
Abstract
This paper describes a headspace solid-phase microextraction (HS-SPME) procedure coupled to gas chromatography with mass spectrometric detection (GC–MS) for the determination of eight PAHs in aquatic species. The influence of various parameters on the PAH extraction efficiency was carefully examined. At 75 °C and for an extraction time of 60 min, a polydimethylsiloxane–divinylbenzene (PDMS/DVB) fiber coating was found to be most suitable. Under the optimized conditions, detection limits ranged from 8 to 450 pg g−1, depending on the compound and the sample matrix. The repeatability varied between 7 and 15% (RSD). Accuracy was tested using the NIST SRM 1974b reference material. The method was successfully applied to different samples, and the studied PAHs were detected in several of the samples.

Headspace SPME sampling followed by GC–MS facilitates routine monitoring of PAHs in aquatic species
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are widespread environmental contaminants. Some of them have been classified as carcinogens by the International Agency for Research on Cancer (IARC) [1] and 16 PAHs have been listed as priority pollutants by the US Environmental Protection Agency (US EPA) [2]. The eight PAHs studied in the present work are included by the International Programme on Chemical Safety (IPCS) in the Environmental Health Criteria document on PAHs [3]. One of the most important sources of human exposure to PAHs is the ingestion of contamined food [4], and so the analysis of PAHs in food samples has become an important subject in analytical chemistry and is under continuous revision [5]. Although environmental PAH concentrations are low, they have a strong tendency to bioaccumulate in organic tissues due to their lipophilic character and electrochemical stability.
Several reviews have been published dealing with the chromatographic determination of PAHs [6–9]. Gas chromatography (GC) and to a lesser extent high-performance liquid chromatography (HPLC) are commonly used for PAH determination in environmental and food samples. Mass spectrometry (MS) [10–26] and fluorescence detection (FLD) [27–33] are the most commonly used detectors for GC and HPLC, respectively.
The complex matrices of fish samples make it necessary to include steps for isolating the analytes from the sample matrix and clean-up stages. In this sense, liquid–liquid extraction (LLE) [10, 14, 16, 17, 20, 22, 28, 31], supercritical fluid extraction (SFE) [12] and matrix solid-phase dispersion [29] have been applied successfully, but with the inherent disadvantages of being rather labor-intensive, time-consuming, and requiring large volumes of solvents and the inclusion of supplementary clean-up steps.
Solid-phase microextraction (SPME) is a relatively straightforward, rapid and efficient alternative for analyzing organic substances present in trace amounts in complex matrices, because it allows extraction and clean-up to be carried out in a single step and eliminates the need for solvents and expensive equipment. The literature describes a few procedures for PAH determination in food samples using SPME [11, 18, 19, 24, 26]. In particular, as far as we know, only one previous work has been reported for fish samples [11]. Therefore, any new procedure for analyzing PAHs in this type of matrix will be of undoubted interest from an analytical and safety point of view.
The aim of this study is to develop a procedure for determining volatile PAHs with up to four rings [naphthalene (NA), acenaphthylene (AcPY), acenaphthene (AcP), fluorene (FL), phenanthrene (PHEN), anthracene (AN), fluoranthene (FLUR) and pyrene (PY)] in aquatic species using headspace SPME sampling followed by GC–MS in the single ion monitoring (SIM) mode.
Experimental
Reagents and materials
PAH standards were provided by Fluka (Steinheim, Germany) (NA, AcP, PHEN and PY), Aldrich (Steinheim, Germany) (AcPY, FL and FLUR) and Dr. Ehrenstorfer (Augsburg, Germany) (AN). All standards were of 97–99.7% purity. Stock solutions at 1000 mg L−1 were prepared in acetonitrile (NA, AcPY, AcP, PHEN and PY), methanol (FL and FLUR) or acetonitrile:dichloromethane 50:50 (v/v) (AN) and stored in the dark at 4 °C. More dilute solutions at 10 mg L−1 were prepared monthly in acetonitrile. A working standard mixed solution was prepared daily by diluting with acetonitrile.
Analytical-reagent-grade acetonitrile, methanol and dichloromethane were supplied by Lab-Scan (Dublin, Ireland). Sodium acetate was obtained from Riedel-de Haën (Steinheim, Germany) and sodium chloride from Panreac (Barcelona, Spain). Deionized water was obtained from a Milli-Q water purification system (Millipore, Bedford, MA, USA).
