
Abstract
A methodology for the extraction and quantification of 16 polycyclic aromatic hydrocarbons (PAHs) based on microwave-assisted extraction coupled with headspace solid-phase microextraction followed by gas chromatography/mass spectroscopy was validated for needles and bark of two pine species (Pinus pinaster Ait. and Pinus pinea L.). The limits of detection were below 0.92 ng g−1 (dry weight) for needles and below 0.43 ng g−1 (dw) for bark. Recovery assays were performed with two sample masses spiked at three levels and the overall mean values were between 70 and 110 % for P. pinaster and 75 and 129 % for P. pinea. In the first species, the increase in sample mass lowered the recoveries slightly for most PAHs, whereas for the second, the recoveries were higher for the needles. Naturally contaminated samples from 4 sites were analysed, with higher levels for urban sites (1,320 and 942 ng g−1 (dw) vs. 272 and 111 ng g−1 (dw) for needles and 696 and 488 ng g−1 (dw) vs. 270 and 103 ng g−1 (dw) for bark) than for rural ones and also for P. pinaster samples over P. pinea. It is also shown that gas-phase PAHs are predominant in the needles (over 65 % of the total PAHs) and that the incidence for particulate material in bark, reaching 40 % as opposed to a maximum below 20 % for the needles. The method has proved to be fit and improved some of the existing approaches, on the assessment of particulate PAHs and bark levels.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ever since its introduction in the early 1990s [1], solid-phase microextraction (SPME) has been firmly established as a valid environmental-friendlier alternative to traditional extraction methods. It has been used to extract a wide range of compounds, with particular incidence given to priority pollutants including polycyclic aromatic hydrocarbons (PAHs) [2–4].
PAHs, petroleum- and combustion-derived pollutants released from natural (forest fires) and anthropogenic sources (traffic, industrial processes, domestic heating and oil spills), were extracted using this technique from matrices such as water [5, 6], seawater [7], wastewater [8], landfill leachates [9], soils [10, 11], sediments [12, 13], air [14, 15], bitumen fumes [16], gasoline soot [17], bilge waste [18], vegetable oils [19], seaweed [20], fish [21] and human samples like blood serum [22] or urine [23]. Headspace (HS) SPME was preferred to fiber immersion when dealing with “dirtier” (particularly solid) materials and gas chromatography with mass spectrometry detection (GC/MS) was predominant in the subsequent quantification step, although liquid chromatography (HPLC) was also used [4]. Although plant species have been used as natural monitors of contaminants since the 1960s [24], to our knowledge SPME was never applied in the biomonitoring of PAHs by these kinds of matrices, and particularly by pine trees. There are studies describing HS-SPME approaches involving pine needles but only to extract the volatile organic constituents of the needles [25, 26].
Pine trees benefit from their worldwide presence and the retention properties exhibited by the waxy layer of their needles [27] and by their very porous bark [28] to be considered a valuable matrix in biomonitoring studies of several priority contaminants, including PAHs. However, the analytical methodologies employed to extract such pollutants involve solvent-consuming and sometimes time-consuming techniques like Soxhlet [29, 30], ultrasonic extraction (USE) [30–32], microwave-assisted extraction (MAE) [30, 33] or pressurised liquid extraction (PLE) [30, 31, 34], followed by intricate clean-up of the numerous unwanted matrix extracts employing different solid-phase extraction (SPE) commercial or laboratory-made silica-based columns [31, 32, 35] or size-exclusion chromatography [36]. A first approach to the application of a solvent-reduced clean-up-free methodology, namely involving hollow-fiber liquid-phase microextraction (HF-LPME), was successfully reported by Ratola et al. [37] in the extraction of PAHs from pine needles. The challenge now is to study a new efficient alternative, applied not only to needles but also to pine bark, which shows a much lower PAH entrapment capacity under conventional extraction methodologies [47].
Hence, the efficiency of microwave-assisted headspace SPME was tested for the first time in the extraction of 16 PAHs from needles and bark from Pinus pinaster Ait. and Pinus pinea L. trees. Similar approaches are reported in literature, but applied to landfill leachate and sediments [16, 21]. For the validation assays, two sample masses (1 and 5 g) and three PAHs spiking levels (10, 50 and 100 ng g−1) were used for needles and bark of both pine species. The PAH quantification was obtained by GC/MS, using deuterated PAHs as internal standards. The proposed methodology was also tested in naturally contaminated samples from four sites in Portugal (two urban and two rural, one of each species), by assessing their respective PAHs concentrations.
