
Abstract
Autophagy and apoptosis are two crucial self-destructive processes that maintain cellular homeostasis, which are characterized by their morphology and regulated through signal transduction mechanisms. These pathways determine the fate of cellular organelle and protein involved in human health and disease such as neurodegeneration, cancer, and cardiovascular disease. Cell death pathways share common molecular mechanisms, such as mitochondrial dysfunction, oxidative stress, calcium ion concentration, reactive oxygen species, and endoplasmic reticulum stress. Some key signaling molecules such as p53 and VEGF mediated angiogenic pathway exhibit cellular and molecular responses resulting in the triggering of apoptotic and autophagic pathways. Herein, based on previous studies, we describe the intricate relation between cell death pathways through their common genes and the role of various stress-causing agents. Further, extensive research on autophagy and apoptotic machinery excavates the implementation of selective biomarkers, for instance, mTOR, Bcl-2, BH3 family members, caspases, AMPK, PI3K/Akt/GSK3β, and p38/JNK/MAPK, in the pathogenesis and progression of neurodegenerative diseases. This molecular phenomenon will lead to the discovery of possible therapeutic biomolecules as a pharmacological intervention that are involved in the modulation of apoptosis and autophagy pathways. Moreover, we describe the potential role of micro-RNAs, long non-coding RNAs, and biomolecules as therapeutic agents that regulate cell death machinery to treat neurodegenerative diseases.
Graphical abstract
Mounting evidence demonstrated that under stress conditions, such as calcium efflux, endoplasmic reticulum stress, the ubiquitin–proteasome system, and oxidative stress intermediate molecules, namely p53 and VEGF, activate and cause cell death. Further, activation of p53 and VEGF cause alteration in gene expression and dysregulated signaling pathways through the involvement of signaling molecules, namely mTOR, Bcl-2, BH3, AMPK, MAPK, JNK, and PI3K/Akt, and caspases. Alteration in gene expression and signaling cascades cause neurotoxicity and misfolded protein aggregates, which are characteristics features of neurodegenerative diseases. Excessive neurotoxicity and misfolded protein aggregates lead to neuronal cell death by activating death pathways like autophagy and apoptosis. However, autophagy has a dual role in the apoptosis pathways, i.e., activation and inhibition of the apoptosis signaling. Further, micro-RNAs and LncRNAs act as pharmacological regulators of autophagy and apoptosis cascade, whereas, natural compounds and chemical compounds act as pharmacological inhibitors that rescue neuronal cell death through inhibition of apoptosis and autophagic cell death.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Accumulation of protein aggregates in the cellular milieu is a major burden for neurons, and it greatly disturbs the nervous system homeostasis. These misfolded and aggregated proteins are hampering the activities and transmission of the neuronal cell. The accumulation of aggregates induces toxicity, which causes memory loss, cognitive decline, and impairment in the maturation of neuronal cells that result in the progression of several neurodegenerative disorders (NDDs), including Alzheimer’s disease (AD), Parkinson’s disease (PD), Amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and Multiple sclerosis (MS) [1, 2]. Excessive accumulation of abnormally aggregated/non-functional proteins in the cytoplasmic region of the cell leads to organelle damage, which is responsible for neuronal death in the central nervous system (CNS) and ultimately leads to cognitive defects and synaptic dysfunction. Apoptosis and autophagy are two degradation mechanisms that are currently known for eliminating the degraded components and quality control of cellular components, which is necessary for maintaining cellular homeostasis. Autophagy is defined as the lysosomal-dependent degradation process of cytoplasmic constituents, whereas, apoptosis is considered as programmed cell death (PCD) of cells. Autophagy is of three types, namely macroautophagy, microautophagy, and chaperone-mediated autophagy that occurs through the formation of autophagosomes followed by association with lysosomes leads to the formation of the autophagolysosomal complex. On the contrary, apoptosis is described as morphological and physiological changes required to maintain cellular homeostasis by inducing nuclear membrane destruction, DNA fragmentation, and generation of apoptotic bodies [3, 4]. Recent studies demonstrated that the perturbation of autophagic machinery causes accumulation of misfolded proteins, and excessive induction of apoptotic mechanism leads to neuronal death that is involved in the pathogenesis of NDDs [5, 6]. Excessive loss of neuronal cells leads to cognitive defects, impaired neurogenesis and neural differentiation, synaptic dysfunction, and memory impairment, which are characteristic features of NDDs [7, 8]. However, the molecular crosstalk between autophagic degradation and apoptotic cell death is a complicated phenomenon and has provided conflicting results but at the same time necessary for determining the fate of the cell. However, under physiological conditions, such as excessive oxidative stress, reactive oxygen species (ROS) production, mitochondrial dysfunction, and endoplasmic reticulum (ER) stress, neuronal cells exhibit defective or incomplete autophagic degradation of misfolded protein aggregates and, therefore, apoptotic machinery that causes neuronal cell death. Extensive investigations identified the potential implementation of epigenetic regulator p53 and pro-angiogenic marker vascular endothelial growth factor (VEGF) in the modulation and regulation of both apoptosis and autophagy machinery.
Moreover, autophagy is known to have a dual effect on apoptosis, which involves inhibition and induction of the apoptosis pathway. Under stress conditions, apart from misfolded protein degradation, autophagic machinery, either itself or through apoptotic induction, causes cell death depending upon the exposure of a stress condition [9, 10]. Both autophagy and apoptosis pathways regulate brain homeostasis through the involvement of downstream targets such as the mammalian target of rapamycin (mTOR), Bcl and BH3 family of proteins, caspases, 5' AMP-activated protein kinase (AMPK), class III phosphatidylinositol 3-kinase (PI3K), and glycogen synthase kinase 3β (GSK3β). Recent studies explored the potential of biomolecules, long non-coding RNAs (LncRNAs), and micro-RNAs (miRNAs) as therapeutic modulators of these pathways involved in the pathogenesis and progression of NDDs.
Herein, we provided a comprehensive story derived from various literature sources to dissect the molecular mechanism between apoptosis and autophagy in NDDs. In the beginning, we have discussed about cell death pathways followed by the shared mechanism between three types of cell death pathways and the dual role of autophagy on apoptosis. The later part of the review discusses the molecular markers of cell death in NDDs with apoptosis and autophagy signaling. Finally, we discuss the potential application of miRNAs, LncRNAs, and biomolecules on different cell death pathways.
Overview of cell death pathways
Autophagic pathway: act as pro-death and pro-survival signaling cascade
Autophagy is a molecular phenomenon used to eliminate damaged organelle and protein aggregates, which is characterized by the formation of autophagosomes and interaction with the lysosome. Cytoplasmic component degradation in the lysosome is divided into three subtypes as follows: macroautophagy, microautophagy, and chaperone-mediated autophagy. The mechanism underlying autophagy includes phagophore membrane formation from the Golgi apparatus, mitochondria, plasma membrane, and ER, where misfolded proteins and degraded cytoplasmic material are wrapped, elongated and forms autophagosome. This autophagosome, through microtubule dynamics, transports to the lysosome, where the formation of autolysosome occurs through the fusion of autophagosome and lysosome [11, 12]. Further, autophagy is a multi-regulatory process initiated by two major clusters of proteins UN51-like Ser/Thr kinases (ULK) complex and PI3K complex. The ULK complex consists of ULK1/2 family, FAK family kinase interacting protein of 200 kDa, autophagy-related protein 13 (Atg13) whereas, PI3K complex consists of vacuolar protein sorting 34 (Vps34), p15 (Vps15), beclin1 (Atg6), and Barkor (Atg14) [13, 14]. Two ubiquitin complexes control the elongation and interaction of autophagosomes. Firstly, a complex Atg5/Atg7/Atg12 is formed due to covalent interaction between Atg5/Atg7 and Atg12. Secondly, this Atg5/Atg7/Atg12 complex interacts with Atg16 to form another complex, Atg5/Atg7/Atg12/Atg16, that is required for autophagosomes elongation. Another complex associated with the molecular marker of autophagosome is formed through the proteolytic cleavage of microtubule-associated protein 1 light chain 3 (LC3) with Atg4B to generate LC3-II [15,16,17,18]. However, autophagosomes require a motor and kinesin protein along with the recruitment of protein complexes known as the soluble NSF attachment protein receptor (SNAREs) for relocation along the microtubule, fusion with the lysosome, and protein degradation [19] (Fig. 1).

Molecular connection between apoptosis and necroptosis as cell death pathways opening a new area of research in the field of neurodegenerative disorders. However, whether autophagy is a pro-death or pro-survival pathway is still a matter of concern. From the past two decades, extensive research in this field has found the connection between three pathways involving different molecular mechanisms and biological processes. Mitochondrial dysfunction, genotoxic stress, oxidative stress, Ca2+ concentration, and ER stress were among the major external factors that activated the signaling cascade leading to cell death. Here, Bax and Bak were two pro-apoptotic proteins that activate the apoptosis pathway, while mTOR, TSC1, and TSC2 were important in regulating the autophagic pathway. Activation of mTOR causes inhibition of ULK1 complex that further inhibits the autophagic pathway. Similarly, phosphorylation of TSC1 and TSC2 causes inhibition of Rheb, which leads to mTOR activation and subsequently inhibition of autophagy pathway. Besides external stress factors, Atg12 leads to activation of autophagosomes, cytochrome C, and caspase, followed by necrosome leads to the autophagic, apoptotic, and necroptosis pathway, respectively
Apoptosis pathway: intrinsic and extrinsic cell death machinery
Apoptosis, an important molecular phenomenon, which is also known as PCD, is involved in the maintenance of tissue homeostasis. Apoptosis is best described as nuclear morphological changes characterized by chromatin regulation, degradation of cytoskeletal proteins, nuclear membrane breakdown, DNA fragmentation, and generation of apoptotic bodies adjacent to the cell surface [20, 21]. The physical execution of apoptosis can be initiated by either the extrinsic or intrinsic apoptotic pathway. Moreover, death receptors and internal stimuli such as DNA damage, activation of pro-apoptotic factors of B-cell lymphoma 2 (Bcl-2) family, and upregulation of p53 play a major function in regulating the apoptotic pathway [22, 23]. Extrinsic apoptotic pathway induces the attachment of tumor necrosis factor (TNF) family receptor on the cell surface, which increases the recruitment of fas-associated death domain protein (FADD) and TNF-related apoptosis-inducing ligand (TRAIL) following the binding of initiator caspases (caspase 8 and caspase 9), which initiate its autoproteolytic processing. Initiation of autoproteolytic processing leads to activation of effector caspases (caspase 3 and caspase 7), resulting in cleavage of Bcl-2 homology region 3 (BH3) protein, which induces pro-apoptotic factors’ activation and alters inner mitochondria membrane permeability [24,25,26]. On the contrary, the intrinsic apoptotic pathway, also called the mitochondrial apoptotic pathway, is a death receptor-independent mechanism and requires Bcl-2 member proteins, consisting of the BH1-3 domain, to decide whether to undergo mitochondrial membrane permeabilization or not. Further, intrinsic apoptotic pathway causes sequestration of pro-apoptotic factors from mitochondria to cytosol, including cytochrome C, a second mitochondria-derived activator of caspases/direct IAP-binding protein with low PI (Smac/DIABLO), HtrA2/Omi, and apoptosis-inducing factors (AIFs), which results in the generation of apoptosome complex.
Bcl-2 associated X protein (Bax) and Bcl-2 homologous antagonist/killer (Bak), which have BH1-3 domain required for the execution of the mitochondrial apoptotic pathways in a caspase-dependent or caspase-independent manner [27,28,29]. Apoptosis is a highly regulated phenomenon controlled by the inhibitor of apoptosis proteins (IAP) and X-linked inhibitor of apoptosis protein (XIAP), which can interfere in the caspase activation process leading to caspase-dependent or caspase-independent apoptosis. Further, ROS, nitrogen–oxygen species (NOS), and DNA damage are considered to be inducers of apoptosis, resulting in the activation of signaling cascade that results in cell death in various disease models of NDDs. These agents lead to activation of janus kinases/signal transducer and activator of transcription protein (JAK/STAT) signaling pathway, through increased activity of cytokines, such as a nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway and PI3K-like kinases, respectively, which promotes cell apoptosis [30,31,32] (Fig. 1).
Necroptosis cell death machinery
Necroptosis is the well-characterized molecular phenomenon of unprogrammed cell death activated by cellular damage or pathogenic infiltration regulating necrosis mediated by receptor-interacting protein kinase 1 (RIPK1) and receptor-interacting protein kinase 3 (RIPK3). Activation of RIPK1 and RIPK3 eventually leads to plasma membrane permeabilization, activation of cytokines and chemokines, sequestering cell content, and exposure of damage-associated molecular patterns (DAMP’s) [33]. RIPK1 initiated a signaling cascade, which phosphorylates and activates RIPK3 that further phosphorylates and activates mixed lineage kinase domain-like (MLKL), forming a complex known as necrosome. Necrosome cause cell rupture because of the pore-forming ability of MLKL aggregates, modulation of ion channels, and the inflammasome formation in some cellular contexts [34,35,36,37]. Inhibition of RIPK1, RIPK3, and MLKL and activation of necrosome in concert with necrosis is the pharmacological feature of necroptosis. The number of literatures suggesting the role of caspase 8 inhibition in transferring the mitochondrial apoptotic pathway to necroptosis cell death pathway due to increased expression of RIPK3 and MLKL and initiation due to immune-based ligands [38]. Necroptosis resembles the apoptotic cell death pathways due to the implication of caspase 8 and death receptors such as TNF alpha, FADD, Tumor necrosis factor receptor type 1-associated DEATH domain protein (TRADD), and TNF receptor-associated factor 2 (TRAF2) and hence is called as alternative cell death signaling pathway [39]. In addition, FADD/RIP3 and FLIP/RIP3 knock out the model interplay between apoptosis and necroptosis due to the absence of FLIP and caspase 8-FLIP heterodimers. In another study function of protein kinase B (Akt) as a molecular switch between apoptosis and necroptosis through phosphorylation, production of TNFα, and blocking of pro-apoptotic factor response was demonstrated [40, 41] (Fig. 1).
Crosslinking autophagy and apoptosis signaling pathway
Calcium efflux and endoplasmic reticulum stress response
Misfolded protein aggregates cause activation of ER stress signaling, which involves the synthesis and degradation of proteins via autophagic pathway and endoplasmic-reticulum-associated protein degradation (ERAD) pathway. Further, eliminating the damaged organelle through apoptotic machinery decides the fate of the cell that depends on the intensity and time-duration of the implied stress condition [42]. During this process, molecular chaperone GRP78/BiP interacts with mechanistic UPR signaling molecules, namely activating transcription factor 6 (ATF6), protein kinase RNA (PKR), ER kinase (PERK), and inositol-requiring protein 1α (IRE1α). The complex between GRP78/BiP and UPR signaling molecules activate respective transducers and assist in the folding of accumulated proteins. However, PERK attenuates mRNA translation and thus inhibits the entry of newly synthesized protein in contact with the ER under stress conditions along with eIF2α activation [43]. Moreover, eIF2α phosphorylation causes protein synthesis inhibition mediated through a dedicated protein translational mechanism.
Under a high-stress environment, ATF4 causes both autophagy and apoptosis induction through regulation of Atg genes, and XIAP interacts with C/EBP homologous proteins (CHOP/GADD153) mediated through increased caspase activation [44]. Moreover, CHOP activates apoptotic pathways through increased expression of pro-apoptotic factors (such as BIM and death receptor 5), decreased expression of anti-apoptotic factors (Bcl members), and increased mitochondrial activity. Further, increased mitochondrial function leads to elevate cytochrome-c release from mitochondrial pores along with EROα and IP3R. Activation of EROα and IP3R causes an increase in mitochondrial calcium influx, which induces the apoptosis pathway [45]. However, under the ER stress environment, JNK mediated Bcl-2 phosphorylation leads to Beclin-1/Bcl-2 dissociation and autophagy activation, while a prolonged stress environment causes activation of the apoptotic pathway [46]. Further, ER stress increases calcium influx, which leads to AMPK activation and inhibits mTOR activity and thus induces the autophagy pathway. Similarly, ER stress also causes mitochondrial dysfunction through increased generation of mitochondrial pores leading to mitochondrial death via apoptotic machinery [47, 48]. Altogether, it may be concluded that ER stress regulates both autophagy and apoptosis machinery through modulating downstream targets and increased calcium ion concentration leading to mitochondrial dysfunction.
The implication of ubiquitin–proteasome system
UPS machinery is the major protein degradation pathway involved in neuronal regeneration and plasticity, whereas apoptosis and autophagy are the major regulatory signaling cascade involved in neuronal cell death that leads to neurodegeneration. Mounting evidence suggests the extensive crosstalk between autophagy, apoptosis, and UPS, which are involved in regulating brain homeostasis [49]. A recent study by Tsai et al., demonstrated that administration of Maackiain (MK) in the SH-SY5Y cell line prevents PD pathology through apoptosis inhibition and autophagic degradation due to increased PINK1/parkin expression and enhanced UPS machinery [50]. Similarly, Mudawal et al. demonstrated that dose-dependent administration of lindane in aged rats at 2.5 mg/kg concentration for 21 days causes alteration in apoptosis and autophagic markers expression. The study concluded that administration of lindane causes significant upregulation of Bax, Bad, caspase 3, caspase 9, ATG5, ATG12, LC-III levels, and causes a decrease in Bcl-2 expression. Thus, the analysis concluded that administration of lindane alters the expression of proteins associated with UPS machinery, autophagic cascade, and apoptotic pathway [51]. In post-traumatic brain injury, UPS machinery, axonal degeneration, apoptosis, and autophagic degradation play an important role, where enhanced expression of UCH-L1 modulates the autophagic pathway and UPS pathway. Congregation of UCH-L1 with TAT promotes neuronal transduction where it causes inhibition of K48-linkage polyubiquitination in the hippocampus but no effects on K65-linkage polyubiquitination. Further, the combination of UCH-L1 and TAT decreases autophagic degradation and neuronal apoptosis through decreased expression of Beclin-1 and LC3-II proteins [52].
Further, Guo et al. demonstrated the involvement of p-p38α as a central mediator of autophagy and apoptosis in response to UPS impairment. Reduced phosphorylation of p-p38α in response to BIRB796 causes a decrease in autophagic flux and neuronal apoptosis [53]. Likewise, the interaction between E3-ubiquitin ligase FBXO32/atrogin-1 and FOXO3A regulates autophagic and apoptotic cascade. Thus, administration of Endophilin-A in cultured neurons downregulates FBXO32 expression, which causes a decrease in neuronal apoptosis and increases autophagosome formation [54]. Similarly, administration of Trehalose in HD patients demonstrated a decrease in ROS levels, ubiquitinated protein expression, caspase 3 expression. Further, administration of Trehalose counteracts the decrease in LC-3 levels induced by Epoxomicin [55].
Moreover, Dietary restriction is known to regulate autophagic and apoptotic cell death through the involvement of UPS machinery. Shruthi et al. demonstrated that dietary restriction increases autophagic degradation in a spontaneous obese rat model and decreases Bax and p53 activity, thus preventing neurodegeneration [56]. Further, Xu et al., in SH-SY5Y cell culture, demonstrated that SIAH silencing through siRNA suppressed apoptosis, promoted cell proliferation, and decreases LC3-II expression [57]. Furthermore, XIAP, a ubiquitin E3 ligase, regulates mitochondrial depolarization, where XIAP in the absence of BH3 protein activates Bax-induced mitochondrial outer membrane potential (MOMP). XIAP targets the dysfunctional mitochondria for the autophagy-lysosomal pathway and delays cytochrome-C release, hence lowering the mitochondrial apoptotic potential [58]. Altogether, it may be concluded that UPS machinery regulates both apoptosis and autophagy signaling cascade through respective downstream targets in case of neurodegeneration.
Dual role of autophagy on the apoptotic signaling cascade
In the above sections, direct and indirect factors have been described through which the relationship between autophagy and apoptosis has been established, for instance, autophagic degradation of active caspases, the interaction between Beclin and proteins of family Bcl, expression activity of autophagic protein Atg, calpain-mediated cleavage of Atg, functional activity of cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein, and p53 mediated regulation. [59,60,61,62]. Autophagy helps in degrading misfolded and unfolded protein structures, but only up to a certain threshold beyond which it may cause cell death either directly or via regulation of apoptosis through common regulators. Several autophagic proteins were regulating apoptotic cascade through direct involvement with apoptotic machinery without activation of the entire autophagic process. Numerous studies demonstrated that genetic manipulation in the autophagic pathway regulates the activation of the Fas-dependent death-inducing signaling complex, which activates pro-apoptotic genes and initiates apoptotic pathways [63]. Moreover, ER stress induced by tunicamycin and thapsigargin regulates caspase 8 ubiquitination, which forms a complex containing caspase 8, Atg5, FADD, and translocation autophagosomal membrane. Further, this complex in the absence of caspase 9, Bax, and Bak leads to the activation of caspase 8 dependent apoptotic cell death. Moreover, knockdown of Atg5 and Atg7 resulted in the deficiency of caspase 8 dependent apoptosis [64, 65]. Different studies performed on the regulatory steps of autophagy concluded that inhibition of late steps of autophagy induced caspase 8 activation, which leads to induction of apoptosis rather than knockdown of Atg5 and Atg7 at early stages. Thus, activation of apoptosis due to early inhibition of autophagy contradicted findings of the experiments performed by Amir et al., 2013, which stated that inhibition of Atg7 leads to caspase-dependent apoptotic cell death [66, 67]. However, the molecular mechanism and factor that trigger autophagosomes to initiate caspase activation and the apoptotic pathway are still poorly understood. Moreover, autophagy is also capable of apoptosis induction by inhibiting the conserved family of cytosolic protein known as IAPs by activating caspases [68]. During stress conditions, Atg5 and Atg12 have been evolved as an important regulator of an apoptotic pathway independent of their specific functions in autophagy machinery, which is cleaved by calpains leading to translocation of its N-terminal fragment in mitochondria where it mediated the release of cytochrome c through pro-survival factors such as BCL and BCLXL. Further, mitophagy is the molecular phenomenon through which autophagy reduces the tendency of the cell to undergo an apoptotic pathway. Mitochondria, as an initiator of apoptosis, release pro-apoptotic factors, namely cytochrome c and SMAC, which cause the failure of mitochondrial bioenergetics due to the rupture of the mitochondrial membrane. Thus, removal of damaged mitochondria by the autophagic phenomenon can increase the threshold for apoptosis induction [69,70,71,72]. Altogether, autophagy is not only capable of attenuating apoptosis through damaged mitochondria but also the expression of caspases. Hou et al., demonstrated that autophagy inhibition mediated by Beclin-1 and Vps34 knockdown causes an increase in catalytic processing of caspase 8 prodomain, the release of cytochrome c, and generation of Annexin V-positive cells’ subpopulation in TRAIL-induced Bax-/-Hct cells and cisplatin-treated caspase 8 deficient mice cells [73]. Autophagy is considered a molecular phenomenon through which cells can evade apoptosis, but the molecular mechanism of such a process is poorly understood. However, different studies demonstrated the synergic effect of autophagy inhibitors and other drugs in estimating the relationship between autophagy and apoptosis. Fitzwalter et al. observed that autophagy regulating FOXO3a due to basal autophagy leads to a potential feedback loop, which on autophagy inhibition increases the expression of pro-apoptotic factors such as Bcl-2 Binding Component 3 (BBC3/PUMA), which sensitize apoptotic pathway [74]. Another study demonstrated that infracted high mobility group box 1 (HMGB1) upregulated autophagy by increasing the expression of proteins, including LC3, Beclin-1, and Atg7, along with the decrease in Bax, Bcl-2, Caspase 3, and mTOR expression activity [75]. Altogether it may be concluded that autophagy and apoptosis are two interconnected molecular phenomena in response to cellular stress. However, the mechanism is still not yet understood. The cytoprotective function of autophagy involves negative regulation of apoptosis and vice-versa. p53 is another important regulator of autophagy and apoptosis, which inhibits mTOR activity followed by downstream targets, regulates cell cycle progression and apoptosis pathway. This study observed that knockdown of p53 or autophagy inducers mediates the proteasomal degradation of p53 through the HDM3/E3 ubiquitin ligase system [76, 77].
