
Abstract
Mitochondrial dysfunction represents a prominent pathological feature in many neurodegenerative diseases, particularly in Parkinson’s disease (PD). Mutations in the genes encoding the proteins Pink1 and Parkin have been identified as genetic risk factors in familiar cases of PD. Research during the last decade has identified both proteins as crucial components of an organellar quality control system that contributes to the maintenance of mitochondrial function in healthy cells. The Pink1/Parkin system acts as a sensor for mitochondrial quality and is activated, in particular, after the loss of the electric potential across the inner mitochondrial membrane. Pink1 molecules accumulate at the surface of damaged mitochondria to recruit and activate Parkin, which, in turn, elicits a signaling pathway eventually leading to the autophagic removal of the damaged organelles. This review summarizes recent advances in our knowledge of the functional role of the Pink1/Parkin system in preventing the accumulation of damaged mitochondria by mitophagy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondria represent essential organelles inside eukaryotic cells. They provide energy in the form of ATP and many nutrients for cellular metabolism but are also involved in signaling mechanisms that monitor general cellular health. Apart from the loss of important metabolic molecules, damaged mitochondria can exert a negative influence on cellular survival by releasing large amounts of reactive oxygen species (ROS) or other molecules that are toxic for the cell (Halliwell 2006). Mitochondria therefore have to be maintained in a functional and active state, a process that is summarized by the term “mitochondrial homeostasis”. Generally, mitochondrial homeostasis can be achieved on three different levels: (1) on the level of individual protein function, (2) on the organellar level by protecting mitochondrial function and integrity as a whole, and (3) on the cellular level, when irreversibly damaged mitochondria accumulate in large numbers, and the cell removes itself from a tissue by regulated cellular suicide. Briefly, mitochondria contain an endogenous system responsible for quality control of proteins (level 1). This system consists of several types of molecular chaperones that cooperate closely with ATP-dependent chambered proteases in the repair or removal of damaged polypeptides (Voos 2013). Based on their endosymbiotic origin, this chaperone-protease network is similar to related processes in bacterial cells. Regulated cell death or “apoptosis” (level 3) can be initiated by internal or external signaling processes that involve, in both cases, mitochondria as a major component (Tait and Green 2010). In general, apoptosis is initiated by a mitochondrial membrane permeability transition that results in the release of cytochrome c and other apoptotic proteins. These, in turn, activate a cytosolic signaling process that leads to the activation of a caspase cascade as the major executor of the cell death reaction. The permeability transition may also be caused by severe mitochondrial damage, although this represents only one out of many diverse ways to induce apoptosis.
In this review, we focus on the role of the Pink1/Parkin system in the context of organellar quality control (OQC), as an example of a homeostasis process acting on the organelle (level 2). Current models of the Pink1/Parkin mechanism during OQC assign them a prominent role in the removal of damaged mitochondria by a variant of autophagy, called “mitophagy”. Notably, mitophagy represents only one process involved in OQC. Indeed, mitophagy is closely linked to another aspect of OQC, the ability of mitochondria to undergo fusion and fission (Ni et al. 2015). The dynamic nature of mitochondrial morphology allows the periodic mixing of the contents of all mitochondria of one cell, resulting in a complementation of occasional defects in single members. On the other hand, a specific removal of individual damaged mitochondria seems to be supported by a fission process that separates healthy from defective organelles (Youle and van der Bliek 2012). Last but not least, several signaling pathways have been identified that involve communication between mitochondria and the gene expression machinery in the nucleus, allowing an adjustment of mitochondrial protein content and accordingly organellar activity to different internal or external stress situations (Quiros et al. 2016). However, the mechanistic details and the physiological relevance of mito-nuclear communication phenomena are still poorly understood. One of the more prominent pathways, with a direct relationship to OQC, is the mitochondrial unfolded protein response (mtUPR). Here, mitochondrial dysfunction, caused by defective protein folding, is communicated to the nucleus, which in response increases the expression of mitochondrial proteins such as chaperones or proteases to deal with the misfolded polypeptides (Haynes and Ron 2010). Although many biochemical processes contribute to mitochondrial homeostasis in a cell, the multitude of environmental insults affecting genome or proteome integrity over the lifetime of an individual human will inevitably result in a progressive decline of mitochondrial function, significantly contributing to the many aspects of neurodegenerative diseases.
Pink1/Parkin system
The proteins Pink1 and Parkin gained widespread notoriety by their prominent involvement in the etiology of Parkinson’s disease (PD). Mutations in the genes encoding both proteins have been identified as genetic risk factors in familiar cases of the neurodegenerative disease (Klein and Westenberger 2012). After its initial identification (Valente et al. 2004), the kinase Pink1 (PTEN-induced putative kinase 1) was quickly associated with mitochondria, thereby allowing, for the first time, the construction of a direct biochemical link between mitochondrial dysfunction in patients and PD. Moreover, at a cellular level, mutations in Pink1 have been found to be associated with general mitochondrial dysfunction (Gautieret al. 2008; Hoepken et al. 2007). Indeed, Pink1-mutant mice develop Parkinson-like pathologies in old age (Gispert et al. 2009), and Pink1 has been shown to be necessary for the long-term survival of human dopaminergic neurons, the cells most affected in PD (Wood-Kaczmar et al. 2008). Subsequently, genetic evidence has indicated a direct functional link between Pink1 and the cytosolic E3 ubiquitin ligase Parkin (Clark et al. 2006; Exner et al. 2007; Park et al. 2006), which has been independently identified as a protein involved in PD (Kitada et al. 1998). Both proteins are thought to collaborate closely in initiating the specific removal of damaged mitochondria by mitophagy (Rüb et al. 2015). A failure of this process together with the progressive accumulation of dysfunctional mitochondria is thought to contribute to or even cause PD.
