
Abstract
Sepsis is a dysregulated response to severe infection characterized by life-threatening organ failure and is the leading cause of mortality worldwide. Multiple organ failure is the central characteristic of sepsis and is associated with poor outcome of septic patients. Ultrastructural damage to the mitochondria and mitochondrial dysfunction are reported in sepsis. Mitochondrial dysfunction with subsequent ATP deficiency, excessive reactive oxygen species (ROS) release, and cytochrome c release are all considered to contribute to organ failure. Consistent mitochondrial dysfunction leads to reduced mitochondrial quality control capacity, which eliminates dysfunctional and superfluous mitochondria to maintain mitochondrial homeostasis. Mitochondrial quality is controlled through a series of processes including mitochondrial biogenesis, mitochondrial dynamics, mitophagy, and transport processes. Several studies have indicated that multiple organ failure is ameliorated by restoring mitochondrial quality control mechanisms and is further amplified by defective quality control mechanisms. This review will focus on advances concerning potential mechanisms in regulating mitochondrial quality control and impacts of mitochondrial quality control on the progression of sepsis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The definition of sepsis has been updated to include infection-induced multiple organ dysfunction. Sepsis is the leading cause of intensive care units (ICU) mortality worldwide, and the mortality varies between 20 and 50% [1, 2]. The incidence of sepsis has increased recently, and an increasing elderly population, cancer, immunosuppression, and antimicrobial resistance contribute to the increased incidence [2,3,4,5]. Although there have been advances in sepsis management, both mortality and morbidity remain high [6]. Most patients displaying an initial overwhelming hyper-inflammatory response die rapidly days or weeks later [7, 8]. Additional investigations demonstrated that immunosuppression plays a central role in the late phase of sepsis and contributes to multiple organ dysfunction [9,10,11]. Immunosuppression is caused by dysfunction and apoptosis of immune cells including CD4+ and CD8+ T cells, B cells, and dendritic cells (DCs). Although there are strategies targeting immunosuppression, the outcome of sepsis is still poor [12, 13]. Therefore, it is critical to understand the specific mechanisms of immunosuppression and subsequent multiple organ failure in sepsis.
Ultrastructural damage to the mitochondria and mitochondrial dysfunction has been reported in immune cells, cardiomyocytes, skeletal muscle, and hepatocytes under septic exposure [14,15,16,17,18]. Furthermore, ATP depletion, depletion of intracellular antioxidant systems, and respiratory chain (electron transport chain) inhibition are all observed in septic patients and experimental models of sepsis [16, 17]. Persistent mitochondrial dysfunction contributes to sepsis-related organ failure and poor outcome of septic patients. Thus, removal of dysfunctional mitochondria and generation of healthy mitochondria improve recovery of organ function in sepsis. However, consistent mitochondrial dysfunction shows defective mitochondrial quality control mechanisms. Recent studies have demonstrated that organ dysfunction is amplified by defective mitochondrial quality control mechanisms and is ameliorated by recovery of mitochondrial quality control mechanisms [19,20,21].
In this review, we illustrate how mitochondrial quality control allows the mitochondrial network to segregate or recognize and eliminate damaged mitochondria, and generate new mitochondria (Fig. 1). Furthermore, we provide an overview of the role of the mitochondrial quality control mechanisms on organ dysfunction under septic exposure (Table 1).

Mitochondrial quality control mechanisms. Mitochondrial biogenesis generates new mitochondria to recover the mitochondrial network. PGC-1α is a central regulator of mitochondrial biogenesis. PGC-1α interacts with various transcription factors including estrogen-related receptor α (ERR-α), forkhead box class-O (FoxO1), hepatocyte nuclear factor 4a (HNF4a), nuclear respiratory factor (NRF1), and NRF2 to activate the transcription of nuclear genes encoding mitochondrial proteins. Tfam is imported into mitochondria and activates mtDNA transcription and replication. Mitochondria undergo fusion and fission to modulate mitochondrial morphology, number, and size. Mitochondrial fission and fusion are regulated by evolutionarily conserved dynamin-related GTPases that include fission protein dynamin-related protein 1 (Drp1) and its receptors mitochondrial fission protein 1 (Fis1), mitochondrial fission factor (Mff), mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51), and the fusion proteins mitofusin (Mfn)1, Mfn2, and optic atrophy 1 (OPA1). Mitophagy is regulated by Pink1/PARK2 pathway and eliminates damage mitochondria via fusing with lysosome
Mitochondrial dysfunction
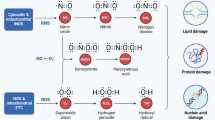
Mitochondria are subcellular organelles that provide energy termed ATP via oxidative phosphorylation (OXPHOS) that are responsible for most oxygen consumption. Mitochondria are also associated with calcium homeostasis, intracellular ROS generation, and cell signaling functions [22, 23]. In response to sepsis, the inflammatory cytokines of the innate immune response including TNF-α, IL-1β, and IL-6 promote mitochondrial permeability transition, inhibit oxidative phosphorylation, and improve ROS production [24, 25]. In addition, oxidative stress caused by the imbalance of ROS generation and antioxidant protective mechanisms leads to peroxidation of the mitochondrial lipids cardiolipin, mtDNA damage, and more ROS formation under septic conditions [26, 27]. Nitric oxide (NO) and ROS also inhibit electron transport chain (complex IV and complex I). High Ca2+, low ATP, mtDNA mutations, mitochondrial permeability pore opening, and oxidative stress are all observed in sepsis. These changes lead to mitochondrial swelling that damages the mitochondrial ultrastructure and results in release of cytochrome c and apoptosis-related markers into the cytoplasm [28]. Mitochondrial dysfunction has been demonstrated to activate cell apoptosis and ultimately results in organ failure. Interestingly, targets for mitochondrial therapy augment multiple organ function during sepsis [29]. For example, L-carnitine, succinate, ATP-MgCl2, cytochrome c, and ubiquinol (coenzyme Q) administration can target the electron transport system (ETS) and all have been reported to increase the survival rate in septic models by restoring ETS function and increasing oxygen consumption and phosphorylation [30,31,32,33,34]. In addition, mitochondrial antioxidants, including MitoQ/PBN/SkQ, SS peptides, glutathione, and melatonin all can ameliorate multiple organ failure during sepsis by decreasing oxidative stress markers [35,36,37].