Molecular phenomenon between apoptosis and autophagy
Involvement of p53 pathway
Tumor suppressor, TP53 gene encodes p53 protein from three transcription factor (TF) subunits such as p53, p63, and p73, which have a central role in transcriptional regulation involved in the pathogenesis of NDDs. P53, a gatekeeper of the cell, is activated by different post-translational modifications, namely acetylation, methylation, and ubiquitination. Further, it is known that p53 responds to a number of cell toxicity conditions, such as genotoxicity, oxidative stress, and metabolic stress [78,79,80,81]. p53 is a well-known regulator of autophagy and apoptotic cell death pathways during the DNA damage response and cell cycle arrest [82, 83]. Moreover, p53 also promotes the activation of both extrinsic and intrinsic apoptotic pathways. In the extrinsic pathway, nuclear p53 accelerates the expression of the APO-1/Fas receptor and the TRAIL receptor, whereas cytoplasmic p53 increases the caspase 3 and caspase 8 activities. In the intrinsic pathway, nuclear p53 is known to upregulate pro-apoptotic factors such as PIDD, BH3 only protein, p53 upregulated modulator of apoptosis (PUMA), Phorbol-12-myristate-13-acetate-induced protein 1 (NOXA), Bax, and BID leads to caspase 9 and caspase 8 activation. Likewise, cytoplasmic p53 translocates towards mitochondria, promoting the activity of Bax and Bak proteins after forming a complex with Bcl-2/Bcl-XL and activation of crucial apoptosome protein APAF1 [84,85,86,87]. Kim et al. demonstrated that depletion of intracellular zinc in N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine (TPEN) induced mouse cortical neuronal cells regulate the apoptosis pathway by p53-induced protein synthesis, where poly(ADP-ribose) polymerase (PARP)-1 acts as an upstream effector of p53 induced neuronal apoptosis [88, 89].
Different studies have also demonstrated the effect of p53 on the autophagic cell death pathway through inhibition of the mTOR complex 1 by transcriptional activation of sestrin proteins and AMPK. Further, p53 induces the expression of damaged-regulated-autophagy-modulator (DRAM) through an unknown molecular mechanism that helps in regulating the expression of crucial autophagic genes such as LKB1 and ULK1/2 along with autophagosome maturation genes such as Atg4, Atg7, and Atg10 [90,91,92]. Moreover, p53 promotes the TFEB/TF binding to IGHM enhancer 3 (TFEB/TFE3) nuclear translocation during the DNA damage response through an increase in TF forkhead box O3a (FOXO3) expression and activity, which regulates upstream effectors of the autophagy pathway [93, 94]. However, further studies need to be done to understand the mechanism of p53 in autophagy. p53 mediated increase in autophagic cell death may be implemented in several neuronal cell death, but the precise mechanism should be defined before any concluding remarks. Lee et al. demonstrated the interrelation between apoptosis and autophagy in mouse embryo fibroblasts, where the deficiency of Atg7 leads to induce p53 dependent apoptosis. Moreover, Robin et al. demonstrated that the absence of p53 in Drosophila results in autophagic flux impairment, caspase activation, and mortality under oxidative stress [95] (Fig. 2A).

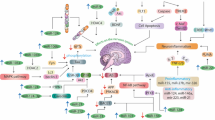
A Molecular mechanism of P53 involvement in the autophagic and apoptosis pathway. In the presence of oxidative stress and DNA damage response activation of MAPK. In the presence of oxidative stress and DNA damage response activation of MAPK, NF-KB, ATR, and ATM genes were carried out, which ultimately leads to the activation of p53. p53 activates m-TOR, HSF1, Sestrin ½, Bax, and NOXA, leading to the activation of different signaling cascades that regulate autophagic and apoptotic cell death pathways. Beclin-1 leads to the activation of PIP3 and Caspase 3, which activates autophagy and apoptosis, respectively. B VEGF is another essential protein that connects autophagy and apoptosis through different signaling molecules and cascades. VEGF leads to activation of VEGFR and VEGFR2, followed by activation of downstream targets such as PI3K/Akt and MAPKT of autophagic pathway and eNOS, BAD, and Bcl-2 of the apoptotic pathway. VEGF also acts on AMPK, HIF-1, Caspase 3, and Integrins, which further regulates downstream targets of signaling cascade such as TSC1, TSC2, PI3P, ATGG, mTOR, and Beclin that leads to the regulation of an autophagic pathway. Similarly, VEGF's interaction with PI3K/Akt, Ras, and EGFR activates pro-apoptotic factors that in turn activate signaling molecules like HSP90, cytochrome c, ASK1, MDM2, and Raf, leading to the initiation of apoptotic death pathway
Angiogenic pathway: role of VEGF
VEGF is involved in biological processes, such as cell proliferation, cell migration, and tube formation, which can induce diseases such as NDDs, cancer, arthritis, and diabetes [96]. Recent studies demonstrated the antiproliferative, apoptotic, and autophagic effects of anti-angiogenic drugs targeting VEGF, which induces cellular and molecular responses during stress conditions. For instance, Liu et al. showed that apatinib, a highly selective inhibitor of vascular endothelial growth factor receptor 2 (VEGFR2) tyrosine kinase that is involved in the alteration of cell cycle arrest, apoptosis, and autophagy. Further, inhibition of autophagy increases apoptotic effect through direct binding between VEGFR2 and signal transducer and activator of transcription 3 (STAT3). Inhibition of VEGFR2 mediated by siRNA resulted in the downregulation of STAT3 and Bcl-2 reinforced autophagy and apoptosis induced by apatinib [97]. Further, Endostatin activates autophagy through decreased Bcl-2 expression and increased Beclin-1 expression in Eahy926 human endothelial cells [98]. Yang et al. 2014 demonstrated the inducing effect of convallatoxin on autophagy and apoptosis through increased cleavage of caspase 3 and PARP along with LC3 conversion. Moreover, convallatoxin inhibits the mTOR/p70S6K signaling pathway, resulting in autophagic induction and exerting anti-angiogenic activity in-vitro and in-vivo [99].
VEGF-B is the neuroprotectant lacking general angiogenic activity that rescues neurons from apoptosis in rat and mouse cell lines. VEGF-B inhibits the expression activity of BH3 proteins along with p53, a member of the caspase family mediated through activation of VEGFR1, thus hampering retinal neovascularization [100]. Similarly, Falk et al. 2009 demonstrated the neuroprotective implication of VEGF-B in the culture model of PD where expression of VEGF-B was upregulated while the activity of VEGF-A remains unaltered [101].
Moreover, the lentiviral-mediated expression of VEGF165 was found to be neuroprotective in both SHSY-5Y and rat primary striatal cultures, which attenuated DARPP-32+ mediated neuronal loss and rescued Exp-Htt aggregation [102]. Religa et al. 2013 studied the effect of VEGF on β-amyloid (Aβ) induced endothelial cells in-vitro. VEGF significantly prevents neuronal apoptosis and restored memory deficit in the transgenic AD mice model [103]. Further, the administration of batroxobin would exhibit neuroprotective effects in the spinal cord injury model mediated through neurotrophic factors and increased expression of VEGF, which reduces apoptosis [104]. Administration of VEGFR2 inhibitor PTK787/ZK222584 on primary cerebellar granule neurons prevented 1-methyl-4-phenylpyridinium ion (MPP+) induced neurotoxicity followed by neuronal apoptosis. Inhibition of VEGFR2 activates PI3K/Akt and ERK pathways, which play the opposite role in MPP+-induced neuronal apoptosis [105]. Studies in the past demonstrated the plausible function and mechanism of VEGF-B in neurodegeneration, altering mitochondrial dysfunction and neuronal cell apoptosis while lacking traditional angiogenic activity, especially in the PD model. VEGF also acts as a therapeutic target in NDDs and can be an interesting topic for crosstalk between oxidative stress and mitochondrial biogenesis [106] (Fig. 2B).
Molecular markers of neuronal cell death
Mammalian target of rapamycin
mTOR is the key signaling mechanism of cell growth and is considered as the master regulator of autophagy, protein synthesis [107], and mRNA translation [108], transcriptional regulation, and phosphorylation of other protein substrates. Inhibition of mTOR with rapamycin acts as an initiator for autophagy induction as mTOR activity inhibits autophagosome formation, which is crucial for the induction of autophagy signaling cascade. Alteration in autophagy cascade, possibly due to mTOR implication, has been observed in different neurological defects [109,110,111]. Further, the mTOR signaling cascade has been linked with the establishment of neuronal plasticity, shape, spine morphology, and axonal development. In an in-vitro study, it was demonstrated that activation of the mTOR signaling pathway induces the growth and branching of dendritic cells along with the reduction of dendritic complexity through mTOR or S6K1 knockdown. Further, in rat hippocampal neurons, it was observed that activation of both mTOR1 and mTOR2 signaling is required for neuronal development and organization along with the change in expression activity of Calcium/calmodulin (Ca2+/CaM) dependent protein kinase II [112,113,114]. Similarly, the mTOR pathway regulates axon outgrowth, as shown in mouse dorsal root ganglia neurons (DRGNs). Further, deletion of TSC2 and association of the mTOR with tuberin and GTP-binding protein Ras homolog enriched in the brain (RHEB) was found to promote axon outgrowth both in the in-vivo and in-vitro mouse model [115, 116]. Likewise, the mTOR signaling cascade modulates excitatory and inhibitory neurotransmission regulating synaptic plasticity as observed in the phosphatase and tensin homolog protein model of the knockout mouse. The mTOR pathway increases synaptic vesicles, synapse response, and the number of synapses both in glutamatergic and GABAergic neurons [117]. Likewise, the mTOR antagonist rapamycin treatment results in hippocampal neurons demonstrated long-term reduced potentiation promoted by high-frequency stimulations, together with inhibition of synaptic potentiation promoted by brain-derived neurotrophic factors (BDNF) [118]. Moreover, rapamycin prevented 3,5-dihydroxyphenylalanine induced metabotropic glutamate receptor (mGluR) mediated long-term potentiation through Akt and mTOR phosphorylation in CA1 hippocampal neurons [119]. Abundant evidence suggests the possible role of mTOR inhibition in the anti-aging effect through cellular senescence relevant to NDDs such as AD, PD, ALS, and HD [120]. In the 3XTg AD and S6K1 knockout mouse model, inhibition of the mTOR downstream signaling pathway resulted in decreased cognitive defects by reducing Aβ and Tau pathology [121]. In-vitro and in-vivo models have demonstrated rapamycin-mediated neuroprotection from synaptic toxicity, tau-induced neuronal cell death, and astrogliosis [122]. Altogether, rapamycin antagonist temsirolimus prevents tau-induced toxicity and the formation of neurofibrillary tangles via enhanced autophagy [123]. Several studies have demonstrated the effect of the increased number of autophagosomes in α-synuclein-induced dopaminergic cell death, suggesting a pivotal role of autophagy pathway induction in the PD model while inhibition of mTOR with rapamycin causes an increase in autophagy, which inhibits the accumulation of ubiquitinated α-synuclein [124, 125] (Fig. 3A).

A mTOR is an anti-autophagic molecule that acts on the ULK1 complex and P70S6K leads to activation of downstream signaling molecules to alter autophagy and apoptosis pathway. mTOR inhibits ULK complex followed by deactivation of autophagy, which inhibits mHTT and alpha-synuclein clearance and increases memory impairment, and cognitive decline. mTOR also increases SOD aggregate and ALS and decreases expression of P70S6K, which increases Aβ aggregation followed by Aβ toxicity, which causes memory impairment and ultimately leads to AD. B Neurotoxins cause oxidative stress, which activates AMPK, decreases phosphorylation of AMPK, activates p53, and increases mitochondrial dysfunction. ER stress increases the calcium influx, which activates calpain, caspase 4, and caspase 12 results in increased neuronal apoptosis. Mitochondrial dysfunction activates cytochrome-c followed by caspase 9 and caspase 3 activation, which increases Tau phosphorylation, causes synaptic loss and cognitive decline, ultimately leading to neuronal apoptosis. Activation of AMPK decreases neuronal autophagy, followed by alpha-synuclein degradation, and leads to neuroprotection. Similarly, deactivation of phosphorylated AMPK decreases P-CREB, which causes the release of inflammatory cytokines followed by activation of inflammation signaling cascade, and leads to neuronal apoptosis
Involvement of Bcl-2 and BH3 family members
With the limitations of the apoptotic pathway in post-mitotic neuronal differentiation and maturation, Bcl-2 member was highly expressed in different forms with proliferating NPCs in the developing brain. However, the differentiated form in post-mitotic neurons, as demonstrated by restricted expression of Bak in post-mitotic neuronal differentiation, depends on Bax to promote neural apoptosis where the genetic knockout of Bax provides neuronal protection in multiple disorders [126,127,128,129,130,131,132,133]. Interestingly, N-Bak, an alternative splicing form of Bak characterized by additional exon and generation of BH3 only proteins due to translation, is expressed in neurons that further interact with anti-apoptotic protein Bcl-XL rather than Bax and induce apoptosis through Bax dependent pathway. Further, apart from neurotoxic function, N-Bak has neuroprotective abilities, as demonstrated in different studies [134,135,136]. For instance, Ginsenoside Re and Alcohol Dehydrogenase 1B suppresses Aβ induced neurotoxicity in SHSY-5Y cell culture and AD mouse model, respectively, through increased Bcl-2/Bax ratio, caspase inactivation, and reduced cytochrome-c release [137, 138]. He et al. demonstrated the potential implication of HECT, UBA, and WWE domain-containing 1 (Huwe1), an E3 ubiquitin ligase, in neuronal apoptosis. It was observed that induction of JNK inhibitor (SP600125) or a p38 mitogen-activated protein kinase (MAPK) inhibitor (SB203508) in pretreated Huwe1 increases caspase 3 cleavage, Bax and Bak expression, and p53 activity involved in the progression of neuronal apoptosis [139].
Moreover, myeloid leukemia cell differentiation protein (Mcl-1), an anti-apoptotic member of the Bcl-2 family, is highly expressed throughout the developing cortex regulating apoptotic pathways in differentiating and post-mitotic neuronal cells. A study concluded that deletion of Mcl-1 results in the induction of apoptosis, where GCN precursor does not depend on Mcl-1 for apoptosis [140, 141]. As compared to Mcl-1, the expression pattern of Bcl-XL is different, which is expressed at a low level in the developing brain and at a high level in post-mitotic differentiating neuronal cells where the genetic knockout of Bcl-XL is not able to induce apoptosis in the developing brain but induces cell death in post-mitotic differentiating cells [142]. Lauren et al., demonstrated the anti-apoptotic function of Mcl-1 and Bcl-XL in mouse embryonic CNS during different stages of neurogenesis promoting cell survival. The authors concluded that the sequential deletion of MCL-1 and BCL-x promotes cell survival during neurogenesis at embryonic day 10 in proliferating NPC and at day 11 within the post-mitotic cell population. The same study observed that in the double knockout mouse model, caspase-dependent apoptosis was initiated in non-proliferating and proliferating cell populations [143]. Bcl-2, another member of the Bcl family, is also widely expressed in developing and mature brain, but unlike Mcl-1, loss of Bcl-2 does not induce apoptosis but the result in progressive degeneration of the peripheral and facial neurons due to excessive accumulation of ROS involved in the regulation of oxidative stress pathways [144, 145]. Moreover, anti-apoptotic Bcl-w, whose expression is restricted during embryonic development but highly increased in post-mitotic differentiating neurons, regulates cell death signaling cascade. However, the deletion of Bcl-w neither induces neuronal apoptosis nor sensitizes hippocampal neurons; rather, Bcl-w plays a neuroprotective function in axons of sensory neurons during axonal degeneration [146,147,148,149,150].
BH3 is a pro-apoptotic protein highly expressed in the embryonic brain. At the same time, the expression reduces in the postnatal brain. However, BH3-interacting domain-containing protein 3 (Hrk/DP5), a neuronal-specific BH3 protein, is significantly expressed in the postnatal brain rather than the embryonic brain [151,152,153,154]. Different experimental studies demonstrated that consistent deletion or inhibition of BH3 proteins hampers neuronal apoptosis. Administration of arsenite causes deletion of PUMA, which causes an upregulated activity of BH3 only protein and leads to neuroprotection [155,156,157,158,159,160,161,162]. Post-translational modifications such as cleavage of Bid and dephosphorylation of Bad along with modifications in Bim, PUMA, NOXA, Bmf, and Hrk/DP5 activated BH3 only proteins transcriptionally induced by apoptotic stimuli. Interestingly, several apoptotic stimuli regulate TFs that activate BH3 only proteins such as Bim, PUMA, Hrk/DP5, and Bmf were transcriptionally activated by nerve growth factor (NGF) deprivation. Further, activation of activator protein 1 and TF c-Jun by phosphorylation result in Bim, PUMA, and Hrk/DP5 induction in response to neurotoxic elements [157, 160, 163,164,165,166,167,168,169,170]. Moreover, after the DNA damage response, the P53 signaling pathway stimulates PUMA and NOXA in response to seizures 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP+) induced neurotoxicity, NGF withdrawal, and Aβ aggregation in the mature brain and neuronal cells [155, 171,172,173,174]. Activation of FoxO1 and FoxO3a downstream targets such as AMPK, tribbles pseudokinase 3, macrophage stimulating 1, and cyclin-dependent kinase 5 (Cdk5/p35) mediate Bim induction in response to external stimuli such as NGF withdrawal, oxidative stress, and Aβ aggregation through nuclear translocation of FoxO TF either by Akt or 14–3-3 mediated inhibition or sequestering of FoxO TFs [175,176,177,178,179,180,181].
Moreover, ER stress induces PUMA and Bim activation through transactivating their promoters through the interaction between CHOP, Cdk4, and FoxO3a TFs in neuronal cells, which upregulates the B-Myb required for Bim activation and neuronal death [182,183,184,185,186]. In healthy neurons, survival pathways, including PI3K/Akt and MEK/ERK, represses the expression of BH3 only proteins through inhibition of FoxO3 or inhibition of Akt and ERK itself, which is involved in the induction of Bim activity via both transcriptional and post-transcriptional mechanism [178, 187, 188]. Further, MEK/ERK survival pathway promoted the proteasomal degradation of Bim via interaction with ubiquitin ligase tripartite motif-containing 2 through phosphorylation on ser65 by ERK1/2 followed by polyubiquitination and proteasomal degradation, which was found to be neuroprotective under stress conditions [189, 190]. ERK5 induces phosphorylation of Bad through CAMP-response element-binding protein (CREB) on ser112, ser136, ser155, and ser170 regulates Bad expression and pro-apoptotic functions in the mature and adult brain. Similarly, phosphorylation of ser112 by MEK/ERK/RSK pathway and on ser136 by Akt dissociates its interaction with Bcl-XL and increases its interaction with 14–3-3 regulatory protein to promote neuronal survival [188, 191,192,193].
AMPK and caspases
Being an essential regulator of neurodevelopment and neuroprotective activities, the mechanism of caspases in neuronal cell death is still not well defined [194]. Although, decreased expression of Caspase 3, an effector caspase, was observed in neuronal cell death caused by neuronal injury in the ischemic brain model. Further, neurodevelopment activity was observed in adults as compared to the neonate rodent model. However, mature neurons reflect both apoptotic and non-apoptotic pathways, but the maturation of neurons is also associated with decreased activity of the caspase family gene. Moreover, the activation of caspase 3 through the copper-induced ROS generation causes increased activity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression leading to neuronal cell death in the P19 cell culture model [195]. Thus, caspase inhibition has the potential to minimize cell death caused by ER stress, oxidative stress, and calcium withdrawal in NDDs both in-vivo and in-vitro conditions through a decrease in expressions of upstream and downstream targets, such as PERK, heat shock 70 kDa protein 5, CHOP, PARP, HIV-1 TAR RNA binding protein (TRBP), PKC, TNFα and protein activator [196, 197]. A study found that downregulation of apoptotic protease activating factor 1 decreases the activation of effector caspases, possibly through apoptosome, leading to impaired neuronal development and reduced synaptic plasticity [198]. Likewise, in PD murine model, the NLR family pyrin domain containing 3 (NLRP3) antagonist kaempferol promoted neuroprotection through decreased expression of caspase 1 along with disruption in NLRP3-PYD and CARD domain-containing (PYCARD)-caspase 1 complex assembly [199]. Further, inhibition of caspase 1 via caspase 6 resulted in downregulating the proteolytic cleavage at D586 of mutant Htt, axon degeneration, and pathological lesions [200, 201] (Fig. 3B).
In different experimental studies, it was demonstrated that inhibition of caspase 1 and caspase 3 signaling pathway in microglia promotes neuroprotection through reduced neuroinflammation in microglia, reduced impaired cognition and regulation of neuronal cell apoptosis, possibly through a decrease in beta-secretase 1 expression and macrophage stimulating 1/JNK signaling cascade [202,203,204,205,206,207]. In the case–control study, two caspases 8 variants, that is p.K148R, and p.I298V are involved in neuronal cell loss, which on interaction with caspase 3 involved in synaptic plasticity, microglia inflammation, and memory impairment [208]. Extracellular adenosine increases the expression level of caspase 9, followed by caspase 3 through activation of two independent pathways. A1 adenosine receptor-mediated adenylate cyclase inhibition and adenosine uptake into cells/conversion to AMP/activation of AMPK are two independent pathways, which leads to astrocytoma cell death through the apoptotic pathway [209]. Moreover, Song et al., demonstrated the crosstalk between autophagy and apoptosis through AMPK and activated caspase. In this study, inhibition of the mTOR and the proteasome with rapamycin and Bortezomib respectively activates AMPK, which phosphorylate downstream target Beclin-1 resulted in autophagic cell death followed by its cleavage through activated caspase resulted in apoptotic cell death through mitochondrial dysfunction [210, 211].
Further, neurotoxins such as 6-hydroxydopamine, oxygen–glucose deprivation, and MPP+ increase oxidative stress, followed by an increase in autophagy and apoptosis. Inhibition of AMPK phosphorylation and the activation of mTOR phosphorylation with antioxidants, such as propofol and alpha-lipoic acid, downregulates autophagic and apoptotic cell death, which causes an increase in synaptic plasticity, cognitive ability, and neuroprotection [212,213,214,215]. Similarly, Meares et al., 2013 observed that in-vitro AMPK expression inhibits gene expression of C–C Motif chemokine ligand 2, TNFα, C-X-C motif chemokine 10 and inducible nitric oxide synthase (iNOS), mediated by IFN-γ through signal transducer and activator of transcription 1 [216]. Further, intraperitoneal treatment of lipopolysaccharide treated with 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) in the disturbed neuronal mouse model demonstrated a reduction in TNFα-mRNA expression level along with increased mRNA expression level of peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC-1α). The same study observed that after 24 h of lipopolysaccharide injection treatment with AICAR decreases glial fibrillary acidic protein (GFAP) activity. However, different studies demonstrated the detrimental effect of AMPK activation as treatment with AICAR increase apoptosis in SHSY-5Y and Neuro 2a cell culture models mediated through an increase in caspase 3 activity [217]. Likewise, it was found that Aβ induced neurotoxicity in human neural stem cells decreases cell viability by decreasing AMPK activation and expression of neuroprotective genes such as Bcl-2 and CREB. The same study also concluded that Aβ neurotoxicity causes an increase in caspase 3, caspase 9, and cytochrome c expression [218, 219]. Altogether, it may conclude that AMPK activation promotes apoptosis mediated through increased expression of pro-apoptotic genes such as caspases and cytochrome c.