The SPME manual holder and fibers coated with 100 μm polydimethylsiloxane (PDMS), 65 μm polydimethylsiloxane-divinylbenzene (PDMS/DVB) and 85 μm polyacrylate (PA) were purchased from Supelco (Bellefonte, PA, USA). The fibers were conditioned prior to use by heating in the injection port of the GC according to the manufacturer’s recommendations. All analyses were performed in 15-mL glass SPME vials, and the solutions were stirred with a magnetic stirrer (RH KT/C IKA Werke, Staufen, Germany) using PTFE-coated magnetic stir bars (10 mm × 6 mm o.d.). To prevent analyte evaporation, the vials were immediately sealed with hole-caps and PTFE/silicone septa. A laboratory-made system consisting of a drilled block equipped with an electronic temperature control system built in the Central Laboratory Service of the University of Murcia was used for heating.
Four canned species (tuna, clam, anchovy and mussel) and fresh and smoked salmon and trout were obtained from a local supermarket.
In order to check the accuracy of the proposed method, a certified reference material was analyzed: Organics in Mussel Tissue (NIST SRM 1974b), obtained from the National Institute of Standards and Technology (Gaithersburg, MA, USA). This material was prepared by weighing 1 g in a 15-mL SPME vial and adding 5 mL of a 24% (w/v) sodium chloride solution.
Solid-phase microextraction procedure
Prior to extraction, the samples were cut and homogenized in a mortar. To carry out the extractions, 4 g of the homogenate were weighed into a 15-mL SPME vial and 5 mL of a 24% (w/v) sodium chloride solution were added. The mixture was homogenized by inserting the vial into the heating block, which had previously been programmed at 75 °C, and then stirring for 1 min at maximum power. After this sample homogenization step, the fiber was exposed to the sample vial headspace for 60 min at 75 °C, while the mixture was magnetically stirred at 730 rpm. The fiber was immediately desorbed in the injection port of the GC in the splitless mode at 270 °C for 5 min.
Gas chromatography–mass spectrometry
GC analyses were performed on an Agilent 6890N (Agilent, Waldbronn, Germany) gas chromatograph coupled to an Agilent 5973 quadrupole mass selective spectrometer equipped with an inert ion source and provided with a split-splitless injection port. The chromatograph was performed on an HP-5MS (Agilent, Waldbronn, Germany) capillary column (5% diphenyl–95% dimethyl polysiloxane) (30 m × 0.25 mm i.d., 0.25 μm film thickness). The carrier gas was helium at a constant pressure of 13 psi. Desorption of the fibers into the injection port was carried out in the splitless mode at 270 °C for 5 min using a SPME direct inlet liner (0.75 mm i.d.) (Supelco). The oven temperature was programmed as follows: from 40 to 120 °C at 10 °C min−1 and then to 210 °C at 3 °C min−1. The ionization was carried out in the electron impact (EI) mode at 70 eV. The temperatures of the ion source and the transfer line were 230 and 325 °C, respectively. The compounds were quantified in the selected ion monitoring (SIM) mode using suitable ions for each compound, in order to improve the detection limits.
Recovery assays
Recovery studies were applied to four different fish samples (canned tuna and clam, fresh salmon and smoked salmon) at two concentration levels, and two replicates were analyzed in each case.
Fish samples were spiked by adding 25 μL of a mixture standard solution prepared in acetonitrile to 4 g of sample placed in a 15-mL SPME vial. Fortification levels were in the range 0.5–10 ng g−1. The spiked samples were homogenized by manual shaking for 5 min and kept at room temperature for 3 h before being analyzed.
Results and discussion
Chromatographic conditions
The capillary column coated with a nonpolar stationary phase, HP-5MS, was selected according to the nonpolar character of PAHs. Several temperature programs and helium pressure values were tested in order to elute the compounds in the lowest time, and the selected conditions are summarized in the “Experimental” section. The retention times for each compound are shown in Table 1. Three specific ions (the quantification ion and two for confirmation purposes) were monitored for each PAH. The quantification ion and the qualifier-to-quantification ion ratios for each compound are indicated in Table 1 too.
