerra RT, Montes de Oca-Luna R, Le Loir Y (2002) Production of human papillomavirus type 16 E7 protein in Lactococcus lactis. Appl Environ Microbiol 68:917–922
Bermúdez-Humarán LG, Langella P, Cortes-Perez NG, Gruss A, Tamez-Guerra RS, Oliveira SC, Saucedo-Cardenas O, Montes de Oca-Luna R, Le Loir Y (2003) Intranasal immunization with recombinant Lactococcus lactis secreting murine interleukin-12 enhances antigen-specific Th1 cytokine production. Infect Immun 71:1887–1896
Boels IC, Ramos A, Kleerebezem M, Devos WM (2001) Functional analysis of the Lactococcus lactis galU and galE genes and their impact on sugar nucleotide and exopolysaccharide biosynthesis. Appl Environ Microbiol 67:3033–3040
Boels IC, Kleerebezem M, de Vos WM (2003) Engineering of carbon distribution between glycolysis and sugar nucleotide biosynthesis in Lactococcus lactis. Appl Environ Microbiol 69:1129–1135
Boels IC, Beerthuyzen MM, Kosters MH, Van Kaauwen MP, Kleerebezem M, De Vos WM (2004) Identification and functional characterization of the Lactococcus lactis rfb operon, required for dTDP–rhamnose biosynthesis. J Bacteriol 186:1239–1248
Bolotin A, Wincker P, Mauger S, Jaillon O, Malarme K, Weissenbach J, Ehrlich SD, Sorokin A (2001) The complete genome sequence of the lactic acid bacterium Lactococcus lactis ssp. lactis IL1403. Genome Res 11:731–753
Bron PA, Benchimol MG, Lambert J, Palumbo E, Deghorain M, Delcour J, Devos WM, Kleerebezem M, Hols P (2002) Use of the alr gene as a food-grade selection marker in lactic acid bacteria. Appl Environ Microbiol 68:5663–5670
Bryan EM, Bae T, Kleerebezem H, Dunny GM (2000) Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid 44:183–190
Burgess C, O'Connell-Motherway M, Sybesma W, Hugenholtz J, van Sinderen D (2004) Riboflavin production in Lactococcus lactis: potential for in situ production of vitamin-enriched foods. Appl Environ Microbiol 70:5769–5777
Campo N, Daveran-Mingot ML, Leenhouts K, Ritzenthaler P, Le Bourgeois P (2002) Cre-loxP recombination system for large genome rearrangements in Lactococcus lactis. Appl Environ Microbiol 68:2359–2367
Chatel JM, Nouaille S, Adelpatient K, Le Loir Y, Boe H, Gruss A, Wal JM, Langella P (2003) Characterization of a Lactococcus lactis strain that secretes a major epitope of bovine beta-lactoglobulin and evaluation of its immunogenicity in mice. Appl Environ Microbiol 69:6620–6627
Chong BF, Nielsen LK (2003) Amplifying the cellular reduction potential of Streptococcus zooepidemicus. J Biotechnol 100:33–41
Christensson C, Bratt H, Collins LJ, Coolbear T, Holland R, Lubbers MW, Otoole PW, Reid JR (2002) Cloning and expression of an oligopeptidase, PepO, with novel specificity from Lactobacillus rhamnosus HN001 (DR20). Appl Environ Microbiol 68:254–262
Cibik R, Tailliez P, Langella P, Chapot-Chartier MP (2001) Identification of Mur, an atypical peptidoglycan hydrolase derived from Leuconostoc citreum. Appl Environ Microbiol 67:858–864
de Ruyter PG, Kuipers OP, Beerthuyzen MM, Alen-Boerrigter I, de Vos WM (1996a) Functional analysis of promoters in the nisin gene cluster of Lactococcus lactis. J Bacteriol 178:3434–3439
de Ruyter PG, Kuipers OP, de Vos WM (1996b) Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Appl Environ Microbiol 62:3662–3667
de Ruyter PG, Kuipers OP, Meijer WC, de Vos WM (1997) Food-grade controlled lysis of Lactococcus lactis for accelerated cheese ripening. Nat Biotechnol 15:976–979
de Vos WD (1987) Gene cloning and expression in lactic streptococci. FEMS Microbiol Lett 46:281–295