PI3K/Akt/GSK3β pathway
GSK3 is ubiquitously expressed in the nervous system and involved in regulating neuronal plasticity, and neurological disorders with GSK3β remain the dominant form compared to GSK3α. Inhibition of GSK3β through Akt-dependent phosphorylation, PI3K activation, and PKC activation implicated in glutamate-induced N-methyl-D-aspartate receptor (NMDAR) dependent neuronal plasticity and facilitates the surface transport of potassium voltage-gated channel subfamily Q member 2 subunits that are involved in the regulation of neuronal excitability [220,221,222,223]. It has been considered that the PI3K/Akt/GSK3β pathway is involved in Aβ induced neurotoxicity, which causes memory impairment and learning deficits. However, the mechanism behind this rationale is poorly defined. Further, Akt-dependent inhibition of GSK3β found to reverse learning and memory deficits [224, 225]. It has been observed that GSK3β activity causes hyperphosphorylation of tau protein and accumulation of amyloid precursor protein, which leads to detachment of tau from microtubule and decreases amyloidogenic processing, respectively, resulting in neurite degeneration. Further, GSK3β has the potential to bind with NMDAR receptors, and modulating their function leads to the accumulation of Ca2+ ions causes degeneration of neurons, ultimately leading to neuronal cell death [226, 227]. Moreover, the active form of GSK3 was enhanced in patients suffering from PD, which is localized with the halo form of α-Synuclein, leading to memory impairment and neural degeneration [228]. Further, inhibition of GSK3 activity decreases the aggregation and phosphorylation of α-Synuclein and increases autophagic flux, while activation of GSK3β leads to impaired autophagy [229]. GSK3β has been found to regulate apoptosis through phosphorylating the downstream targets such as p53, Bax, p21, and initiate caspase cascade, which is regulated by many signaling events involved in the modification of mitochondrial activity [230, 231]. A study demonstrated that overexpression of inactive GSK3 mutant prevents apoptosis, which was later confirmed by studies using specific GSK3 inhibitors. Altogether, reduction in GSK3β serine 9 phosphorylation causes increased cytochrome c release and caspase 3 activity and direct involvement in cell death induced by PI3K/mTOR inhibitor and histone deacetylase inhibitor such as Trichostatin A in different cell lines [232,233,234]. Mcl-2, another Bcl-2 family member, stabilizes mitochondrial outer membrane permeabilization through Bim and Bid, followed by phosphorylation activity at serine 159 recognized for ubiquitination and degradation. [235, 236]. In neuronal cells, GSK3β dependent phosphorylation of Bcl-2 family member Bax at serine 163 induces its mitochondrial translocation exerting pro-apoptotic function [237, 238]. Mitochondria being the major producer and center of oxidative stress, undergo mitochondrial permeability transition resulting in apoptotic cell death due to GSK3 activation, which causes hyperphosphorylation of different downstream targets, namely oxidative damage associated cellular defense protein nuclear factor erythroid 2-related factor 2 (Nrf2) [239,240,241,242] (Fig. 4A).

A PI3K/Akt is a molecular marker that activates apoptosis and autophagy, which regulates neurodegenerative disorders. PI3K/Akt activates GSK3β, which acts on downstream signaling molecules involved in neurodegenerative diseases. TSC1 and TSC2 activate mTOR, decreasing neuronal autophagy, followed by an increase in neuronal toxicity, while activated GSK3β decreases NRF2 expression and activates neuroinflammation signaling cascade. Activation of P-CRMP2 and NMDAR mediated through GSK3β increases caspase 3 activations, and the calcium influx respectively lead to an increase in neuronal apoptosis, ultimately increases memory impairment and neuronal cell death. B Rotenone and MPTP activate P38 MAPK, which leads to activation of downstream signaling molecules such as JNK, ROS, and iNOS, followed by activation of the signaling mechanism of neurodegenerative disorders. Activation of JNK activates BIM, increases the release of inflammatory cytokines, and decreases expression of GSK3β, which further activates Cytochrome-C, inflammation signaling cascade, and neuronal toxicity, respectively, ultimately leads to neurodegeneration. P38 MAPK increases ROS causes oxidative stress leads to activation of caspase 1 and caspase 2, which increases neuronal apoptosis followed by memory impairment and cognitive decline involved in the pathogenesis of neurodegenerative disorders. Similarly, activation of iNOS releases NO causes mitochondrial dysfunction, which increases neuronal toxicity leads to neuronal cell death followed by neurodegeneration
Moreover, the implication of GSK3 has been extensively studied in manipulating autophagy from the last few years. GSK3β inhibits autophagy by activating mTOR complex 1 through phosphorylation of mTOR associated scaffold protein raptor on serine 859. Inhibition of GSK3β activity inhibits mTOR complex 1 and raptor interaction and reduced phosphorylation of ULK1, followed by increased autophagic flux [243, 244]. Similarly, inhibition of GSK3β leads to an increase in AMP/ATP cause AMPK activation followed by autophagic induction through sequential phosphorylation of tuberin by AMPK and GSK3β, which causes mTOR inhibition [245,246,247]. Apart from its inhibition GSK3β in the absence of growth factors, activates acetyltransferase KAT/TIP60, followed by activation of the ULK1 complex to induce autophagy [248]. Inhibiting GSK3β expression through enhancing mTOR activity through overexpression of Aurora A kinase induces resistance to autophagic cell death while activation of GSK3β signal transduction pathway mediated by cadmium promotes autophagic cell death in ROS elevated conditions [249,250,251]. Further, pharmacological and genetic knockdown of GSK3β expression and Akt activation significantly alleviate autophagic cell death in a neuronal cell, while GSK3β mediated phosphorylation of MCL1 has been observed to induce axonal autophagy and axonal degeneration [252,253,254]. Inhibiting the activity of calpain, Akt, and GSK3β reduces the autophagosome number and increases microtubule stability in paeoniflorin-treated okadaic acid-induced tau hyperphosphorylated SH-SY5Y cell model [255]. Also, the Wnt3a ligand promotes AMPK activation, followed by GSK3β inhibition modulating the autophagic phenomenon in hippocampal neurons [256]. These data suggested that GSK3 has potential relevance in autophagic and apoptotic cell death and maybe a potential therapeutic target in NDDs.
p38 and JNK MAPK pathway
MAPK, due to its tremendous application in different cellular functions such as apoptosis, cell survival and proliferation, cell differentiation, inflammatory activities, and external ROS, has been considered as a potential therapeutic target against NDDs. p38 MAPK inhibitors have been considered as potential therapeutic agents against chronic inflammatory diseases, including AD, PD, ALS, and HD. MAPK causes phosphorylation of its downstream targets, including P38, c-Jun, and JNK signaling, which is linked with neuronal apoptosis, where c-Jun activation is required for NGF withdrawal-induced apoptosis. In contrast, inhibition of c-Jun activity protects neuronal cell death.
Moreover, MAP3K-ASK1 has been associated with JNK’s activation and promotes neuronal apoptosis in PC12 cells. However, different studies concluded that standalone JNK signaling was associated with reducing apoptotic cell death [257,258,259,260,261,262]. A series of experiments demonstrated the functional effect of MAPK inhibitors on HMGB1-induced neuronal apoptosis [263]. A study demonstrated that activator protein 1 and c-Jun act as both anti and pro-apoptotic factors depending on the level of stress and suggesting the implication of defective mitophagy in MAPK/c-Jun-induced apoptosis [264]. Further, activation of the JNK and P38 MAPK pathway leads to activation of NF-κB-induced phosphorylation activity, which leads to proteasome degradation. On the contrary, inhibition of p38 MAPK leads to impaired proinflammatory NF-κB transcriptional activity without altering its DNA binding activity. It downregulates the expression of inducible NO synthase through acetylation activity of p65 rather than phosphorylation activity [265]. An in-vitro study performed by Papademetrio et al. demonstrated the autophagy inhibition and apoptosis induction in both caspase-dependent and caspase-independent patterns in MIA PaCa-2 and PANC-1 cells. Although, administration of caffeic acid phenethyl ester reverses autophagic degradation and apoptotic cell death by inhibiting MAPK and NF-κB pathways. [266]. Recently, several studies concluded the protective effect of inhibitors, namely doxycycline, steppogenin, neferine, alantolactone, and indirubin, against lipopolysaccharide-induced primary microglial cells through inhibition of MAPK phosphorylation and NF-κB nuclear translocation. Altogether inhibition of MAPK and NF-κB pathways through the action of inhibitors lowers the expression of microglial activation markers, including IBA1, reduced ROS, NOS, and activation of proinflammatory cytokines [267,268,269,270,271]. The MAPK-activated protein kinase 2 complexes are known to regulate the phenomenon of inflammation through the production and activation of inflammatory mediators. It has been observed that MAPK-activated protein kinase 2 knockout mice are resistant to endotoxic stress and involved in the regulation of TNFα, Interleukin 6, Interleukin 8, and other regulatory cytokines involved in the process of neuroinflammation [272,273,274] (Fig. 4B).
Pharmacological intervention targeting apoptotic and autophagic machinery
Implementation of microRNAs in the regulation of cell-death pathway
The microRNAs are a family of 23–25 nucleotide sequences involved in transcriptional regulation that can be used as potential biomarkers in various diseases, including NDDs. miRNAs modulate several biological processes, such as cell cycle progression, apoptosis, autophagy, and inflammation [275, 276]. Various studies demonstrated the role of miRNA in neuronal cell death, regulating apoptosis and autophagy. However, the functional mechanism of miRNAs in these processes must be elucidated. Table 1 lists the miRNA that regulates autophagy and apoptosis cascade in the pathogenesis and progression of NDDs. For instance, H. Jia et al. demonstrated the effect of the miR-499-5p hypoxic-ischemic encephalopathy rat model, where it was found that the administration of miRNA significantly reduced the expression of C-reactive protein followed by a reduction in neuronal apoptosis. Further, the study indicated that miR-499-5p increases spatial learning ability, spatial memory, and locomotor functions [277]. Similarly, miR-217/138-5p, miR-15a, and miR-129-5p regulate the expression of sirtuin 1, TNFα, IL-1β, BDNF, and SOX6 through oxidative stress, inflammatory pathway, and Akt/GSK3β signaling cascade, which resulted in decreased neuronal apoptosis in MPP+-induced SH-SY5Y cells, oxygen–glucose deprivation neurons of rats, and AD rat model, respectively [278,279,280]. Likewise, miR-93 regulates the expression activity of the TLR4/NF-κB signaling pathway through inhibition of TNFα, IL-6, IL-1β, and VEGF, along with the decrease in pro-apoptotic molecules expression [281]. Further, H. Ge et al., demonstrated the neuroprotective effect of miR-410 in 6-hydroxydopamine-induced SH-SY5Y and PC12 cellular PD model through inhibition PTEN/Akt/mTOR signaling cascade. At the same time, Wang et al. studied that miR-124 exerts neuroprotective effects in the MPTP-induced PD model through the hedgehog signaling pathway targeting endothelin 2. Both studies demonstrated that induction of miRNA causes a reduction in apoptosis, caspase 3 expressions, and ROS activity [282, 283]. Similarly, Chen et al. demonstrated that miR-98 reduces Aβ aggregation and improves oxidative stress and mitochondrial dysfunction through a notch signaling pathway targeting Hes‑related with YRPW motif protein 2 and decreases hippocampal neuronal apoptosis in the AD mice model [284]. Moreover, in the SH-SY5Y cell line, miR-764 protected the neuronal cell from hydrogen peroxide-induced neuronal apoptosis through regulating ninjurin-2 expression and motor neuron functions [285]. Likewise, miR-429 and miR-34a regulate neuronal damage by inhibiting apoptotic expression in mouse cortical neurons and MPP-induced SH-SY5Y cells, respectively [286, 287]. Moreover, miRNA was also found to regulate the ER stress-induced apoptotic pathway. miR-211 inhibits ER stress and upregulates H3K27 methylation of the CHOP promoter leads to cell survival [288]. miR-378 and miR-155 regulate caspase -3 activity resulted in decreased apoptotic expression, whereas, miR-106b attenuates apoptotic pathway targeting caspase 7 expressions [289,290,291].
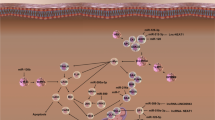
Further, miRNA also modulates the autophagic pathway by regulating different proteins and complexes involved in the signaling cascade. It was reported that miR-20a, miR-106b, miR-372, miR-26b, and miR-93 involved in the regulation of autophagy-mediated through ULK1 and ULK2 complex situated at the beginning of autophagic cascade [292,293,294]. Similarly, miR-338-5p, miR-30a, miR-376b, miR-216a, miR-630, miR-374a, and miR-17-5p suppress the autophagic pathway through negative regulation of class III PI3K complex [295,296,297,298,299,300] (Fig. 5).

microRNAs have been implemented to regulate autophagy and apoptosis signaling in the pathogenesis of neurological defects. Both overexpression and downregulation of different microRNAs known to regulate the expression of apoptotic and autophagic proteins by activating or inhibiting different signaling pathways that ultimately lead to the pathogenesis of NDDs
Moreover, miR-101, miR-376b, miR-17, and miR-495 modulate ATG4D, ATG4, ATG7, and ATG3 expression, which resulted in autophagy inhibition [298, 301,302,303]. Several studies indicated the potential of miRNA as therapeutic agents in neuronal autophagy. A study conducted by Wang et al. demonstrated that overexpression of miR-9a-5p reverses neurological deficits in MACO rat and SH-SY5Y cell lines through decreased autophagy and ATG5 expression [304]. miR-96 and miR-204 alleviate cognitive impairment by suppressing autophagic signaling cascade and exerts neuroprotective effects through decreased expression of LC3, Beclin-1, and mTOR [305, 306]. Likewise, in the MPTP induced SH-SY5Y and PC-12 PD model, miR-124, miR-185, and miR-181b rescue memory deficits and cognitive decline through AMPK/mTOR and PTEN/Akt/mTOR pathway. In addition, Gong et al., 2016 demonstrated that miR-124 suppression significantly increased cell apoptosis and LC3-II/LC3-I ratio, whereas, overexpression of miR-124 decreases the percentage of apoptotic cells and LC3-II/LC3-I ratio. Similarly, overexpression of miR-185 and miR-181b significantly downregulates the LC3-II/LC3-I ratio and apoptosis [307,308,309]. Moreover, miR-212-5p prevents dopaminergic cell death in the MPTP induced PD mouse model (C57BL/6 mice) through SIRT2 inhibition resulting in increased p53 acetylation and reduced autophagy [310]. Similarly, miR-124 in MPTP induced SH-SY5Y PD cell culture model regulates p62/p38, Bim, and Bax expression level resulted in increased autophagy and decreased neuroinflammation [311, 312]. Additionally, Zhao et al., demonstrated that miR-326 inhibits NOS expression and promotes autophagy degradation through the JNK signaling cascade. miR-326 interacts with X‐box binding protein 1, resulting in increased expression of LC3-II, c-jun, and p-α-synuclein [313]. Similarly, miR-27a and miR-23b in post-traumatic brain injury attenuates neuronal deficits and improves cognitive impairment and neurological functions through altered neuronal autophagy by FOXO3a and ATG12 regulation, respectively [314, 315].
Long non-coding RNAs as a pharmacological target
LncRNAs are a set of RNAs having more than 200 nucleotides that regulate gene expression, transcriptional activity, epigenetic modifications, and translational control. Different studies indicate the involvement of altered LncRNAs expression in the progression and pathogenesis of neurological defects such as AD, PD, ischemic stroke, HD, traumatic brain injury, spinal cord injury, and ALS through the regulation of cell death pathways, namely apoptosis and autophagy. Table 2 discusses the different potential LncRNAs, which regulate the expression of both apoptosis and autophagy. For instance, LncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), referred to as non-coding nuclear-enriched abundant transcript 2 (NEAT2), was found to be expressed in the in vitro model of ischemic stroke. Guo et al., 2017 demonstrated that down-regulation of MALAT1 suppresses neuronal apoptosis through downregulating Beclin-1 dependent autophagy degradation. In the same study, the authors concluded that the downregulation of autophagy is through regulation of the MALAT1-miR30a-Beclin-1 axis [359]. Similarly, Wu and Yi 2018 concluded that downregulation of MALAT1 reverses neurological defects by inhibiting excessive autophagy and apoptosis through regulating PI3K/Akt signaling pathway [360]. Further, LncRNA colorectal neoplasia differentially expressed (CRNDE) also regulates apoptosis and autophagy in different neurological defects. For instance, Chun-Hua et al. 2020 demonstrated that in hypoxic-ischemic (HI) brain damage (HIBD), silencing of CRNDE promotes autophagy and inhibits neuronal apoptosis both in-vivo and in-vitro conditions [361]. Likewise, downregulation of CRNDE in traumatic brain injury inhibits autophagy and apoptosis through regulation of GFAP, BrdU, NGF, and Nestin [362]. Wei-Lan et al. 2019 concluded that LncRNA small nucleolar RNA host gene 12 (SNHG12) inhibits miR-199a, which upregulated the activity of SIRT1 through activation of AMPK. Activation of AMPK leads to increased autophagy and decreases neuronal apoptosis [363]. Another member of the SNHG family known as SNHG14 was considered to be associated with the progression and pathogenesis of cerebral ischemia–reperfusion injury. Deng et al. 2020 in HT22 mouse hippocampal neuronal cells demonstrated that SNHG14 promotes neuronal injury through excessive mitophagy and neuronal apoptosis by regulating the miR-182-5p/BINP3 axis [338]. Likewise, Cao et al., 2020 concluded that LncRNA SNHG3 promotes autophagic degradation and neuronal cell apoptosis through increased activity of miR-485 and increased expression of ATG7 [364]. Thus, despite having several evidence, which concluded the potential role of LncRNAs in the regulation of apoptosis and autophagy, simultaneously in the pathogenesis and progression of neurological defects, there will be a need for in vivo studies (Fig. 6A).

A long non-coding RNAs have been implemented to regulate autophagic and apoptosis signaling cascade through modulation of different signaling cascades. For example, BACE1-AS, HAGLROS, MIAT, 17A, and MEG3 through miR-214-3p/ATG5 axis, PI3K/Akt/mTOR pathway, CUL4A-DDB1-REDD1 axis, GABAB signaling, and Beclin-1 signaling, respectively, increase autophagy and apoptosis simultaneously. Similarly, LncRNAs, such as HOTAIR, RMRP, and TCTN2 through miR-874-5p/ATG10 axis, PI3K/Akt/mTOR and miR-216b-Beclin-1 axis, respectively, lead to an increase in autophagy and decrease in the apoptosis pathway. R2Q1 B natural biomolecules act as a potential therapeutic agent in modulating autophagy and apoptosis pathway in the pathogenesis and progression of neurological disorders. For instance, Flavones and flavanols modulate mTOR, Akt, NF-κB signaling, and caspase 3, whereas phenolic acids and alkaloids modulate the expression of Atg3 and Beclin-1. Similarly, Flavanols, Flavanones, Isoflavones, Alkaloids, and Flavones regulate the activity of Bcl-2, whereas Lignines, Flavones, Flavanols, and Flavanones regulates the expression of caspase 9 and Atg3
Small-molecule inhibitors in autophagy and apoptosis pathways in NDDs
Recent studies implicated the potential of cell death pathways, including the autophagic pathway and apoptosis pathway, in the progression and pathogenesis of various diseases such as cancer, cardiovascular, and NDDs. These emerging discoveries led to expanding the pharmacological interventions targeting PCD pathways and provided the opportunities for development and prosecutions of known drugs or novel compounds as a therapeutic approach. Autophagy and apoptosis were commonly involved in NDD progression mediated through different signaling cascades and molecules. Oxidative stress, calcium imbalance, mitochondrial dysfunction, AMPK signaling, inflammatory response, and ER stress are commonly involved pathways in the autophagic degradation of accumulated toxic proteins and neuronal apoptosis due to aggregated misfolded proteins. Further, recent studies have shown that upregulation of autophagy through autophagy inducers causes a decrease in the accumulation of misfolded proteins and delays the progression of NDDs. Likewise, inhibition of pro-apoptotic proteins and activating anti-apoptotic proteins through synthetic or natural molecules delay the progression of NDDs. Thus, induction of autophagic degradation and inhibition of apoptosis signaling cascade can be used as a therapeutic strategy for NDDs. Table 3 discusses the drugs that undergo clinical trials for induction of autophagy in the pathogenesis of NDDs.
Another study indicated that Apelin-13 reverses amyloid-induced memory deficits by inhibiting apoptosis and autophagy, whereas administration of malathion in N2a neuroblastoma cells increases neuronal apoptosis and decreases autophagic flux through inducing lysosomal membrane permeabilization [375, 376]. Apart from NDDs, modulation of autophagy and apoptosis pathways could be protective in other neurological diseases, such as spinal cord injury, sleep deprivation, traumatic brain injury, ischemic stroke, and epilepsy. For instance, Modafinil protects hippocampal neurons by inhibiting autophagy and apoptosis pathway in the mice model, whereas metformin protects neuronal cells against spinal cord injury through inhibition of autophagy and apoptosis cascade by regulating mTOR/p70S6K signaling pathway [377, 378]. Similarly, Ganoderma lucidum extracts reverse MPTP-induced neurodegeneration by inhibiting excessive autophagy and apoptosis by regulating oxidative stress and mitochondrial function [379]. Further, recent studies concluded the potential of flavanols, flavonols, flavones, flavones, and flavanones as therapeutic agents in the treatment of NDDs through reversing the effects of dysregulated autophagic degradation and apoptosis. For instance, Singh et al., demonstrated that administration of fisetin, a natural flavonol compound in D-galactosidase aged rats decreased the activity of pro-oxidants and increased the activity of antioxidants. Further, fisetin causes a decrease in neuronal cell apoptosis and upregulates the expression of autophagic genes, such as Atg-3 and Beclin-1 [380]. Likewise, Yang et al., demonstrated that administration of fisetin improves synaptic dysfunction through the decrease in neuronal apoptosis and neuroinflammation by inducing autophagy and activation of AMPK [381]. Further, administration of rapamycin leads to increased autophagy and protects the neuronal cell from oxidative stress and apoptotic cell death [382]. Further, catechin can protect hippocampal neuronal cell apoptosis by inhibiting the JNK/MLCK pathway and microglial activation [383, 384]. Further, theaflavin decreases neuronal apoptosis by inhibiting the inflammatory response and ROS-induced oxidative stress [385, 386]. Naringenin, a dietary flavanone, reduces apoptotic cell death, inhibits oxidative stress, and improves mitochondrial function through Nrf2/ARE signaling pathway [387, 388], whereas, naringin inhibits neuronal apoptosis through inhibiting oxido-nitrosative stress and neuroinflammatory response [389]. Meng et al. 2021 in a mouse model of AD, demonstrated that naringin could improve cognitive function through decreased neuronal cell death by MAPK/p38 pathway [390]. Further, Guo et al. 2020 demonstrated that administration of genistein promotes neuroprotection against Aβ-induced neuronal cell death through PI3K/Akt/Nrf2 signaling pathway, whereas Jiang et al., 2017 concluded that genistein attenuates isoflurane-induced neurotoxicity and improves spatial learning and memory abilities through cAMP/CREB and BDNF/PI3K/Akt pathway [391, 392]. Similarly, equol, a dietary daidzein attenuates neuronal cell death and promotes neuroprotection through inhibiting microglial activation and cell cycle reentry [393, 394]. Moreover, apigenin also promotes neuroprotection through inhibition of neuroinflammatory response and oxidative stress-induced neuronal apoptosis [395, 396]. Kim et al., 2021 concluded that administration of apigenin repressed scopolamine-induced neuronal damage and reduced cognitive impairment. The authors also concluded that neuronal protection by apigenin is the result of enhanced BDNF activity, which decreases neuronal apoptosis and amyloidogenesis [397]. Similarly, luteolin promotes neuroprotection through reduced neuronal cell apoptosis by regulating SIRT3/AMPK/mTOR and p62/Keap1/Nrf2 signaling pathway [398, 399]. In addition, administration of luteolin and apigenin causes activation of autophagic degradation through HMOX1 and mTOR/AMPK/ULK1 complex, respectively, which promotes neuroprotection [400]. Peruru and Dodoala in 2021 concluded that diosmin, a citrus flavonoid, promotes neuroprotection by suppressing NOX4 and its subunits [401]. Moreover, apart from the above-mentioned polyphenol compounds, studies demonstrated the protective effects of lignins and phenolic acid against neuronal apoptosis and autophagic cell death in NDDs and other neurological diseases. For example, caffeic acid phenethyl ester, a phenolic compound, prevented neuronal cell apoptosis against Aβ1-42 through the modulation of GSK3β in the mice model of AD, whereas, gallic acid protects from 6-OHDA induced neurotoxicity and cell apoptosis through inhibition of oxidative stress [402, 403]. Similarly, geraniin protected neuronal cells from apoptosis in PC12 cell culture against Aβ25-35 toxicity through the modulation of the NF-κB pathway, whereas arctigenin protected PC12 cell culture against ethanol‑induced nerve damage [404, 405]. Furthermore, recent studies demonstrated the protective effects of natural alkaloids in preventing neuronal cell viability [406,407,408,409]. For instance, tricyclic pyridine, an alkaloid from Fusarium lateritium SSF2, prevents neuronal cell apoptosis against glutamate-induced oxidative stress in the HT22 hippocampal neuronal cell line by inhibiting caspase 9 and caspase 3 [410]. Similarly, dendrobium alkaloids enhanced neural function through reduced neuronal cell death by modulating the expression of inflammatory cytokines [411]. Thus, from the evidence mentioned above, it might be concluded that targeting apoptosis or autophagy pathways could be beneficial for reverses neurological defects. Table 4 lists the natural and synthetic biomolecules in the regulation of autophagy and apoptosis machinery (Fig. 6B).