General mechanism of autophagy/mitophagy
Autophagy is a general mechanism in eukaryotic cells and is employed to degrade intracellular components, ranging from parts of the cytosol to protein aggregates and organelles. Generally, three different types of autophagy are distinguished: chaperone-mediated autophagy (CMA), microautophagy, and macroautophagy. In the process of macroautophagy, the respective cellular components are surrounded by a double-membrane structure, called an autophagosome. The autophagosome then fuses with a lysosome and delivers its cargo for degradation by lysosomal enzymes. This non-selective degradation of substrates is mainly induced under starvation conditions to provide the essential nutrients for the cell by recycling intracellular components (Wang and Klionsky 2011; Youle and Narendra 2011). As in the case of mitophagy, macroautophagy can also be a highly selective process that removes superfluous or damaged organelles. So far, three prominent examples of mitophagy have been identified: (1) the removal of mitochondria during reticulocyte development, (2) the selective elimination of paternal mitochondria from fertilized oocytes, and (3) the degradation of damaged mitochondria (Ashrafi and Schwarz 2013). All three pathways of mitophagy use the same core components of the autophagic machinery; however, the events, which lead to the initiation of mitophagy, are distinct. Typically, the initiation of the mitophagy process requires a selectivity factor, acting as a “sensor” to mark the organelles destined for removal. In a second step, the autophagic membranes are recruited with the help of “adaptor” proteins that also interact with general autophagy components. In a final step, the autophagosome fuses with a lysosome and its contents are degraded. The principal biochemical mechanisms and components underlying mitophagy have been established in the yeast model system. Several studies have identified specific mitophagy selectivity factors, in particular, the autophagy protein Atg32 (Kanki and Klionsky 2010). This protein, which is localized on the outer mitochondrial membrane (OMM), can bind the isolation membrane protein Atg8 either directly or indirectly through the adaptor protein Atg11, which bridges mitochondria and isolation membrane by binding both Atg32 and Atg8 (Okamoto et al. 2009). The interaction between Atg32 and Atg8 recruits mitochondria into autophagosomes and facilitates their subsequent degradation by fusion with the lysosome. Atg8 is a member of a larger protein family also including several mammalian homologs that are divided into three subfamilies: MAP1LC3 (microtubule-associated protein 1 light-chain 3; further called LC3), GABARAP (γ-aminobutyric acid receptor-associated protein), and GATE-16 (Golgi-associated ATPase enhancer of 16 kDa; Slobodkin and Elazar 2013). So far, no homologs of ATG11 and ATG32 have been identified in higher eukaryotic organisms. However, several proteins in mammals execute the same function, e.g., the mitochondrial outer membrane protein NIP3-like protein X (NIX), which, like Atg32 and Atg11, is able to bind LC3, an Atg8 homolog, on the isolation membrane (Schweers et al. 2007). The expression of NIX is increased in the development of red blood cells mediating the removal of mitochondria during their differentiation. In case of the specific elimination of damaged mitochondria, mitophagy requires a specific labeling and autophagosome recruitment system that is represented by the Pink1/Parkin signaling system in mammalian cells (Youle and Narendra 2011).
Biochemical properties of Pink1
Like most mitochondrial proteins, the 581-amino-acid (aa) Pink1 polypeptide is encoded in the nucleus and synthesized at cytosolic ribosomes. Its N-terminal portion resembles a mitochondrial targeting signal (Valente et al. 2004) typically found in precursor proteins destined for the matrix or inner membrane compartment (Fig. 1a). In the canonical import pathway into the matrix compartment, mitochondrial preproteins are first recognized by cytosol-exposed TOM receptors of the OMM via this cleavable N-terminal signal sequence. The TOM and TIM23 translocase complexes then enable the passage of the preprotein through the outer and inner membranes, respectively. Insertion of the preprotein into the TIM23 channel implicitly requires the presence of a mitochondrial inner membrane potential (Δψ). Whereas inner membrane proteins can be laterally released from the TIM23 complex, the import motor complex at the inner face of the TIM23 complex drives complete translocation of matrix-destined preproteins in an ATP-dependent reaction. When the preprotein crosses the IMM, it is usually cleaved by the matrix-processing peptidase (MPP) to remove the targeting signal sequence. This processing reaction gives rise to the mature protein, which is released into the matrix compartment. Several other import routes exist to guide mitochondrial proteins to their respective sub-organellar destinations (Schulz et al. 2015).

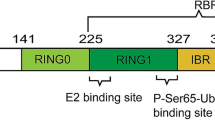
Schematic representation of Pink1 and Parkin domain structures. (a) Domain structure and cleavage sites within the Pink1 sequence. The N-terminal mitochondrial targeting signal is followed by a hydrophobic transmembrane domain that comprises residues 85 to 110 and acts as an inner mitochondrial membrane (IMM) stop-transfer signal. Residues 156 to 509 constitute the Ser/Thr-kinase domain, followed by a C-terminal domain that may act as an outer mitochondrial membrane (OMM) retention signal. Protease cleavage sites for matrix-processing peptidase (MPP) and Presinilin-associated rhomboid-like protein (PARL) and the resulting Pink1 fragments are indicated. Note that the unknown MPP cleavage site was estimated from the molecular mass of the MPP processing product. (b) Domain structure of Parkin. The N-terminal ubiquitin-like (Ubl) domain is followed by a linker region and the RING0 domain. Three more zinc-finger domains, namely RING1, IBR, and RING2, form the RBR (RING In-between RING) motif (RING really interesting new gene domain, IBR in between RING domain; modified after Trempe et al. 2013)