de Vos WM (1999) Safe and sustainable systems for food-grade fermentations by genetically modified lactic acid bacteria. Int Dairy J 9:3–10
de Vos WM, Hugenholtz J (2004) Engineering metabolic highways in lactococci and other lactic acid bacteria. Trends Biotechnol 22:72–79
Dieye Y, Usai S, Clier F, Gruss A, Piard JC (2001) Design of a protein-targeting system for lactic acid bacteria. J Bacteriol 183:4157–4166
Dodd HM, Gasson MJ (1994) Bacteriocins of lactic acid bacteria. In: Gasson MJ, de Vos WM (eds) Genetics and biotechnology of lactic acid bacteria. Blackie, London
Duwat P, Sourice S, Cesselin B, Lamberet G, Vido K, Gaudu P, Le Loir Y, Violet F, Loubière P, Gruss A (2001) Respiration capacity of the fermenting bacterium Lactococcus lactis and its positive effects on growth and survival. J Bacteriol 183:4509–4516
Eichenbaum Z, Federle MJ, Marra D, de Vos WM, Kuipers OP, Kleerebezem M, Scott JR (1998) Use of the lactococcal nisA promoter to regulate gene expression in gram-positive bacteria: comparison of induction level and promoter strength. Appl Environ Microbiol 64:2763–2769
Enouf V, Langella P, Commissaire J, Cohen J, Corthier G (2001) Bovine rotavirus nonstructural protein 4 produced by Lactococcus lactis is antigenic and immunogenic. Appl Environ Microbiol 67:1423–1428
Fernández L, Beerthuyzen MM, Brown J, Siezen RJ, Coolbear T, Holland R, Kuipers OP (2000) Cloning, characterization, controlled overexpression, and inactivation of the major tributyrin esterase gene of Lactococcus lactis. Appl Environ Microbiol 66:1360–1368
Francia MV, Clewell DB (2002) Transfer origins in the conjugative Enterococcus faecalis plasmids pAD1 and pAM373: identification of the pAD1 nic site, a specific relaxase and a possible TraG-like protein. Mol Microbiol 45:375–395
Franke CM, Tiemersma J, Venema G, Kok J (1999) Membrane topology of the lactococcal bacteriocin ATP-binding cassette transporter protein LcnC. Involvement of LcnC in lactococcin A maturation. J Biol Chem 274:8484–8490
Fuglsang A (2003) Lactic acid bacteria as prime candidates for codon optimization. Biochem Biophys Res Commun 312:285–291
Gasson MJ (1983) Plasmid complements of Streptococcus lactis NCDO 712 and other lactic streptococci after protoplast-induced curing. J Bacteriol 154:1–9
Gasson MJ, de Vos WM (1994) Genetics and biotechnology of lactic acid bacteria. Blackie, London
Guillot A, Gitton C, Anglade P, Mistou MY (2003) Proteomic analysis of Lactococcus lactis, a lactic acid bacterium. Proteomics 3:337–354
Gupta SK, Bhattacharyya TK, Ghosh TC (2004) Synonymous codon usage in Lactococcus lactis: mutational bias versus translational selection. J Biomol Struct Dyn 21:527–536
Hagting A, Knol J, Hasemeier B, Streutker MR, Fang G, Poolman B, Konings WN (1997) Amplified expression, purification and functional reconstitution of the dipeptide and tripeptide transport protein of Lactococcus lactis. Eur J Biochem 247:581–587
Hanniffy S, Wiedermann U, Repa A, Mercenier A, Daniel C, Fioramonti J, Tlaskolova H, Kozakova H, Israelsen H, Madsen S, Vrang A, Hols P, Delcour J, Bron P, Kleerebezem M, Wells J (2004) Potential and opportunities for use of recombinant lactic acid bacteria in human health. Adv Appl Microbiol 56:1–64
Hasper HE, de Kruijff B, Breukink E (2004) Assembly and stability of nisin-lipid II pores. Biochemistry 43:11567–11575
Henrich B, Klein JR, Weber B, Delorme C, Renault P, Wegmann U (2002) Food-grade delivery system for controlled gene expression in Lactococcus lactis. Appl Environ Microbiol 68:5429–5436