Conclusion and future perspectives
This review displayed the intricacies between two major cell death pathways, viz. apoptosis and autophagy in NDDs, which provide a great avenue for therapeutics. These two pathways have several common mechanisms, such as initiator and effector molecules, genes and proteins, and signaling pathways that form a connection. With the development of research technologies and specific inhibitors, our understanding of cell death pathways is ready to be executed. Herein, we tried to elaborate the knowledge about molecular phenomena between the two death pathways involved in NDDs, for instance, interactions between targets and pathological mechanisms of molecular targets involved in cell death pathways and autophagy. However, many critical issues must be resolved while targeting cell death pathways concerning autophagy as a therapeutic approach in NDDs. Investigating molecular targets, regulatory mechanisms, and signaling cascade is a matter of extensive research to maximize the potential of cell death pathways. Moreover, regulation of PCD and NDDs through miRNAs is a new direction for research in this field, where miRNA may target more than one component of the cell death pathways or sometimes may target more than one death pathway. In this review, we also discussed the molecular mechanism of autophagy and apoptosis in NDD’s while focusing on the molecular markers, signaling cascades, and shared mechanisms such as ER stress and Ca2+ concentration. Both autophagy and apoptosis can regulate each other mediated by inhibition of activation of apoptosis-associated caspases. However, to maximize the potential of cell death pathways as a therapeutic approach, further in-vitro and in-vivo studies are required.
Availability of data and materials
Not applicable.
Abbreviations
- NDD’s:
-
Neurodegenerative diseases
- AD:
-
Alzheimer’s disease
- PD:
-
Parkinson’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- HD:
-
Huntington’s disease
- MS:
-
Multiple sclerosis
- UPR:
-
Unfolded protein response
- miRNA:
-
Micro RNAs
- ULK:
-
UN51-like Ser/Thr kinases
- Atg13:
-
Autophagy-related protein 13
- PI3K:
-
Phosphatidylinositol-3-kinase
- Vps34:
-
Vacuolar protein sorting 34
- VPS15:
-
P15
- Atg6:
-
Beclin-1
- Atg14:
-
Barkor
- LC3:
-
Light chain 3
- SNAREs:
-
Soluble NSF attachment protein receptor
- PCD:
-
Programmed cell death
- Bcl-2:
-
B-cell lymphoma 2
- TNF:
-
Tumor necrosis factor
- FADD:
-
Fas-associated death domain protein
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- BH3:
-
Bcl-2 homology region 3
- Smac/DIABLO:
-
Second Mitochondria-derived Activator of Caspases/ Direct IAP-Binding protein with Low PI
- AIFs:
-
Apoptosis-inducing factors
- BAX:
-
Bcl-2 associated X protein
- BAK:
-
Bcl-2 homologous antagonist/killer
- IAP:
-
Inhibitor of apoptosis
- XIAP:
-
X-linked inhibitor of apoptosis
- ROS:
-
Reactive oxygen species
- NOS:
-
Nitrogen oxygen species
- JAK/STAT:
-
Janus kinases/ Signal Transducer and Activator of Transcription proteins
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- RIPK1:
-
Receptor interacting protein kinase 1
- RIPK3:
-
Receptor interacting protein kinase 3
- DAMP’s:
-
Damage-associated molecular patterns
- MLKL:
-
Mixed lineage kinase domain-like
- TRADD:
-
Tumor necrosis factor receptor type 1-associated DEATH domain protein
- TRAF2:
-
TNF receptor associated factor 2
- Akt:
-
Protein kinase B
- ERAD:
-
Endoplasmic-reticulum-associated protein degradation
- PKR:
-
Protein kinase RNA
- PERK:
-
Protein kinase RNA like ER kinase
- IRE1α:
-
Inositol-requiring protein 1α
- ATF6:
-
Activating transcription factor 6
- CHOP/GADD153:
-
X-linked inhibitor of apoptosis protein and co-operating with C/EBP homologous protein
- ASK1:
-
Apoptosis signal regulating kinase 1
- JNK:
-
Jun N-terminal kinase
- PKC:
-
Protein kinase C
- PUMA:
-
P53 upregulated modulator of apoptosis
- NOXA:
-
Phorbol-12-myristate-13-acetate-induced protein 1
- TPEN:
-
N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine
- PARP:
-
Poly (ADP-ribose) polymerase-1
- AMPK:
-
5' AMP-activated protein kinase
- m-TORC1:
-
Mammalian target of rapamycin complex 1
- DRAM:
-
Damaged regulated autophagy modulator
- TFEB/TFE3:
-
The transcription factor EB/Transcription Factor Binding to IGHM Enhancer 3
- FOXO3a:
-
Transcription factor forkhead box O3a
- VEGF:
-
Vascular endothelial growth factor
- VEGF2:
-
Vascular endothelial growth factor receptor 2
- STAT3:
-
Signal transducer and activator of transcription 3
- Aβ:
-
β-Amyloid
- MPP+ :
-
1-Methyl-4-phenylpyridinium ion
- FLICE:
-
FADD-like IL-1β-converting enzyme
- BBC3:
-
Bcl-2 Binding Component 3
- HMGB1:
-
High mobility group box 1
- DRGNs:
-
Dorsal root ganglial neurons
- RHEB:
-
Ras homolog enriched in brain
- MAPK:
-
Mitogen activated protein kinase
- Mcl-1:
-
Myeloid leukemia cell differentiation protein
- Hrk/DP5:
-
Activator of apoptosis Harakiri/ death protein 5
- MPTP+ :
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- Cdk5:
-
Cyclin dependent kinase 5
- MEK/ERK:
-
Mitogen-activated protein kinase 1
- ERK1/2:
-
Extracellular signal-regulated kinase 1
- CREB:
-
CAMP-response element-binding protein
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- APAF1:
-
Apoptotic protease activating factor 1
- NLRP3:
-
NLR family pyrin domain containing 3
- PYCARD:
-
PYD and CARD domain containing
- iNOS:
-
Inducible nitric oxide synthase
- AICAR:
-
5-Aminoimidazole-4-carboxamide ribonucleotide
- GSK3:
-
Glycogen synthase kinase 3
- GSK3β:
-
Glycogen synthase kinase 3 β
- GSK3α:
-
Glycogen synthase kinase 3 α
- NMDAR:
-
N-methyl-D-aspartate receptor
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- NLRP1:
-
NACHT-LRR-PYD domains-containing protein 1
- NLRP3:
-
NACHT-LRR-PYD domains-containing protein 3
- NLRC4:
-
NLR Family CARD Domain Containing 4
- ALR’s:
-
Augmenter of liver regeneration
- PAMP and DAMP:
-
Pathogen and damage associated molecular patterns
- SNpc:
-
Substantia nigra pars compacta
- PBBI:
-
Penetrating ballistic-like brain injury
- ROF:
-
Roflupram
- CNS:
-
Central nervous system
- ER:
-
Endoplasmic reticulum
- MALAT1:
-
Metastasis-associated lung adenocarcinoma transcript 1
- NEAT2:
-
Nuclear-enriched transcript 2
- CRNDE:
-
Colorectal neoplasia differentially expressed
- GFAP:
-
Glial fibrillary acidic protein
- BrdU:
-
Bromodeoxyuridine
- SNHG12:
-
Small nucleolar host gene 12
References
Ribeiro A, Lopes M, Monteiro R et al (2017) Neurodegenerative Disease. Nanomed Inflamm Dis. https://doi.org/10.1201/9781315152356-17
Winner B, Kohl Z, Gage FH (2011) Neurodegenerative disease and adult neurogenesis. Eur J Neurosci 33:1139–1151. https://doi.org/10.1111/J.1460-9568.2011.07613.X
Martinez-Vicente M (2015) Autophagy in neurodegenerative diseases: From pathogenic dysfunction to therapeutic modulation. Semin Cell Dev Biol 40:115–126. https://doi.org/10.1016/J.SEMCDB.2015.03.005
Ghavami S, Shojaei S, Yeganeh B et al (2014) Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol 112:24–49. https://doi.org/10.1016/J.PNEUROBIO.2013.10.004
Nah J, Yuan J, Jung Y-K (2015) Autophagy in Neurodegenerative Diseases: From Mechanism to Therapeutic Approach. Mol Cells 38:381–389. https://doi.org/10.14348/molcells.2015.0034
Okouchi M, Ekshyyan O, Maracine M, Aw TY (2007) Neuronal Apoptosis in Neurodegeneration. https://home.liebertpub.com/ars 9:1059–1096. https://doi.org/10.1089/ARS.2007.1511
Soto C, Estrada LD (2008) Protein misfolding and neurodegeneration. Arch Neurol 65:184–189. https://doi.org/10.1001/ARCHNEUROL.2007.56
Xiang C, Wang Y, Zhang H, Han F (2017) The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis. https://doi.org/10.1007/S10495-016-1296-4
Radi E, Formichi P, Battisti C, Federico A (2014) Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimer’s Dis 42:S125–S152. https://doi.org/10.3233/JAD-132738
Mukhopadhyay S, Panda PK, Sinha N et al (2014) (2014) Autophagy and apoptosis: where do they meet? Apoptosis 194(19):555–566. https://doi.org/10.1007/S10495-014-0967-2
Loos B, du Toit A, Hofmeyr J-HS (2014) Defining and measuring autophagosome flux—concept and reality. Autophagy 10:2087–2096. https://doi.org/10.4161/15548627.2014.973338
Moloudizargari M, Asghari MH, Ghobadi E et al (2017) Autophagy, its mechanisms and regulation: Implications in neurodegenerative diseases. Ageing Res Rev 40:64–74. https://doi.org/10.1016/J.ARR.2017.09.005
Jung CH, Jun CB, Ro S-H et al (2009) ULK-Atg13-FIP200 complexes mediate mTOR Signaling to the autophagy machinery. Mol Biol Cell 20:1992. https://doi.org/10.1091/MBC.E08-12-1249
Fan W, Nassiri A, Zhong Q (2011) Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). Proc Natl Acad Sci 108:7769–7774. https://doi.org/10.1073/pnas.1016472108
Kabeya Y, Mizushima N, Ueno T et al (2003) Erratum: LC3, a mammalian homolog of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728
Shao Y, Gao Z, Feldman T, Jiang X (2007) Stimulation of ATG12-ATG5 conjugation by ribonucleic acid. Autophagy. https://doi.org/10.4161/auto.3270
Fujita N, Hayashi-Nishino M, Fukumoto H et al (2008) An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. https://doi.org/10.1091/mbc.e08-03-0312
Salminen A, Kaarniranta K, Kauppinen A (2013) Beclin 1 interactome controls the crosstalk between apoptosis, autophagy and inflammasome activation: Impact on the aging process. Ageing Res Rev 12:520–534. https://doi.org/10.1016/J.ARR.2012.11.004
Ravikumar B, Acevedo-Arozena A, Imarisio S et al (2005) Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. https://doi.org/10.1038/ng1591
Toné S, Sugimoto K, Tanda K et al (2007) Three distinct stages of apoptotic nuclear condensation revealed by time-lapse imaging, biochemical and electron microscopy analysis of cell-free apoptosis. Exp Cell Res. https://doi.org/10.1016/j.yexcr.2007.06.018
Tower J (2015) Programmed cell death in aging. Ageing Res Rev 23:90–100. https://doi.org/10.1016/J.ARR.2015.04.002
Kumar R, Herbert PE, Warrens AN (2005) An introduction to death receptors in apoptosis. Int J Surg 3:268–277. https://doi.org/10.1016/J.IJSU.2005.05.002
Roos WP, Kaina B (2006) DNA damage-induced cell death by apoptosis. Trends Mol Med 12:440–450. https://doi.org/10.1016/J.MOLMED.2006.07.007
Micheau O, Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114:181–190. https://doi.org/10.1016/S0092-8674(03)00521-X
Fuchs Y, Steller H (2011) Programmed cell death in animal development and disease. Cell 147:742. https://doi.org/10.1016/J.CELL.2011.10.033
Zhang JH, Zhang Y, Herman B (2003) Caspases, apoptosis and aging. Ageing Res Rev 2:357–366. https://doi.org/10.1016/S1568-1637(03)00026-6
Cheng EH-Y, Wei MC, Weiler S et al (2001) BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8:705–711. https://doi.org/10.1016/S1097-2765(01)00320-3
Wei MC, Zong W-X, Cheng EH-Y et al (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727. https://doi.org/10.1126/SCIENCE.1059108
Du H, Wolf J, Schafer B et al (2011) BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem 286:491. https://doi.org/10.1074/JBC.M110.167148
Okouchi M, Ekshyyan O, Maracine M et al (2007) Neuronal apoptosis in neurodegeneration. Antioxidants Redox Signal. https://doi.org/10.1089/ars.2007.1511
Bakkenist CJ, Kastan MB (2004) Initiating cellular stress responses. Cell 118:9–17. https://doi.org/10.1016/J.CELL.2004.06.023
Chandra J, Samali A, Orrenius S (2000) Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med 29:323–333. https://doi.org/10.1016/S0891-5849(00)00302-6
Krysko O, Aaes TL, Kagan VE et al (2017) Necroptotic cell death in anti-cancer therapy. Immunol Rev 280:207–219. https://doi.org/10.1111/IMR.12583
Conrad M, Angeli JPF, Vandenabeele P (2016) Stockwell BR (2016) Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 155(15):348–366. https://doi.org/10.1038/nrd.2015.6
Galluzzi L, Kepp O, Krautwald S et al (2014) Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol 35:24–32. https://doi.org/10.1016/J.SEMCDB.2014.02.006
Murphy JM, Garnier J-M, Babon JJ et al (2014) Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1408987111
Xia B, Fang S, Chen X et al (2016) MLKL forms cation channels. Cell Res. https://doi.org/10.1038/cr.2016.26
Choi ME, Price DR, Ryter SW, Choi AMK (2019) Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight. https://doi.org/10.1172/JCI.INSIGHT.128834
Han J, Zhong CQ, Zhang DW (2011) Programmed necrosis: Backup to and competitor with apoptosis in the immune system. Nat Immunol 12:1143–1149. https://doi.org/10.1038/NI.2159
Dillon CP, Oberst A, Weinlich R et al (2012) Survival function of the FADD-CASPASE-8-cFLIPL complex. Cell Rep 1:401–407. https://doi.org/10.1016/J.CELREP.2012.03.010
McNamara CR, Ahuja R, Osafo-Addo AD et al (2013) Akt regulates TNFα synthesis downstream of RIP1 kinase activation during necroptosis. PLoS ONE 8:e56576. https://doi.org/10.1371/JOURNAL.PONE.0056576
Iurlaro R, Muñoz-Pinedo C (2016) Cell death induced by endoplasmic reticulum stress. FEBS J 283:2640–2652. https://doi.org/10.1111/FEBS.13598
Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 132(13):89–102. https://doi.org/10.1038/nrm3270
Zhu G, Lee AS (2015) Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J Cell Physiol. https://doi.org/10.1002/jcp.24923
Rodriguez DA, Zamorano S, Lisbona F et al (2012) BH3-only proteins are part of a regulatory network that control the sustained signalling of the unfolded protein response sensor IRE1α. EMBO J. https://doi.org/10.1038/emboj.2012.84
Zalckvar E, Berissi H, Eisenstein M, Kimchi A (2009) Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 5(5):720–722. https://doi.org/10.4161/auto.5.5.8625
Mariño G, Niso-Santano M, Baehrecke EH (2014) Kroemer G (2014) Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 152(15):81–94. https://doi.org/10.1038/nrm3735
Smaili S, Pereira SG, Costa M et al (2013) The role of calcium stores in apoptosis and autophagy. Curr Mol Med. https://doi.org/10.2174/1566524011313020003
He GQ, Chen Y, Liao HJ et al (2020) Associations between Huwe1 and autophagy in rat cerebral neuron oxygen-glucose deprivation and reperfusion injury. Mol Med Rep. https://doi.org/10.3892/mmr.2020.11611
Tsai RT, Tsai CW, Liu SP et al (2020) Maackiain ameliorates 6-hydroxydopamine and snca pathologies by modulating the pink1/parkin pathway in models of parkinson’s disease in caenorhabditis elegans and the sh-sy5y cell line. Int J Mol Sci. https://doi.org/10.3390/ijms21124455
Mudawal A, Srivastava A, Singh A et al (2018) Proteomic approaches to investigate age related vulnerability to lindane induced neurodegenerative effects in rats. Food Chem Toxicol. https://doi.org/10.1016/j.fct.2018.03.049
Liu H, Rose ME, Ma X et al (2017) In vivo transduction of neurons with TAT-UCHL1 protects brain against controlled cortical impact injury. PLoS ONE. https://doi.org/10.1371/journal.pone.0178049
Guo F, He XB, Li S, Le W (2017) A central role for phosphorylated p38α in linking proteasome inhibition-induced apoptosis and autophagy. Mol Neurobiol. https://doi.org/10.1007/s12035-016-0260-1
Murdoch JD, Rostosky CM, Gowrisankaran S et al (2016) Endophilin-A deficiency induces the Foxo3a-Fbxo32 network in the brain and causes dysregulation of autophagy and the ubiquitin-proteasome system. Cell Rep. https://doi.org/10.1016/j.celrep.2016.09.058
Fernandez-Estevez MA, Casarejos MJ, Sendon JL et al (2014) Trehalose reverses cell malfunction in fibroblasts from normal and huntington’s disease patients caused by proteosome inhibition. PLoS ONE. https://doi.org/10.1371/journal.pone.0090202
Shruthi K, Reddy SS, Reddy PY et al (2016) Amelioration of neuronal cell death in a spontaneous obese rat model by dietary restriction through modulation of ubiquitin proteasome system. J Nutr Biochem. https://doi.org/10.1016/j.jnutbio.2016.03.008
Xu J, Zhang X-Z, Zhang Y-J et al (2015) Silencing of SIAH1 in SH-SY5Y affects α-synuclein degradation pathway. Int J Clin Exp Pathol 8:12885
Hamacher-Brady A, Choe SC, Krijnse-Locker J, Brady NR (2014) Intramitochondrial recruitment of endolysosomes mediates Smac degradation and constitutes a novel intrinsic apoptosis antagonizing function of XIAP E3 ligase. Cell Death Differ. https://doi.org/10.1038/cdd.2014.101
Ojha R, Ishaq M, Singh S (2015) Caspase-mediated crosstalk between autophagy and apoptosis: Mutual adjustment or matter of dominance. J Cancer Res Ther. https://doi.org/10.4103/0973-1482.163695
Oral O, Oz-Arslan D, Itah Z et al (2012) Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis. https://doi.org/10.1007/s10495-012-0735-0
Mulay SR, Desai J, Kumar SV et al (2016) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Nat Commun 25:15077–15082
Suresh SN, Chakravorty A, Giridharan M et al (2020) Pharmacological tools to modulate autophagy in neurodegenerative diseases. J Mol Biol 432:2822–2842. https://doi.org/10.1016/J.JMB.2020.02.023
Song S, Tan J, Miao Y et al (2017) Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J Cell Physiol 232:2977–2984. https://doi.org/10.1002/JCP.25785
Deegan S, Saveljeva S, Logue SE et al (2014) Deficiency in the mitochondrial apoptotic pathway reveals the toxic potential of autophagy under ER stress conditions. Autophagy. https://doi.org/10.4161/15548627.2014.981790
Tomar D, Prajapati P, Sripada L et al (2013) TRIM13 regulates caspase-8 ubiquitination, translocation to autophagosomes and activation during ER stress induced cell death. Biochim Biophys Acta - Mol Cell Res. https://doi.org/10.1016/j.bbamcr.2013.08.021
Amir M, Zhao E, Fontana L et al (2013) Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ. https://doi.org/10.1038/cdd.2013.21
Young MM, Takahashi Y, Khan O et al (2012) Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem 287:12455–12468. https://doi.org/10.1074/jbc.M111.309104
Hiramatsu N, Messah C, Han J et al (2014) Translational and posttranslational regulation of XIAP by eIF2 and ATF4 promotes ER stress-induced cell death during the unfolded protein response. Mol Biol Cell. https://doi.org/10.1091/mbc.e13-11-0664
Galluzzi L, Kepp O (2012) Kroemer G (2012) Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol 1312(13):780–788. https://doi.org/10.1038/nrm3479
Youle RJ, Narendra DP (2010) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 121(12):9–14. https://doi.org/10.1038/nrm3028
Ding W-X, Yin X-M (2012) Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 393:547. https://doi.org/10.1515/HSZ-2012-0119
Rezzani R, Stacchiotti A, Rodella LF (2012) Morphological and biochemical studies on aging and autophagy. Ageing Res Rev 11:10–31. https://doi.org/10.1016/J.ARR.2011.09.001
Hou W, Han J, Lu C et al (2010) Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy. https://doi.org/10.4161/auto.6.7.13038
Fitzwalter BE, Towers CG, Sullivan KD et al (2018) Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev Cell. https://doi.org/10.1016/j.devcel.2018.02.014
Foglio E, Puddighinu G, Germani A et al (2017) HMGB1 Inhibits apoptosis following mi and induces autophagy via mTORC1 inhibition. J Cell Physiol. https://doi.org/10.1002/jcp.25576
Liu J, Xia H, Kim M et al (2011) Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. https://doi.org/10.1016/j.cell.2011.08.037
Maiuri MC, Galluzzi L, Morselli E et al (2010) Autophagy regulation by p53. Curr Opin Cell Biol 22:181–185. https://doi.org/10.1016/J.CEB.2009.12.001
Labuschagne CF, Zani F, Vousden KH (2018) Control of metabolism by p53 – cancer and beyond. Biochim Biophys Acta Rev Cancer 1870:32–42. https://doi.org/10.1016/J.BBCAN.2018.06.001
Brooks CL, Gu W (2003) Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol 15:164–171. https://doi.org/10.1016/S0955-0674(03)00003-6
Brooks CL, Gu W (2011) The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2:456. https://doi.org/10.1007/S13238-011-1063-9
Berkers CR, Maddocks ODK, Cheung EC et al (2013) Metabolic regulation by p53 family members. Cell Metab 18:617–633. https://doi.org/10.1016/J.CMET.2013.06.019
Matlashewski G, Banks L, Pim D, Crawford L (1986) Analysis of human p53 proteins and mRNA levels in normal and transformed cells. Eur J Biochem 154:665–672. https://doi.org/10.1111/J.1432-1033.1986.TB09449
Crighton D, Woiwode A, Zhang C et al (2003) p53 represses RNA polymerase III transcription by targeting TBP and inhibiting promoter occupancy by TFIIIB. EMBO J 22:2810. https://doi.org/10.1093/EMBOJ/CDG265
Wu H-J, Pu J-L, Krafft PR et al (2014) The molecular mechanisms between autophagy and apoptosis: potential role in central nervous system disorders. Cell Mol Neurobiol 351(35):85–99. https://doi.org/10.1007/S10571-014-0116-Z