Experimental
Chemicals and reagents
The 16 PAHs in study (naphthalene (Naph), acenaphthylene (Acy), acenaphthene (Ace), fluorene (Fluo), phenanthrene (Phen), anthracene (Ant), fluoranthene (Flt), pyrene (Pyr), benzo[a]anthracene (BaA), chrysene (Chry), benzo[b]fluoranthene (BbF), benzo[k]fluoranthene (BkF), benzo[a]pyrene (BaP), indeno[1,2,3-cd]pyrene (IcdP), dibenzo[a,h]anthracene (DahA) and benzo[ghi]perylene (BghiP)) and surrogate deuterated PAHs (naphthalene-d8, acenaphthene-d10, phenanthrene-d10, chrysene-d12 and perylene-d12) were 2,000 μg mL−1 standard solutions in hexane:dichloromethane (1:1) from Supelco (Bellefonte, PA, USA). The calibration standards (0.1, 0.5, 2.5, 5.0, 7.5 and 10.0 μg L−1, in a 1.8 % ethanol aqueous solution) were prepared from working solutions of target and deuterated PAHs (200 μg L−1 in a 36 % ethanol solution in water). Ethanol SupraSolv was from Merck (Darmstadt, Germany) and water was distilled on site. Nitrogen (99.995 % purity) for drying and helium (99.9999 % purity) for gas chromatography were from Air Liquide (Maia, Portugal). All glassware used was silanised in a 15 % dichlorodimethylsilane solution in toluene, both from Aldrich (Milwaukee, WI, USA), to prevent adsorption onto the glass.
Sample collection and handling
Samples of pine bark and needles used in the method validation assays were collected from the two most common pine species in Portugal: P. pinaster Ait. and P. pinea L. One tree of each species located in Porto (urban site) was chosen. The same trees were also used for the survey of naturally contaminated samples, together with other two from Loulé and Estrela, two rural sites. Bark samples from the external layer were collected all around the trunk at a height of 1 to 1.5 m, while needles were removed whole from the bottom and outer branches of the pine trees. All samples were wrapped in aluminium foil and sealed in plastic bags. Those used in the method validation assays were immediately analysed, whereas the naturally contaminated samples were frozen and stored away from light. Prior to analysis they were defrosted at ambient temperature.
To allow the expression of the results on a dry weight basis, the water content of both matrices was measured by drying triplicate 5 g samples of bark and needles at 80 °C until constant weight. For both pine species, the water percentages were the same: 11 % for bark and 59 % for needles.
Method validation
The linearity of the 16 PAHs in study was checked using six calibration standards (0.1, 0.5, 2.5, 5.0, 7.5 and 10.0 μg L−1, in a 1.8 % ethanol aqueous solution) extracted using the same method as for pine samples. The limits of detection (LODs) were calculated by the signal-to-noise (S/N) ratio of three rule, employing the least concentrated calibration standard.
Repeatability was checked with three consecutive extractions of the 0.5 μg L−1 standard.
To perform the recovery assays, two sample masses (1 and 5 g) and three spiking levels of target PAHs (10, 50 and 100 ng g−1) were used. Quadruplicate analyses were performed in each case, together with two blank assays. The surrogate deuterated PAHs were added to all samples at 50 ng g−1. On the other hand, for the naturally contaminated samples only 2 g samples were used (in duplicate), adding the same concentration of deuterated PAHs. Procedural blanks were performed periodically to account for possible external PAH contamination.
Microwave-assisted headspace solid-phase microextraction
The extraction of PAHs was accomplished employing a microwave-assisted headspace solid-phase microextractions (MA-HS-SPME) methodology based on a previous work by Herbert et al. [9] to determine semi-volatile pollutants from leachates and sediments. A WP700P17-3 domestic microwave oven from Electric Co. (2,450 MHz, China) was adapted to allow the introduction of a 250-mL flask containing the samples attached to a condenser and a SPME fibre and holder from Supelco (Bellefonte, PA, USA). The fibre coatings tested were 100-μm polydimethylsiloxane (PDMS) and 65 μm polydimethylsiloxane /divinylbenzene (DVB), also from Supelco. The set-up was assembled under a fume hood and a microwave radiation detector (MS-M128, Meet Int., Hong Kong) checked for leaks during operation. The water bath, at 16 °C, was an F34 model from Julabo (Seelbach, Germany). According to the case, one or five gram samples of bark (ground to <1 cm2 pieces with pestle and mortar) or needles (cut into 1 cm portions) were placed in 250-mL round-bottomed glass flasks with 50 mL H2O, 900 μL ethanol and 50 ng g−1 of the surrogate deuterated PAHs. After extraction for 60 min at 513 W, the SPME fibre was removed, inserted in the gas chromatography with mass spectroscopy (GC-MS) injector and desorbed for at least 15 min (10 min in splitless mode).