Whereas the N-terminal targeting signal of Pink1 was reported to be sufficient for its localization to mitochondria, clarification as to which particular import route was followed by Pink1 was not without complications. First, ambiguous sub-mitochondrial localizations of the protein were reported. Although initial studies proposed its localization to the IMM (Muqit et al. 2006; Silvestri et al. 2005) or the intramembrane space (IMS; Plun-Favreau et al. 2007; Silvestri et al. 2005), Pink1 was subsequently shown to localize predominantly to the OMM with the kinase domain facing the cytosol (Becker et al. 2012; Zhou et al. 2008). Second, Pink1 was proposed to be sequentially processed by several mitochondrial proteases (Deas et al. 2010; Greene et al. 2012). Intriguingly, in vitro import experiments showed that both the 64-kDa full-length form of Pink1 and the 53-kDa major processing product have a final localization in the OMM (Becker et al. 2012). Taken together, these observations are inconsistent with a canonical import pathway for Pink1. Third, although the mitochondrial membrane potential seems to affect the localization, processing, and arguably stability of Pink1 (Jin et al. 2010), the dependence of the Pink1 import reaction on the membrane potential is not as absolute as other preproteins following the canonical pathway to the IMM or matrix (Becker et al. 2012). The results of the latter and other studies suggest that, in the presence of an inner membrane potential, the Pink1 polypeptide is partially inserted into the IMM through the TOM and TIM23 complexes (Fig. 2). A hydrophobic stop-transfer signal, adjacent to the presequence, prevents full translocation through the IMM. Only the very N-terminal segment reaches the matrix and is cleaved by MPP, giving rise to a 60-kDa Pink processing intermediate (Greene et al. 2012). The inner membrane-resident protease PARL (Presinilin-associated rhomboid-like protein) then catalyzes a second cleavage between residues 103 and 104 within the Pink1 sequence, generating the 53-kDa processed Pink1 (Pink1-PF; Deas et al. 2010). Pink1 then associates with the outer face of the OMM via its C-terminus or is released into the cytosol. In depolarized mitochondria, full-length Pink1 associates with the OMM, possibly through an interaction of its presequence-like N-terminal segment with cytosol-exposed TOM receptors, and is stabilized by the hydrophobic C-terminal domain (Fig. 2). In any case, the OMM association is independent of the state of the inner membrane potential. Results from ourselves and other groups showed that Pink1 is present in at least one high molecular weight complex of apparently 700 kDa, notably when it accumulates at mitochondria upon dissipation of the membrane potential (Becker et al. 2012; Lazarou et al. 2012; Okatsu et al. 2012). Whereas this complex potentially comprises Pink1 together with other interaction partners, the functional relevance and the specific composition of this complex have not been clarified so far. Lazzarou et al. (2012) have proposed that the complex represents an association of Pink1 with the TOM receptor complex. However, our own observations have not confirmed this co-migration, as the fully assembled TOM complex in native polyacrylamide gel electrophoresis experiments runs at about 400 kDa (Becker et al. 2012). In addition, Okatsu et al. (2012) have reported the presence of two Pink1 molecules in a slightly higher molecular weight complex of apparently 850 kDa.

Model of Pink1 import in the presence or absence of Δψ. In depolarized mitochondria (right), the Pink1 precursor (Pink1p64) associates with the OMM, possibly via TOM components. Accumulating Pink1 recruits Parkin, in turn inducing mitophagy (1a). In the presence of Δψ (left), the presequence-like N-terminal segment of the Pink1 precursor (Pink1p64) drives translocation of Pink1 across the OMM via the TOM complex and its insertion into the IMM via the TIM23 complex (1b). The inner membrane stop-transfer signal prevents complete translocation of Pink1 over the IMM. The N-terminus of Pink1 reaches the matrix, allowing cleavage of the mitochondrial targeting signal by the matrix processing peptidase (MPP) (2). The IMM protease PARL cleaves Pink1p64 at position 104, generating the processed form Pink1f53, which is released from the import machinery (3). Pink1f53 either associates with the OMM as a peripheral membrane protein, possibly via its C-terminal domain (4) or is degraded by the proteasome (OMM outer mitochondrial membrane, IMS intramembrane space, IMM inner mitochondrial membrane)
Pink1 belongs to the enzyme family of Ser/Thr-protein kinases. Apart from N- and C-terminal sequences involved in targeting and localization, the largest part of the protein is represented by the kinase domain (Fig. 1a). Among the more than 60 PD-related mutations in Pink1, those affecting or abolishing its kinase activity are by far the most frequent (Klein and Westenberger 2012). This observation unambiguously implies a function of the catalytic activity of Pink1 in the etiology of PD. Results from in vitro experiments have revealed that Pink1 is capable of phosphorylating itself (Silvestri et al. 2005). In detail, autophosphorylation of Ser228 and Ser402 within the Pink1 kinase domain upon loss of the membrane potential has been postulated to positively regulate its catalytic activity (Aerts et al. 2015; Okatsu et al. 2012). Several other phosphorylation substrates of Pink1 have been proposed, including the mitochondrial Hsp90 chaperone TRAP1 (Pridgeon et al. 2007), mitofusin 2 (Chen and Dorn 2013), the Rho-GTPase Miro1 (Wang et al. 2011), and the inner membrane-resident mitochondrial protease HtrA2/Omi (Plun-Favreau et al. 2007). However, the functional significance of most of these phosphorylation reactions is not established so far. Pink1-mediated phosphorylation and activation of the E3 protein-ubiquitin ligase Parkin have been the focus of extensive research (Kondapalli et al. 2012; Shiba-Fukushima et al. 2012). Most remarkably, Pink1 phosphorylates not only Parkin, but also ubiquitin in the context of Parkin activation (Kane et al. 2014; Kazlauskaite et al. 2014; Koyano et al. 2014). This recent discovery makes Pink1 the first only described ubiquitin kinase. The way in which Pink1 and Parkin interact in damage-related mitophagy will be described below.
Biochemical properties of Parkin
The 465-aa cytosolic protein Parkin, encoded by the PARK2 gene, is an E3 ubiquitin-protein ligase (Winklhofer 2014). In general, E3 enzymes, in collaboration with E1 and E2 class enzymes, mediate the covalent binding of the small 76-aa protein ubiquitin (Ub) to substrate proteins. In a catalytic cascade, ubiquitin is first activated by an ubiquitin-activating enzyme (E1) and then transferred to an ubiquitin-conjugating enzyme (E2). The subsequent transfer of the ubiquitin moiety to a substrate amino group, typically a lysine side chain, is catalyzed by an ubiquitin ligase (E3). Ubiquitin modifications can either be mono-ubiquitinations or polyubiquitin chains, which are formed by conjugating one ubiquitin to any of the seven lysine residues or the N-terminal amino group of a second ubiquitin molecule (Ciechanover 2005). Depending on the ubiquitin chain length and linkage type, a substrate protein is tagged for signaling processes or turnover by either of the major cellular degradation machineries, namely the proteasome or the lysosome via autophagy (Clague and Urbe 2010).