Engeland K (2018) Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ 251(25):114–132. https://doi.org/10.1038/cdd.2017.172
Singh R, Letai A, Sarosiek K (2019) Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol 20:175–193. https://doi.org/10.1038/s41580-018-0089-8
Reuter S, Eifes S, Dicato M et al (2008) Modulation of anti-apoptotic and survival pathways by curcumin as a strategy to induce apoptosis in cancer cells. Biochem Pharmacol 76:1340–1351. https://doi.org/10.1016/J.BCP.2008.07.031
Kim HL, Ra H, Kim KR, et al (2015) Poly(ADP-ribosyl)ation of p53 contributes to tpen-induced neuronal apoptosis. Mol Cells. https://doi.org/10.14348/molcells.2015.2142
Okuda A, Kurokawa S, Takehashi M et al (2017) Poly(ADP-ribose) polymerase inhibitors activate the p53 signaling pathway in neural stem/progenitor cells. BMC Neurosci. https://doi.org/10.1186/s12868-016-0333-0
Chantranupong L, Wolfson RL, Orozco JM et al (2014) The sestrins interact with gator2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. https://doi.org/10.1016/j.celrep.2014.09.014
Kenzelmann Broz D, Mello SS, Bieging KT et al (2013) Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. https://doi.org/10.1101/gad.212282.112
Lionaki E, Markaki M, Tavernarakis N (2013) Autophagy and ageing: Insights from invertebrate model organisms. Ageing Res Rev 12:413–428. https://doi.org/10.1016/J.ARR.2012.05.001
Brady OA, Jeong E, Martina JA et al (2018) The transcription factors TFE3 and TFEB amplify p53 dependent transcriptional programs in response to DNA damage. Elife. https://doi.org/10.7554/ELIFE.40856
Fischer M (2017) Census and evaluation of p53 target genes. Oncogene 3628(36):3943–3956. https://doi.org/10.1038/onc.2016.502
Robin M, Issa AR, Santos CC et al (2019) Drosophila p53 integrates the antagonism between autophagy and apoptosis in response to stress. Autophagy 15:771. https://doi.org/10.1080/15548627.2018.1558001
Carmeliet P, Jain RK (2011) Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 10(6):417–427. https://doi.org/10.1038/nrd3455
Cross MJ, Claesson-Welsh L (2001) FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci 22:201–207. https://doi.org/10.1016/S0165-6147(00)01676-X
Chau YP, Lin SY, Chen JHC, Tai MH (2003) Endostatin induces autophagic cell death in EAhy926 human endothelial cells. Histol Histopathol https://doi.org/10.14670/HH-18.715
Yang SY, Kim NH, Cho YS et al (2014) Convallatoxin, a dual inducer of autophagy and apoptosis, inhibits angiogenesis in vitro and in vivo. PLoS ONE. https://doi.org/10.1371/journal.pone.0091094
Li Y, Zhang F, Nagai N et al (2008) VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. J Clin Invest. https://doi.org/10.1172/JCI33673
Falk T, Zhang S, Sherman SJ (2009) Vascular endothelial growth factor B (VEGF-B) is up-regulated and exogenous VEGF-B is neuroprotective in a culture model of Parkinson’s disease. Mol Neurodegener. https://doi.org/10.1186/1750-1326-4-49
Ellison SM, Trabalza A, Tisato V et al (2013) Dose-dependent neuroprotection of VEGF165 in Huntington’s disease striatum. Mol Ther. https://doi.org/10.1038/mt.2013.132
Religa P, Cao R, Religa D et al (2013) VEGF significantly restores impaired memory behavior in Alzheimer’s mice by improvement of vascular survival. Sci Rep. https://doi.org/10.1038/srep02053
Yu H, Lin B, He YZ et al (2015) Batroxobin protects against spinal cord injury in rats by promoting the expression of vascular endothelial growth factor to reduce apoptosis. Exp Ther Med. https://doi.org/10.3892/etm.2015.2368
Cui W, Li W, Han R et al (2011) PI3-K/Akt and ERK pathways activated by VEGF play opposite roles in MPP+-induced neuronal apoptosis. Neurochem Int 59:945–953. https://doi.org/10.1016/j.neuint.2011.07.005
Caballero B, Sherman SJ, Falk T (2017) Insights into the mechanisms involved in protective effects of VEGF-B in dopaminergic neurons. Parkinsons Dis. https://doi.org/10.1155/2017/4263795
Ma XM, Blenis J (2009) Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 105(10):307–318. https://doi.org/10.1038/nrm2672
Ben-Sahra I, Howell JJ, Asara JM, Manning BD (2013) Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339:1323. https://doi.org/10.1126/SCIENCE.1228792
Ravikumar B, Sarkar S, Davies JE et al (2010) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90:1383–1435. https://doi.org/10.1152/PHYSREV.00030.2009
Fitzwalter B, Thorburn A (2015) Recent insights into cell death and autophagy. FEBS J 282:4279. https://doi.org/10.1111/FEBS.13515
Pazoki-Toroudi H, Amani H, Ajami M et al (2016) Targeting mTOR signaling by polyphenols: A new therapeutic target for ageing. Ageing Res Rev 31:55–66. https://doi.org/10.1016/J.ARR.2016.07.004
Urbanska M, Gozdz A, Swiech LJ, Jaworski J (2012) Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) control the dendritic arbor morphology of hippocampal neurons. J Biol Chem. https://doi.org/10.1074/jbc.M112.374405
Rüegg MA (2013) In vivo evidence for mTORC2-mediated actin cytoskeleton rearrangement in neurons. BioArchitecture. https://doi.org/10.4161/bioa.26497
Sosanya NM, Cacheaux LP, Workman ER et al (2015) Mammalian target of rapamycin (mTOR) tagging promotes dendritic branch variability through the capture of Ca2+/calmodulin-dependent protein kinase II α (CaMKIIα) mRNAs by the RNA-binding protein HuD. J Biol Chem 290:16357–16371. https://doi.org/10.1074/jbc.M114.599399
Abe N, Borson SH, Gambello MJ et al (2010) Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J Biol Chem. https://doi.org/10.1074/jbc.M110.125336
Nie D, Di Nardo A, Han JM et al (2010) Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci. https://doi.org/10.1038/nn.2477
Weston MC, Chen H, Swann JW (2012) Multiple Roles for Mammalian target of rapamycin signaling in both glutamatergic and GABAergic synaptic transmission. J Neurosci. https://doi.org/10.1523/jneurosci.1283-12.2012
Briz V, Zhu G, Wang Y et al (2015) Activity-dependent rapid local RhoA synthesis is required for hippocampal synaptic plasticity. J Neurosci. https://doi.org/10.1523/jneurosci.2302-14.2015
Hou L (2004) Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. https://doi.org/10.1523/jneurosci.0995-04.2004
Richardson A, Galvan V, Lin AL, Oddo S (2015) How longevity research can lead to therapies for Alzheimer’s disease: The rapamycin story. Exp Gerontol. https://doi.org/10.1016/j.exger.2014.12.002
Caccamo A, Branca C, Talboom JS et al (2015) Reducing Ribosomal Protein S6 Kinase 1 Expression Improves Spatial Memory and Synaptic Plasticity in a Mouse Model of Alzheimer’s Disease. J Neurosci. https://doi.org/10.1523/jneurosci.2781-15.2015
Siman R, Cocca R, Dong Y (2015) The mTOR inhibitor rapamycin mitigates perforant pathway neurodegeneration and synapse loss in a mouse model of early-stage Alzheimer-type tauopathy. PLoS ONE. https://doi.org/10.1371/journal.pone.0142340
Frederick C, Ando K, Leroy K et al (2015) Rapamycin ester analog CCI-779/Temsirolimus alleviates tau pathology and improves motor deficit in mutant tau transgenic mice. J Alzheimer’s Dis. https://doi.org/10.3233/JAD-142097
Pan T, Kondo S, Le W, Jankovic J (2008) The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 131:1969–1978. https://doi.org/10.1093/BRAIN/AWM318
Spencer B, Potkar R, Trejo M et al (2009) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of parkinson’s and lewy body diseases. J Neurosci 29:13578. https://doi.org/10.1523/JNEUROSCI.4390-09.2009
Sarosiek KA, Fraser C, Muthalagu N et al (2017) Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell 31:142–156. https://doi.org/10.1016/J.CCELL.2016.11.011
Polster BM, Basañez G, Young M et al (2003) Inhibition of bax-induced cytochrome c release from neural cell and brain mitochondria by dibucaine and propranolol. J Neurosci 23:2735. https://doi.org/10.1523/JNEUROSCI.23-07-02735.2003
Zhu C, Wang X, Xu F et al (2005) (2004) The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia–ischemia. Cell Death Differ 122(12):162–176. https://doi.org/10.1038/sj.cdd.4401545
Shimohama S, Fujimoto S, Sumida Y, Tanino H (1998) Differential expression of rat brain Bcl-2 Family proteins in development and aging. Biochem Biophys Res Commun 252:92–96. https://doi.org/10.1006/BBRC.1998.9577
Ke F, Bouillet P, Kaufmann T et al (2013) Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell Death Dis. https://doi.org/10.1038/cddis.2013.176
Sun W, Gould TW, Vinsant S et al (2018) Neuromuscular development after the prevention of naturally occurring neuronal death by bax deletion. J Neurosci. https://doi.org/10.1523/jneurosci.23-19-07298.2003
Miller TM, Moulder KL, Knudson CM et al (1997) Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. https://doi.org/10.1083/jcb.139.1.205
Kudo W, Lee HP, Smith MA et al (2012) Inhibition of Bax protects neuronal cells from oligomeric Aβ neurotoxicity. Cell Death Dis. https://doi.org/10.1038/cddis.2012.43
Jakobson M, Jakobson M, Llano O et al (2013) Multiple mechanisms repress N-Bak mRNA translation in the healthy and apoptotic neurons. Cell Death Dis. https://doi.org/10.1038/cddis.2013.297
Uo T, Kinoshita Y, Morrison RS (2005) Neurons exclusively express N-Bak, a BH3 domain-only Bak isoform that promotes neuronal apoptosis. J Biol Chem. https://doi.org/10.1074/jbc.M413030200
Fannjiang Y, Kim CH, Huganir RL et al (2003) BAK alters neuronal excitability and can switch from anti- to pro-death function during postnatal development. Dev Cell. https://doi.org/10.1016/S1534-5807(03)00091-1
Wang Y, Zhang Y, Zhang X et al (2019) Alcohol dehydrogenase 1B suppresses β-amyloid-induced neuron apoptosis. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2019.00135
Liu M, Bai X, Yu S et al (2019) Ginsenoside re inhibits ROS/ASK-1 dependent mitochondrial apoptosis pathway and activation of Nrf2-antioxidant response in beta-amyloid-challenged SH-SY5Y cells. Molecules. https://doi.org/10.3390/MOLECULES24152687
He G-Q, Xu W-M, Liao H-J et al (2019) Silencing Huwe1 reduces apoptosis of cortical neurons exposed to oxygen-glucose deprivation and reperfusion. Neural Regen Res 14:1977–1985. https://doi.org/10.4103/1673-5374.259620
Arbour N, Vanderluit JL, Le Grand JN et al (2008) Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J Neurosci. https://doi.org/10.1523/jneurosci.4940-07.2008
Crowther AJ, Gama V, Bevilacqua A et al (2013) Tonic activation of bax primes neural progenitors for rapid apoptosis through a mechanism preserved in medulloblastoma. J Neurosci. https://doi.org/10.1523/jneurosci.2602-13.2013
Nakamura A, Swahari V, Plestant C et al (2016) Bcl-xL is essential for the survival and function of differentiated neurons in the cortex that control complex behaviors. J Neurosci. https://doi.org/10.1523/jneurosci.4247-15.2016
Fogarty LC, Flemmer RT, Geizer BA et al (2018) Mcl-1 and Bcl-xL are essential for survival of the developing nervous system. Cell Death Differ. https://doi.org/10.1038/s41418-018-0225-1
Michaelidis TM, Sendtner M, Cooper JD et al (1996) Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron. https://doi.org/10.1016/S0896-6273(00)80282-2
Hochman A, Sternin H, Gorodin S et al (2002) Enhanced oxidative stress and altered antioxidants in brains of Bcl-2-deficient mice. J Neurochem. https://doi.org/10.1046/j.1471-4159.1998.71020741.x
Middleton G, Wyatt S, Ninkina N, Davies AM (2001) Reciprocal developmental changes in the roles of Bcl-w and Bcl-x(L) in regulating sensory neuron survival. Development 128:447–457. https://doi.org/10.1242/DEV.128.3.447
Murphy B, Dunleavy M, Shinoda S et al (2007) Bcl-w protects hippocampus during experimental status epilepticus. Am J Pathol. https://doi.org/10.2353/ajpath.2007.070269
Courchesne SL, Karch C, Pazyra-Murphy MF, Segal RA (2011) Sensory Neuropathy Attributable to Loss of Bcl-w. J Neurosci. https://doi.org/10.1523/jneurosci.3347-10.2011
Simon DJ, Pitts J, Hertz NT et al (2016) Axon degeneration gated by retrograde activation of somatic pro-apoptotic signaling. Cell. https://doi.org/10.1016/j.cell.2016.01.032
Henshall DC, Engel T (2013) Contribution of apoptosis-associated signaling pathways to epileptogenesis: lessons from Bcl-2 family knockouts. Front Cell Neurosci. https://doi.org/10.3389/fncel.2013.00110
Krajewska M, Mai JK, Zapata JM, et al (2002) Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ. https://doi.org/10.1038/sj.cdd.4400934
Madden SD, Donovan M, Cotter TG (2007) Key apoptosis regulating proteins are down-regulated during postnatal tissue development. Int J Dev Biol. https://doi.org/10.1387/ijdb.062263sm
Imaizumi K, Tsuda M, Imai Y et al (1997) Molecular cloning of a novel polypeptide, DP5, induced during programmed neuronal death. J Biol Chem. https://doi.org/10.1074/jbc.272.30.18842
Shimohama S, Fujimoto S, Sumida Y, Tanino H (1998) Differential expression of rat brain Bcl-2 family proteins in development and aging. Biochem Biophys Res Commun. https://doi.org/10.1006/bbrc.1998.9577
Akhter R, Sanphui P, Biswas SC (2014) The essential role of p53-up-regulated modulator of apoptosis (Puma) and its regulation by FoxO3a transcription factor in β-amyloid-induced neuron death. J Biol Chem. https://doi.org/10.1074/jbc.M113.519355
Kole AJ, Swahari V, Hammond SM, Deshmukh M (2011) miR-29b is activated during neuronal maturation and targets BH3-only genes to restrict apoptosis. Genes Dev. https://doi.org/10.1101/gad.1975411
Akhter R, Saleem S, Saha A, Biswas SC (2018) The pro-apoptotic protein Bmf co-operates with Bim and Puma in neuron death induced by β-amyloid or NGF deprivation. Mol Cell Neurosci. https://doi.org/10.1016/j.mcn.2018.02.011
Concannon CG, Tuffy LP, Weisová P et al (2010) AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stressinduced apoptosis. J Cell Biol. https://doi.org/10.1083/jcb.200909166
Harder JM (2013) Libby RT (2012) Deficiency in Bim, Bid and Bbc3 (Puma) do not prevent axonal injury induced death. Cell Death Differ 201(20):182–182. https://doi.org/10.1038/cdd.2012.119
Fernandes KA, Harder JM, Kim J, Libby RT (2013) JUN regulates early transcriptional responses to axonal injury in retinal ganglion cells. Exp Eye Res. https://doi.org/10.1016/j.exer.2013.04.021
Ren D, Tu HC, Kim H et al (2010) BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science (80-). https://doi.org/10.1126/science.1190217
Wong HK, Fricker M, Wyttenbach A et al (2005) Mutually exclusive subsets of BH3-only proteins are activated by the p53 and c-Jun N-terminal kinase/c-Jun signaling pathways during cortical neuron apoptosis induced by arsenite. Mol Cell Biol 25:8732–8747. https://doi.org/10.1128/MCB.25.19.8732-8747.2005
Kuan C-Y, Whitmarsh AJ, Yang DD et al (2003) A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.2336254100
Towers E, Gilley J, Randall R et al (2009) The proapoptotic dp5 gene is a direct target of the MLK-JNK-c-Jun pathway in sympathetic neurons. Nucleic Acids Res. https://doi.org/10.1093/nar/gkp175
Ma C, Ying C, Yuan Z et al (2007) dp5/HRK is a c-Jun target gene and required for apoptosis induced by potassium deprivation in cerebellar granule neurons. J Biol Chem 282:30901–30909. https://doi.org/10.1074/jbc.M608694200
Kristiansen M, Menghi F, Hughes R et al (2011) Global analysis of gene expression in NGF-deprived sympathetic neurons identifies molecular pathways associated with cell death. BMC Genom. https://doi.org/10.1186/1471-2164-12-551
Ambacher KK, Pitzul KB, Karajgikar M et al (2012) The JNK- and AKT/GSK3beta- signaling pathways converge to regulate Puma induction and neuronal apoptosis induced by trophic factor deprivation. PLoS ONE 7:e46885. https://doi.org/10.1371/journal.pone.0046885
Akhter R, Sanphui P, Das H et al (2015) The regulation of p53 up-regulated modulator of apoptosis by JNK/c-Jun pathway in beta-amyloid-induced neuron death. J Neurochem 134:1091–1103. https://doi.org/10.1111/jnc.13128
Biswas SC, Shi Y, Sproul A, Greene LA (2007) Pro-apoptotic Bim induction in response to nerve growth factor deprivation requires simultaneous activation of three different death signaling pathways. J Biol Chem. https://doi.org/10.1074/jbc.M702634200
Harder JM, Libby RT (2013) Deficiency in Bim, Bid and Bbc3 (Puma) do not prevent axonal injury induced death. Cell Death Differ 20(1):182
Maor-Nof M, Romi E, Shalom HS et al (2016) Axonal degeneration is regulated by a transcriptional program that coordinates expression of pro- and anti-degenerative factors. Neuron. https://doi.org/10.1016/j.neuron.2016.10.061
Engel T, Murphy BM, Hatazaki S et al (2010) Reduced hippocampal damage and epileptic seizures after status epilepticus in mice lacking proapoptotic Puma. FASEB J. https://doi.org/10.1096/fj.09-145870
Qi X, Davis B, Chiang YH et al (2016) Dopaminergic neuron-specific deletion of p53 gene is neuroprotective in an experimental Parkinson’s disease model. J Neurochem. https://doi.org/10.1111/jnc.13706
Cregan SP (2004) p53 Activation domain 1 Is essential for PUMA upregulation and p53-mediated neuronal cell death. J Neurosci. https://doi.org/10.1523/jneurosci.2114-04.2004
Sanphui P, Biswas SC (2013) FoxO3a is activated and executes neuron death via Bim in response to β-amyloid. Cell Death Dis. https://doi.org/10.1038/cddis.2013.148
Li D, Luo L, Xu M et al (2017) AMPK activates FOXO3a and promotes neuronal apoptosis in the developing rat brain during the early phase after hypoxia-ischemia. Brain Res Bull. https://doi.org/10.1016/j.brainresbull.2017.05.001
Davila D, Connolly NMC, Bonner H et al (2012) Two-step activation of FOXO3 by AMPK generates a coherent feed-forward loop determining excitotoxic cell fate. Cell Death Differ. https://doi.org/10.1038/cdd.2012.49
Zareen N, Biswas SC, Greene LA (2013) A feed-forward loop involving Trib3, Akt and FoxO mediates death of NGF-deprived neurons. Cell Death Differ https://doi.org/10.1038/cdd.2013.128
Saleem S, Biswas SC (2017) Tribbles pseudokinase 3 induces both apoptosis and autophagy in amyloid-β-induced neuronal death. J Biol Chem. https://doi.org/10.1074/jbc.M116.744730
Yuan Z, Lehtinen MK, Merlo P et al (2009) Regulation of neuronal cell death by MST1-FOXO1 signaling. J Biol Chem. https://doi.org/10.1074/jbc.M900461200
Shi C, Viccaro K, Lee H, Shah K (2016) Cdk5–Foxo3 axis: initially neuroprotective, eventually neurodegenerative in Alzheimer’s disease models. J Cell Sci. https://doi.org/10.1242/jcs.185009
Ghosh AP, Klocke BJ, Ballestas ME, Roth KA (2012) CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS ONE. https://doi.org/10.1371/journal.pone.0039586
Galehdar Z, Swan P, Fuerth B et al (2010) Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci. https://doi.org/10.1523/jneurosci.1598-10.2010
Matus S, Lopez E, Valenzuela V et al (2013) Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS ONE. https://doi.org/10.1371/journal.pone.0066672
Morishima Y, Gotoh Y, Zieg J et al (2018) β-Amyloid Induces Neuronal Apoptosis Via a Mechanism that Involves the c-Jun N-Terminal Kinase Pathway and the Induction of Fas Ligand. J Neurosci. https://doi.org/10.1523/jneurosci.21-19-07551.2001
Biswas SC, Shi Y, Vonsattel J-PG et al (2007) Bim is elevated in alzheimer’s disease neurons and is required for -amyloid-induced neuronal apoptosis. J Neurosci. https://doi.org/10.1523/jneurosci.3524-06.2007
Linseman DA, Phelps RA, Bouchard RJ et al (2002) Insulin-like growth factor-i blocks bcl-2 interacting mediator of cell death (Bim) induction and intrinsic death signaling in cerebellar granule neurons. J Neurosci. https://doi.org/10.1523/jneurosci.22-21-09287.2002
Finegan KG, Wang X, Lee EJ et al (2009) Regulation of neuronal survival by the extracellular signal-regulated protein kinase 5. Cell Death Differ. https://doi.org/10.1038/cdd.2008.193
Canals JM, Checa N, Marco S et al (2018) Expression of brain-derived neurotrophic factor in cortical neurons is regulated by striatal target area. J Neurosci. https://doi.org/10.1523/jneurosci.21-01-00117.2001
Almeida S, Laço M, Cunha-Oliveira T et al (2009) BDNF regulates BIM expression levels in 3-nitropropionic acid-treated cortical neurons. Neurobiol Dis. https://doi.org/10.1016/j.nbd.2009.06.006
Kamada H, Nito C, Endo H, Chan PH (2007) Bad as a converging signaling molecule between survival PI3-K/Akt and death JNK in neurons after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab 27:521–533. https://doi.org/10.1038/sj.jcbfm.9600367
Roberts ML, Virdee K, Sampson CPB et al (2000) The combination of Bcl-2 expression and NGF-deprivation facilitates the selective destruction of BAD protein in living sympathetic neurons. Mol Cell Neurosci. https://doi.org/10.1006/mcne.2000.0867