Heuberger EHML, Smits E, Poolman B (2001) Xyloside transport by XylP, a member of the galactoside–pentoside–hexuronide family. J Biol Chem 276:34465–34472
Hickey RM, Twomey DP, Ross RP, Hill C (2003) Potential of the enterocin regulatory system to control expression of heterologous genes in Enterococcus. J Appl Microbiol 95:390–397
Hickey RM, Ross RP, Hill C (2004) Controlled autolysis and enzyme release in a recombinant lactococcal strain expressing the metalloendopeptidase enterolysin A. Appl Environ Microbiol 70:1744–1748
Hoefnagel MH, Starrenburg MJ, Martens DE, Hugenholtz J, Kleerebezem M, Van S II, Bongers R, Westerhoff HV, Snoep JL (2002) Metabolic engineering of lactic acid bacteria, the combined approach: kinetic modelling, metabolic control and experimental analysis. Microbiology 148:1003–1013
Hols P, Kleerebezem M, Schanck AN, Ferain T, Hugenholtz J, Delcour J, de Vos WM (1999) Conversion of Lactococcus lactis from homolactic to homoalanine fermentation through metabolic engineering. Nat Biotechnol 17:588–592
Hugenholtz J, Kleerebezem M, Starrenburg M, Delcour J, de Vos W, Hols P (2000) Lactococcus lactis as a cell factory for high-level diacetyl production. Appl Environ Microbiol 66:4112–4114
Hugenholtz J, Sybesma W, Groot MN, Wisselink W, Ladero V, Burgess K, van Sinderen D, Piard JC, Eggink G, Smid EJ, Savoy G, Sesma F, Jansen T, Hols P, Kleerebezem M (2002) Metabolic engineering of lactic acid bacteria for the production of nutraceuticals. Antonie Van Leeuwenhoek 82:217–235
Hughes MJ, Moore JC, Lane JD, Wilson R, Pribul PK, Younes ZN, Dobson RJ, Everest P, Reason AJ, Redfern JM, Greer FM, Paxton T, Panico M, Morris HR, Feldman RG, Santangelo JD (2002) Identification of major outer surface proteins of Streptococcus agalactiae. Infect Immun 70:1254–1259
Jensen PR, Hammer K (1993) Minimal requirements for exponential growth of Lactococcus lactis. Appl Environ Microbiol 59:4363–4366
Klaenhammer T, Altermann E, Arigoni F, Bolotin A, Breidt F, Broadbent J, Cano R, Chaillou S, Deutscher J, Gasson M, van de Guchte M, Guzzo J, Hartke A, Hawkins T, Hols P, Hutkins R, Kleerebezem M, Kok J, Kuipers O, Lubbers M, Maguin E, McKay L, Mills D, Nauta A, Overbeek R, Pel H, Pridmore D, Saier M, van Sinderen D, Sorokin A, Steele J, O'Sullivan D, de Vos W, Weimer B, Zagorec M, Siezen R (2002) Discovering lactic acid bacteria by genomics. Antonie Van Leeuwenhoek 82:29–58
Kleerebezem M, Hugenholtz J (2003) Metabolic pathway engineering in lactic acid bacteria. Curr Opin Biotechnol 14:232–237
Kleerebezem M, Quadri LE (2001) Peptide pheromone-dependent regulation of antimicrobial peptide production in Gram-positive bacteria: a case of multicellular behavior. Peptides 22:1579–1596
Kleerebezem M, Beerthuyzen MM, Vaughan EE, de Vos WM, Kuipers OP (1997a) Controlled gene expression systems for lactic acid bacteria: transferable nisin-inducible expression cassettes for Lactococcus, Leuconostoc, and Lactobacillus spp. Appl Environ Microbiol 63:4581–4584
Kleerebezem M, Quadri LE, Kuipers OP, de Vos WM (1997b) Quorum sensing by peptide pheromones and two-component signal-transduction systems in Gram-positive bacteria. Mol Microbiol 24:895–904
Kleerebezem M, de Vos WM, Kuipers OP (1999) The lantibiotics nisin and subtilin act as extracellular regulators of their own biosynthesis. In: Dunny GM, Winams SC (eds) Cell–cell signalling in bacteria. American Society for Microbiology, Washington, D.C.