Mitochondrial biogenesis
Mitochondrial biogenesis is regulated by multiple molecular signals in response to energy demand and contributes to an increase in mitochondrial mass and recovery of the mitochondrial network. These molecular signals include several DNA-binding transcriptional factors, such as peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), nuclear respiratory factor 1 (NRF1) and NRF2, and mitochondrial transcription factor A (TFAM) [20, 38]. PGC-1α is a central regulator of mitochondrial biogenesis. PGC-1α interacts with various transcription factors including estrogen-related receptor α (ERR-α), forkhead box class-O (FoxO1), hepatocyte nuclear factor 4a (HNF4a), nuclear respiratory factor (NRF1), and NRF2 to regulate nuclear gene expression [39]. In addition, PGC-1α and NRF1 coactive the expression of TFAM, TFB1M, and TFB2M that mediate mtDNA transcription and replication [40]. NRF1 and NRF2 are two key regulators of mitochondrial biogenesis and are activated by PGC-1α. Activated NRF1 and NRF2 reportedly regulate the expression of proteins related with mitochondrial respiratory function and mitochondrial translation encoded by nuclear genes [41]. Following activation by PGC-1α and NRF1, TFAM and TFB2M are translocated from the cytosol into mitochondria. In mitochondria, TFAM binds to mitochondrial DNA and regulates mtDNA transcription and replication [42].
It has been shown that the activation of mitochondrial biogenesis transcription factors is earlier in survivors than non-survivors, which indicates the earliest recovery of mitochondrial function is crucial in sepsis. A deep investigation found that mRNA expression of PGC-1α is only elevated in survivors and the decrease in transcription of respiratory chain subunits is less in survivors than non-survivors [20]. In the liver, key regulators of mitochondrial biogenesis such as PGC-1α, NRF1, NRF2, and TFAM are markedly increased in experimental models following septic exposure [43, 44]. In an age-dependent model, the results show that young mice exhibit a time-dependent increase in nuclear levels of PGC-1α, whereas mature mice exhibit a decrease in nuclear levels of PGC-1α after sepsis [45]. In the lung, PGC-1α expression is increased more than sixfold and TFAM expression is increased fourfold during murine sepsis [46]. Furthermore, previous studies have shown that expression levels of PGC-1α, NRF1, NRF2, and TFAM are all increased in the heart and in astrocytes under septic conditions [47, 48]. Several pharmacological therapies can prevent organ failure partly through activation of mitochondrial biogenesis in sepsis. Septic models with inhaled CO show rescue of liver failure from sepsis through activation of mitochondrial biogenesis [49]. Additionally, the lipophilic antioxidant CoQ10 can prevent LPS-induced mitochondrial dysfunction in multiple organs by improving mitochondrial biogenesis [50]. Conversely, mice with lipopolysaccharide (LPS)-induced acute kidney injury (AKI) show a decrease in PGC-1α mRNA in the renal cortex [51, 52]. The overexpression of PGC-1α promotes recovery from LPS-induced acute kidney injury, and both global and tubule-specific PGC-1α-knockout mice suffer persistent impaired kidney function [51]. These results indicate that key factors of mitochondrial biogenesis are mostly increased in sepsis, and therapies targeting mitochondrial biogenesis could partly reverse organ failure under septic conditions.
In the initial stage of sepsis, inflammatory cytokines including TNF-α, IL-1β, and IL-6 are increased and the increased cytokine production ultimately leads to an inflammatory cytokine storm, which is a main cause of death in the initial stage of sepsis. Global ablation of the PGC-1α gene in mice increases the expression levels of IL-6 and TNF-α, and the subsequent chronic elevation of circulating IL-6 [53]. Activation of PGC-1α protects endothelial cells from LPS-induced loss of mitochondrial function and decreases LPS-induced high IL-6 concentration [54]. The mechanisms linking PGC-1α activation to the downregulation of inflammation genes may be related to recovery of mitochondrial function and reduction of ROS generation by PGC-1α activation. Recent studies have shown that overexpression of PGC-1α inhibits intracellular and mitochondrial ROS production [55]. Mice with muscle-specific PGC-1α-knockout show decreased expression of anti-ROS genes, which makes a substantial contribution to the observed increase of cytokine expression [56].
Activation of mitochondrial biogenesis by pharmacological agents emerges as therapeutic strategies in sepsis. Clinical data showed that patients with dyslipidemias treatment presented a better clinical outcome during sepsis [57, 58]. Bezafibrate, an agonist of peroxisome proliferator-activated receptor (PPAR), is a commonly used drug in dyslipidemias, which has been reported to increase PGC-1α expression [59]. A recent study demonstrated that bezafibrate treatment presented anti-inflammatory effects in experimental sepsis [60]. However, whether the protective effects of bezafibrate on sepsis is probably through activating mitochondrial biogenesis has not been evaluated so far. Another pharmacological agent which has been reported to activate mitochondrial biogenesis is metformin, a commonly used drug in diabetes [61]. Several studies have demonstrated that metformin treatment ameliorated induced brain and cardiac injury in experimental sepsis [62, 63]. However, clinical data showed that pretreatment with metformin in both diabetes and sepsis patients presented no difference in the outcome of sepsis [64].