Huang HY, Lin SZ, Kuo JS et al (2007) G-CSF protects dopaminergic neurons from 6-OHDA-induced toxicity via the ERK pathway. Neurobiol Aging. https://doi.org/10.1016/j.neurobiolaging.2006.05.037
Mukherjee A, Williams DW (2017) More alive than dead: Non-apoptotic roles for caspases in neuronal development, plasticity and disease. Cell Death Differ 24(8):1411–1421. https://doi.org/10.1038/cdd.2017
Radovanović V, Vlainić J, Hanžić N et al (2019) Neurotoxic effect of ethanolic extract of propolis in the presence of copper ions is mediated through enhanced production of ROS and stimulation of caspase-3/7 activity. Toxins 11:273
Hou S, Wang L, Zhang G (2019) Mitofusin-2 regulates inflammation-mediated mouse neuroblastoma N2a cells dysfunction and endoplasmic reticulum stress via the Yap-Hippo pathway. J Physiol Sci. https://doi.org/10.1007/s12576-019-00685-6
Burnett SB, Vaughn LS, Strom JM et al (2019) A truncated PACT protein resulting from a frameshift mutation reported in movement disorder DYT16 triggers caspase activation and apoptosis. J Cell Biochem. https://doi.org/10.1002/jcb.29223
Hollville E, Romero SE, Deshmukh M (2019) Apoptotic cell death regulation in neurons. FEBS J. https://doi.org/10.1111/febs.14970
Han X, Sun S, Sun Y et al (2019) Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: implications for Parkinson disease. Autophagy. https://doi.org/10.1080/15548627.2019.1596481
Martin DDO, Schmidt ME, Nguyen YT et al (2018) Identification of a novel caspase cleavage site in huntingtin that regulates mutant huntingtin clearance. FASEB J. https://doi.org/10.1096/fj.201701510rrr
Wang X-J, Cao Q, Zhang Y, Su X-D (2014) Activation and regulation of caspase-6 and its role in neurodegenerative diseases. Annu Rev Pharmacol Toxicol. https://doi.org/10.1146/annurev-pharmtox-010814-124414
Yu T, Yu H, Zhang B et al (2019) Promising neuroprotective function for M2 microglia in kainic acid-induced neurotoxicity via the down-regulation of NF-κB and caspase 3 signaling pathways. Neuroscience. https://doi.org/10.1016/j.neuroscience.2019.03.002
Thawkar BS, Kaur G (2019) Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J Neuroimmunol 326:62–74. https://doi.org/10.1016/J.JNEUROIM.2018.11.010
Khan M, Rutten BPF, Kim MO (2019) MST1 regulates neuronal cell death via JNK/Casp3 signaling pathway in HFD mouse brain and HT22 cells. Int J Mol Sci 20:2504. https://doi.org/10.3390/IJMS20102504
Flores J, Noël A, Foveau B et al (2018) Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat Commun. https://doi.org/10.1038/s41467-018-06449-x
Bihaqi SW, Alansi B, Masoud AM et al (2018) Influence of early life lead (pb) exposure on α-synuclein, GSK-3β and caspase-3 mediated tauopathy: implications on alzheimer’s disease. Curr Alzheimer Res. https://doi.org/10.2174/1567205015666180801095925
Erekat NS, Al-Jarrah MD (2018) Association of Parkinson Disease Induction with Cardiac Upregulation of Apoptotic Mediators P53 and Active Caspase-3: An Immunohistochemistry Study. Med Sci Monit Basic Res. https://doi.org/10.12659/msmbr.910307
Rehker J, Rodhe J, Nesbitt RR et al (2017) Caspase-8, association with Alzheimer’s Disease and functional analysis of rare variants. PLoS ONE. https://doi.org/10.1371/journal.pone.0185777
Sai K, Yang D, Yamamoto H et al (2006) A1 adenosine receptor signal and AMPK involving caspase-9/-3 activation are responsible for adenosine-induced RCR-1 astrocytoma cell death. Neurotoxicology 27:458–467. https://doi.org/10.1016/j.neuro.2005.12.008
Song X, Kim SY, Zhang L et al (2014) Role of AMP-activated protein kinase in cross-talk between apoptosis and autophagy in human colon cancer. Cell Death Dis. https://doi.org/10.1038/cddis.2014.463
Salminen A, Kaarniranta K (2012) AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev 11:230–241. https://doi.org/10.1016/J.ARR.2011.12.005
Sun B, Ou H, Ren F et al (2018) Propofol inhibited autophagy through Ca 2+ /CaMKKβ/AMPK/mTOR pathway in OGD/R-induced neuron injury. Mol Med. https://doi.org/10.1186/s10020-018-0054-1
Zhao M, Chen J, Mao K et al (2019) Mitochondrial calcium dysfunction contributes to autophagic cell death induced by MPP + via AMPK pathway. Biochem Biophys Res Commun. https://doi.org/10.1016/j.bbrc.2018.12.148
Domise M, Sauvé F, Didier S et al (2019) Neuronal AMP-activated protein kinase hyper-activation induces synaptic loss by an autophagy-mediated process. Cell Death Dis. https://doi.org/10.1038/s41419-019-1464-x
Zhou L, Cheng Y (2019) Alpha-lipoic acid alleviated 6-OHDA-induced cell damage by inhibiting AMPK/mTOR mediated autophagy. Neuropharmacology 155:98–103. https://doi.org/10.1016/j.neuropharm.2019.04.009
Meares GP, Qin H, Liu Y et al (2013) AMP-activated protein kinase restricts IFN-γ signaling. J Immunol. https://doi.org/10.4049/jimmunol.1202390
Peixoto CA, Oliveira WH, Araújo SMR, Nunes AKS (2017) AMPK activation: Role in the signaling pathways of neuroinflammation and neurodegeneration. Exp Neurol 298:31–41. https://doi.org/10.1016/J.EXPNEUROL.2017.08.013
Chiang MC, Cheng YC, Chen SJ et al (2016) Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against Amyloid-beta-induced mitochondrial dysfunction. Exp Cell Res. https://doi.org/10.1016/j.yexcr.2016.08.013
Salminen A, Kaarniranta K, Kauppinen A (2016) Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res Rev 28:15–26. https://doi.org/10.1016/J.ARR.2016.04.003
Shahab L, Plattner F, Irvine EE et al (2014) Dynamic range of GSK3α not GSK3β is essential for bidirectional synaptic plasticity at hippocampal CA3-CA1 synapses. Hippocampus. https://doi.org/10.1002/hipo.22362
Jiang L, Kosenko A, Yu C et al (2015) Activation of m1 muscarinic acetylcholine receptor induces surface transport of KCNQ channels through a CRMP-2-mediated pathway. J Cell Sci. https://doi.org/10.1242/jcs.175547
Peineau S, Taghibiglou C, Bradley C et al (2007) LTP inhibits LTD in the hippocampus via regulation of GSK3β. Neuron. https://doi.org/10.1016/j.neuron.2007.01.029
Miras-Portugal MT, Gomez-Villafuertes R, Gualix J et al (2016) Nucleotides in neuroregeneration and neuroprotection. Neuropharmacology 104:243–254. https://doi.org/10.1016/J.NEUROPHARM.2015.09.002
Morroni F, Sita G, Tarozzi A et al (2016) Early effects of Aβ1-42 oligomers injection in mice: Involvement of PI3K/Akt/GSK3 and MAPK/ERK1/2 pathways. Behav Brain Res. https://doi.org/10.1016/j.bbr.2016.08.002
Yi JH, Baek SJ, Heo S et al (2018) Direct pharmacological Akt activation rescues Alzheimer’s disease like memory impairments and aberrant synaptic plasticity. Neuropharmacology. https://doi.org/10.1016/j.neuropharm.2017.10.028
Deng Y, Xiong Z, Chen P et al (2014) β-Amyloid impairs the regulation of N-methyl-D-aspartate receptors by glycogen synthase kinase 3. Neurobiol Aging. https://doi.org/10.1016/j.neurobiolaging.2013.08.031
Zhang Z-X, Zhao R-P, Wang D-S, Wang A-N (2016) Fuzhisan ameliorates Aβ production and tau phosphorylation in hippocampal of 11month old APP/PS1 transgenic mice: A Western blot study. Exp Gerontol 84:88–95. https://doi.org/10.1016/j.exger.2016.09.003
Noh MY, Koh SH, Kim Y et al (2009) Neuroprotective effects of donepezil through inhibition of GSK-3 activity in amyloid-β-induced neuronal cell death. J Neurochem. https://doi.org/10.1111/j.1471-4159.2008.05837.x
Yuan Y, Yan W, Sun J et al (2015) The molecular mechanism of rotenone-induced α-synuclein aggregation: Emphasizing the role of the calcium/GSK3β pathway. Toxicol Lett 233:163–171. https://doi.org/10.1016/j.toxlet.2014.11.029
Bijur GN, Jope RS (2001) proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3β. J Biol Chem. https://doi.org/10.1074/jbc.M105725200
Anuradha R, Saraswati M, Kumar KG, Rani SH (2014) Apoptosis of beta cells in diabetes mellitus. DNA Cell Biol. https://doi.org/10.1089/dna.2014.2352
Giampietri C, Petrungaro S, Coluccia P et al (2010) c-Flip overexpression affects satellite cell proliferation and promotes skeletal muscle aging. Cell Death Dis. https://doi.org/10.1038/cddis.2010.17
Rahmani M, Aust MM, Benson EC et al (2014) PI3K/mTOR inhibition markedly potentiates HDAC inhibitor activity in NHL cells through BIM- and MCL-1-Dependent mechanisms in vitro and in vivo. Clin Cancer Res. https://doi.org/10.1158/1078-0432.CCR-14-0034
Alao JP, Stavropoulou AV, Lam EWF, Coombes RC (2006) Role of glycogen synthase kinase 3 beta (GSK3β) in mediating the cytotoxic effects of the histone deacetylase inhibitor trichostatin A (TSA) in MCF-7 breast cancer cells. Mol Cancer. https://doi.org/10.1186/1476-4598-5-40
Lin CF, Tsai CC, Huang WC et al (2016) Glycogen synthase kinase-3β and caspase-2 mediate ceramide-And etoposide-induced apoptosis by regulating the lysosomal-mitochondrial axis. PLoS ONE. https://doi.org/10.1371/journal.pone.0145460
Maurer U, Charvet C, Wagman AS et al (2006) Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. https://doi.org/10.1016/j.molcel.2006.02.009
Huang L, Wu S, Xing D (2011) Glycogen synthase kinase-3β facilitates cell apoptosis induced by high fluence low-power laser irradiation through acceleration of Bax translocation. SPIE 7887:788708. https://doi.org/10.1117/12.874138
Linseman DA, Butts BD, Precht TA et al (2004) Glycogen synthase kinase-3β phosphorylates bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci 24:9993. https://doi.org/10.1523/JNEUROSCI.2057-04.2004
Rasola A, Bernardi P (2011) Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50:222–233. https://doi.org/10.1016/j.ceca.2011.04.007
Johri A, Beal MF (2012) Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther 342:619. https://doi.org/10.1124/JPET.112.192138
Luca A, Calandra C, Luca M (2016) Gsk3 signalling and redox status in bipolar disorder: evidence from lithium efficacy. Oxid Med Cell Longev. https://doi.org/10.1155/2016/3030547
Rojo AI, Medina-Campos ON, Rada P et al (2012) Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: Role of glycogen synthase kinase-3. Free Radic Biol Med. https://doi.org/10.1016/j.freeradbiomed.2011.11.003
Azoulay-Alfaguter I, Elya R, Avrahami L et al (2015) Combined regulation of mTORC1 and lysosomal acidification by GSK-3 suppresses autophagy and contributes to cancer cell growth. Oncogene. https://doi.org/10.1038/onc.2014.390
Stretton C, Hoffmann TM, Munson MJ et al (2015) GSK3-mediated raptor phosphorylation supports amino-acid-dependent mTORC1-directed signalling. Biochem J. https://doi.org/10.1042/bj20150404
Windsperger A, Yang J, Chen R et al (2013) 1326 GSK-3β controls autophagic response by modulating LKB1-AMPK pathway in prostate cancer cells. J Urol. https://doi.org/10.1016/j.juro.2013.02.2680
Sun A, Li C, Chen R et al (2016) GSK-3β controls autophagy by modulating LKB1-AMPK pathway in prostate cancer cells. Prostate. https://doi.org/10.1002/pros.23106
Inoki K, Ouyang H, Zhu T et al (2006) TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. https://doi.org/10.1016/j.cell.2006.06.055
Lin S-Y, Li TY, Liu Q et al (2012) Protein phosphorylation-acetylation cascade connects growth factor deprivation to autophagy. Autophagy 8:1385. https://doi.org/10.4161/AUTO.20959
Xu L-Z, Long Z-J, Peng F, et al (2014) Aurora kinase a suppresses metabolic stress-induced autophagic cell death by activating mTOR signaling in breast cancer cells. Oncotarget. https://doi.org/10.18632/oncotarget.2241
Wang SH, Shih YL, Kuo TC et al (2009) Cadmium toxicity toward autophagy through ROS-Activated GSK-3β in mesangial cells. Toxicol Sci. https://doi.org/10.1093/toxsci/kfn266
Lin CJ, Chen TH, Yang LY, Shih CM (2014) Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. https://doi.org/10.1038/cddis.2014.123
Liu L, Li CJ, Lu Y et al (2015) Baclofen mediates neuroprotection on hippocampal CA1 pyramidal cells through the regulation of autophagy under chronic cerebral hypoperfusion. Sci Rep. https://doi.org/10.1038/srep14474
Ha S, Ryu HY, Chung KM et al (2015) Regulation of autophagic cell death by glycogen synthase kinase-3β in adult hippocampal neural stem cells following insulin withdrawal. Mol Brain. https://doi.org/10.1186/s13041-015-0119-9
Wakatsuki S, Tokunaga S, Shibata M, Araki T (2017) GSK3B-mediated phosphorylation of MCL1 regulates axonal autophagy to promote Wallerian degeneration. J Cell Biol. https://doi.org/10.1083/jcb.201606020
Ma XH, Duan WJ, Mo YS et al (2018) Neuroprotective effect of paeoniflorin on okadaic acid-induced tau hyperphosphorylation via calpain/Akt/GSK-3β pathway in SH-SY5Y cells. Brain Res. https://doi.org/10.1016/j.brainres.2018.03.022
Ríos JA, Godoy JA, Inestrosa NC (2018) Wnt3a ligand facilitates autophagy in hippocampal neurons by modulating a novel GSK-3β-AMPK axis. Cell Commun Signal. https://doi.org/10.1186/s12964-018-0227-0
Li D, Li X, Wu J et al (2015) Involvement of the JNK/FOXO3a/Bim pathway in neuronal apoptosis after hypoxic-ischemic brain damage in neonatal rats. PLoS ONE. https://doi.org/10.1371/journal.pone.0132998
Kanamoto T, Mota M, Takeda K et al (2000) Role of apoptosis signal-regulating kinase in regulation of the c-Jun N-terminal kinase pathway and apoptosis in sympathetic neurons. Mol Cell Biol. https://doi.org/10.1128/mcb.20.1.196-204.2000
Venugopal SK, Chen J, Zhang Y et al (2007) Role of MAPK phosphatase-1 in sustained activation of JNK during ethanol-induced apoptosis in hepatocyte-like VL-17A cells. J Biol Chem. https://doi.org/10.1074/jbc.M703729200
Chen Y, Ba L, Huang W et al (2017) Role of carvacrol in cardioprotection against myocardial ischemia/reperfusion injury in rats through activation of MAPK/ERK and Akt/eNOS signaling pathways. Eur J Pharmacol. https://doi.org/10.1016/j.ejphar.2016.11.053
Chang YL, Chen CL, Kuo CL et al (2010) Glycyrrhetinic acid inhibits ICAM-1 expression via blocking JNK and NF-B pathways in TNF-α-activated endothelial cells. Acta Pharmacol Sin. https://doi.org/10.1038/aps.2010.34
Shklover J, Mishnaevski K, Levy-Adam F, Kurant E (2015) JNK pathway activation is able to synchronize neuronal death and glial phagocytosis in Drosophila. Cell Death Dis. https://doi.org/10.1038/cddis.2015.27
Sun Y, Liu W-Z, Liu T et al (2015) Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct 35:600–604. https://doi.org/10.3109/10799893.2015.1030412
Ryan TA, Roper KM, Bond J et al (2018) A MAPK/c-Jun-mediated switch regulates the initial adaptive and cell death responses to mitochondrial damage in a neuronal cell model. Int J Biochem Cell Biol 104:73–86. https://doi.org/10.1016/j.biocel.2018.09.008
Saha RN, Jana M, Pahan K (2007) MAPK p38 regulates transcriptional activity of NF-κB in primary human astrocytes via acetylation of p65. J Immunol. https://doi.org/10.4049/jimmunol.179.10.7101
Papademetrio DL, Lompardía SL, Simunovich T et al (2016) Inhibition of survival pathways MAPK and NF-kB triggers apoptosis in pancreatic ductal adenocarcinoma cells via suppression of autophagy. Target Oncol. https://doi.org/10.1007/s11523-015-0388-3
Santa-Cecília FV, Socias B, Ouidja MO et al (2016) Doxycycline suppresses microglial activation by inhibiting the p38 MAPK and NF-kB signaling pathways. Neurotox Res. https://doi.org/10.1007/s12640-015-9592-2
Lai J, Liu Y, Liu C et al (2017) Indirubin inhibits LPS-induced inflammation via TLR4 abrogation mediated by the NF-kB and MAPK signaling pathways. Inflammation. https://doi.org/10.1007/s10753-016-0447-7
Xing J, Li R, Li N et al (2015) Anti-inflammatory effect of procyanidin B1 on LPS-treated THP1 cells via interaction with the TLR4–MD-2 heterodimer and p38 MAPK and NF-κB signaling. Mol Cell Biochem. https://doi.org/10.1007/s11010-015-2457-4
Kim DC, Quang TH, Oh H, Kim YC (2017) Steppogenin isolated from Cudrania tricuspidata shows antineuroinflammatory effects via NF-κB and MAPK pathways in LPS-stimulated BV2 and primary rat microglial cells. Molecules. https://doi.org/10.3390/molecules22122130
Guolan D, Lingli W, Wenyi H et al (2018) Anti-inflammatory effects of neferine on LPS-induced human endothelium via MAPK, and NF-κβ pathways. Pharmazie 73:541–544. https://doi.org/10.1691/PH.2018.8443
Kotlyarov A, Neininger A, Schubert C et al (1999) (1999) MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis. Nat Cell Biol 12(1):94–97. https://doi.org/10.1038/10061
Fyhrquist N, Matikainen S, Lauerma A (2010) MK2 signaling: lessons on tissue specificity in modulation of inflammation. J Invest Dermatol 130:342–344. https://doi.org/10.1038/JID.2009.372
Neininger A, Kontoyiannis D, Kotlyarov A et al (2002) MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels *. J Biol Chem 277:3065–3068. https://doi.org/10.1074/JBC.C100685200
Neault M, Couteau F, Bonneau É et al (2017) Molecular Regulation of Cellular Senescence by MicroRNAs: Implications in Cancer and Age-Related Diseases. Int Rev Cell Mol Biol 334:27–98. https://doi.org/10.1016/bs.ircmb.2017.04.001
Singh T, Yadav S (2020) Role of microRNAs in neurodegeneration induced by environmental neurotoxicants and aging. Ageing Res Rev 60:101068. https://doi.org/10.1016/J.ARR.2020.101068
Jia H, Qu M, Fan G et al (2020) miR-499-5p suppresses C-reactive protein and provides neuroprotection in hypoxic-ischemic encephalopathy in neonatal rat. Neurosci Res 161:44–50. https://doi.org/10.1016/J.NEURES.2019.12.002
Wang M, Sun H, Yao Y et al (2019) MicroRNA-217/138-5p downregulation inhibits inflammatory response, oxidative stress and the induction of neuronal apoptosis in MPP+-induced SH-SY5Y cells. Am J Transl Res 11:6619
Hu J-J, Qin L-J, Liu Z-Y et al (2020) miR-15a regulates oxygen glucose deprivation/reperfusion (OGD/R)-induced neuronal injury by targeting BDNF. Kaohsiung J Med Sci 36:27–34. https://doi.org/10.1002/KJM2.12136
Zeng Z, Liu Y, Zheng W et al (2019) MicroRNA-129-5p alleviates nerve injury and inflammatory response of Alzheimer’s disease via downregulating SOX6. Cell Cycle 18:3095. https://doi.org/10.1080/15384101.2019.1669388
Shang Y, Dai S, Chen X et al (2019) MicroRNA-93 regulates the neurological function, cerebral edema and neuronal apoptosis of rats with intracerebral hemorrhage through TLR4/NF-κB signaling pathway. Cell Cycle 18:3160–3176. https://doi.org/10.1080/15384101.2019.1670509
Wang J, Wang W, Zhai H (2019) MicroRNA-124 enhances dopamine receptor expression and neuronal proliferation in mouse models of parkinson’s disease via the hedgehog signaling pathway by targeting EDN2. NeuroImmunoModulation. https://doi.org/10.1159/000501339
Ge H, Yan Z, Zhu H, Zhao H (2019) MiR-410 exerts neuroprotective effects in a cellular model of Parkinson’s disease induced by 6-hydroxydopamine via inhibiting the PTEN/AKT/mTOR signaling pathway. Exp Mol Pathol. https://doi.org/10.1016/j.yexmp.2019.05.002
Chen FZ, Zhao Y, Chen HZ (2019) MicroRNA-98 reduces amyloid β-protein production and improves oxidative stress and mitochondrial dysfunction through the Notch signaling pathway via HEY2 in Alzheimer’s disease mice. Int J Mol Med. https://doi.org/10.3892/ijmm.2018.3957
Jing D, Yinzhu L, Jinjing P et al (2018) Targeting ninjurin 2 by miR-764 regulates hydrogen peroxide (H2O2)-induced neuronal cell death. Biochem Biophys Res Commun 505:1180–1188. https://doi.org/10.1016/j.bbrc.2018.09.184
Fu S, Zhang J, Zhang S (2018) Knockdown of miR-429 attenuates Aβ-induced neuronal damage by targeting SOX2 and BCL2 in mouse cortical neurons. Neurochem Res. https://doi.org/10.1007/s11064-018-2643-3
Shanesazzade Z, Peymani M, Ghaedi K, Nasr Esfahani MH (2018) miR-34a/BCL-2 signaling axis contributes to apoptosis in MPP+-induced SH-SY5Y cells. Mol Genet Genomic Med 6:975–981. https://doi.org/10.1002/mgg3.469
Chitnis NS, Pytel D, Bobrovnikova-Marjon E et al (2012) MiR-211 Is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol Cell. https://doi.org/10.1016/j.molcel.2012.08.025
Wang HQ, Yu XD, Liu ZH et al (2011) Deregulated miR-155 promotes Fas-mediated apoptosis in human intervertebral disc degeneration by targeting FADD and caspase-3. J Pathol. https://doi.org/10.1002/path.2931
Hudson RS, Yi M, Esposito D et al (2013) MicroRNA-106b-25 cluster expression is associated with early disease recurrence and targets caspase-7 and focal adhesion in human prostate cancer. Oncogene. https://doi.org/10.1038/onc.2012.424
Fang J, Song X-W, Tian J et al (2012) Overexpression of microRNA-378 attenuates ischemia-induced apoptosis by inhibiting caspase-3 expression in cardiac myocytes. Apoptosis 17:410–423. https://doi.org/10.1007/s10495-011-0683-0
Wu H, Wang F, Hu S et al (2012) MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell Signal. https://doi.org/10.1016/j.cellsig.2012.07.001