Kleerebezem M, Boels IC, Groot MN, Mierau I, Sybesma W, Hugenholtz J (2002) Metabolic engineering of Lactococcus lactis: the impact of genomics and metabolic modelling. J Biotechnol 98:199–213
Kleerebezem M, Bongers R, Rutten G, de Vos WM, Kuipers OP (2004) Autoregulation of subtilin biosynthesis in Bacillus subtilis: the role of the spa-box in subtilin-responsive promoters. Peptides 25:1415–1424
Koebmann BJ, Nilsson D, Kuipers OP, Jensen PR (2000) The membrane-bound H(+)-ATPase complex is essential for growth of Lactococcus lactis. J Bacteriol 182: 4738–4743
Kotelnikova EA, Gelfand MS (2002) Bacteriocin production by Gram-positive bacteria and the mechanisms of transcriptional regulation. Russ J Genet 38:628–641
Kuipers OP, Beerthuyzen MM, Siezen RJ, De Vos WM (1993) Characterization of the nisin gene cluster nisABTCIPR of Lactococcus lactis. Requirement of expression of the nisA and nisI genes for development of immunity. Eur J Biochem 216:281–291
Kuipers OP, Beerthuyzen MM, de Ruyter PG, Luesink EJ, de Vos WM (1995) Autoregulation of nisin biosynthesis in Lactococcus lactis by signal transduction. J Biol Chem 270:27299–27304
Kuipers OP, de Ruyter PGGA, Kleerebezem M, de Vos WM (1998) Quorum sensing-controlled gene expression in lactic acid bacteria. J Biotechnol 64:15–21
Kunji ER, Mierau I, Hagting A, Poolman B, Konings WN (1996) The proteolytic systems of lactic acid bacteria. Antonie Van Leeuwenhoek 70:187–221
Kunji ER, Slotboom DJ, Poolman B (2003) Lactococcus lactis as host for overproduction of functional membrane proteins. Biochim Biophys Acta 1610:97–108
Le Loir Y, Nouaille S, Commissaire J, Brétigny L, Gruss A, Langella P (2001a) Signal peptide and propeptide optimization for heterologous protein secretion in Lactococcus lactis. Appl Environ Microbiol 67:4119–4127
Le Loir Y, Nouaille S, Ribeiro L, Commissaire J, Corthier G, Gilbert S, Chatel J, L'Haridon R, Gruss A, Langella P (2001b) Secretion of heterologous proteins of therapeutical interest in Lactococcus lactis. Lait 81:217–226
Leroy F, Devuyst L (2004) Lactic acid bacteria as functional starter cultures for the food fermentation industry. Trends Food Sci Technol 15:67–78
Lindholm A, Smeds A, Palva A (2004) Receptor binding domain of Escherichia coli F18 fimbrial adhesin FedF can be both efficiently secreted and surface displayed in a functional form in Lactococcus lactis. Appl Environ Microbiol 70:2061–2071
Looijesteijn PJ, Boels IC, Kleerebezem M, Hugenholtz J (1999) Regulation of exopolysaccharide production by Lactococcus lactis subsp. cremoris by the sugar source. Appl Environ Microbiol 65:5003–5008
Lopez de Felipe F, Hugenholtz J (1999) Pyruvate flux distribution in NADH-oxidase-overproducing Lactococcus lactis strain as a function of culture conditions. FEMS Microbiol Lett 179:461–466
Lopez de Felipe F, Kleerebezem M, de Vos WM, Hugenholtz J (1998) Cofactor engineering: a novel approach to metabolic engineering in Lactococcus lactis by controlled expression of NADH oxidase. J Bacteriol 180:3804–3808
Luoma S, Peltoniemi K, Joutsjoki V, Rantanen T, Tamminen M, Heikkinen I, Palva A (2001) Expression of six peptidases from Lactobacillus helveticus in Lactococcus lactis. Appl Environ Microbiol 67:1232–1238
Madsen PL, Johansen AH, Hammer K, Brøndsted L (1999) The genetic switch regulating activity of early promoters of the temperate lactococcal bacteriophage TP901-1. J Bacteriol 181:7430–7438
Margolles A, Putman M, van Veen HW, Konings WN (1999) The purified and functionally reconstituted multidrug transporter LmrA of Lactococcus lactis mediates the transbilayer movement of specific fluorescent phospholipids. Biochemistry 38:16298–16306
Mathiesen G, Sørvig E, Blatny J, Naterstad K, Axelsson L, Eijsink VGH (2004) High-level gene expression in Lactobacillus plantarum using a pheromone-regulated bacteriocin promoter. Lett Appl Microbiol 39:137–143
McCormick JK, Hirt H, Waters CM, Tripp TJ, Dunny GM, Schlievert PM (2001) Antibodies to a surface-exposed, N-terminal domain of aggregation substance are not protective in the rabbit model of Enterococcus faecalis infective endocarditis. Infect Immun 69:3305–3314