Chromatographic analysis
Analysis of PAHs was done with a Varian CP-3800 gas chromatograph (Lake Forest, CA, USA) equipped with a split/splitless injector (model 1079) and coupled to a Varian 4000 mass spectrometer detector working in electron impact mode (70 eV). The capillary column was a Factor Four VF-5MS from Varian coated with 5 % diphenyl-polydimethylsiloxane (30 m × 0.25 mm I.D, film thickness 0.25 μm). SPME fibres injection was done in splitless mode and they stayed desorbing for at least 15 min to minimise carryover. Meanwhile, the split valve was open at 10 min. The column oven temperature programme was as follows: start at 60 °C for 2 min, raised at 50 °C min−1 to 160 °C, then at 2.5 °C min−1 until 200 °C, at 50 °C min−1 to 250 °C, then 2.5 °C min−1 until 270 °C and finally at 50 °C min−1 to 300 °C, completing a total runtime of 57 min. Helium (at 1.0 mL min−1) acted as the carrier gas and the temperatures for the injector, transfer line and ion source were 290, 280 and 200 °C, respectively. Acquisition was done in selected ion storage (SIS) mode using five retention time windows (one of the deuterated PAH acting as internal standard per window). The target PAHs were identified and quantified with the Mass Spectrometry Workstation 6.6 software from Varian, using the retention times and three ions, as shown in Table 1.
Results and discussion
Preliminary assays
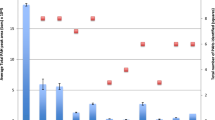
Before performing the actual validation of the method, two available common SPME fibres with different coatings were tested: 100 μm PDMS and 65 μm polydimethylsiloxane /divinylbenzene (DVB). In literature either fibre is also used successfully [5, 15] and Wang et al. [38] considered them the best for PAHs extraction among five fibres studied. It could be seen that in some cases, the differences were not significant, on others, depending on the PAHs, their performance was not quite the same. This could be due to their different coating structure and extraction mechanism. PDMS is a liquid and non-porous and non-polar polymer and DVB/PDMS is a porous phase where DVB microspheres are immobilised by PDMS [39]. These characteristics favour the simultaneous extraction of both the low and high molecular volume PAHs with similar efficiencies, but the difference in the extraction mechanism (absorption for PDMS and adsorption for DVB) suggest a higher affinity of the latter towards the lighter PAHs and of the former towards the heavier [38]. Again, this is not always the case, as seen in Fig. 1. In complex matrices, some unexpected behaviours may occur and since the PDMS fibres tend to be more resistant to matrix composition and also considering the tests done for both bark and needles of the two pine species it was decided to select PDMS fibres for this study. Figure 1 shows the results obtained for P. pinaster needles, which represent the overall behaviour of both fibre types.

Performance of PBDS and DVB fibres in the extraction of PAHs from P. pinaster needles (5-g needles spiked with 50 ng g−1)
Parameters affecting the sampling efficiency, such as extraction time, microwave power and sample volume, were already optimised in a previous study involving landfill leachate [9]. In brief, extraction times of 5, 10, 20, 30, 40, 50 and 60 min were tested and the best results were obtained with 60 min; microwave irradiation powers of 163, 327, 397, 513, 560 and 700 W were studied, with 560 W producing the higher extraction rates; and also the extraction solvent volume was varied from 25 to 50 and to 100 mL, with 50 mL yielding the best performances.
Calibration and repeatability
The calibration curves showed linear behaviour from 0.01 to 1 mg L−1 for all PAHs except Fluo (0.01–0.5 mg L−1) and Chry (0.01–0.75 mg L−1), and correlation coefficients (r 2) between 0.973 and 0.999. Good chromatographic resolution was achieved as well as low instrumental and method LODs, calculated by S/N ratio of 3. Table 2 presents the results obtained for LODs and repeatability (mean of three consecutive samples spiked at 10 ng g−1).