Mitochondrial dynamics
Mitochondria are highly dynamic organelles and frequently undergo fission and fusion to modulate mitochondrial morphology, number, and size. In physiological conditions, mitochondrial fusion and fission is balanced to maintain mitochondrial and cellular homeostasis. During sepsis, mitochondrial dysfunction results in activation of mitochondrial fission and inactivation of mitochondrial fusion, which promotes dysfunctional mitochondrial fragmentation and ultimately results in organ failure [65]. Mitochondrial fragmentation contributes to BAX activation, outer membrane permeabilization (MOMP), remodeling of mitochondrial cristae, and increased ROS production and ATP depletion that results in cell death immediately and ultimately organ failure [66, 67]. Mitochondrial fission and fusion are regulated by evolutionarily conserved dynamin-related GTPases that include fission protein dynamin-related protein 1 (Drp1) and its receptors mitochondrial fission protein 1 (Fis1), mitochondrial fission factor (Mff), mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51), and the fusion proteins mitofusin (Mfn)1, Mfn2, and optic atrophy 1 (OPA1) [68]. Mfn1 and Mfn2 are located on the mitochondrial outer membrane, whereas OPA1 is located on the mitochondrial inner membrane. During the process of mitochondrial outer membrane fusion, Mfn1 and Mfn2 form homo-oligomeric and hetero-oligomeric structures to tether two adjacent mitochondria for fusion [69]. Unlike Mfns, OPA1 does not require the oligomerization of OPA1 to mediate inner membrane fusion. The coordinated actions of Mfns and OPA1 starts with outer membrane fusion and ends with mitochondrial inner membrane fusion [68]. During mitochondrial fission, Drp1 is transported from the cytosol to the mitochondrial outer membrane and forms oligomeric Drp1 complexes. The Drp1 protein oligomeric structure wraps around and constricts the mitochondrial tubule, which dissects the parent organelle into two daughters [70, 71]. The signaling mechanism of Drp1-recruitment to the mitochondria has been proposed to depend on several mitochondrial outer membrane proteins such as Fis1, Mff, MiD49, and MiD51. However, the exact functions of these proteins in mitochondrial fission remain unclear.
There are studies that monitored mitochondrial morphology and conserved dynamin-related GTPases in sepsis, but the number of studies is very limited. It has been suggested that fragmented mitochondria and Drp1 expression levels are increased in skeletal muscle under septic conditions [72]. Treatment with the de novo sphingolipid biosynthesis inhibitor myriocin ameliorates dysfunction of skeletal muscle by reverting mitochondrial morphology and decreasing Drp1 expression levels induced by sepsis [72]. Gonzalez et al. demonstrate that Mfn2 is decreased at 4–6 h, and mitochondrial fragmentation is increased at 6 h in liver after LPS treatment [21]. In contrast, Mfn2 is decreased at 12–18 h and Drp1 is increased at 4 h in liver after cecal ligation and puncture (CLP) surgery [21]. Additionally, TNF-α induces the expression levels of mitochondrial Drp1, increases phosphorylated Drp1, and enhances mitochondrial fragmentation in H9C2 cardiomyocytes [73]. Notably, Mfn2 is decreased in CD4 + T cells after HMGB1 treatment [74, 75]. The upregulation of Mfn2 protects CD4 + T cells from immune dysfunction and apoptosis induced by HMGB1. It still unclear whether the upregulation of Mfn2 to prevent the dysfunction lymphocyte apoptosis is associated with improved mitochondrial dynamics. However, mitochondrial fusion and fission rates of human PBMC obtained from a control group and sepsis group showed an insignificant difference [76].
Mdivi-1 is a pharmacological inhibitor of Drp1 and has been reported to revert sepsis-induced fragmented mitochondria to tubular mitochondria. Recent studies have shown that Mdivi-1 reduces the inhibition of mitochondrial ETC complex and hepatocyte apoptosis induced by CLP [21]. Mdivi-1 administration also ameliorates LPS-induced brain injury. Mechanically, Mdivi-1 inhibits the Drp1 increase and attenuates the OPA1 decrease in hippocampus under septic exposure [77]. There are other nonspecific pharmacological therapies that attenuate organ damage through regulating mitochondrial dynamics in sepsis. For example, heme oxygenase (HO)-1/carbon monoxide attenuates acute lung injury in sepsis partly through balancing mitochondrial fusion and fission [78].
Mitochondrial autophagy
Autophagy is a central process in cell survival under stress and non-selectively or selectively eliminates damaged proteins and organelles by formation of a double-membrane autophagosome, which then fuses with lysosomes [79]. Mitophagy is the selective autophagy of mitochondria and requires receptors to recognize damaged mitochondria for degradation. Mitophagy is regulated by several pathways including the PTEN-induced putative protein kinase 1 (Pink1) and the E3 ubiquitin ligase (Park2/Parkin), FUN14 domain-containing protein 1 (FUNDC1), NIX/BNIP3, and BCL2L1.
The Pink1/Park2 pathway is the main regulator of mitophagy and has become a topic of scientific research due to its involvement in the pathogenesis of Parkinson’s disease (PD) [80, 81]. In healthy mitochondria, Pink1 translocates on the outer mitochondrial membrane (OMM) via the TOM components, and then inserts into the inner mitochondrial membrane (IMM) through the TIM23 complex. When the N-terminal mitochondrial targeting sequence (MTS) of Pink1 that drives the translocation reaches the matrix, a matrix processing peptidase (MPP) recognizes and cleaves Pink1 [82]. Subsequently, the IMM protease PARL cleaves Pink1 p64 and generates the Pink1 p53, which then is released to the cytosol and is degraded by the proteasome [83]. In sepsis, the dysfunctional mitochondria prevent the import of Pink1 through TIM23 and this results in accumulation of Pink1 on the outer mitochondrial membrane. PINK1 recruits Park2 to translocate to the outer mitochondrial membrane from the cytosol and activates Parkin by phosphorylation of Ser65 on Park2 [84]. In the cytosol, Park2 remains “closed,” which means the repressor element of Park2 (REP) blocks E2-binding site in RING1 and RING0 shields catalytic C431 [85, 86]. Under stress conditions, Pink1 undergoes autophosphorylation at Ser228 and Ser402 and then phosphorylates basal OMM ubiquitin, which can attract Park2 to accumulate on the OMM [87]. When Park2 binds to Ps65-Ub, the constructs of Park2 are changed and this includes replacement of the inhibitory N-terminal ubiquitin-like (UBL) domain and segmentation of REP, which means that Park2 is “open” [85, 88]. The activated Park2 is further phosphorylated by Pink1 and induces more Park2 recruitment.