McGrath S, Fitzgerald GF, van Sinderen D (2002) Identification and characterization of phage-resistance genes in temperate lactococcal bacteriophages. Mol Microbiol 43:509–520
Mierau I, Leij P, van Swam I, Blommestein B, Floris E, Mond J, Smid EJ (2005a) Industrial-scale production and purification of a heterologous protein in Lactococcus lactis using the nisin-controlled gene expression system NICE: the case of lysostaphin. Microb Cell Fact 4:15
Mierau I, Olieman K, Mond J, Smid EJ (2005b) Optimization of the Lactococcus lactis nisin-controlled gene expression system NICE for industrial applications. Microb Cell Fact 4:16
Miyoshi A, Poquet I, Azevedo V, Commissaire J, Bermudez-Humaran L, Domakova E, Le Loir Y, Oliveira SC, Gruss A, Langella P (2002) Controlled production of stable heterologous proteins in Lactococcus lactis. Appl Environ Microbiol 68:3141–3146
Neu T, Henrich B (2003) New thermosensitive delivery vector and its use to enable nisin-controlled gene expression in Lactobacillus gasseri. Appl Environ Microbiol 69:1377–1382
Neves AR, Ramos A, Costa H, van Swan II, Hugenholtz J, Kleerebezem M, de Vos W, Santos H (2002) Effect of different NADH oxidase levels on glucose metabolism by Lactococcus lactis: kinetics of intracellular metabolite pools determined by in vivo nuclear magnetic resonance. Appl Environ Microbiol 68:6332–6342
Nielsen J (2001) Metabolic engineering. Appl Microbiol Biotechnol 55:263–283
Nouaille S, Ribeiro LA, Miyoshi A, Pontes D, Le Loir Y, Oliveira SC, Langella P, Azevedo V (2003) Heterologous protein production and delivery systems for Lactococcus lactis. Genet Mol Res 2:102–111
Nouaille S, Commissaire J, Gratadoux JJ, Ravn P, Bolotin A, Gruss A, Leloir Y, Langella P (2004) Influence of lipoteichoic acid D-alanylation on protein secretion in Lactococcus lactis as revealed by random mutagenesis. Appl Environ Microbiol 70:1600–1607
Novotny R, Scheberl A, Giry-Laterriere M, Messner P, Schäffer C (2005) Gene cloning, functional expression and secretion of the S-layer protein SgsE from Geobacillus stearothermophilus NRS 2004/3a in Lactococcus lactis. FEMS Microbiol Lett 242:27–35
Pavan S, Hols P, Delcour J, Geoffroy MC, Grangette C, Kleerebezem M, Mercenier A (2000) Adaptation of the nisin-controlled expression system in Lactobacillus plantarum: a tool to study in vivo biological effects. Appl Environ Microbiol 66:4427–4432
Pedersen MB, Koebmann BJ, Jensen PR, Nilsson D (2002) Increasing acidification of nonreplicating Lactococcus lactis delta-thyA mutants by incorporating ATPase activity. Appl Environ Microbiol 68:5249–5257
Platteeuw C, van Alen-Boerrigter I, van Schalkwijk S, de Vos WM (1996) Food-grade cloning and expression system for Lactococcus lactis. Appl Environ Microbiol 62:1008–1013
Poolman B, Konings WN (1988) Relation of growth of Streptococcus lactis and Streptococcus cremoris to amino acid transport. J Bacteriol 170:700–707
Poquet I, Saint V, Seznec E, Simoes N, Bolotin A, Gruss A (2000) HtrA is the unique surface housekeeping protease in Lactococcus lactis and is required for natural protein processing. Mol Microbiol 35:1042–1051
Ramos A, Neves AR, Ventura R, Maycock C, López P, Santos H (2004) Effect of pyruvate kinase overproduction on glucose metabolism of Lactococcus lactis. Microbiology 150:1103–1111
Ravn P, Arnau J, Madsen SM, Vrang A, Israelsen H (2003) Optimization of signal peptide SP310 for heterologous protein production in Lactococcus lactis. Microbiology 149:2193–2201
Ribardo DA, McIver KS (2003) amrA encodes a putative membrane protein necessary for maximal exponential phase expression of the Mga virulence regulon in Streptococcus pyogenes. Mol Microbiol 50:673–685
Ribeiro LA, Azevedo V, Le Loir Y, Oliveira SC, Dieye Y, Piard JC, Gruss A, Langella P (2002) Production and targeting of the Brucella abortus antigen L7/L12 in Lactococcus lactis: a first step towards food-grade live vaccines against brucellosis. Appl Environ Microbiol 68:910–916
Riesenberg D, Guthke R (1999) High-cell-density cultivation of microorganisms. Appl Microbiol Biotechnol 51:422–430