The instrumental LODs were below 10 pg L−1 for all PAHs except IcdP and DahA. These values decrease the limits found for more classic approaches such as USE [30] or Soxhlet extraction followed by HPLC [40] by tenfold, although with slightly higher repeatabilities in the first case. The method LODs varied from 21 to 915 pg g−1 (dw) for needles and from 10 to 421 pg g−1 (dw) for bark. For pine needles, the LOD values are similar to those found in literature for PAHs extraction from pine needles by USE [30, 33], PLE [36] or HF-LPME [37]. Regarding pine bark, the results are slightly better than those previously reported for USE and simple MAE [33], although again with higher repeatabilities. In fact, these were better for needles in P. pinaster and for bark in P. pinea, which in turn yielded better results than the same matrix for the other pine species. Ratola et al. [33] found similar differences using USE and MAE, but in this case, needles performed better than bark in both species. The dissimilar behaviour of needles of different species towards PAHs uptake is already documented and justified by some morphological and physiological particularities [32, 41]. There are no such comparisons for bark of more than one pine species, but it is reasonable to assume that the same explanation is valid as well.
Recovery
As mentioned before, the recovery assays were performed using three spiking levels of target PAHs (10, 50 and 100 ng g−1), applied to two sample masses (1 and 5 g). Given the uptake behaviour differences shown between P. pinaster and P. pinea needles using a ultrasonic/solid-phase extraction type methodology [41], it was decided to separate both species and show the comparison between needles and bark in each case. Results will be presented as the mean of the three spiking levels for each of the two sample masses.
P. pinaster
The mean recoveries for needles and bark of P. pinaster species are presented in Table 3, for 1-and 5 g samples.
For most of the PAHs, the results are between 70 and 110 %. In general, the recoveries do not seem to display particular patterns according to the number of aromatic rings of the PAH molecules. This behaviour is somewhat different to that found for USE and MAE followed by SPE clean-up, where there was a clear decrease on the recoveries for the heavier PAHs [33]. This suggests that MAE-HS-SPME may contribute to enhance the quantification of these particulate phase PAHs. The values between both sample masses and even for the two matrices are not so different, but in these cases there are some particular evidences and exceptions that are worthy of mention. For instance, for some PAHs, the recoveries are not so good with a higher amount of sample. This is more visible in needle samples and especially for Flt and Pyr. In these two cases, the chromatographic interferences for this pine species, already reported in a previous work [33], may have resulted in the low values showed for the 5 g samples. It is expected that the increase of the sample mass will improve the peak areas of both the target compounds and matrix-related coeluting compounds, potentially affecting chromatographic resolutions and the recovery percentages. In this case, it can be seen that the recoveries tend to decrease for the needles, with higher sample mass, for the lighter gas phase and particularly for the gas/particulate phase PAHs (Fig. 2). Gas phase corresponds to two and three rings (Naph, Acy, Ace, Fluo, Phen and Ant), particulate phase to five and six rings (BbF, BkF, BaP, DahA, BghiP and IcdP) and four-ring PAHs (Flt, Pyr, BaA and Chry) can appear as a mix of the two phases (gas/particulate) [42].

Mean recovery patterns of three spiking levels of PAHs (10, 50 and 100 ng g−1) according to their aromatic rings for MAE-HS-SPME extraction of 1 and 5 g samples of P. pinaster and P. pinea needles and bark. Gas phase—two and three rings; gas/particulate phases—four rings; particulate phase—five and six rings
Boden and Reiner [43] stated that when analysing PAHs, the ions formed by GC-MS in EI mode have similar masses as those of other matrix compounds (humic acids, sulphur, fats, waxes or oils). In the case of pine needles, the waxy layer they possess is crucial in the entrapment of organic contaminants [27], especially in the gaseous phase. Compounds such as fatty acids, polyesters or paraffins [44] that form this layer are effective in pollutants entrapment. Hence, more matrix interferences can make peak identification more difficult, as well as inefficient ionisation. Since the particle-bound PAHs are mainly deposited in the surface of both needles and bark, the increase of sample mass will not carry as much matrix-derived interferences, and the results are even slightly better, reaching mean values of 80 to 100 %, as seen in Fig. 2. Bark has a very porous and roughly inert in the presence of organic compounds [45], with pollutants mainly accumulating by atmospheric deposition in the outer layer [28], suggesting a stronger affinity towards the particulate materials. The recoveries found for the heavier PAHs reflect this pattern (Fig. 2) since bark shows slightly better results compared with the needles. Still, according to Fradinho et al. [46], P. pinaster bark is formed by lignin and polyphenolics (ca. 44 %), polysaccharides (ca. 39 %), dichloromethane, ethanol and water extractives (ca. 17 %), as well as by 1 % of ash materials. Lignin and cellulose contents are 33.2 and 24%, respectively. This means that bark contains elements which may also trap the gaseous fraction by several mechanisms, namely the hemicellulose and the extractives, by ionic exchange [47]. Suberin, a waxy highly hydrophobic substance, was reported as favouring the uptake of organochlorine pollutants onto bark [48].