As shown in Fig. 1b, Parkin consists of an N-terminal ubiquitin-like (Ubl) domain, followed by a 60-aa linker region and four zinc-finger domains. The last three zinc-finger domains form a RING1 In-Between RING2 (RBR) motif. Accordingly, Parkin belongs to the RBR family of E3 ligases (Trempe and Fon 2013). RBR ligases are unusual in that they utilize a hybrid mechanism, combining characteristics of the two other classes of E3 enzymes, RING and HECT (Smit et al. 2012; Stieglitz et al. 2012; Wenzel et al. 2011). Whereas RING-type E3 ligases catalyze the direct transfer of Ub from an E2 enzyme to the substrate protein, HECT E3 ligases form a thioester-linked intermediate with Ub via a catalytic cysteine. This intermediate enables the subsequent transfer of Ub to a primary amino group of the substrate protein, forming an isopeptide bond (Winklhofer 2014). Similar to RING enzymes, RBR enzymes first bind an Ub-charged E2 via a specific site in the RING1 domain. In a reaction resembling the mechanism described for HECT enzymes, Ub is then transferred to a catalytic cysteine within the RING2 domain (Cys431 in Parkin) forming a thioester-bound intermediate (Wenzel et al. 2011). High-resolution crystal structures of Parkin have revealed that, under steady-state conditions, the enzyme is maintained in an autoinhibited state by two mechanisms (Riley et al. 2013; Trempe et al. 2013; Wauer and Komander 2013). First, the RING0 domain shields the Ub-acceptor site Cys431. Second, the so-called repressor element of parkin (REP), an α-helix located between the IBR and RING2 domain, blocks the E2-binding site within the RING1 domain. Upon mitochondrial damage, Parkin undergoes steric changes that relax the inhibitory RING0 and REP domains. The catalytic Cys431 becomes exposed, and Parkin is converted into an active E3 enzyme (Koyano et al. 2014). The role of Pink1 in Parkin activation is described below. In its active state, Parkin ubiquitinates cytosolic protein substrates and various cytosol-exposed proteins of the OMM. A quantitative analysis of the various linkage types has revealed that Parkin forms mainly Lys48- and Lys63-linked polyubiquitin chains, which represent common ubiquitin modifications, but also noncanonical Lys6- and Lys11-linked chains are formed (Ordureau et al. 2014).
Pink1/Parkin reaction pathway
A biochemical analysis of Pink1 function was initially hampered by the lack of antibodies sensitive enough to detect endogenous levels of the protein. This led to the effect that almost all observations of the behavior of Pink1 in cells were based on the analysis of ectopically expressed Pink1 fusion proteins containing some type of protein tag. Under normal growth conditions, cellular Pink1 levels are so low that it is virtually undetectable. Endogenous Pink1 levels are only high enough to be detected by western blot or microscopic visualization under certain mitochondrial stress conditions. This observation specifies the main property of Pink1 in the context of OQC: a significant rise in cellular protein amounts and its accumulation at dysfunctional mitochondria. The main hallmark of dysfunctional mitochondria is represented by a loss of the electric inner membrane potential (Δψ) generated by the respiratory chain. Indeed, treatment of cells with the protonophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP), which leads to an uncoupling of the respiratory chain, is generally used to simulate mitochondrial dysfunction. The mitochondrial accumulation of Pink1 has also been associated with other types of mitochondrial defects, such as elevated levels of ROS (Priyadarshini et al. 2013) or an accumulation of misfolded proteins in the mitochondrial matrix (Jin and Youle 2013). However, it is likely that ROS treatment, proteotoxic stress, or any other type of mitochondrial damage to indirectly result in a depletion of the Δψ similar to the activity of the uncoupling toxin CCCP.
The major observation leading to the conclusion that Pink1 was a prominent sensor for mitochondrial damage was its significant accumulation at potential-deficient mitochondria. After the addition of CCCP to cells, endogenous Pink1 levels become barely detectable after 3 h of incubation and reach a maximum level after 12–16 h. Under these conditions, only the full-length polypeptide is observed, and all Pink1 molecules co-purify with mitochondria. After removal of the uncoupling toxin, Pink1 amounts return relatively quickly to very low levels when the ribosomal synthesis of new polypeptides is blocked (Matsuda et al. 2010; Narendra et al. 2010). A slight upshift of the endogenous full-length Pink1 band under CCCP induction conditions compared with exogenously expressed Pink1 indicates that the accumulated molecules might be phosphorylated (Okatsu et al. 2012).
A major question concerning the regulation of mitophagy is: what is the cause for the observed strong increase in the cellular amounts of Pink1 in response to mitochondrial perturbations? The initial hypothesis postulated an import-related mitochondrial degradation mechanism to explain this phenomenon (Jin et al. 2010). This model proposed that, under normal conditions, Pink1 would be taken up by healthy mitochondria with a functional inner membrane potential Δψ via the usual import pathway of presequence-containing proteins and be quickly degraded inside the mitochondria. In dysfunctional mitochondria with a depleted Δψ, the import reaction would be blocked, and full-length Pink1 polypeptides would accumulate at the mitochondrial outer membrane, consistent with the observations in many different cellular systems. However, this model has serious problematic points. First, various experiments indicate an exclusive localization of Pink1, both full-length and processed forms, at the mitochondrial outer membrane facing the cytosol (Becker et al. 2012; Zhou et al. 2008). Even in the original publication of Jin et al. (2010), a close look at the data shows that Pink1 exhibits an identical sensitivity to external proteases such as the outer membrane import receptor protein Tom20. All internal mitochondrial proteins, even in the IMS, are completely protected against protease treatment by the membranes, even at very high protease concentrations. In addition, the mitochondrial protease that is responsible for the degradation of any imported Pink1 molecules remains unclear. Direct experiments have established that the processing step during the import reaction is performed by the presenilin-associated rhomboid-like protease (PARL), a proteolytic enzyme of the inner membrane (Deas et al. 2010; Jin et al. 2010). Several other mitochondrial proteases have been claimed to be involved in Pink1 turnover (Greene et al. 2012; Thomas et al. 2014). However, these conclusions were mainly based on RNAi knock-down experiments and carried the danger of an indirect accumulation of general mitochondrial defects, in particular, as the identified proteases (MPP, LON, or mAAA) are important components of mitochondrial protein biogenesis and quality control pathways. Depletions of these proteases typically lead to a defect in respiratory activity and breakdown of the Δψ, which in turn results in all cases in the accumulation of Pink1, exactly as observed. On the other hand, Pink1 has clearly been demonstrated to underlie proteolytic turnover in the cytosol by the proteasome. Inhibition of proteasomal degradation by the compound MG132 results in an accumulation of the 53-kDa processed form of Pink1 under normal culture conditions (Lin and Kang 2008; Takatori et al. 2008). As a release of processed Pink1 molecules from the OMM to the cytosol has been observed (Fedorowicz et al. 2014), a proteasomal degradation reaction seems to be possible, even for a protein principally targeted to mitochondria.