SPME conditions
Selection of the fiber type and SPME mode
These SPME parameters were optimized using 2 g of a canned tuna sample (spiked with the PAHs at concentration levels ranging from 10 to 20 ng g−1, depending on the compound), suspended in 5 mL of water for each analysis. All experiments were carried out in duplicate. The nonpolar PDMS, medium-polar PDMS/DVB and polar PA coatings were investigated and the highest extraction efficiencies were attained for most compounds with the medium-polar PDMS-DVB fiber, due to the π–π interactions between DVB and the aromatic rings of the analytes. As expected, because of the nonpolar character of the analytes, the lowest adsorptions were obtained using the PA fiber.
Both headspace (HS) and direct immersion (DI) SPME extraction modes were checked by exposing the fiber to the sample for 60 min at 75 °C and 40 °C, respectively. The sample solution was not stirred when the DI mode was tested because the suspended particles might damage the fiber. The high background obtained with DI, especially in the 128 ion chromatogram, hindered the integration of peaks when the fiber was immersed in the extraction solution, and so HS mode was selected. In this way, fibers could be used for 95–100 analyses under the selected conditions.
Addition of sodium chloride
The addition of salt might increase the PAH extraction efficiency in HS by reducing their solubility in the extraction solution. Therefore, the effect of the ionic strength was studied by varying the sodium chloride concentration between 0 and 36% (w/v). PHEN, AN, FLUR and PY signals increased up to a 24% (w/v) salt concentration and then decreased. However, the other compounds showed slightly lower signals with increasing salt concentration. A 24% (w/v) sodium chloride concentration was finally selected in order to enhance the sensitivity of the most strongly retained and less sensitive compounds.
Extraction temperature and time
The influence of the adsorption temperature was tested in the 55–85 °C range. As expected, the extraction efficiency increased with temperature for all of the compounds, as shown in Fig. 1a. This effect was highly marked for four-ring PAHs, for which a tenfold increase in peak area was obtained in the temperature range studied. Nevertheless, the repeatability was poor (RSD>30%) when temperatures higher than 75 °C were used, and so this value was selected.

Influence of adsorption temperature (a) and adsorption time (b) on the peak area
The extraction time was studied between 20 and 90 min (Fig. 1b). As can be seen, maximum sensitivity was attained at 60 min for all of the compounds, after which time the signals decreased. An adsorption time of 60 min was therefore adopted.
Stirring speed
When the stirring speed was varied between 100 to 2000 rpm, the highest sensitivity was obtained at 730 rpm for all compounds, and so this speed was adopted (see Fig. 2).

Effect of stirring speed on the peak area of PAHs. NA (filled circles), AcPY (unfilled circles), AcP (filled triangles), FL (unfilled triangles), PHEN (filled squares), AN (unfilled squares), FLUR (filled rhombi) and PY (unfilled rhombi)
Extraction solution volume and sample mass
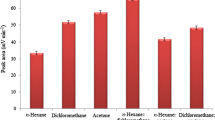
Spiked samples (2 g) were suspended in different volumes (2–6 mL) of 24% (w/v) sodium chloride solution. Higher volumes were not used to avoid the partial immersion of the fiber. Figure 3a shows the influence of this variable on the extraction efficiency. Since most compounds showed the highest sensitivity at 5 mL (except for FLUR and PY, whose areas increased with increasing solution volumes), this volume was selected.

Effect of extraction solution volume (a) and sample mass (b) on the sensitivities of the PAHs
The influence of sample mass was checked in the 2–4 g range, using a blank sample and maintaining the PAH concentrations in the final extraction solution constant. Figure 3b illustrates that no significant differences in sensitivity were observed with sample mass, and so a mass of 4 g was used since it provided lower detection limits. Masses above 4 g were not tested because of poor homogenization of the sample solutions.
Desorption conditions
The fiber was submitted to different temperatures, ranging between 200 and 270 °C (the highest allowable coating temperature), in the injection port of the GC. The sensitivities of all of the compounds increased with temperature, and so 270 °C was selected. As regards the desorption time, 5 min was sufficient to desorb the analytes and to prevent carryover.