P. pinea
Table 3 shows the mean recoveries for pine needles and bark samples from P. pinea trees. The overall recoveries for this species fall between 75 and 120 % and show a small improvement comparing to those of P. pinaster, particularly for the needles. As mentioned before, the chromatographic analysis of P. pinea needles proved to be easier than P. pinaster’s and this can be an explanation for this better yield. Furthermore, and contrarily to the observed for P. pinaster, the increase in sample mass from 1 to 5 g does not affect the recovery values, except for Flt and Pyr, which maintained such a trend. It is noteworthy that the results for the higher molecular PAHs are again considerably better that for the USE-SPE and MAE-SPE methodologies [33], for both needles and bark. However, Fig. 2 shows that bark in this case presents some problems in the recoveries of the gas-bound lighter PAHs such as Acy, Phen of Ant, with mean values not reaching 70 %. Since to the author’s best knowledge there are no studies comparing the physical properties, morphology or constituents of the barks of the two species in study, this evidence may be related to a different composition of the P. pinea bark, probably less prone to retain gaseous organic pollutants. According to Fig. 2, it can be said that in terms of PAHs recoveries, P. pinea trees show slightly better performance than P. pinaster.
Since this methodology has never been tested for pine bark and needles it is difficult to have terms of comparison. Still, it can be said that the recoveries obtained for the heavier PAHs are better that those found for USE-SPE and MAE-SPE approaches. For similar approaches employed in the PAHs quantification on other types of matrices, Herbert et al. [9] found overall lower recoveries (between 8 and 121 %) for 12 PAHs in landfill leachate, which can probably be considered an even more complicated matrix to analyse. Better results were reported by Wei et al. [15] for eight predominantly gas-phase PAHs (between 80 and 108 %), but in this case for much “cleaner” matrices as are air samples collected in XAD-2 adsorbents.
Overall, the validation of MAE-HS-SPME using spiked samples revealed that needles and bark from both pine species are suitable for the extraction of quantification of PAHs, and may enhance the assessment of the particulate-phase PAHs relatively to other approaches.
Validation with naturally contaminated samples
To complete the method validation, its application was studied for naturally contaminated samples from four sites in Portugal, two urban (Porto) and two rural (Estrela and Loulé). Both pine species were represented by an urban and a rural site. Given the recovery results it was decided to analyse 2 g samples, an intermediate value between the masses used in the spiked samples. The concentrations of individual and total PAHs are presented in Table 4. All target PAHs were identified except for some of the particulate-bound PAHs in the rural sites.
Some essential evidences can be seen in the results. Considering the total PAHs concentration, it is clear that for both needles and bark the urban sites have a stronger incidence than the rural ones and that the P. pinaster sites have higher levels than the P. pinea’s considering the same site type. These data were within the expected, taking into account previous works in literature [32, 41, 49–51]. But if the differences between needles and bark in the same site are considered, then two things occur: for the rural sites, the values are similar for the two matrices whereas for the urban areas the total concentration in the needles almost doubles that of the bark. This fact suggest that bark probably has a limit to the amount of PAHs uptake that is lower that for needles, but only in the most contaminated this limit is reached and this difference can be acknowledged. Given the number of sites considered, this assumption must be taken with care. In the previous study by Ratola et al. [33] using USE-SPE and MAE-SPE and involving the same pine species, a considerable disparity was found between needles and bark for the same site (from 2 to 17 times higher total PAH concentration in the needles). This can also mean that the proposed MAE-HS-SPME approach allows not only a better quantification of the PAHs trapped in the bark but also an enhancement of the needles assessment, since the concentrations found for P. pinaster in Porto are also higher than those found in the aforementioned study (1,320 vs. 655 ng g−1, dry weight). The concentrations for bark also surpass those obtained by other authors using diverse extraction procedures but also different pine species [40, 45].