Interestingly, the article of Narendra et al. (2010) contained data showing that the inhibition of protein synthesis by cycloheximide (CHX) almost completely prevented the CCCP-driven accumulation of both Pink1 and Parkin at mitochondria, indicating that the increase in protein levels might be attributable to the induced biosynthesis of new polypeptides. Indeed, several recent experiments indicate that both Pink1 and Parkin levels in the cell underlie a transcriptional regulation mechanism. Growth factor deprivation, starvation, or other conditions of severe cellular damage have been shown to lead to the induction of Pink1 and Parkin transcription (Klinkenberg et al. 2012; Mei et al. 2009; Murata et al. 2015; Sakurai et al. 2009). One can argue, that, under these conditions resulting in a general autophagy reaction, Pink1 and Parkin are activated by a different mechanism than under acute mitochondrial damage resulting in specific mitophagy. However, Pink1 mRNA levels have also been observed to increase under CCCP treatment (Gomez-Sanchez et al. 2014), again arguing for a transcriptional induction mechanism. Indeed, an analysis of the Pink1 promoter has revealed that it is under the control of the transcription factor NF-kB (Duan et al. 2014). These observations indicate that a transcriptional induction mechanism cannot be ruled out as the process responsible for the strong accumulation of Pink1 at damaged mitochondria. However, the specific details of this potential mito-nuclear communication pathway remain to be established.
Although both Pink1 and Parkin had previously been implicated in PD, the first hint of their cooperative role in maintaining mitochondrial quality came from Drosophila knockout models. Similar mitochondrial defects, including muscle degeneration and abnormal mitochondrial morphology were observed upon loss of either protein. Whereas overexpression of Parkin partially rescued the Pink1-deficient phenotype, conversely, Pink1 could not complement Parkin deficiency. Hence, Pink1 was proposed to function upstream of Parkin in a common pathway to maintain mitochondrial quality (Clark et al. 2006; Park et al. 2006). In human cells exposed to CCCP, Parkin was subsequently shown to translocate from the cytosol to damaged mitochondria upon loss of Δψ and to mediate the autophagic removal of the organelle (Narendra et al. 2008). Providing an explanation for their genetic interaction, Pink1 was finally demonstrated to be responsible for Parkin translocation to depolarized mitochondria (Geisler et al. 2010b; Narendra et al. 2010; Vives-Bauza et al. 2010; Ziviani et al. 2010). In addition, the usually repressed ubiquitin ligase function of Parkin was shown to be activated upon its Pink1-mediated translocation to mitochondria (Narendra et al. 2010).
So, how does Pink1 recruit and activate Parkin when it accumulates at the surface of damaged mitochondria? After initial experiments had revealed that Parkin translocation to mitochondria and the subsequent induction of mitophagy require the kinase function of Pink1 (Geisler et al. 2010a; Matsuda et al. 2010; Narendra et al. 2010), it became clear that several Pink1-mediated phosphorylation events contribute to this process. First, upon loss of Δψ, Pink1 undergoes autophosphorylation at Ser228 and Ser402, which is required for Parkin recruitment (Okatsu et al. 2012). Second, Pink1 directly phosphorylates Parkin at Ser65 within the Ubl domain (Kondapalli et al. 2012; Shiba-Fukushima et al. 2012). Intriguingly, Parkin carrying a Ser65Ala mutation to abolish its phosphorylation and a Parkin mutant lacking the Ubl domain are still translocated to mitochondria in a Pink1-kinase dependent manner (Kane et al. 2014). An explanation for this observation lies within a recently discovered novel mechanism in which Pink1 phosphorylates ubiquitin at Ser65 (homologous to Ser65 in the Parkin Ubl domain) and, in turn, phospho-Ub activates the E3 ligase activity of Parkin (Kane et al. 2014; Kazlauskaite et al. 2014; Koyano et al. 2014). The extent to which the phosphorylation of Parkin, on the one hand, and ubiquitin, on the other, contribute to Parkin activation remains controversial. Whereas Kane et al. (2014) have proposed that phospho-Ub is sufficient for Parkin activation, Koyano and colleagues (2014) have come to the conclusion that the phosphorylation of Ser65 in both Parkin and ubiquitin is required to fully activate the E3 ligase. According to a recently proposed feed-forward model (Fig. 3), Parkin is first activated through Pink1-dependent phosphorylation and, in turn, ubiquitinates proteins on the mitochondrial surface. Pink1 then phosphorylates these newly formed polyubiquitin chains, generating phospho-Ub, which further promotes Parkin activity (Ordureau et al. 2014). As to the mechanism underlying the phospho-Ub mediated activation of Parkin, phosphorylated or phosphomimetic ubiquitin has been shown to promote the release of E2-bound unmodified ubiquitin in vitro, and this reaction requires the catalytic Cys431 of Parkin (Koyano et al. 2014). Hence, the authors conclude that phospho-Ub is a strictly allosteric effector of Parkin, rather than a preferred Parkin substrate (the substrate in this assay is unmodified ubiquitin). Whereas the proposed allosteric mechanism presumes the binding of phospho-Ub to Parkin, the respective binding site on Parkin remains to be identified. One candidate is a binding site for phospho-peptides within the RING0 domain, previously identified in the crystal structure of Parkin (Zheng and Hunter 2014). Apart from activating Parkin, phospho-Ub may also be involved in Parkin recruitment from the cytosol to the outer mitochondrial membrane. Although the exact mechanism is not clear, Pink1 has been proposed to phosphorylate ubiquitin moieties previously conjugated to OMM proteins, such as mitofusin 1 (Mfn1). The newly generated phospho-Ub would then serve as a receptor for Parkin (Pickrell and Youle 2015).