Evaluation of the matrix effect
The matrix effect was studied by comparing the slopes of aqueous standards and standard additions calibration graphs for four different samples. In all cases, four spiking levels were performed and peak areas were used for calibration purposes. The presence of suspended biological matter significantly reduced the extraction efficiency, clear differences in sensitivity being obtained between aqueous standards in the absence and in the presence of the sample. A paired t-test was applied and the “p” values obtained were lower than 0.05 in most cases, and so the standard additions method was deemed to be necessary for quantitation purposes.
Analytical characteristics of the method
As expected, the analytical characteristics of the method differed for each sample analyzed. Good linearity was observed, with correlation coefficients (r 2) of more than 0.99 being obtained for all of the compounds in each of the sample matrices analyzed. Detection limits using a signal-to-noise ratio of 3 varied between 8 and 170 pg g−1, corresponding to AcP and PY, respectively, for the canned clam sample, selected as a representative sample for low-fat matrix content. Quantitation limits using a signal-to-noise ratio of 10 varied between 25 and 560 pg g−1, depending on the compound. The repeatability was calculated by using the relative standard deviation from a series of ten HS-SPME consecutive analyses of a spiked sample at concentration levels ranging from 0.5 to 5 ng g−1, depending on the compound. The obtained values ranged between 7 and 15%. Detection limits, calculated as detailed above, for smoked salmon varied between 15 and 450 pg g−1, for NA and PHEN, respectively, with the corresponding quantification limits being 50 and 1500 pg g−1.
Analysis of samples and validation of the method
The proposed method was applied to eight different samples. Figure 4 shows the extracted ion chromatograms obtained by HS–SPME–GC–MS under the SIM mode for a spiked canned clam sample under the selected conditions. The results obtained (Table 2) revealed that all of the samples contained one or more of the PAHs, except for the canned anchovy and fresh trout samples. PY was not detected in any sample. As expected, higher PAH concentrations were found in smoked samples with respect to unsmoked counterparts, since PAHs may also be formed as a result of certain food preparation methods, including smoking [4].

Total ion chromatograms (TIC) obtained by HS–SPME–GC–MS under the SIM mode from a canned clam sample, spiked with a standard mixture of the PAHs at concentration levels of: (1) NA, 0.5 ng g−1; (2) AcPY, 0.5 ng g−1; (3) AcP, 0.5 ng g−1; (4) FL, 0.5 ng g−1; (5) PHEN, 1.0 ng g−1; (6) AN, 1.0 ng g−1; (7) FLUR, 1.0 ng g−1; (8) PY, 1.0 ng g−1
To check the accuracy of the proposed method, recovery studies and an analysis of the standard reference mussel tissue (SRM 1974b) were carried out. Recovery studies involved spiking four different samples (canned tuna and clam, fresh salmon and smoked salmon) at two different concentration levels between 0.5 and 10 ng g−1, depending on the compound. The PAH recoveries from spiked samples varied from 88 to 111%, with an average recovery±SD (n = 128) of 99 ± 4.5, and were calculated by subtracting the corresponding analyte contents. The reference material was prepared by weighing 1 g in a 15 mL vial and adding 5 mL of a 24% (w/v) sodium chloride solution. A sample mass of 1 g was used to analyze this material in order to obtain analytical signals within the linearity range of all of the certified PAHs. It was also checked that 4 g of sample provided good results for the four PAHs with lower contents. Good agreement between the results obtained and the reference values was attained, as shown in Table 3.
Conclusions
The combination of HS-SPME and GC–MS represents a simple, reliable and sensitive alternative for the routine monitoring of volatile PAHs in aquatic species. The proposed method provides very low detection limits for the samples analyzed. Nevertheless, the complexity of the matrices of the samples makes it necessary to quantify the analytes by means of the standard additions method. The analytical characteristics of the proposed method make it a useful tool for the routine monitoring of PAHs in aquatic species.