Rigoulay C, Poquet I, Madsen SM, Gruss A (2004) Expression of the Staphylococcus aureus surface proteins Htra(1) and Htra(2) in Lactococcus lactis. FEMS Microbiol Lett 237:279–288
Sakamoto K, Margolles A, van Veen HW, Konings WN (2001) Hop resistance in the beer spoilage bacterium Lactobacillus brevis is mediated by the ATP-binding cassette multidrug transporter HorA. J Bacteriol 183:5371–5375
Schiraldi C, Adduci V, Valli V, Maresca C, Giuliano M, Lamberti M, Carteni M, De Rosa M (2003) High cell density cultivation of probiotics and lactic acid production. Biotechnol Bioeng 82:213–222
Simões-Barbosa A, Abreu H, Silva Neto A, Gruss A, Langella P (2004) A food-grade delivery system for Lactococcus lactis and evaluation of inducible gene expression. Appl Microbiol Biotechnol 65:61–67
Smid EJ, Molenaar D, Hugenholtz J, de Vos WM, Teusink B (2005) Functional ingredient production: application of global metabolic models. Curr Opin Biotechnol 16:190–197
Steen A, Buist G, Leenhouts KJ, El Khattabi M, Grijpstra F, Zomer AL, Venema G, Kuipers OP, Kok J (2003) Cell wall attachment of a widely distributed peptidoglycan binding domain is hindered by cell wall constituents. J Biol Chem 278:23874–23881
Sybesma W, Starrenburg M, Kleerebezem M, Mierau I, de Vos WM, Hugenholtz JR (2003a) Increased production of folate by metabolic engineering of Lactococcus lactis. Appl Environ Microbiol 69:3069–3076
Sybesma W, van den Born E, Starrenburg M, Mierau I, Kleerebezem M, de Vos WM, Hugenholtz J (2003b) Controlled modulation of folate polyglutamyl tail length by metabolic engineering of Lactococcus lactis. Appl Environ Microbiol 69:7101–7107
Terzaghi BE, Sandine WE (1975) Improved medium for lactic streptococci and their bacteriophages. Appl Microbiol 29:807–813
Teuber M (1995) The genus Lactococcus. In: Wood BJB, Holzapfel WH (eds) The genera of lactic acid bacteria. Blackie, London, pp 173–234
Ton-That H, Marraffini LA, Schneewind O (2004) Protein sorting to the cell wall envelope of Gram-positive bacteria. Biochim Biophys Acta 11:269–278
van Asseldonk M, de Vos WM, Simons G (1993) Functional analysis of the Lactococcus lactis usp45 secretion signal in the secretion of a homologous proteinase and a heterologous alpha-amylase. Mol Gen Genet 240:428–434
van Kraaij C, de Vos WM, Siezen RJ, Kuipers OP (1999) Lantibiotics: biosynthesis, mode of action and applications. Nat Prod Rep 16:575–587
Waters CM, Wells CL, Dunny GM (2003) The aggregation domain of aggregation substance, not the RGD motifs, is critical for efficient internalization by HT-29 enterocytes. Infect Immun 71:5682–5689
Waters CM, Hirt H, McCormick JK, Schlievert PM, Wells CL, Dunny GM (2004) An amino-terminal domain of Enterococcus faecalis aggregation substance is required for aggregation, bacterial internalization by epithelial cells and binding to lipoteichoic acid. Mol Microbiol 52:1159–1171
Wegmann U, Klein JR, Drumm I, Kuipers OP, Henrich B (1999) Introduction of peptidase genes from Lactobacillus delbrueckii subsp. lactis into Lactococcus lactis and controlled expression. Appl Environ Microbiol 65:4729–4733
Wood BJB, Warner PJ (2003) Genetics of lactic acid bacteria. Kluwer Academic/Plenum, New York
Wouters JA, Kamphuis HH, Hugenholtz J, Kuipers OP, de Vos WM, Abee T (2000) Changes in glycolytic activity of Lactococcus lactis induced by low temperature. Appl Environ Microbiol 66:3686–3691
Zhou L, Manias DA, Dunny GM (2000) Regulation of intron function: efficient splicing in vivo of a bacterial group II intron requires a functional promoter within the intron. Mol Microbiol 37:639–651
Zúñiga M, Franke-Fayard B, Venema G, Kok J, Nauta A (2002) Characterization of the putative replisome organizer of the lactococcal bacteriophage r1t. J Virol 76:10234–10244
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mierau, I., Kleerebezem, M. 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis . Appl Microbiol Biotechnol 68, 705–717 (2005). https://doi.org/10.1007/s00253-005-0107-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-005-0107-6