Current model of Pink1/Parkin-mediated mitophagy. Under steady-state conditions, Pink1 levels are very low. Mitochondrial stress conditions may lead to mitochondrial damage, accompanied by a decrease or loss of the mitochondrial membrane potential (Δψ; 1). In the absence of Δψ, Pink1 accumulates at the outer mitochondrial membrane (OMM; 2). Pink1 recruits and activates the usually cytosolic E3 ubiquitin ligase Parkin in a process involving Pink1-mediated phosphorylation of Parkin at Ser65 (3). Parkin conjugates ubiquitin (Ub) to various OMM proteins (4). Pink1 phosphorylates Ub attached to OMM proteins. The resulting phospho-Ub further activates Parkin (5). Adaptor proteins (red) that bind both Ub and the autophagic protein LC3 (light blue) mediate sequestration of the organelle in an autphagosomal membrane (gray lines) (6). The autophagosome then fuses with a lysosome, delivering its complete content to degradation by lysosomal hydrolases (green; 7)
Apart from activating Parkin upon mitochondrial damage, Pink1 has also been proposed to repress the induction of mitophagy under steady-state conditions (Fedorowicz et al. 2014). Using an immunoprecipitation approach, the authors found that, after being generated inside mitochondria, the 53-kDa major Pink1 cleavage product (Pink1-PF) translocates to the cytosol and binds Parkin, thereby preventing Parkin translocation to mitochondria and the concomitant induction of mitophagy. Importantly, in assigning a distinct function to the processed Pink1 species, the proposed model challenges the idea that Pink1-PF is merely a degradation intermediate (Greene et al. 2012).
Following its mitochondrial recruitment and activation, Parkin is thought to nonspecifically ubiquitinate proteins of the OMM. As a broad spectrum of substrates for ubiquitination via Parkin has been revealed by proteomic experiments, the observed global ubiquitination pattern has been suggested to be more important than the ubiquitination of specific substrates in order to induce mitophagy (Sarraf et al. 2013). The ubiquitination of proteins of the OMM by Parkin initiates further mitophagy reactions by marking the entire organelle for elimination via autophagy. The formation of the isolation membrane around damaged mitochondria is mediated by autophagy receptors that are able to bind to ubiquitinated proteins through their ubiquitin-binding domain and to LC3 on autophagosomal membranes through a LC3-interaction region (LIR; Pankiv et al. 2007). So far, several receptors playing a role in PINK1/Parkin-mediated mitophagy have been identified (Pickrell and Youle 2015; Yoshii and Mizushima 2015), including SQSTM1/p62 (sequestosome 1; Geisler et al. 2010a), NBR1 (neighbor of BRCA1 gene 1; Kirkin et al. 2009), NDP52 (calcium binding and coiled-coil domain 2; Lazarou et al. 2015), and optineurin (Wong and Holzbaur 2014). Studies suggest that optineurin is sufficient to activate mitophagy, whereas p62 and NBR1 are not (Lazarou et al. 2015; Shi et al. 2015; Wong and Holzbaur 2014). Optineurin and NDP52, but not p62, have been shown to be recruited by PINK1 via its generation of phospho-ubiquitin, which serves as an autophagy signal, independent of Parkin (Lazarou et al. 2015).
An effect upstream of mitophagy is the PINK1/Parkin-mediated ubiquitination of the OMM fusion proteins Mfn1 and Mitofusin 2 (Mfn2), which are GTPases that mediate mitochondrial fusion (Gegg et al. 2010). This ubiquitination targets the proteins for degradation by the proteasome, a process that is mediated by the AAA+ ATPase p97 (Tanaka et al. 2010). Degradation of Mfn1 and Mfn2 impedes the fusion of damaged mitochondria within the network of healthy organelles and may thereby contribute to the fragmentation and subsequent degradation of defective mitochondria. This mechanism is proposed to contribute to the protection of a healthy mitochondrial population (Ashrafi and Schwarz 2013; Ni et al. 2015; Youle and van der Bliek 2012). Taken together, these data suggest that mitochondrial dynamics via the fusion/fission machineries and mitophagy of dysfunctional mitochondria are closely linked (Fig. 4). For example, phosphorylation of Mfn2 by Pink1 has been shown to mediate Parkin recruitment (Chen and Dorn 2013). Moreover, in Drosophila models, inactivation of the fusion components Mfn1/2 and Opa1 and the overexpression of the fission component Drp1 have been demonstrated to be able to rescue Pink1 or Parkin mutations (Deng et al. 2008).

Graphic representation of the Pink1/Parkin interaction network. Identified protein-protein interaction partners of the Pink1-Parkin system and their (in part, hypothetical) functional involvement in the various cell biological processes
Conversely, several lines of evidence indicate a potential regulatory influence of the Pink1/Parkin system on the equilibrium between fusion and fission reactions. Pink1 and/or Parkin mutants and a downregulation of these proteins increase Drp1-dependent mitochondrial fragmentation (Lutz et al. 2009). Very recently, Pink1 has been shown to be able to signal an increased fission reaction by affecting mitochondria-associated protein kinase A, which normally inhibits Drp1 (Pryde et al. 2016). Hence, the pathological relations of the Pink1/Parkin system might go beyond “simple” mitophagy processes such as, in Caenorhabditis elegans, the inhibition of mitochondrial fusion by another PD-related mutant protein, namely α-synuclein, has been shown to be rescued by Pink1 and Parkin (Kamp et al. 2010). Furthermore, the PINK1/Parkin-mediated proteasomal degradation of the mitochondrial Rho GTPase 1 (Miro), which anchors kinesin motor proteins to the mitochondrial surface (Glater et al. 2006), results in the arrest of mitochondrial motility (Wang et al. 2011). An antagonist of Parkin is the deubiquitinase USP30, which removes ubiquitin molecules from proteins of the OMM and therefore inhibits mitophagy. The inhibition of USP30 could be relevant for therapeutic PD approaches that enhance mitochondrial quality control by supporting the mitophagy of damaged mitochondria via the PINK1/Parkin pathway (Bingol et al. 2014).
Regarding the vast number of publications focusing on the role of Pink1 and Parkin in mediating mitophagy, attention should also be paid to another concept that assigns the two proteins a function in regulating the translation of specific mitochondrial proteins. As has recently been reported, Pink1 and Parkin regulate the localized translation of nuclear-encoded respiratory chain subunits at the mitochondrial surface (Gehrke et al. 2015). In the proposed model, Pink1 mediates the translocation of specific translationally repressed mRNAs from the cytosol to the outer mitochondrial membrane. This process has further been shown to involve Tom20, a constituent of the outer membrane translocase complex (TOM). In collaboration with Parkin, Pink1 would then mediate the displacement of translation inhibitors, thereby enabling the translation of the respective respiratory chain components. By the proposed mechanism Pink1 and Parkin might promote the repair of less severely damaged mitochondria (Gehrke et al. 2015).