In individual terms, Phen was the most abundant PAH in all needle samples except for bark in Porto, where Pyr showed a slightly higher concentration. The stability and abundance of Phen is reported in literature and most similar studies confirm this evidence [52]. But it would be interesting to study the possibility of a similitude of patterns according to the number of aromatic rings and, consequently, to the predominant phase of each PAH. The results are shown in Fig. 3.

PAHs patterns in terms of predominant existing phase in naturally contaminated pine needles and bark samples from urban (Porto) and rural (Estrela and Loulé) sites. Gas phase—two and three rings; gas/particulate phases—four rings; particulate phase—five and six rings
It is clear that the bark samples trap higher percentages of the heavier (particulate) PAHs than the needles and these are even predominant in the P. pinaster samples from Porto (almost 40 %). On the other hand, the needles maintain a relatively constant pattern between sites, with a predominance of the gas phase PAHs (over 65 %) and there are also no significant differences between P. pinaster and P. pinea. This reinforces the proneness of needles to uptake predominantly the lighter PAHs onto their waxy layer and the higher affinity for the deposition of particulate material into the porous surface of bark. Bark shows, however, a less consistent behaviour between each site, which may be explained by the fact that meteorological parameters such as wind or rain may contribute to the easier removal of the particulate material from the surface of the respective matrix [53].
Conclusions
Microwave assisted extraction coupled with headspace solid-phase microextraction (MAE-HS-SPME) followed by GC-MS proved its suitability for biomonitoring assessment of PAHs in pine needles and bark from two different species. Validation was performed mainly by recovery assays at three different spiking levels and two sample masses and revealed some improvement comparing to the current techniques, especially for bark extraction and the particulate-bound PAHs. Naturally contaminated samples from two urban and two rural sites revealed that the needles have a predominance of gas-phase PAHs and that bark has a higher tendency to trap the particulate PAHs. For the sites with higher total PAH levels, needles concentrations surpassed those of bark by twofold and P. pinaster samples yielded higher values than P. pinea’s in all cases.
References
Arthur CL, Pawliszyn J (1990) Anal Chem 62:2145–2148
Augusto F (2002) Antonio Valente ALP. Trends Anal Chem 21:428–438
Namieśnik J, Bożena Z, Kot-Wasik A, Partyka M, Wasik A (2005) Anal Bioanal Chem 381:279–301
Tang B, Isacsson U (2008) Energy Fuel 22:1425–1438
Doong R-A, Chang S-M, Sun Y-C (2000) J Chromatogr A 879:177–188
Psillakis E, Ntelekos A, Mantzavinos D, Nikolopoulos E, Kalogerakis N (2003) J Environ Monit 5:135–140
Li Q, Xu X, Sen-Chun LF, Wang X (2006) Sci China B Chem 49:481–491
Negrão MR, Alpendurada MF (1998) J Chromatogr A 823:211–218
Herbert P, Silva AL, João MJ, Santos L, Alves A (2006) Anal Bioanal Chem 386:324–331
Hageman KJ, Mazeas L, Grabanski CB, Miller DJ, Hawthorne SB (1998) Anal Chem 68:3892–3898
Havenga WJ, Rohwer ER (1999) J Chromatogr A 848:279–295
Pino V, Ayala JH, Afonso AM, González V (2003) Anal Chim Acta 477:81–91
Zuazagoitia D, Millán E, Garcia-Arrona R (2009) Chromatographia 69:175–178
Vaz JM (2003) Talanta 60:687–693
Wei M-C, Chang W-T, Jen J-G (2007) Anal Bioanal Chem 387:999–1005
Agozzino P, Avellone G, Boscaino G, Miceli S (1999) J Mass Spectrom 34:1383–1384