Activated Park2 ubiquitinates several mitochondrial proteins on the outer membrane without stringent selection, including mitochondrial fusion proteins Mfn1/2, Miro, and voltage-dependent anion channel (VDAC) 1/2/3 [89, 90]. Mfn1/2 and Miro belong to K48-linked chains, and K48 modification by Park2 induces degradation of proteins by the proteasome. The degradation of Mfn1/2 prevents mitochondrial fusion that thereby promotes mitochondrial fragmentation and mitophagy [91, 92]. When the mitochondrial transport protein Miro is degraded, all mitochondrial mobility is arrested, and this arrest may segregate damaged mitochondria before mitophagy [93, 94]. The VDAC1/2/3 are K63 ubiquitin chains and K63 is associated with the recruitment of mitophagy receptors [95]. In addition, Park2 builds ubiquitin chains on the outer mitochondrial membrane that could be sufficient to recruit autophagic receptors such as optineurin, nuclear dot protein 52 (NDP52), p62 (SQSTM1), and Tax1-binding protein 1 (TAX1BP1) [96, 97]. Optineurin and NDP52 are the primary receptors in this process, and following recruitment of optineurin and NDP52, ULK1, DFCP1, and WIPI1 are independently recruited to focal spots proximal to initiate autophagosome formation [96]. At late stages of mitophagy, LC3 bind to the autophagosome membrane to form a complete autophagosome that fuses with lysosomes for degradation.
In sepsis, mitochondrial dysfunction induces a loss of mitochondrial membrane potential that triggers mitophagy. In the heart, mitophagy is induced during sepsis and this is evidenced by the presence of several double-membrane autophagosomes, increased LC3B-II and p62 expression, and mitochondrial recruitment of Park2, BNIP3L, and BNIP3 [98, 99]. Park2-deficient mice exhibit impaired recovery of cardiac function and constant cardiac mitochondrial dysfunction in the heart during sepsis [98]. Cardiac-specific overexpression of Beclin-1 promotes PINK1/Park2-dependent mitophagy, improves cardiac function, and reduces cardiac inflammation in response to LPS [99]. LC3B-II expression in the lung is increased and LC3 colocalizes with citrate synthase in the mitochondria, which indicates mitophagy occurs during sepsis [46]. MKK3-deficient mice increase PINK1/Park2-dependent mitophagy, and this reduces lethality of mice and lowers the levels of lung and mitochondrial injury in sepsis [100]. The data also show that heterozygous PINK1 null mice are more susceptible to death and heterozygous PINK1, MKK3 null mice have higher survival than heterozygous PINK1 null mice in sepsis. The findings demonstrate the inhibition of mitophagy increases mortality of mice in sepsis. Compared with non-septic patients, both higher MMK3 and PINK1 activation are detected in PBMCs from septic patients [100]. Hepatocyte exposure to LPS induces translocation of PINK1 and Park2 to the mitochondria and colocalization of LC3 to mitochondria [101]. In the kidney, increased autophagosomes co-localize with damage and citrate in mitochondria during sepsis [102]. Collectively, these studies support that mitophagy is induced in several organs during sepsis.
Autophagy
Although several studies have demonstrated that mitophagy is induced in major organs in sepsis, the effect of mitophagy in sepsis remains unclear. Studies examining the role of mitophagy in sepsis are very limited, and therefore, we discuss the role of autophagy in sepsis. Several studies have shown that impaired autophagy contributes to amplifying organ failure in sepsis.
In the CLP model, the whole autophagic process (autophagic flux) is characterized by increased levels of LC3B-II, ATG5, ATG7, Beclin1, and p62; the presence of autophagosomes; and further increased levels of LC3B-II and p62 after chloroquine treatment early in liver. These changes are followed by incomplete autophagic flux and even suppression of autophagy at the late stage of sepsis [103,104,105]. Liver failure and apoptosis occur as soon as autophagic flux is suppressed, which indicates that suppression of autophagy is associated with liver failure after CLP. A previous study reported autophagy deficiency by downregulation of ATG7, an essential gene for autophagosome formation, aggravates hepatic mitochondrial dysfunction and liver damage and apoptosis after CLP [106]. Additionally, liver injury and apoptosis are accelerated in liver-specific ATG5-deficient mice under sepsis [19]. Chloroquine is a pharmacologic inhibitor of autophagic flux and contributes to amplifying liver failure and increases mortality during sepsis [104]. One pharmacologic therapy is the autophagic flux enhancer carbamazepine, which improves survival rates of septic mice and attenuates liver injury, inflammation, and apoptosis in the CLP septic model [105]. In the LPS-treated model, complete autophagic response is induced in hepatocytes [107, 108]. The suppression of autophagy by chloroquine and knockout out of ATG7 sensitizes mice to LPS-induced liver injury [107, 108]. In the heart, studies have shown that autophagic flux is induced in two prototype sepsis models: endotoxemia and cecal ligation and puncture (CLP) [99, 109]. Increasing Beclin-1-dependent autophagy improves cardiac function and decreases cytokine levels and protects myocardium from fibrosis following LPS challenge [99]. Moreover, rapamycin is the best-known pharmacologic agent to induce autophagy and CLP mice treated with rapamycin increase ATP generation and decrease cytokine levels [109]. Similarly, the protective effect of autophagy in sepsis can also be found in lung, kidney, skeletal muscle, and immune cells [110,111,112,113,114,115]. Collectively, these studies demonstrated that the induction of autophagy ameliorates organ failure and suppression of autophagy amplifies organ failure in sepsis.
An interesting finding showed that critically ill patients receiving over or standard feeding presented worse outcome than those subjecting to underfeeding [116]. Starvation is a main inducer of autophagy, and reduction of sepsis mortality in critically ill patients with underfeeding may be probably related to sustained induction of autophagy. Therefore, pharmacological induction of autophagy is a potential therapeutic strategy in sepsis. Rapamycin, a commonly used pharmacological agent for autophagy induction, has been reported to protect against sepsis-induced organ injury and improve the survival rate of septic mice [110, 117]. However, rapamycin treatment also affects many other metabolic pathways, and therefore clinical use of it for sepsis has little progress. There are other pharmacological agents for autophagy induction without affecting many signaling pathways, including carbamazepine, sodium valproate, and lithium, but fewer studies have demonstrated the effect of these agents on sepsis.