Chen H, Zhang Z, Lu Y et al (2017) Downregulation of ULK1 by microRNA-372 inhibits the survival of human pancreatic adenocarcinoma cells. Cancer Sci 108:1811–1819. https://doi.org/10.1111/cas.13315
Li W, Yang Y, Ba Z et al (2017) MicroRNA-93 regulates hypoxia-induced autophagy by targeting ULK1. Oxid Med Cell Longev. https://doi.org/10.1155/2017/2709053
Ju JA, Huang CT, Lan SH et al (2013) Characterization of a colorectal cancer migration and autophagy-related microRNA miR-338-5p and its target gene PIK3C3. Biomarkers Genomic Med. https://doi.org/10.1016/j.bgm.2013.07.006
Zhu H, Wu H, Liu X et al (2009) Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy. https://doi.org/10.4161/auto.9064
Xu R, Liu S, Hong Chen H, Lao L (2016) MicroRNA-30a downregulation contributes to chemoresistance of osteosarcoma cells through activating Beclin-1-mediated autophagy. Oncol Rep. https://doi.org/10.3892/or.2015.4497
Korkmaz G, Le Sage C, Tekirdag KA et al (2012) miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy. https://doi.org/10.4161/auto.8.2.18351
Menghini R, Casagrande V, Marino A et al (2014) MiR-216a: A link between endothelial dysfunction and autophagy. Cell Death Dis. https://doi.org/10.1038/cddis.2013.556
Hou W, Song L, Zhao Y et al (2017) Inhibition of beclin-1-mediated autophagy by microRNA-17-5p enhanced the radiosensitivity of glioma cells. Oncol Res. https://doi.org/10.3727/096504016X14719078133285
Mikhaylova O, Stratton Y, Hall D et al (2012) VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell. https://doi.org/10.1016/j.ccr.2012.02.019
Frankel LB, Wen J, Lees M et al (2011) MicroRNA-101 is a potent inhibitor of autophagy. EMBO J. https://doi.org/10.1038/emboj.2011.331
Li W, Yang Y, Hou X et al (2016) MicroRNA-495 regulates starvation-induced autophagy by targeting ATG3. FEBS Lett. https://doi.org/10.1002/1873-3468.12108
Wang N, Yang L, Zhang H et al (2018) MicroRNA-9a-5p alleviates ischemia injury after focal cerebral ischemia of the Rat by targeting ATG5-mediated autophagy. Cell Physiol Biochem. https://doi.org/10.1159/000486224
Yan L, Shi E, Jiang X et al (2019) Inhibition of MicroRNA-204 conducts neuroprotection against spinal cord ischemia. Ann Thorac Surg. https://doi.org/10.1016/j.athoracsur.2018.07.082
Liu P, Liu P, Wang Z et al (2018) Inhibition of MicroRNA-96 ameliorates cognitive impairment and inactivation autophagy following chronic cerebral hypoperfusion in the rat. Cell Physiol Biochem. https://doi.org/10.1159/000492844
Gong X, Wang H, Ye Y et al (2016) miR-124 regulates cell apoptosis and autophagy in dopaminergic neurons and protects them by regulating AMPK/mTOR pathway in Parkinson’s disease. Am J Transl Res 8:2127
Li W, Jiang Y, Wang Y et al (2018) MiR-181b regulates autophagy in a model of Parkinson’s disease by targeting the PTEN/Akt/mTOR signaling pathway. Neurosci Lett 675:83–88. https://doi.org/10.1016/J.NEULET.2018.03.041
Wen Z, Zhang J, Tang P et al (2018) Overexpression of miR-185 inhibits autophagy and apoptosis of dopaminergic neurons by regulating the AMPK/mTOR signaling pathway in Parkinson’s disease. Mol Med Rep 17:131. https://doi.org/10.3892/MMR.2017.7897
Sun S, Han X, Li X et al (2018) MicroRNA-212-5p prevents dopaminergic neuron death by inhibiting SIRT2 in MPTP-induced mouse model of parkinson’s disease. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2018.00381
Yao L, Zhu Z, Wu J et al (2019) MicroRNA-124 regulates the expression of p62/p38 and promotes autophagy in the inflammatory pathogenesis of Parkinson’s disease. FASEB J 33:8648–8665. https://doi.org/10.1096/fj.201900363R
Wang H, Ye Y, Zhu Z et al (2016) MiR-124 regulates apoptosis and autophagy process in MPTP model of Parkinson’s disease by targeting to bim. Brain Pathol. https://doi.org/10.1111/bpa.12267
Zhao XH, Wang YB, Yang J et al (2019) MicroRNA-326 suppresses iNOS expression and promotes autophagy of dopaminergic neurons through the JNK signaling by targeting XBP1 in a mouse model of Parkinson’s disease. J Cell Biochem. https://doi.org/10.1002/jcb.28761
Sun L, Zhao M, Wang Y et al (2017) Neuroprotective effects of miR-27a against traumatic brain injury via suppressing FoxO3a-mediated neuronal autophagy. Biochem Biophys Res Commun. https://doi.org/10.1016/j.bbrc.2016.12.001
Sun L, Liu A, Zhang J et al (2018) miR-23b improves cognitive impairments in traumatic brain injury by targeting ATG12-mediated neuronal autophagy. Behav Brain Res. https://doi.org/10.1016/j.bbr.2016.09.020
Ruan Z, Li Y, He R, Li X (2021) Inhibition of microRNA-10b-5p up-regulates HOXD10 to attenuate Alzheimer’s disease in rats via the Rho/ROCK signalling pathway. J Drug Target. https://doi.org/10.1080/1061186X.2020.1864739
Tian Z, Dong Q, Wu T, Guo J (2021) MicroRNA-20b-5p aggravates neuronal apoptosis induced by β-Amyloid via down-regulation of Ras homolog family member C in Alzheimer’s disease. Neurosci Lett. https://doi.org/10.1016/j.neulet.2020.135542
Li J, Li D, Zhou H, et al (2020) MicroRNA-338-5p alleviates neuronal apoptosis via directly targeting BCL2L11 in APP/PS1 mice. Aging (Albany NY) 12:20728–20742. https://doi.org/10.18632/aging.104005
Liang C, Mu Y, Tian H et al (2021) MicroRNA-140 silencing represses the incidence of Alzheimer’s disease. Neurosci Lett. https://doi.org/10.1016/j.neulet.2021.135674
Chen M-L, Hong C-G, Yue T et al (2021) Inhibition of miR-331-3p and miR-9-5p ameliorates Alzheimer’s disease by enhancing autophagy. Theranostics 11:2395–2409. https://doi.org/10.7150/thno.47408
Zhang Y, Lv X, Liu C et al (2016) MiR-214-3p attenuates cognition defects via the inhibition of autophagy in SAMP8 mouse model of sporadic Alzheimer’s disease. Neurotoxicology. https://doi.org/10.1016/j.neuro.2016.07.004
Zhang Y, Liu C, Wang J et al (2016) MiR-299-5p regulates apoptosis through autophagy in neurons and ameliorates cognitive capacity in APPswe/PS1dE9 mice. Sci Rep 6:24566. https://doi.org/10.1038/srep24566
Lv Q, Zhong Z, Hu B et al (2021) MicroRNA-3473b regulates the expression of TREM2/ULK1 and inhibits autophagy in inflammatory pathogenesis of Parkinson disease. J Neurochem. https://doi.org/10.1111/jnc.15299
Zhou T, Lin D, Chen Y et al (2019) α-synuclein accumulation in SH-SY5Y cell impairs autophagy in microglia by exosomes overloading miR-19a-3p. Epigenomics 11:1661–1677. https://doi.org/10.2217/epi-2019-0222
Qian C, Ye Y, Mao H et al (2019) Downregulated lncRNA-SNHG1 enhances autophagy and prevents cell death through the miR-221/222 /p27/mTOR pathway in Parkinson’s disease. Exp Cell Res 384:111614. https://doi.org/10.1016/j.yexcr.2019.111614
Qazi TJ, Lu J, Duru L et al (2021) Upregulation of mir-132 induces dopaminergic neuronal death via activating SIRT1/P53 pathway. Neurosci Lett 740:135465. https://doi.org/10.1016/j.neulet.2020.135465
Choi DC, Yoo M, Kabaria S, Junn E (2018) MicroRNA-7 facilitates the degradation of alpha-synuclein and its aggregates by promoting autophagy. Neurosci Lett. https://doi.org/10.1016/j.neulet.2018.05.009
Li W, Jiang Y, Wang Y et al (2018) MiR-181b regulates autophagy in a model of Parkinson’s disease by targeting the PTEN/Akt/mTOR signaling pathway. Neurosci Lett. https://doi.org/10.1016/j.neulet.2018.03.041
Feng Z, Zhang L, Wang S, Hong Q (2020) Circular RNA circDLGAP4 exerts neuroprotective effects via modulating miR-134-5p/CREB pathway in Parkinson’s disease. Biochem Biophys Res Commun 522:388–394. https://doi.org/10.1016/j.bbrc.2019.11.102
Chiu C-C, Yeh T-H, Chen R-S et al (2019) Upregulated expression of MicroRNA-204-5p leads to the death of dopaminergic cells by targeting DYRK1A-mediated apoptotic signaling cascade. Front Cell Neurosci 13:399. https://doi.org/10.3389/fncel.2019.00399
Liu Y, Song Y, Zhu X (2017) MicroRNA-181a regulates apoptosis and autophagy process in Parkinson’s disease by inhibiting p38 mitogen-activated protein kinase (MAPK)/c-Jun N-terminal kinases (JNK) signaling pathways. Med Sci Monit https://doi.org/10.12659/MSM.900218
Wen Z, Zhang J, Tang P et al (2018) Overexpression of miR-185 inhibits autophagy and apoptosis of dopaminergic neurons by regulating the AMPK/mTOR signaling pathway in Parkinson’s disease. Mol Med Rep. https://doi.org/10.3892/mmr.2017.7897
Zhao J, Yang M, Li Q et al (2020) MiR-132-5p regulates apoptosis and autophagy in MPTP model of Parkinson’s disease by targeting ULK1. NeuroReport. https://doi.org/10.1097/WNR.0000000000001494
Li C, Chen Y, Chen X et al (2017) Downregulation of microRNA-193b-3p promotes autophagy and cell survival by targeting TSC1/mTOR signaling in NSC-34 cells. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2017.00160
Yu SJ, Yu MJ, Bu ZQ et al (2021) MicroRNA-670 aggravates cerebral ischemia/reperfusion injury via the Yap pathway. Neural Regen Res. https://doi.org/10.4103/1673-5374.300455
Dai Q, Ma Y, Xu Z et al (2021) Downregulation of circular RNA HECTD1 induces neuroprotection against ischemic stroke through the microRNA-133b/TRAF3 pathway. Life Sci 264:118626. https://doi.org/10.1016/j.lfs.2020.118626
Liu W, Miao Y, Zhang L et al (2020) MiR-211 protects cerebral ischemia/reperfusion injury by inhibiting cell apoptosis. Bioengineered 11:189–200. https://doi.org/10.1080/21655979.2020.1729322
Deng Z, Ou H, Ren F et al (2020) LncRNA SNHG14 promotes OGD/R-induced neuron injury by inducing excessive mitophagy via miR-182-5p/BINP3 axis in HT22 mouse hippocampal neuronal cells. Biol Res 53:38. https://doi.org/10.1186/s40659-020-00304-4
Li B, Huang Z, Meng J et al (2020) MiR-202-5p attenuates neurological deficits and neuronal injury in MCAO model rats and OGD-induced injury in Neuro-2a cells by targeting eIF4E-mediated induction of autophagy and inhibition of Akt/GSK-3β pathway. Mol Cell Probes 51:101497. https://doi.org/10.1016/j.mcp.2019.101497
Chen X, Lin S, Gu L et al (2019) Inhibition of miR-497 improves functional outcome after ischemic stroke by enhancing neuronal autophagy in young and aged rats. Neurochem Int. https://doi.org/10.1016/j.neuint.2019.01.005
Wang P, Liang J, Li Y et al (2014) Down-regulation of miRNA-30a alleviates cerebral ischemic injury through enhancing Beclin 1-mediated autophagy. Neurochem Res. https://doi.org/10.1007/s11064-014-1310-6
Wu Y, Gao Z, Zhang J (2020) Transcription factor E2F1 aggravates neurological injury in ischemic stroke via microRNA-122-targeted sprouty2. Neuropsychiatr Dis Treat 16:2633–2647. https://doi.org/10.2147/NDT.S271320
Tian F, Yuan C, Yue H (2018) MiR-138/SIRT1 axis is implicated in impaired learning and memory abilities of cerebral ischemia/reperfusion injured rats. Exp Cell Res. https://doi.org/10.1016/j.yexcr.2018.03.042
Zhou J, Wu J-S, Yan Y, et al (2020) MiR-199a modulates autophagy and inflammation in rats with cerebral infarction via regulating mTOR expression. Eur Rev Med Pharmacol Sci 24:6338–6345. https://doi.org/10.26355/eurrev_202006_21532
Wang D, Chen F, Fang B et al (2020) MiR-128-3p alleviates spinal cord ischemia/reperfusion injury associated neuroinflammation and cellular apoptosis via SP1 suppression in rat. Front Neurosci. https://doi.org/10.3389/fnins.2020.609613
Zhou Z, Hu B, Lyu Q et al (2020) miR-384-5p promotes spinal cord injury recovery in rats through suppressing of autophagy and endoplasmic reticulum stress. Neurosci Lett 727:134937. https://doi.org/10.1016/j.neulet.2020.134937
Wang J, Rong Y, Ji C et al (2020) MicroRNA-421-3p-abundant small extracellular vesicles derived from M2 bone marrow-derived macrophages attenuate apoptosis and promote motor function recovery via inhibition of mTOR in spinal cord injury. J Nanobiotechnology 18:72. https://doi.org/10.1186/s12951-020-00630-5
Li X, Lou X, Xu S et al (2018) Knockdown of miR-372 inhibits nerve cell apoptosis induced by spinal cord ischemia/reperfusion injury via enhancing autophagy by up-regulating beclin-1. J Mol Neurosci. https://doi.org/10.1007/s12031-018-1179-y
Li D, Huang S, Yin Z et al (2019) Increases in miR-124-3p in microglial exosomes confer neuroprotective effects by targeting FIP200-mediated neuronal autophagy following traumatic brain injury. Neurochem Res. https://doi.org/10.1007/s11064-019-02825-1
Li D, Huang S, Zhu J, et al (2019) Exosomes from MiR-21–5p-increased neurons play a role in neuroprotection by suppressing rab11a-mediated neuronal autophagy in vitro after traumatic brain injury. Med Sci Monit. https://doi.org/10.12659/MSM.915727
Wang L, Song LF, Chen XY et al (2019) MiR-181b inhibits P38/JNK signaling pathway to attenuate autophagy and apoptosis in juvenile rats with kainic acid-induced epilepsy via targeting TLR4. CNS Neurosci Ther. https://doi.org/10.1111/cns.12991
Wen X, Han XR, Wang YJ et al (2018) MicroRNA-421 suppresses the apoptosis and autophagy of hippocampal neurons in epilepsy mice model by inhibition of the TLR/MYD88 pathway. J Cell Physiol. https://doi.org/10.1002/jcp.26498
Fei S, Cao L, Li S (2021) microRNA-139-5p alleviates neurological deficit in hypoxic-ischemic brain damage via HDAC4 depletion and BCL-2 activation. Brain Res Bull. https://doi.org/10.1016/j.brainresbull.2020.12.020
Du L, Jiang Y, Sun Y (2021) Astrocyte-derived exosomes carry microRNA-17-5p to protect neonatal rats from hypoxic-ischemic brain damage via inhibiting BNIP-2 expression. Neurotoxicology. https://doi.org/10.1016/j.neuro.2020.12.006
Li R, Jin Y, Li Q et al (2018) MiR-93-5p targeting PTEN regulates the NMDA-induced autophagy of retinal ganglion cells via AKT/mTOR pathway in glaucoma. Biomed Pharmacother. https://doi.org/10.1016/j.biopha.2018.01.044
Wang B, Li Y, You C (2021) miR-129-3p targeting of MCU protects against glucose fluctuation-mediated neuronal damage via a mitochondrial-dependent intrinsic apoptotic pathway. Diabetes Metab Syndr Obes Targets Ther. https://doi.org/10.2147/dmso.s285179
Xu J, Sun M, Li X et al (2021) MicroRNA expression profiling after recurrent febrile seizures in rat and emerging role of miR-148a-3p/SYNJ1 axis. Sci Rep. https://doi.org/10.1038/s41598-020-79543-0
Wen B, He C, Zhang Q et al (2020) Overexpression of microRNA-221 promotes the differentiation of stem cells from human exfoliated deciduous teeth to neurons through activation of Wnt/β-catenin pathway via inhibition of CHD8. Cell Cycle 19:3231–3248. https://doi.org/10.1080/15384101.2020.1816308
Guo D, Ma J, Yan L et al (2017) Down-Regulation of Lncrna MALAT1 Attenuates Neuronal Cell Death Through Suppressing Beclin1-Dependent Autophagy by Regulating Mir-30a in Cerebral Ischemic Stroke. Cell Physiol Biochem 43:182–194. https://doi.org/10.1159/000480337
Wu Q, Yi X (2018) Down-regulation of long noncoding RNA MALAT1 protects hippocampal neurons against excessive autophagy and apoptosis via the PI3K/Akt signaling pathway in rats with epilepsy. J Mol Neurosci 65:234–245. https://doi.org/10.1007/s12031-018-1093-3
Fu C-H, Lai F-F, Chen S et al (2020) Silencing of long non-coding RNA CRNDE promotes autophagy and alleviates neonatal hypoxic-ischemic brain damage in rats. Mol Cell Biochem 472:1–8. https://doi.org/10.1007/s11010-020-03754-2
Yi M, Dai X, Li Q et al (2019) Downregulated lncRNA CRNDE contributes to the enhancement of nerve repair after traumatic brain injury in rats. Cell Cycle 18:2332–2343. https://doi.org/10.1080/15384101.2019.1647024
Yin W-L, Yin W-G, Huang B-S, Wu L-X (2019) LncRNA SNHG12 inhibits miR-199a to upregulate SIRT1 to attenuate cerebral ischemia/reperfusion injury through activating AMPK signaling pathway. Neurosci Lett 690:188–195. https://doi.org/10.1016/j.neulet.2018.08.026
Cao Y, Pan L, Zhang X et al (2020) LncRNA SNHG3 promotes autophagy-induced neuronal cell apoptosis by acting as a ceRNA for miR-485 to up-regulate ATG7 expression. Metab Brain Dis 35:1361–1369. https://doi.org/10.1007/s11011-020-00607-1
Guo X, Wang Y, Zheng D et al (2021) LncRNA-MIAT promotes neural cell autophagy and apoptosis in ischemic stroke by up-regulating REDD1. Brain Res. https://doi.org/10.1016/j.brainres.2021.147436
Zhou Y, Ge Y, Liu Q et al (2021) LncRNA BACE1-AS promotes autophagy-mediated neuronal damage through the miR-214-3p/ATG5 signalling axis in alzheimer’s disease. Neuroscience 455:52–64. https://doi.org/10.1016/j.neuroscience.2020.10.028
Zhao J, Li H, Chang N (2020) LncRNA HOTAIR promotes MPP+-induced neuronal injury in Parkinson’s disease by regulating the miR-874–5p/ATG10 axis. EXCLI J 19:1141–1153. https://doi.org/10.17179/excli2020-2286
Fan Y, Zhao X, Lu K, Cheng G (2020) LncRNA BDNF-AS promotes autophagy and apoptosis in MPTP-induced Parkinson’s disease via ablating microRNA-125b-5p. Brain Res Bull 157:119–127. https://doi.org/10.1016/j.brainresbull.2020.02.003
Wang X, Zhang M, Liu H (2019) LncRNA17A regulates autophagy and apoptosis of SH-SY5Y cell line as an in vitro model for Alzheimer’s disease. Biosci Biotechnol Biochem 83:609–621. https://doi.org/10.1080/09168451.2018.1562874
Li Z, Hao S, Yin H et al (2016) Autophagy ameliorates cognitive impairment through activation of PVT1 and apoptosis in diabetes mice. Behav Brain Res 305:265–277. https://doi.org/10.1016/j.bbr.2016.03.023
Zhou Z, Xu H, Liu B et al (2019) Suppression of lncRNA RMRP ameliorates oxygen-glucose deprivation/re-oxygenation-induced neural cells injury by inhibiting autophagy and PI3K/Akt/mTOR-mediated apoptosis. Biosci Rep. https://doi.org/10.1042/BSR20181367
Ren X-D, Wan C-X, Niu Y-L (2019) Overexpression of lncRNA TCTN2 protects neurons from apoptosis by enhancing cell autophagy in spinal cord injury. FEBS Open Bio 9:1223–1231. https://doi.org/10.1002/2211-5463.12651
Sun W, Li Y-N, Ye J-F, et al (2018) MEG3 is involved in the development of glaucoma through promoting the autophagy of retinal ganglion cells. Eur Rev Med Pharmacol Sci 22:2534–2540. https://doi.org/10.26355/eurrev_201805_14942
Peng T, Liu X, Wang J et al (2019) Long noncoding RNA HAGLROS regulates apoptosis and autophagy in Parkinson’s disease via regulating miR-100/ATG10 axis and PI3K/Akt/mTOR pathway activation. Artif Cells Nanomed Biotechnol 47:2764–2774. https://doi.org/10.1080/21691401.2019.1636805
Aminyavari S, Zahmatkesh M, Farahmandfar M et al (2019) Protective role of Apelin-13 on amyloid β25–35-induced memory deficit; Involvement of autophagy and apoptosis process. Prog Neuro-Psychopharmacol Biol Psychiatry 89:322–334. https://doi.org/10.1016/j.pnpbp.2018.10.005
Venkatesan R, Park YU, Ji E et al (2017) Malathion increases apoptotic cell death by inducing lysosomal membrane permeabilization in N2a neuroblastoma cells: a model for neurodegeneration in Alzheimer’s disease. Cell Death Discov 3:17007. https://doi.org/10.1038/cddiscovery.2017.7
Cao Y, Li Q, Liu L et al (2019) Modafinil protects hippocampal neurons by suppressing excessive autophagy and apoptosis in mice with sleep deprivation. Br J Pharmacol 176:1282–1297. https://doi.org/10.1111/bph.14626
Guo Y, Wang F, Li H et al (2018) Metformin protects against spinal cord injury by regulating autophagy via the mTOR signaling pathway. Neurochem Res 43:1111–1117. https://doi.org/10.1007/s11064-018-2525-8
Ren Z, Wang C, Wang T et al (2019) Ganoderma lucidum extract ameliorates MPTP-induced parkinsonism and protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis. Acta Pharmacol Sin. https://doi.org/10.1038/s41401-018-0077-
Singh S, Singh AK, Garg G, Rizvi SI (2018) Fisetin as a caloric restriction mimetic protects rat brain against aging induced oxidative stress, apoptosis and neurodegeneration. Life Sci 193:171–179. https://doi.org/10.1016/j.lfs.2017.11.004
Yang W, Tian Z-K, Yang H-X et al (2019) Fisetin improves lead-induced neuroinflammation, apoptosis and synaptic dysfunction in mice associated with the AMPK/SIRT1 and autophagy pathway. Food Chem Toxicol 134:110824. https://doi.org/10.1016/j.fct.2019.110824
Singh AK, Singh S, Tripathi VK et al (2018) Rapamycin confers neuroprotection against aging-induced oxidative stress, mitochondrial dysfunction, and neurodegeneration in old rats through activation of autophagy. Rejuvenation Res 22:60–70. https://doi.org/10.1089/rej.2018.2070
Farkhondeh T, Pourbagher-Shahri AM, Ashrafizadeh M et al (2020) Green tea catechins inhibit microglial activation which prevents the development of neurological disorders. Neural Regen Res 15:1792. https://doi.org/10.4103/1673-5374.280300
Xu Y, Liu S, Zhu L et al (2021) Green tea protects against hippocampal neuronal apoptosis in diabetic encephalopathy by inhibiting JNK/MLCK signaling. Mol Med Rep 24:1–13. https://doi.org/10.3892/MMR.2021.12214
Fu G, Wang H, Cai Y et al (2018) Theaflavin alleviates inflammatory response and brain injury induced by cerebral hemorrhage via inhibiting the nuclear transcription factor kappa β-related pathway in rats. Drug Des Devel Ther 12:1609. https://doi.org/10.2147/DDDT.S164324