References
World Health Organization International Agency for Research on Cancer (1998) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, vol 32 (online). http://monographs.iarc.fr/ENG/Monographs/vol32/volume32.pdf. Last accessed Sept 2007
US Environmental Protection Agency (2007) Semivolatile target compound list and corresponding CRQLs. http://www.epa.gov/superfund/programs/clp/svtarget.htm. Last accessed Sept 2007
UN International Programme on Chemical Safety (1998) Environmental Health Criteria 202: Selected non-heterocyclic polycyclic aromatic hydrocarbons. http://www.inchem.org/documents/ehc/ehc/ehc202.htm. Last accessed Sept 2007
EC Scientific Commitee on Food (2002) Opinion of the Scientific Commitee on Food on the risks to human health of polycyclic aromatic hydrocarbons in food. http://ec.europa.eu/food/food/chemicalsafety/contaminants/out153_en.pdf. Last accessed Sept 2007
Wenzl T, Simon R, Kleiner J, Anklam E (2006) Trends Anal Chem 25:716–725
Namieśnik J, Zygmunt B (2002) Chromatographia 56:9–18
Santos FJ, Galceran MT (2002) Trends Anal Chem 21:672–685
Stolyhwo A, Sikorski ZE (2005) Food Chem 91:303–311
Poster DL, Schantz MM, Sander LC, Wise SA (2006) Anal Bioanal Chem 386:859–881
Pointet K, Milliet A (2000) Chemosphere 40:293–299
Guillén MD, Errecalde MC (2002) J Sci Food Agric 82:945–952
Ali MY, Cole RB (2002) Anal Bioanal Chem 374:923–931
Easton MDL, Luszniak D, Von der Geest (2002) Chemosphere 46:1053–1074
Vives I, Grimalt JO (2002) J Chromatogr B 768:247–254
King AJ, Readman JW, Zhou JL (2004) Anal Chim Acta 523:259–267
Filipkowska A, Lubecki L, Kowalewska G (2005) Anal Chim Acta 547:243–254
Anyakora C, Ogbeche A, Palmer P, Coker H (2005) J Chromatogr A 1073:323–330
Vichi S, Písale L, Conte LS, Buxaderas S, López-Tamames E (2005) J Chromatogr A 1090:146–154
Guillén MD, Sopelana P (2005) J Dairy Sci 88:13–20
Yurchenko S, Mölder U (2005) J Food Comp Anal 18:857–869
Navarro P, Cortazar E, Bartolomé L, Deusto M, Raposo JC, Zuloaga O, Arana G, Etxebarria N (2006) J Chromatogr A 1128:10–16
Jones RP, Millward RN, Karn RA, Harrison AH (2006) Chemosphere 62:1795–1805
Herbert P, Silva AL, Joao MJ, Santos L, Alves A (2006) Anal Bioanal Chem 386:324–331
Martin D, Ruiz J (2007) Talanta 71:751–757
Wei MC, Chang WT, Jen JF (2007) Anal Bioanal Chem 387:999–1005
Aguinaga N, Campillo N, Viñas P, Hernández-Córdoba M (2007) Anal Chim Acta 596:285–290
Popp P, Bauer C, Moeder M, Paschke A (2000) J Chromatogr A 897:153–159
Jánská M, Tomaniová M, Hajslová J, Kocourek V (2004) Anal Chim Acta 520:93–103
Pensado L, Casais MC, Mejuto MC, Cela R (2005) J Chromatogr A 1077:103–109
Busseti F, Heitz A, Cuomo M, Badoer S, Traverso P (2006) J Chromatogr A 1102:104–115
Pena T, Pensado L, Casais C, Mejuto C, Phan-Tan-Luu R, Cela R (2006) J Chromatogr A 1121:163–169
Viñas P, Campillo N, Aguinaga N, Pérez-Cánovas E, Hernández-Córdoba M (2007) J Chromatogr A 1164:10–17
Perugini M, Visciano P, Manera M, Turno G, Lucisano A, Amorena M (2007) J Agric Food Chem 55:2049–2054
Acknowledgements
The authors are grateful to Comunidad Autónoma de la Región de Murcia (CARM, Fundación Séneca, Project 02993/PI/05) and to the Spanish MEC (Project CTQ2006-08037/BQU) for financial support. N. Aguinaga also acknowledges a fellowship from CajaMurcia, Spain.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aguinaga, N., Campillo, N., Viñas, P. et al. Evaluation of solid-phase microextraction conditions for the determination of polycyclic aromatic hydrocarbons in aquatic species using gas chromatography. Anal Bioanal Chem 391, 1419–1424 (2008). https://doi.org/10.1007/s00216-008-1862-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-1862-x