Conclusion
Both septic patients and animal septic models suggest that mitochondrial quality control mechanisms including mitochondrial biogenesis, dynamics, and mitophagy are induced in the early stage of sepsis (Fig. 2). In sepsis, mitochondrial dysfunction is amplified as soon as LPS/CLP-induced mitochondrial biogenesis and autophagy return to baseline level. Deficient mitochondrial biogenesis and autophagy result in healthy mitochondria depletion and increased mitochondrial fragmentation caused by mitochondrial fission. Moreover, persistent mitochondrial dysfunction leads to multiple organ failure during sepsis.


Changes in mitochondrial quality control mechanisms under sepsis. a In the initial stage of sepsis, the upregulation of PGC-1α and transcription factors promotes mitochondrial biogenesis. And a decrease in Mfn1/2 and OPA1 expression and an increase in DRP1 expression induce that the balance of mitochondrial fusion and fission shifts to mitochondrial fission. Furthermore, the accumulation of PINK1 on mitochondria induces PARK translocation from cytoplasm into mitochondrial outer membrane, which triggers mitophagy. b The mitochondrial functions are recovered in the survival of sepsis. Changes in mitochondrial quality control mechanisms are presented as following. Sustained high levels of PGC-1α and transcription factors continuously activate mitochondrial biogenesis to provide new and healthy mitochondria. The balance of mitochondrial fusion and fission is recovered. Additionally, sustained induction of mitophagy can also be found in the survival of sepsis. c In the progression of sepsis, injured mitochondria cannot be restored and turn into mitochondrial dysfunction. Changes in mitochondrial quality control mechanisms are presented as follows. The decreased levels of PGC-1α and transcription factors prevent mitochondrial biogenesis. And persistent decreased Mfn1/2 and OPA1 expression levels and increased DRP1 expression levels results in more mitochondrial fragmentation generation. Additionally, expression levels of PINK1, PARK2, p62, and LC3 are all decreased, which indicates mitophagy deficiency
Several studies have shown that impaired mitochondrial quality control mechanisms contribute to and amplify organ failure in sepsis. Using pharmacologic therapeutic agents to activate mitochondrial biogenesis or mitochondrial autophagy or decrease mitochondrial fission in septic model can ameliorate organ mitochondrial dysfunction and organ failure. However, further investigations are required to monitor the changes of mitochondrial quality control mechanisms in septic patients and translate these animal therapies into clinical therapies. Furthermore, the role of other potential mitochondrial quality control mechanisms in sepsis requires investigation.
References
Rodriguez A, Lisboa T, Blot S (2009) Mortality in ICU patients with bacterial community-acquired pneumonia: when antibiotics are not enough. Intensive Care Med 35(3):430–438
Esteban A, Frutos-Vivar F, Ferguson ND, Penuelas O, Lorente JA, Gordo F, Honrubia T, Algora A, Bustos A, Garcia G, Diaz-Reganon IR, de Luna RR (2007) Sepsis incidence and outcome: contrasting the intensive care unit with the hospital ward. Crit Care Med 35:1284–1289
Stoller J, Halpin L, Weis M, Aplin B, Qu W, Georgescu C, Nazzal M (2016) Epidemiology of severe sepsis: 2008-2012. J Crit Care 31:58–62
Gaieski DF, Edwards JM, Kallan MJ, Carr BG (2013) Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med 41:1167–1174
Suarez De La Rica A, Gilsanz F, Maseda E (2016) Epidemiologic trends of sepsis in western countries. Ann Transl Med 4:325
Finfer S (2010) The surviving sepsis campaign: robust evaluation and high-quality primary research is still needed. Intensive Care Med 36:187–189
RS Hotchkiss IEK (2003) The pathophysiology and treatment of sepsis. N Engl J Med 348:138–150
Nesseler N, Defontaine A, Launey Y, Morcet J, Malledant Y, Seguin P (2013) Long-term mortality and quality of life after septic shock: a follow-up observational study. Intensive Care Med 39:881–888
Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, Moldawer LL, Moore FA (2012) Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg 72:1491–1501
Delano MJ, Ward PA (2016) The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev 274:330–353
Fattahi F, Ward PA (2017) Understanding immunosuppression after sepsis. Immunity 47:3–5
Hotchkiss RS, Monneret G, Payen D (2013) Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13:862–874
Venet F, Monneret G (2018) Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol 14:121–137
Larche J, Lancel S, Hassoun SM, Favory R, Decoster B, Marchetti P, Chopin C, Neviere R (2006) Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J Am Coll Cardiol 48:377–385
Crouser ED (2004) Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion 4:729–741
Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, Smolenski RT, Singer M (2004) Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Phys Regul Integr Comp Phys 286:R491–R497
Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M (2002) Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360:219–223
Park DW, Zmijewski JW (2017) Mitochondrial dysfunction and immune cell metabolism in sepsis. Infection & Chemotherapy 49:10–21
Oami T, Watanabe E, Hatano M, Teratake Y, Fujimura L, Sakamoto A, Ito C, Toshimori K, Swanson PE, Oda S (2017) Blocking liver autophagy accelerates apoptosis and mitochondrial injury in hepatocytes and reduces time to mortality in a murine sepsis model. Shock 1:427–434
Carre JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P, Stotz M, Singer M (2010) Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med 182:745–751