Wu C-H, Chen C-L, Huang C-T, Lee M-R, Huang C-M (2004) Anal Lett 37:1373–1384
Nievas ML, Commendatore MG, Olivera NL, Esteves JL, Bucalá V (2006) Bioresource Technol 97:2280–2290
Purcaro G, Morrison P, Moret S, Conte LS, Mariott PJ (2007) J Chromatogr A 1161:284–291
Cam D, Gagni S, Lombardi N, Punin MO (2004) J Chromatogr Sci 42:329–335
Aguinaga N, Campillo N, Viñas P, Hernández-Córdoba M (2008) Anal Bioanal Chem 391:1419–1424
Poon K-F, Lam PKS, Lam MHW (1999) Anal Chim Acta 396:303–308
Waidyanatha S, Zheng Y, Rappaport SM (2003) Chem Biol Interact 145:165–174
Hellström A, Kylin H, Strachan WMJ, Jensen S (2004) Environ Pollut 128:29–48
Isidorov VA, Vinogorova VT, Rafałowski K (2003) Atmos Environ 37:4645–4650
Mateus E, Barata RC, Zrostlíková J, Gomes da Silva MDR, Paiva MR (2010) J Chromatogr A 1217:1845–1855
Simonich S, Hites R (1995) Environ Sci Technol 29:2905–2914
Harju L, Saarela K-E, Rajander J, Lill J-O, Lindroos A, Heselius S-J (2002) Nucl Instr And Meth B 189:163–167
Barriada-Pereira M, Concha-Graña E, González-Castro MJ, Muniategui-Lorenzo S, López-Mahía P, Prada-Rodríguez D, Fernández-Fernández E (2003) J Chromatogr A 1008:115–122
Ratola N, Lacorte S, Alves A, Barceló D (2006) J Chromatogr A 1114:198–204
Wenzel KD, Hubert A, Manz M, Weissflog L, Engewald W, Schüürmann G (1998) Anal Chem 70:4827–4835
Piccardo MT, Pala M, Bonaccurso B, Stella A, Redaelli A, Paola G, Valerio F (2005) Environ Pollut 133:293–301
Ratola N, Lacorte S, Barceló D, Alves A (2009) Talanta 77:1120–1128
Lehndorff E, Schwark L (2004) Atmos Environ 38:3793–3808
Tremolada P, Burnett V, Calamari D, Jones KC (1996) Environ Sci Technol 30:3570–3577
Hubert A, Popp P, Wenzel KD, Engewald W, Schüürmann G (2003) Anal Bioanal Chem 376:53–60
Ratola N, Alves A, Kalogerakis N, Psillakis E (2008) Anal Chim Acta 618:70–78
Wang Y, Zhang J, Ding Y, Zhou J, Ni L, Sun C (2009) J Sep Sci 32:3951–3957
Pawliszyn J (1997) Solid phase microextraction. Theory and practice. Wiley, New York
Di Lella LA, Loppi S, Protano G, Riccobono F (2006) Atmos Environ 40:225–237
Ratola N, Amigo JM, Oliveira MSN, Araújo R, Silva JA, Alves A (2011) Environ Exper Bot 72:339–347
Wang W, Simonich SLM, Wang W, Giri B, Zhao J, Xue M, Cao J, Lu X, Tao S (2011) Atmos Res 99:197–206
Boden AR, Reiner EJ (2004) Polycyclic Aromat Compd 24:309–323
Kylin H, Grimvall E, Oestman C (1994) Environ Sci Technol 28:1320–1324
Schulz H, Popp P, Huhn G, Stärk H-J, Schüürmann G (1999) Sci Total Environ 232:49–58
Fradinho DM, Pascoal Neto C, Evtuguin D, Jorge FC, Irle MA, Gil MH, Pedrosa de Jesus J (2002) Ind Crop Prod 16:23–32
Han JS (1999) In: Proceedings of the 2nd Inter-Regional Conference on Environment- Water, Lausanne—Switzerland, 1–3 September 1999
Meredith ML, Hites RA (1987) Environ Sci Technol 21:709–712
Ratola N, Amigo JM, Alves A (2010) Arch Environ Contam Toxicol 58:631–647
Ratola N, Amigo JM, Alves A (2010) Chemosphere 81:1517–1525
Amigo JM, Ratola N (2011) Alves A 45:5988–5996
Hwang H-M, Wade TL, Sericano JL (2003) Atmos Environ 37:2259–2267
Srogi K (2007) Environ Chem Lett 5:169–195
Acknowledgements
The authors wish to thank Fundação para a Ciência e a Tecnologia (Portugal) for the post-doctoral grant SFRH/BPD/67088/2009 and the project PTDC/AGR-CFL/102597/2008.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ratola, N., Herbert, P. & Alves, A. Microwave-assisted headspace solid-phase microextraction to quantify polycyclic aromatic hydrocarbons in pine trees. Anal Bioanal Chem 403, 1761–1769 (2012). https://doi.org/10.1007/s00216-012-5962-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-5962-2