Zhou R, Li X, Li L, Zhang H (2019) Theaflavins alleviate sevoflurane-induced neurocytotoxicity via Nrf2 signaling pathway. Int J Neurosci 130:1–8. https://doi.org/10.1080/00207454.2019.1667788
Wang K, Chen Z, Huang J et al (2017) Naringenin prevents ischaemic stroke damage via anti-apoptotic and anti-oxidant effects. Clin Exp Pharmacol Physiol 44:862–871. https://doi.org/10.1111/1440-1681.12775
Wang K, Chen Z, Huang L et al (2017) Naringenin reduces oxidative stress and improves mitochondrial dysfunction via activation of the Nrf2/ARE signaling pathway in neurons. Int J Mol Med 40:1582–1590. https://doi.org/10.3892/IJMM.2017.3134
Cui J, Wang G, Kandhare AD et al (2018) Neuroprotective effect of naringin, a flavone glycoside in quinolinic acid-induced neurotoxicity: Possible role of PPAR-γ, Bax/Bcl-2, and caspase-3. Food Chem Toxicol 121:95–108. https://doi.org/10.1016/J.FCT.2018.08.028
Meng X, Fu M, Wang S et al (2021) Naringin ameliorates memory deficits and exerts neuroprotective effects in a mouse model of Alzheimer’s disease by regulating multiple metabolic pathways. Mol Med Rep 23:1–13. https://doi.org/10.3892/MMR.2021.11971
Guo J, Yang G, He Y et al (2020) Involvement of α7nAChR in the protective effects of genistein against β-amyloid-induced oxidative stress in neurons via a PI3K/Akt/Nrf2 pathway-related mechanism. Cell Mol Neurobiol 412(41):377–393. https://doi.org/10.1007/S10571-020-01009-8
Jiang T, Wang X, Ding C, Du X (2017) Genistein attenuates isoflurane-induced neurotoxicity and improves impaired spatial learning and memory by regulating cAMP/CREB and BDNF-TrkB-PI3K/Akt signaling. Korean J Physiol Pharmacol 21:579–589. https://doi.org/10.4196/KJPP.2017.21.6.579
Subedi L, Ji E, Shin D et al (2017) Equol, a dietary daidzein gut metabolite attenuates microglial activation and potentiates neuroprotection in vitro. Nutr 9:207. https://doi.org/10.3390/NU9030207
Tsai M-C, Lin S-H, Hidayah K, Lin C-I (2019) Equol pretreatment protection of SH-SY5Y cells against Aβ (25–35)-induced cytotoxicity and cell-cycle reentry via sustaining estrogen receptor alpha expression. Nutr 11:2356. https://doi.org/10.3390/NU11102356
Anusha C, Sumathi T, Joseph LD (2017) Protective role of apigenin on rotenone induced rat model of Parkinson’s disease: Suppression of neuroinflammation and oxidative stress mediated apoptosis. Chem Biol Interact 269:67–79. https://doi.org/10.1016/J.CBI.2017.03.016
Han Y, Zhang T, Su J et al (2017) Apigenin attenuates oxidative stress and neuronal apoptosis in early brain injury following subarachnoid hemorrhage. J Clin Neurosci 40:157–162. https://doi.org/10.1016/J.JOCN.2017.03.003
Kim Y, Kim J, He M et al (2021) Apigenin ameliorates scopolamine-induced cognitive dysfunction and neuronal damage in mice. Mol 26:5192. https://doi.org/10.3390/MOLECULES26175192
Liu S, Su Y, Sun B et al (2020) Luteolin protects against CIRI, potentially via regulation of the SIRT3/AMPK/mTOR signaling pathway. Neurochem Res 4510(45):2499–2515. https://doi.org/10.1007/S11064-020-03108-W
Tan X, Yang Y, Xu J et al (2020) Luteolin exerts neuroprotection via modulation of the p62/Keap1/Nrf2 pathway in intracerebral hemorrhage. Front Pharmacol. https://doi.org/10.3389/FPHAR.2019.01551
Li L, Zhou R, Lv H et al (2021) Inhibitive effect of luteolin on sevoflurane-induced neurotoxicity through activation of the autophagy pathway by HMOX1. ACS Chem Neurosci. https://doi.org/10.1021/ACSCHEMNEURO.1C00157
Peruru DS (2021) Therapeutic potential of diosmin, a citrus flavonoid against arsenic-induced neurotoxicity via suppression of NOX 4 and its subunits. Indian J Pharmacol 53:132. https://doi.org/10.4103/IJP.IJP_837_19
Morroni F, Sita G, Graziosi A, et al (2018) Neuroprotective effect of caffeic acid phenethyl ester in a mouse model of alzheimer’s disease involves Nrf2/HO-1 pathway. Aging Dis 9:605. https://doi.org/10.14336/AD.2017.0903
Chandrasekhar Y, Phani Kumar G, Ramya EM, Anilakumar KR (2018) Gallic acid protects 6-OHDA induced neurotoxicity by attenuating oxidative stress in human dopaminergic cell line. Neurochem Res 43:1150–1160. https://doi.org/10.1007/S11064-018-2530-Y
Youn K, Jun M (2020) Geraniin Protects PC12 Cells Against Aβ25–35-Mediated Neuronal Damage: Involvement of NF-κB and MAPK Signaling Pathways. https://home.liebertpub.com/jmf 23:928–937. https://doi.org/10.1089/JMF.2019.4613
Huang J, Xiao L, Wei J et al (2017) Protective effect of arctigenin on ethanol-induced neurotoxicity in PC12 cells. Mol Med Rep 15:2235–2240. https://doi.org/10.3892/MMR.2017.6222
Liang S, Zheng Y, Lei L et al (2020) Corydalis edulis total alkaloids (CETA) ameliorates cognitive dysfunction in rat model of Alzheimer disease through regulation of the antioxidant stress and MAP2/NF-κB. J Ethnopharmacol 251:112540. https://doi.org/10.1016/J.JEP.2019.112540
Li Z-Q, Zhang F, Shi J-S (2022) Potential neuroprotection by Dendrobium nobile Lindl alkaloid in Alzheimer’s disease models. Neural Regen Res 17:972. https://doi.org/10.4103/1673-5374.324824
Zheng M, Chen M, Liu C et al (2021) Alkaloids extracted from Uncaria rhynchophylla demonstrate neuroprotective effects in MPTP-induced experimental parkinsonism by regulating the PI3K/Akt/mTOR signaling pathway. J Ethnopharmacol 266:113451. https://doi.org/10.1016/J.JEP.2020.113451
Liu J, Zhu T, Niu Q et al (2020) Dendrobium nobile alkaloids protects against H2O2-induced neuronal injury by suppressing JAK–STATs pathway activation in N2A cells. Biol Pharm Bull 43:716–724. https://doi.org/10.1248/BPB.B19-01083
Lee D, Choi HG, Hwang JH et al (2020) Neuroprotective effect of tricyclic pyridine alkaloids from fusarium lateritium SSF2, against glutamate-induced oxidative stress and apoptosis in the HT22 hippocampal neuronal cell line. Antioxidants 9:1115. https://doi.org/10.3390/ANTIOX9111115
Liu D, Dong Z, Xiang F et al (2019) (2019) Dendrobium alkaloids promote neural function after cerebral ischemia-reperfusion injury through inhibiting pyroptosis induced neuronal death in both in vivo and in vitro models. Neurochem Res 452(45):437–454. https://doi.org/10.1007/S11064-019-02935-W
Li Z, Jiang T, Lu Q et al (2019) Berberine attenuated the cytotoxicity induced by t-BHP via inhibiting oxidative stress and mitochondria dysfunction in PC-12 cells. Cell Mol Neurobiol. https://doi.org/10.1007/s10571-019-00756-7
Wang X, Zhang R, Lin Y, Shi P (2020) Inhibition of NF-kappaB might enhance the protective role of roflupram on SH-SY5Y cells under amyloid beta stimulation via PI3K/AKT/mTOR signaling pathway. Int J Neurosci. https://doi.org/10.1080/00207454.2020.1759588
Qi Y, Cheng X, Gong G et al (2020) Synergistic neuroprotective effect of schisandrin and nootkatone on regulating inflammation, apoptosis and autophagy via the PI3K/AKT pathway. Food Funct 11:2427–2438. https://doi.org/10.1039/c9fo02927c
Gugliandolo A, Pollastro F, Bramanti P, Mazzon E (2020) Cannabidiol exerts protective effects in an in vitro model of Parkinson’s disease activating AKT/mTOR pathway. Fitoterapia 143:104553. https://doi.org/10.1016/j.fitote.2020.104553
Li P, Li X, Yao L et al (2020) Soybean isoflavones prevent atrazine-induced neurodegenerative damage by inducing autophagy. Ecotoxicol Environ Saf 190:110065. https://doi.org/10.1016/J.ECOENV.2019.110065
Farmer K, Abd-Elrahman KS, Derksen A et al (2020) mGluR5 allosteric modulation promotes neurorecovery in a 6-OHDA-toxicant model of parkinson’s disease. Mol Neurobiol. https://doi.org/10.1007/s12035-019-01818-z
Abd-Elrahman KS, Ferguson SSG (2019) Modulation of mTOR and CREB pathways following mGluR5 blockade contribute to improved Huntington’s pathology in zQ175 mice. Mol Brain. https://doi.org/10.1186/s13041-019-0456-1
Bai H, Ding Y, Li X et al (2020) Polydatin protects SH-SY5Y in models of Parkinson’s disease by promoting Atg5-mediated but parkin-independent autophagy. Neurochem Int. https://doi.org/10.1016/j.neuint.2020.104671
Salama RM, Abdel-Latif GA, Abbas SS et al (2020) Neuroprotective effect of crocin against rotenone-induced Parkinson’s disease in rats: Interplay between PI3K/Akt/mTOR signaling pathway and enhanced expression of miRNA-7 and miRNA-221. Neuropharmacology. https://doi.org/10.1016/j.neuropharm.2019.107900
Sun Y, Jiang X, Pan R et al (2020) Escins isolated from aesculus chinensis bge promote the autophagic degradation of mutant huntingtin and inhibit its induced apoptosis in HT22 cells. Front Pharmacol. https://doi.org/10.3389/fphar.2020.00116
Wang M, Hua X, Niu H et al (2019) Cornel iridoid glycoside protects against white matter lesions induced by cerebral ischemia in rats via activation of the brain-derived neurotrophic factor/neuregulin-1 pathway. Neuropsychiatr Dis Treat 15:3327–3340. https://doi.org/10.2147/NDT.S228417
Wang P, Lu Y, Han D et al (2019) Neuroprotection by nicotinamide mononucleotide adenylyltransferase 1 with involvement of autophagy in an aged rat model of transient cerebral ischemia and reperfusion. Brain Res. https://doi.org/10.1016/j.brainres.2019.146391
Bellozi PMQ, Gomes GF, De Oliveira LR et al (2019) NVP-BEZ235 (dactolisib) has protective effects in a transgenic mouse model of Alzheimer’s disease. Front Pharmacol. https://doi.org/10.3389/fphar.2019.01345
Cao R, Li L, Ying Z et al (2019) A small molecule protects mitochondrial integrity by inhibiting mTOR activity. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1911246116
Suresh SN, Manjithaya R (2019) A small molecule autophagy inducer exerts cytoprotection against α-synuclein toxicity. Eur J Pharmacol. https://doi.org/10.1016/j.ejphar.2019.172635
Yin P, Wang X, Wang S et al (2019) Maresin 1 improves cognitive decline and ameliorates inflammation in a mouse model of alzheimer’s disease. Front Cell Neurosci. https://doi.org/10.3389/FNCEL.2019.00466
Fan L, Qiu X, Zhu Z et al (2019) Nitazoxanide, an anti-parasitic drug, efficiently ameliorates learning and memory impairments in AD model mice. Acta Pharmacol Sin. https://doi.org/10.1038/s41401-019-0220-1
Zhou H, Shao M, Guo B et al (2019) Tetramethylpyrazine analogue T-006 promotes the clearance of alpha-synuclein by enhancing proteasome activity in parkinson’s disease models. Neurotherapeutics. https://doi.org/10.1007/s13311-019-00759-8
Schreiber KH, Arriola Apelo SI, Yu D et al (2019) A novel rapamycin analog is highly selective for mTORC1 in vivo. Nat Commun. https://doi.org/10.1038/s41467-019-11174-0
Wang L, Jin G, Yu H et al (2019) Protective effect of tenuifolin against alzheimer’s disease. Neurosci Lett 705:195–201. https://doi.org/10.1016/j.neulet.2019.04.045
Chen J, Long Z, Li Y et al (2019) Alteration of the Wnt/GSK3β/β-catenin signalling pathway by rapamycin ameliorates pathology in an Alzheimer’s disease model. Int J Mol Med. https://doi.org/10.3892/ijmm.2019.4198
Sun Q, Wei LL, Zhang M et al (2019) Rapamycin inhibits activation of AMPK-mTOR signaling pathway-induced Alzheimer’s disease lesion in hippocampus of rats with type 2 diabetes mellitus. Int J Neurosci. https://doi.org/10.1080/00207454.2018.1491571
Tramutola A, Lanzillotta C, Barone E et al (2018) Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl Neurodegener. https://doi.org/10.1186/s40035-018-0133-9
Singh AK, Kashyap MP, Tripathi VK et al (2017) Neuroprotection through rapamycin-induced activation of autophagy and PI3K/Akt1/mTOR/CREB signaling against amyloid-β-induced oxidative stress, synaptic/neurotransmission dysfunction, and neurodegeneration in adult rats. Mol Neurobiol. https://doi.org/10.1007/s12035-016-0129-3
Liu YC, Gao XX, Chen L, You X (2017) Rapamycin suppresses Aβ25–35- or LPS-induced neuronal inflammation via modulation of NF-κB signaling. Neuroscience. https://doi.org/10.1016/j.neuroscience.2017.05.005
Chen S, Cai F, Wang J et al (2019) Salidroside protects SHSY5Y from pathogenic alphasynuclein by promoting cell autophagy via mediation of mTOR/p70S6K signaling. Mol Med Rep 20:529–538. https://doi.org/10.3892/mmr.2019.10285
Chen Y, Xu S, Wang N et al (2019) Dynasore suppresses mTORC1 activity and induces autophagy to regulate the clearance of protein aggregates in neurodegenerative diseases. Neurotox Res. https://doi.org/10.1007/s12640-019-00027-9
Lee HJ, Lee JO, Lee YW et al (2019) Lif, a novel myokine, protects against amyloid-beta-induced neurotoxicity via akt-mediated autophagy signaling in hippocampal cells. Int J Neuropsychopharmacol. https://doi.org/10.1093/ijnp/pyz016
Chu Q, Yu L, Zheng Z et al (2019) Apios americana Medik flowers extract protects PC12 cells against H 2 O 2 induced neurotoxicity via regulating autophagy. Food Chem Toxicol. https://doi.org/10.1016/j.fct.2018.12.003
Zhang Z, Wang X, Zhang D, et al (2019) Geniposide-mediated protection against amyloid deposition and behavioral impairment correlates with downregulation of mTOR signaling and enhanced autophagy in a mouse model of Alzheimer’s disease. Aging (Albany NY) https://doi.org/10.18632/aging.101759
Zhao Y, Wang Q, Wang Y et al (2019) Glutamine protects against oxidative stress injury through inhibiting the activation of PI3K/Akt signaling pathway in parkinsonian cell model. Environ Health Prev Med 24:4. https://doi.org/10.1186/s12199-018-0757-5
Shan SR, Jiang F, Xu SM (2019) Effects of H102 on the memory recognition ability and AMPK-mTOR autophagy-related pathway in AD mice]. Zhongguo Ying Yong Sheng Li Xue Za Zhi 35:1–4. https://doi.org/10.12047/j.cjap.5749.2019.001
Yang C-C, Lin C-C, Hsiao L-D, Yang C-M (2018) Galangin inhibits thrombin-induced MMP-9 expression in SK-N-SH cells via protein kinase-dependent NF-kappaB phosphorylation. Int J Mol Sci. https://doi.org/10.3390/ijms19124084
Polis B, Srikanth KD, Elliott E et al (2018) L-norvaline reverses cognitive decline and synaptic loss in a murine model of alzheimer’s disease. Neurotherapeutics. https://doi.org/10.1007/s13311-018-0669-5
Zhu J, Dou S, Jiang Y et al (2019) Apelin-36 exerts the cytoprotective effect against MPP(+)-induced cytotoxicity in SH-SY5Y cells through PI3K/Akt/mTOR autophagy pathway. Life Sci 224:95–108. https://doi.org/10.1016/j.lfs.2019.03.047
Gu HF, Li N, Tang YL et al (2019) Nicotinate-curcumin ameliorates cognitive impairment in diabetic rats by rescuing autophagic flux in CA1 hippocampus. CNS Neurosci Ther. https://doi.org/10.1111/cns.13059
Zhou T, Zhuang J, Wang Z et al (2019) Glaucocalyxin A as a natural product increases amyloid β clearance and decreases tau phosphorylation involving the mammalian target of rapamycin signaling pathway. NeuroReport. https://doi.org/10.1097/WNR.0000000000001202
Song HL, Demirev AV, Kim NY et al (2019) Ouabain activates transcription factor EB and exerts neuroprotection in models of Alzheimer’s disease. Mol Cell Neurosci. https://doi.org/10.1016/j.mcn.2018.12.007
Huang L, Lin M, Zhong X et al (2019) Galangin decreases p-tau, Aβ 42 and β-secretase levels, and suppresses autophagy in okadaic acid-induced PC12 cells via an Akt/GSK3β/mTOR signaling-dependent mechanism. Mol Med Rep. https://doi.org/10.3892/mmr.2019.9824
Song GL, Chen C, Wu QY et al (2018) Selenium-enriched yeast inhibited β-amyloid production and modulated autophagy in a triple transgenic mouse model of Alzheimer’s disease. Metallomics. https://doi.org/10.1039/c8mt00041g
Shao Q, Zhang X, Chen Y et al (2018) Anti-neuroinflammatory effects of 20C from Gastrodia elata via regulating autophagy in LPS-activated BV-2 cells through MAPKs and TLR4/Akt/mTOR signaling pathways. Mol Immunol. https://doi.org/10.1016/j.molimm.2018.04.0144
Arabit JGJ, Elhaj R, Schriner SE et al (2018) Rhodiola rosea improves lifespan, locomotion, and neurodegeneration in a drosophila melanogaster model of huntington’s disease. Biomed Res Int. https://doi.org/10.1155/2018/6726874
Kong F-J, Wu J-H, Sun S-Y et al (2018) Liraglutide ameliorates cognitive decline by promoting autophagy via the AMP-activated protein kinase/mammalian target of rapamycin pathway in a streptozotocin-induced mouse model of diabetes. Neuropharmacology 131:316–325. https://doi.org/10.1016/j.neuropharm.2018.01.001
Ou Z, Kong X, Sun X et al (2018) Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav Immun. https://doi.org/10.1016/j.bbi.2017.12.009
Qu Y, Liu Y, Chen L et al (2018) Nobiletin prevents cadmium-induced neuronal apoptosis by inhibiting reactive oxygen species and modulating JNK/ERK1/2 and Akt/mTOR networks in rats. Neurol Res. https://doi.org/10.1080/01616412.2018.1424685
Guo X, Lv J, Lu J et al (2018) Protopanaxadiol derivative DDPU improves behavior and cognitive deficit in AD mice involving regulation of both ER stress and autophagy. Neuropharmacology. https://doi.org/10.1016/j.neuropharm.2017.11.033
Kim YD, Il JE, Nah J et al (2017) Pimozide reduces toxic forms of tau in TauC3 mice via 5′ adenosine monophosphate-activated protein kinase-mediated autophagy. J Neurochem. https://doi.org/10.1111/jnc.14109
Li Z, Chen X, Lu W et al (2017) Anti-oxidative stress activity is essential for amanita caesarea mediated neuroprotection on glutamate-induced apoptotic HT22 cells and an Alzheimer’s disease mouse model. Int J Mol Sci. https://doi.org/10.3390/ijms18081623
Xiao H, Zhang Q, Peng Y et al (2017) 7-(4-Hydroxy-3-methoxyphenyl)-1-phenyl-4E-hepten-3-one alleviates Abeta1-42 induced cytotoxicity through PI3K-mTOR pathways. Biochem Biophys Res Commun 484:365–371. https://doi.org/10.1016/j.bbrc.2017.01.125
Zhang ZH, Wu QY, Zheng R et al (2017) Selenomethionine mitigates cognitive decline by targeting both tau hyperphosphorylation and autophagic clearance in an Alzheimer’s disease mouse model. J Neurosci. https://doi.org/10.1523/JNEUROSCI.3229-16.2017
Li C, Guo XD, Lei M et al (2017) Thamnolia vermicularis extract improves learning ability in APP/PS1 transgenic mice by ameliorating both Aβ and Tau pathologies. Acta Pharmacol Sin. https://doi.org/10.1038/aps.2016.94
Zhang R, Zhang N, Zhang H et al (2017) Celastrol prevents cadmium-induced neuronal cell death by blocking reactive oxygen species-mediated mammalian target of rapamycin pathway. Br J Pharmacol. https://doi.org/10.1111/bph.13655
Xie L, Yu S, Yang K et al (2017) Hydrogen sulfide inhibits autophagic neuronal cell death by reducing oxidative stress in spinal cord ischemia reperfusion injury. Oxid Med Cell Longev. https://doi.org/10.1155/2017/8640284
Deng M, Huang L, Ning B et al (2016) β-asarone improves learning and memory and reduces Acetyl Cholinesterase and Beta-amyloid 42 levels in APP/PS1 transgenic mice by regulating Beclin-1-dependent autophagy. Brain Res. https://doi.org/10.1016/j.brainres.2016.10.008
Guo XD, Sun GL, Zhou TT et al (2016) Small molecule LX2343 ameliorates cognitive deficits in AD model mice by targeting both amyloid β production and clearance. Acta Pharmacol Sin. https://doi.org/10.1038/aps.2016.80
Liu J, Su H, Qu QM (2016) Carnosic acid prevents beta-amyloid-induced injury in human neuroblastoma SH-SY5Y cells via the induction of autoph agy. Neurochem Res. https://doi.org/10.1007/s11064-016-1945-6
Walter C, Clemens LE, Müller AJ et al (2016) Activation of AMPK-induced autophagy ameliorates Huntington disease pathology in vitro. Neuropharmacology. https://doi.org/10.1016/j.neuropharm.2016.04.041
Tseng YT, Chen CS, Jong YJ et al (2016) Loganin possesses neuroprotective properties, restores SMN protein and activates protein synthesis positive regulator Akt/mTOR in experimental models of spinal muscular atrophy. Pharmacol Res. https://doi.org/10.1016/j.phrs.2016.05.023
Zhou Q, Chen B, Wang X et al (2016) Sulforaphane protects against rotenone-induced neurotoxicity in vivo: Involvement of the mTOR, Nrf2, and autophagy pathways. Sci Rep. https://doi.org/10.1038/srep32206
Acknowledgements
The authors would like to thank the senior management of Delhi Technological University for their constant support and guidance.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
RG, RK and PK conceived and designed this project. Data collected and analyzed by RG and RK. Art work is done by RG and PK. Manuscript has been written by RG and PK.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare no conflict of interest.
Ethics approval and consent to participate
Not applicable.
Consent for publication
The authors consent to publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gupta, R., Ambasta, R.K. & Pravir Kumar Autophagy and apoptosis cascade: which is more prominent in neuronal death?. Cell. Mol. Life Sci. 78, 8001–8047 (2021). https://doi.org/10.1007/s00018-021-04004-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-04004-4