Gonzalez AS, Elguero ME, Finocchietto P, Holod S, Romorini L, Miriuka SG, Peralta JG, Poderoso JJ, Carreras MC (2014) Abnormal mitochondrial fusion-fission balance contributes to the progression of experimental sepsis. Free Radic Res 48:769–783
Galley HF (2011) Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth 107:57–64
Rocha M, Herance R, Rovira S, Hernandez-Mijares A, Victor VM (2012) Mitochondrial dysfunction and antioxidant therapy in sepsis. Infect Disord Drug Targets 12:161–178
Siskind LJ, Kolesnick RN, Colombini M (2002) Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J Biol Chem 277:26796–26803
Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC (1997) Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem 272:11369–11377
Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552:335–344
Suliman HB, Carraway MS, Piantadosi CA (2003) Postlipopolysaccharide oxidative damage of mitochondrial DNA. Am J Respir Crit Care Med 167:570–579
Crompton M (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem J 341(Pt 2):233–249
Dare AJ, Phillips ARJ, Hickey AJR, Mittal A, Loveday B, Thompson N, Windsor JA (2009) A systematic review of experimental treatments for mitochondrial dysfunction in sepsis and multiple organ dysfunction syndrome. Free Radic Biol Med 47:1517–1525
Takeyama N, Takagi D, Matsuo N, Kitazawa Y, Tanaka T (1989) Altered hepatic fatty acid metabolism in endotoxicosis: effect of L-carnitine on survival. Am J Phys 256:E31–E38
Protti A, Carré J, Frost MT, Taylor V, Stidwill R, Rudiger A, Singer M (2007) Succinate recovers mitochondrial oxygen consumption in septic rat skeletal muscle. Crit Care Med 35:2150–2155
Meldrum DR, Ayala A, Chaudry IH (1994) Energetics of lymphocyte “burnout” in late sepsis: adjuvant treatment with ATP-MgCl2 improves energetics and decreases lethality. J Surg Res 56:537–542
Abd el-gawad HM, Khalifa AE (2001) Quercetin, coenzyme Q10, and l-canavanine as protective agents against lipid peroxidation and nitric oxide generation in endotoxin-induced shock in rat brain. Pharmacol Res 43:257–263
Piel DA, Deutschman CS, Levy RJ (2008) Exogenous cytochrome C restores myocardial cytochrome oxidase activity into the late phase of sepsis. Shock 29:612–616
Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF (2008) The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic Biol Med 45:1559–1565
Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH (2004) Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279:34682–34690
Şener G, Toklu H, Kapucu C, Ercan F, Erkanlı G, Kaçmaz A, Tilki M, Yeğen BÇ (2004) Melatonin protects against oxidative organ injury in a rat model of sepsis. Surg Today 35:52–59
de Oliveira MR, Jardim FR, Setzer WN, Nabavi SM, Nabavi SF (2016) Curcumin, mitochondrial biogenesis, and mitophagy: exploring recent data and indicating future needs. Biotechnol Adv 34:813–826
Fernandez-Marcos PJ, Auwerx J (2011) Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93:884S-890
Piantadosi CA, Suliman HB (2012) Transcriptional control of mitochondrial biogenesis and its interface with inflammatory processes. Biochim Biophys Acta Gen Subj 1820:532–541
Dhar SS, Ongwijitwat S, Wong-Riley MT (2008) Nuclear respiratory factor 1 regulates all ten nuclear-encoded subunits of cytochrome c oxidase in neurons. J Biol Chem 283:3120–3129
Campbell CT, Kolesar JE, Kaufman BA (2012) Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim Biophys Acta 1819:921–929
Haden DW, Suliman HB, Carraway MS, Welty-Wolf KE, Ali AS, Shitara H, Yonekawa H, Piantadosi CA (2007) Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am J Respir Crit Care Med 176:768–777
Carchman EH, Whelan S, Loughran P, Mollen K, Stratamirovic S, Shiva S, Rosengart MR, Zuckerbraun BS (2013) Experimental sepsis-induced mitochondrial biogenesis is dependent on autophagy, TLR4, and TLR9 signaling in liver. FASEB J 27:4703–4711
Inata Y, Kikuchi S, Samraj RS, Hake PW, O’Connor M, Ledford JR, O’Connor J, Lahni P, Wolfe V, Piraino G, Zingarelli B (2018) Autophagy and mitochondrial biogenesis impairment contribute to age-dependent liver injury in experimental sepsis: dysregulation of AMP-activated protein kinase pathway. FASEB J 32:728–741
Chang AL, Ulrich A, Suliman HB, Piantadosi CA (2015) Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic Biol Med 78:179–189
Vanasco V, Saez T, Magnani ND, Pereyra L, Marchini T, Corach A, Vaccaro MI, Corach D, Evelson P, Alvarez S (2014) Cardiac mitochondrial biogenesis in endotoxemia is not accompanied by mitochondrial function recovery. Free Radic Biol Med 77:1–9
Suliman H, Weltywolf K, Carraway M, Tatro L, Piantadosi C (2004) Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc Res 64:279–288
MacGarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE, Piantadosi CA (2012) Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am J Respir Crit Care Med 185:851–861
Bullon P, Roman-Malo L, Marin-Aguilar F, Alvarez-Suarez JM, Giampieri F, Battino M, Cordero MD (2015) Lipophilic antioxidants prevent lipopolysaccharide-induced mitochondrial dysfunction through mitochondrial biogenesis improvement. Pharmacol Res 91:1–8
Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, David S, Burstein D, Karumanchi SA, Stillman IE, Arany Z, Parikh SM (2011) PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 121:4003–4014
Smith JA, Stallons LJ, Collier JB, Chavin KD, Schnellmann RG (2015) Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury. J Pharmacol Exp Ther 352:346–357
Handschin C, Spiegelman BM (2008) The role of exercise and PGC1α in inflammation and chronic disease. Nature 454:463–469