Outlook
Although the foremost function of the kinase Pink1 in the initiation of mitophagy via Parkin recruitment and activation has been well established, the connection between mitochondrial damage and cellular Pink1 accumulation itself is not entirely clarified. As described above, the mitochondrial import/turnover model has its shortcomings, whereas a transcriptional regulation mechanism cannot be completely excluded. However, the question regarding the way in which Pink1 gene expression is specifically up-regulated by mitochondrial damage will require further intensive research.
As most information about mitophagy pathways has been established in non-neural cell lines, controversy remains as to whether the damage-induced mitophagy mediated by PINK1 and Parkin can be applied to neurons (Ashrafi et al. 2014). Mitochondria in neurons have a very important role in supplying energy to sustain neuronal activity and are highly exposed to stress conditions such as high Ca2+ influx and oxidative stress over time (Grenier et al. 2013). The majority of mitochondria in neurons is located at distal axons, whereas lysosomes occur in the soma. So far, whether damaged mitochondria have to be translocated back to the soma to be degraded remains unclear. They might be locally sequestered in autophagosomes before their retrograde transport to the soma or may even be degraded outside the soma (Ashrafi and Schwarz 2013).
The biochemical and cell biological characterization of the pathological mechanisms in PD during the last few years has been an extraordinary success story of modern life science. However, as the identified PD-related genetic defects only represent a minority of all disease cases, the relevance of the information already obtained for sporadic cases has to be scrutinized. Many open questions remain as to how exactly age-related mitochondrial dysfunction, mitophagy, and organellar quality control are correlated to cause the specific pathological situation represented by PD, in particular, in patients with no genetic predisposition.
References
Aerts L, Craessaerts K, De Strooper B, Morais VA (2015) PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J Biol Chem 290:2798–2811
Ashrafi G, Schwarz TL (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20:31–42
Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL (2014) Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 206:655–670
Becker D, Richter J, Tocilescu MA, Przedborski S, Voos W (2012) Pink1 kinase and its membrane potential (Deltapsi)-dependent cleavage product both localize to outer mitochondrial membrane by unique targeting mode. J Biol Chem 287:22969–22987
Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M (2014) The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510:370–375
Chen Y, Dorn GW 2nd (2013) PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340:471–475
Ciechanover A (2005) Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol 6:79–87
Clague MJ, Urbe S (2010) Ubiquitin: same molecule, different degradation pathways. Cell 143:682–685
Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441:1162–1166
Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, Abramov AY, Wood NW (2010) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20:867–879
Deng H, Dodson MW, Huang H, Guo M (2008) The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A 105:14503–14508
Duan X, Tong J, Xu Q, Wu Y, Cai F, Li T, Song W (2014) Upregulation of human PINK1 gene expression by NF-kB signalling. Mol Brain 7:57–67
Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, Kruger R, Winklhofer KF, Vogel F, Reichert AS, Auburger G, Kahle PJ, Schmid B, Haass C (2007) Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci 27:12413–12418
Fedorowicz MA, de Vries-Schneider RLA, Rüb C, Becker D, Huang Y, Zhou C, Wolken DMA, Voos W, Liu YH, Przedborski S (2014) Cytosolic cleaved PINK1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep 15:86–93
Gautier CA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 105:11364–11369
Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19:4861–4870
Gehrke S, Wu Z, Klinkenberg M, Sun Y, Auburger G, Guo S, Lu B (2015) PINK1 and Parkin control localized translation of respiratory chain component mRNAs on mitochondria outer membrane. Cell Metab 21:95–108
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010a) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12:119–131
Geisler S, Holmstrom KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, Springer W (2010b) The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 6:871–878
Gispert S, Ricciardi F, Kurz A, Azizov M, Hoepken HH, Becker D, Voos W, Leuner K, Muller WE, Kudin AP, Kunz WS, Zimmermann A, Roeper J, Wenzel D, Jendrach M, Garcia-Arencibia M, Fernandez-Ruiz J, Huber L, Rohrer H, Barrera M, Reichert AS, Rüb U, Chen A, Nussbaum RL, Auburger G (2009) Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 4:e5777
Glater EE, Megeath LJ, Stowers RS, Schwarz TL (2006) Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol 173:545–557
Gomez-Sanchez R, Gegg ME, Bravo-San Pedro JM, Niso-Santano M, Alvarez-Erviti L, Pizarro-Estrella E, Gutierrez-Martin Y, Alvarez-Barrientos A, Fuentes JM, Gonzalez-Polo RA, Schapira AHV (2014) Mitochondrial impairment increases FL-PINK1 levels by calcium-dependent gene expression. Neurobiol Dis 62:426–440
Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 13:378–385
Grenier K, McLelland GL, Fon EA (2013) Parkin- and PINK1-dependent mitophagy in neurons: will the real pathway please stand up? Front Neurol 4:100
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634–1658
Haynes CM, Ron D (2010) The mitochondrial UPR—protecting organelle protein homeostasis. J Cell Sci 123:3849–3855
Hoepken HH, Gispert S, Morales B, Wingerter O, Del Turco D, Mulsch A, Nussbaum RL, Muller K, Drose S, Brandt U, Deller T, Wirth B, Kudin AP, Kunz WS, Auburger G (2007) Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol Dis 25:401–411
Jin SM, Youle RJ (2013) The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 9:1750–1757
Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191:933–942
Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, Eimer S, Winklhofer KF, Haass C (2010) Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J 29:3571–3589
Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205:143–153
Kanki T, Klionsky DJ (2010) The molecular mechanism of mitochondria autophagy in yeast. Mol Microbiol 75:795–800
Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MMK (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser(65). Biochem J 460:127–139
Kirkin V, Lamark T, Johansen T, Dikic I (2009) NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 5:732–733
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile Parkinsonism. Nature 392:605–608
Klein C, Westenberger A (2012) Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2:a008888
Klinkenberg M, Gispert S, Dominguez-Bautista JA, Braun I, Auburger G, Jendrach M (2012) Restriction of trophic factors and nutrients induces PARKIN expression. Neurogenetics 13:9–21
Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, Knebel A, Alessi DR, Muqit MM (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2:120080
Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510:162–166
Lazarou M, Jin SM, Kane LA, Youle RJ (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22:320–333
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314
Lin W, Kang UJ (2008) Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem 106:464–474
Lutz AK, Exner N, Fett ME, Schlehe JS, Kloos K, Lammermann K, Brunner B, Kurz-Drexler A, Vogel F, Reichert AS, Bouman L, Vogt-Weisenhorn D, Wurst W, Tatzelt J, Haass C, Winklhofer KF (2009) Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem 284:22938–22951
Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189:211–221
Mei Y, Zhang Y, Yamamoto K, Xie W, Mak TW, You H (2009) FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc Natl Acad Sci U S A 106:5153–5158
Muqit MM, Abou-Sleiman PM, Saurin AT, Harvey K, Gandhi S, Deas E, Eaton S, Payne Smith MD, Venner K, Matilla A, Healy DG, Gilks WP, Lees AJ, Holton J, Revesz T, Parker PJ, Harvey RJ, Wood NW, Latchman DS (2006) Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem 98:156–169
Murata H, Takamatsu H, Liu S, Kataoka K, Huh NH, Sakaguchi M (2015) NRF2 regulates PINK1 expression under oxidative stress conditions. PLoS ONE 10:e0142438
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298
Ni HM, Williams JA, Ding WX (2015) Mitochondrial dynamics and mitochondrial quality control. Redox Biol 4:6–13
Okamoto K, Kondo-Okamoto N, Ohsumi Y (2009) Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 17:87–97
Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, Shiba-Fukushima K, Sato S, Shimizu H, Fukunaga Y, Taniguchi H, Komatsu M, Hattori N, Mihara K, Tanaka K, Matsuda N (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 3:1016
Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, Wells JA, Gygi SP, Schulman BA, Harper JW (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56:360–375
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145
Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441:1157–1161
Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85:257–273
Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, Wood NW, Martins LM, Downward J (2007) The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol 9:1243–1252
Pridgeon JW, Olzmann JA, Chin L-S, Li L (2007) PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol 5:1494–1503
Priyadarshini M, Orosco LA, Panula PJ (2013) Oxidative stress and regulation of Pink1 in zebrafish (Danio rerio). PLoS ONE 8:e81851
Pryde KR, Smith HL, Chau KY, Schapira AH (2016) PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol 213:163–171
Quiros PM, Mottis A, Auwerx J (2016) Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol 17:213–226
Riley BE, Lougheed JC, Callaway K, Velasquez M, Brecht E, Nguyen L, Shaler T, Walker D, Yang Y, Regnstrom K, Diep L, Zhang Z, Chiou S, Bova M, Artis DR, Yao N, Baker J, Yednock T, Johnston JA (2013) Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun 4:1982
Rüb C, Schröder N, Voos W (2015) Biochemical properties of the kinase PINK1 as sensor protein for mitochondrial damage signalling. Biochem Soc Trans 43:287–291
Sakurai M, Kawamura T, Nishimura H, Suzuki H, Tezuka F, Abe K (2009) Induction of Parkinson disease-related proteins in motor neurons after transient spinal cord ischemia in rabbits. J Cereb Blood Flow Metab 29:752–758
Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496:372–376
Schulz C, Schendzielorz A, Rehling P (2015) Unlocking the presequence import pathway. Trends Cell Biol 25:265–275
Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA (2007) NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A 104:19500–19505
Shi J, Fung G, Deng H, Zhang J, Fiesel FC, Springer W, Li X, Luo H (2015) NBR1 is dispensable for PARK2-mediated mitophagy regardless of the presence or absence of SQSTM1. Cell Death Dis 6:e1943
Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N (2012) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2:1002
Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G (2005) Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 14:3477–3492
Slobodkin MR, Elazar Z (2013) The Atg8 family: multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem 55:51–64
Smit JJ, Monteferrario D, Noordermeer SM, van Dijk WJ, van der Reijden BA, Sixma TK (2012) The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J 31:3833–3844
Stieglitz B, Morris-Davies AC, Koliopoulos MG, Christodoulou E, Rittinger K (2012) LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep 13:840–846
Tait SW, Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11:621–632
Takatori S, Ito G, Iwatsubo T (2008) Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci Lett 430:13–17
Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191:1367–1380
Thomas RE, Andrews LA, Burman JL, Lin WY, Pallanck LJ (2014) PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet 10:e1004279
Trempe JF, Fon EA (2013) Structure and function of Parkin, PINK1, and DJ-1, the Three Musketeers of Neuroprotection. Front Neurol 4:38
Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, Nagar B, Fon EA, Gehring K (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340:1451–1455
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160
Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107:378–383
Voos W (2013) Chaperone-protease networks in mitochondrial protein homeostasis. Biochim Biophys Acta 1833:388–399
Wang K, Klionsky DJ (2011) Mitochondria removal by autophagy. Autophagy 7:297–300
Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147:893–906
Wauer T, Komander D (2013) Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J 32:2099–3112
Wenzel DM, Lissounov A, Brzovic PS, Klevit RE (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474:105–108
Winklhofer KF (2014) Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol 24:332–341
Wong YC, Holzbaur EL (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A 111:E4439–E4448
Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AS, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E, Downward J, Mansfield L, Jat P, Taylor J, Heales S, Duchen MR, Latchman D, Tabrizi SJ, Wood NW (2008) PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 3:e2455
Yoshii SR, Mizushima N (2015) Autophagy machinery in the context of mammalian mitophagy. Biochim Biophys Acta 1853:2797–2801
Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337:1062–1065
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12:9–14
Zheng XD, Hunter T (2014) Pink1, the first ubiquitin kinase. EMBO J 33:1621–1623
Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S (2008) The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A 105:12022–12027
Ziviani E, Tao RN, Whitworth AJ (2010) Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A 107:5018–5023
Author information
Authors and Affiliations
Corresponding author
Additional information
The work was supported by a grant from the Deutsche Forschungsgemeinschaft (to W. V., VO657/5-2).
Rights and permissions
About this article

Cite this article
Rüb, C., Wilkening, A. & Voos, W. Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res 367, 111–123 (2017). https://doi.org/10.1007/s00441-016-2485-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-016-2485-8