McCreath G, Scullion MMF, Lowes DA, Webster NR, Galley HF (2016) Pharmacological activation of endogenous protective pathways against oxidative stress under conditions of sepsis. Br J Anaesth 116:131–139
Kim H-J, Park K-G, Yoo E-K, Kim Y-H, Kim Y-N, Kim H-S, Kim H-T, Park J-Y, Lee K-U, Jang W-G, Kim J-G, Kim B-W, Lee I-K (2007) Effects of PGC-1α on TNF-α–induced MCP-1 and VCAM-1 expression and NF-κB activation in human aortic smooth muscle and endothelial cells. Antioxid Redox Signal 9:301–307
Handschin C, Choi CS, Chin S, Kim S, Kawamori D, Kurpad AJ, Neubauer N, Hu J, Mootha VK, Kim YB, Kulkarni RN, Shulman GI, Spiegelman BM (2007) Abnormal glucose homeostasis in skeletal muscle–specific PGC-1α knockout mice reveals skeletal muscle–pancreatic β cell crosstalk. J Clin Investig 117:3463–3474
Falagas ME, Makris GC, Matthaiou DK, Rafailidis PI (2008) Statins for infection and sepsis: a systematic review of the clinical evidence. J Antimicrob Chemother 61:774–785
Wan YD, Sun TW, Kan QC, Guan FX, Zhang SG (2014) Effect of statin therapy on mortality from infection and sepsis: a meta-analysis of randomized and observational studies. Crit Care 18:R71
Hondares E, Pineda-Torra I, Iglesias R, Staels B, Villarroya F, Giralt M (2007) PPARdelta, but not PPARalpha, activates PGC-1alpha gene transcription in muscle. Biochem Biophys Res Commun 354:1021–1027
Wegner A, Pavlovic D, Haussmann-Vopel S, Lehmann C (2018) Impact of lipid modulation on the intestinal microcirculation in experimental sepsis. Microvasc Res 120:41–46
Kang H, Khang R, Ham S, Jeong GR, Kim H, Jo M, Lee BD, Lee YI, Jo A, Park C (2017) Activation of the ATF2/CREB-PGC-1alpha pathway by metformin leads to dopaminergic neuroprotection. Oncotarget 8:48603–48618
Vaez H, Rameshrad M, Najafi M, Barar J, Barzegari A, Garjani A (2016) Cardioprotective effect of metformin in lipopolysaccharide-induced sepsis via suppression of toll-like receptor 4 (TLR4) in heart. Eur J Pharmacol 772:115–123
Tang G, Yang H, Chen J, Shi M, Ge L, Ge X, Zhu G (2017) Metformin ameliorates sepsis-induced brain injury by inhibiting apoptosis, oxidative stress and neuroinflammation via the PI3K/Akt signaling pathway. Oncotarget 8:97977–97989
van Vught LA, Scicluna BP, Hoogendijk AJ, Wiewel MA, Klein Klouwenberg PM, Cremer OL, Horn J, Nurnberg P, Bonten MM, Schultz MJ (2016) Association of diabetes and diabetes treatment with the host response in critically ill sepsis patients. Crit Care 20:252. https://doi.org/10.1186/s13054-016-1429-8
Zhan M, Brooks C, Liu F, Sun L, Dong Z (2013) Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int 83:568–581
Brooks C, Dong Z (2007) Regulation of mitochondrial morphological dynamics during apoptosis by Bcl-2 family proteins: a key in Bak? Cell Cycle 6:3043–3047
Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126:177–189
Hoppins S, Lackner L, Nunnari J (2007) The machines that divide and fuse mitochondria. Annu Rev Biochem 76:751–780
Chan DC (2012) Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46:265–287
Loson OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24:659–667
Ong SB, Hausenloy DJ (2016) Mitochondrial dynamics as a therapeutic target for treating cardiac diseases. Handb Exp Pharmacol 240:251–279
Hansen ME, Simmons KJ, Tippetts TS, Thatcher MO, Saito RR, Hubbard ST, Trumbull AM, Parker BA, Taylor OJ, Bikman BT (2015) Lipopolysaccharide disrupts mitochondrial physiology in skeletal muscle via disparate effects on sphingolipid metabolism. Shock 44:585–592
Shen YL, Shi YZ, Chen GG, Wang LL, Zheng MZ, Jin HF, Chen YY (2018) TNF-alpha induces Drp1-mediated mitochondrial fragmentation during inflammatory cardiomyocyte injury. Int J Mol Med 41:2317–2327
Zhao GJ, Yao YM, Lu ZQ, Hong GL, Zhu XM, Wu Y, Wang DW, Dong N, Yu Y, Sheng ZY (2012) Up-regulation of mitofusin-2 protects CD4+ T cells from HMGB1-mediated immune dysfunction partly through Ca(2+)-NFAT signaling pathway. Cytokine 59:79–85
Wu ZS, Yao YM, Hong GL, Xu XP, Liu Y, Dong N, Zheng JY, Lu ZQ, Zhao GJ, Zhu XM, Zhang QH, Sheng ZY (2014) Role of mitofusin-2 in high mobility group box-1 protein-mediated apoptosis of T cells in vitro. Cell Physiol Biochem 33:769–783
Jang DH, Greenwood JC, Owiredu S, Ranganathan A, Eckmann DM (2017) Mitochondrial networking in human blood cells with application in acute care illnesses. Mitochondrion 44:27–34
Deng S, Ai Y, Gong H, Feng Q, Li X, Chen C, Liu Z, Wang Y, Peng Q, Zhang L (2018) Mitochondrial dynamics and protective effects of a mitochondrial division inhibitor, Mdivi-1, in lipopolysaccharide-induced brain damage. Biochem Biophys Res Commun 496:865–871
Yu J, Shi J, Wang D, Dong S, Zhang Y, Wang M, Gong L, Fu Q, Liu D (2016) Heme oxygenase-1/carbon monoxide-regulated mitochondrial dynamic equilibrium contributes to the attenuation of endotoxin-induced acute lung injury in rats and in lipopolysaccharide-activated macrophages. Anesthesiology 125:1190–1201
Jiang P, Mizushima N (2014) Autophagy and human diseases. Cell Res 24:69–79
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 13:378–385
Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, Abramov AY, Wood NW (2011) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20:867–879
Rub C, Wilkening A, Voos W (2017) Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res 367:111–123
Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, Nagar B, Fon EA, Gehring K (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340:1451–1455
Nguyen TN, Padman BS, Lazarou M (2016) Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol 26:733–744
Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510:162–166
Wauer T, Simicek M, Schubert A, Komander D (2015) Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524:370–374
Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496:372–376
Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC (2011) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20:1726–1737
Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191:1367–1380
Glauser L, Sonnay S, Stafa K, Moore DJ (2011) Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J Neurochem 118:636–645
Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147:893–906
Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T, Takahashi R, Hattori N, Imai Y, Lu B (2012) Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet 8:e1002537. https://doi.org/10.1371/journal.pgen.1002537
Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ (2014) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6:1090–1106
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314
Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1-PARKIN mitochondrial Ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60:7–20
Piquereau J, Godin R, Deschenes S, Bessi VL, Mofarrahi M, Hussain SN, Burelle Y (2013) Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy 9:1837–1851
Sun Y, Yao X, Zhang QJ, Zhu M, Liu ZP, Ci B, Xie Y, Carlson D, Rothermel BA, Sun Y, Levine B, Hill JA, Wolf SE, Minei JP, Zang QS (2018) Beclin-1-dependent autophagy protects the heart during sepsis. Circulation 138:2247–2262
Mannam P, Shinn AS, Srivastava A, Neamu RF, Walker WE, Bohanon M, Merkel J, Kang MJ, Cruz CSD, Ahasic AM, Pisani MA, Trentalange M, West AP, Shadel GS, Elias JA, Lee PJ (2014) MKK3 regulates mitochondrial biogenesis and mitophagy in sepsis-induced lung injury. Am J Phys Lung Cell Mol Phys 306:L604–L619
Zhang X, Yuan D, Sun Q, Xu L, Lee E, Lewis AJ, Zuckerbraun BS, Rosengart MR (2017) Calcium/calmodulin-dependent protein kinase regulates the PINK1/Parkin and DJ-1 pathways of mitophagy during sepsis. FASEB J 31:4382–4395
Takasu O, Gaut JP, Watanabe E, To K, Fagley RE, Sato B, Jarman S, Efimov IR, Janks DL, Srivastava A, Bhayani SB, Drewry A, Swanson PE, Hotchkiss RS (2013) Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med 187:509–517
Chien W-S, Chen Y-H, Chiang P-C, Hsiao H-W, Chuang S-M, Lue S-I, Hsu C (2011) Suppression of autophagy in rat liver at late stage of polymicrobial sepsis. Shock 35:506–511
Takahashi W, Watanabe E, Fujimura L, Watanabe-Takano H, Yoshidome H, Swanson PE, Tokuhisa T, Oda S, Hatano M (2013) Kinetics and protective role of autophagy in a mouse cecal ligation and puncture-induced sepsis. Crit Care 17:R160
Lin CW, Lo S, Perng DS, Wu DB, Lee PH, Chang YF, Kuo PL, Yu ML, Yuan SS, Hsieh YC (2014) Complete activation of autophagic process attenuates liver injury and improves survival in septic mice. Shock 41:241–249
Thiessen SE, Derese I, Derde S, Dufour T, Pauwels L, Bekhuis Y, Pintelon I, Martinet W, Van den Berghe G, Vanhorebeek I (2017) The role of autophagy in critical illness-induced liver damage. Sci Rep 7:14150
Chung KW, Kim KM, Choi YJ, An HJ, Lee B, Kim DH, Lee EK, Im E, Lee J, Im DS, Yu BP, Chung HY (2017) The critical role played by endotoxin-induced liver autophagy in the maintenance of lipid metabolism during sepsis. Autophagy 13:1–17
Lalazar G, Ilyas G, Malik SA, Liu K, Zhao E, Amir M, Lin Y, Tanaka KE, Czaja MJ (2016) Autophagy confers resistance to lipopolysaccharide-induced mouse hepatocyte injury. Am J Physiol Gastrointest Liver Physiol 311:G377–G386
Hsieh CH, Pai PY, Hsueh HW, Yuan SS, Hsieh YC (2011) Complete induction of autophagy is essential for cardioprotection in sepsis. Ann Surg 253:1190–1200
Yen YT, Yang HR, Lo HC, Hsieh YC, Tsai SC, Hong CW, Hsieh CH (2013) Enhancing autophagy with activated protein C and rapamycin protects against sepsis-induced acute lung injury. Surgery 153:689–698
Lo S, Yuan SS, Hsu C, Cheng YJ, Chang YF, Hsueh HW, Lee PH, Hsieh YC (2013) Lc3 over-expression improves survival and attenuates lung injury through increasing autophagosomal clearance in septic mice. Ann Surg 257:352–363
Sunahara S, Watanabe E, Hatano M, Swanson PE, Oami T, Fujimura L, Teratake Y, Shimazui T, Lee C, Oda S (2018) Influence of autophagy on acute kidney injury in a murine cecal ligation and puncture sepsis model. Sci Rep 8:1050
Hsiao HW, Tsai KL, Wang LF, Chen YH, Chiang PC, Chuang SM, Hsu C (2012) The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock 37:289–296
Stana F, Vujovic M, Mayaki D, Leduc-Gaudet JP, Leblanc P, Huck L, Hussain SNA (2017) Differential regulation of the autophagy and proteasome pathways in skeletal muscles in sepsis. Crit Care Med 45:e971–e979
Oami T, Watanabe E, Hatano M, Sunahara S, Fujimura L, Sakamoto A, Ito C, Toshimori K, Oda S (2017) Suppression of T cell autophagy results in decreased viability and function of T cells through accelerated apoptosis in a murine sepsis model. Crit Care Med 45:e77–e85
Arabi YM, Aldawood AS, Haddad SH, Al-Dorzi HM, Tamim HM, Jones G, Mehta S, McIntyre L, Solaiman O, Sakkijha MH (2015) Permissive underfeeding or standard enteral feeding in critically ill adults. N Engl J Med 372:2398–2408
Liu W, Guo J, Mu J, Tian L, Zhou D (2017) Rapamycin protects sepsis-induced cognitive impairment in mouse hippocampus by enhancing autophagy. Cell Mol Neurobiol 37:1195–1205
Funding
This work was supported, in part, by grants from the National Natural Science Foundation (grant numbers 81571937 and 81772112).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, Y., Yao, YM. & Lu, ZQ. Mitochondrial quality control mechanisms as potential therapeutic targets in sepsis-induced multiple organ failure. J Mol Med 97, 451–462 (2019). https://doi.org/10.1007/s00109-019-01756-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-019-01756-2