
Abstract
Autophagy and the ubiquitin proteasome system (UPS), as two major protein degradation pathways, coordinate with each other in regulating programmed cell death. Autophagy can compensate for the UPS impairment-induced cell dysfunction and apoptosis. However, it is not clear how cells maintain the delicate balance between UPS-related apoptosis and autophagy. Here, we showed that proteasome inhibition-mediated UPS impairment can activate the phosphorylated p38α (p-p38α)-dependent apoptotic pathway and autophagy pathway in both neuroblastoma cell line N2a and primary cortical neuronal cells. Multiple indices were utilized for the autophagy detection including LC3II transition, acidic vesicle formation, lysosomal accumulation, and p62 reduction. Blockade of autophagy flux with autophagy inhibitor 3-methyladenine or bafilomycin A1 resulted in further phosphorylation of p38α, polyubiquitinated protein aggregation, and greater apoptotic cell death. On the contrary, enhancement of autophagy by rapamycin attenuated the cell loss by lowering p-p38α level and degrading protein aggregates, indicating a protective role of autophagy in cell stress and apoptosis. Moreover, de-activation of p38α with pharmaceutical p38α inhibitor BIRB796 greatly increased autophagy activation, reduced protein aggregates, and attenuated cell loss, suggesting a bidirectional regulation between p-p38α and autophagy. In addition, manipulation of p-p38α by BIRB796 or p38α knockdown decreased the phosphorylation of key components of the mammalian target of rapamycin (mTOR)-dependent pathway, indicating that the mTOR pathway mediates the p-p38α regulation on autophagy. Overall, our data emphasize p-p38α as a key mediator in the antagonistic interaction between apoptosis and autophagy in response to UPS impairment. Centering p-p38α as a potential regulatory target may provide a dual advantage of proteostasis maintenance and cell survival for simultaneous inhibition of apoptosis and activation of autophagy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The ubiquitin proteasome system (UPS) is responsible for a highly selective degradation of short-lived proteins to maintain the proteostasis under basal metabolic conditions [1, 2]. This system recruits proteasome to selectively recognize, unfold, and degrade ubiquitinated proteins. When UPS is impaired, accumulation of misfolded proteins causes severe cell damage leading to activation of multiple apoptotic signaling pathways and eventual cell death. Neuronal cells are extremely vulnerable to toxic protein aggregations [3–5]. Impairment of the UPS and the subsequent accumulation of misfolded protein and inclusion body formation have been strongly implicated in the pathology of neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) [5–7].
Autophagy is another major protein degradation pathway for mammalian cells to keep intracellular proteostasis. Different from UPS, autophagy is attributed to the degradation of long-lived proteins and injured organelles through the autophagosome-lysosome pathway (ALP) [8]. Autophagy is regulated by diverse autophagy-related genes [9–11]. The mature process of autophagy is characterized as an “autophagy flux” including autophagosome formation, fusion of autophagosome and lysosome, and eventual protein degradation in autolysosome [12]. Several studies have reported that autophagy plays important roles in the process of neuronal cell survival, proliferation, and differentiation and in the pathogenesis of many neurodegenerative disorders [13, 14].
Accumulating evidence has shown broad and diverse interactions between UPS and autophagy [15–18]. UPS dysfunction can lead to activation of autophagy in cell cultures in vitro and animal models in vivo [17, 19–21]. ALP can compensate hampered proteasome degradation, contributing to toxic signal clearance and subsequent cell survival [16, 22]. Several mediators such as p53 and HDAC6 have been predicted to link UPS and ALP [23, 24]. However, key mediators insighting disease mechanisms and targeting drug discovery still remain largely unknown.
In this study, we identified phosphorylated mitogen-activated protein kinase (MAPK) p38α as a critical mediator linking UPS and ALP pathways. p38α is involved in numerous biological processes, including cell differentiation, proliferation, and apoptosis [25–29]. Activation of p38α induced by stress stimuli, including oxidative stress, inflammation, and cytokines, leads to activation of downstream caspase cascade and subsequent cell death [26, 30–32]. It has been reported that proteasome inhibition in PD-related animal models can increase the level of phosphorylated p38α (p-p38α) [33]. And in such case, activation of p38α can lead to multiple downstream signalings [33–36]. Recent studies have demonstrated that p38α is also involved in basal autophagy and starvation-induced autophagy [25, 37–42]. However, little is known about the diverse faces of p-p38α and its circumstantial contribution to the cellular behavior under specific pathological conditions. We report here that p-p38α has dual functions as an apoptotic initiator for UPS impairment and as an autophagy antagonist, making it a perfect target for modulation of neuronal cell fate.
Results
UPS Impairment Induces Neuronal Cell Death and Activates p-p38α and Autophagy
To study the neuronal cell response to UPS impairment, we employed the 20S proteasome specific inhibitor MG132 to generate UPS impairment neuronal cell models in mouse neuroblastoma cell line N2a and primary neuronal cells derived from embryonic cortical neural precursor cells (NPCs) (Fig. S1a). In both models, treatment of 10 μM MG132 resulted in an increased aggregation of polyubiquitinated proteins in a time-dependent manner (Fig. S1b). More than half of the N2a cells and primary neuronal cells underwent apoptotic cell death within 12 h of treatment with MG132, as shown by an increased transferase-mediated deoxyuridine triphosphate-biotin nick end-labeling (TUNEL) staining (Fig. 1a–c) and the activation of the apoptotic effectors cleaved poly-ADP-ribose polymerase (PARP) and active-caspase 3 (Fig. 1d, e). These cells after MG132 treatment illustrated typical blebbing and cell shrinkage morphology of apoptosis in bright field imaging (Fig. S2a). Strikingly, we found that p38α phosphorylation was significantly increased after MG132 treatment (Fig. 1d, e). Then, we applied a p38α-specific inhibitor BIRB796 to block p38α phosphorylation (Fig. 1f). We found that BIRB796 can greatly attenuate the activation of apoptotic molecules cleaved PARP and active-caspase 3 (Fig. 1f, g) and rescue the MG132-induced apoptotic cell death (Figs. 1h–j and S2b). These results suggest that p-p38α is a positive mediator of proteasome inhibition-induced apoptosis.

MG132 induced cell death and p38α activation in N2a cells and primary neurons, and de-activation of p38α alleviated the apoptosis. The N2a and primary neurons derived from cortical NPCs were treated with MG132 at a concentration of 10 μM for various times. All the following experiments were done at the same concentration if not specifically mentioned. TUNEL staining showing time-dependent reduction of cell number (a, b) and induction of TUNEL-positive apoptotic cells when treated with MG132 (a, c). Representative western blot images (d) and relative quantitation (e) of the apoptosis-related proteins cleaved PARP (C-PARP), cleaved caspase 3 (C-CASP3), phosphorylated-p38α (p-p38α), and p38α along with different periods of MG132 treatment. Actin was used as the reference. N2a cells and primary neurons were pre-treated with 20 μM BIRB796 1 h before MG132 treatment. P38α-specific inhibitor BIRB796 reduced phosphorylated p38α in both cells and apoptotic signaling (f, g) induced by MG132. Representative images (h) and quantification of two cell models showing that TUNEL-positive apoptotic cell number was greatly reduced in BIRB796-treated cells, and the total cell number was rescued (i, j). *P < 0.05; **P < 0.01; ***P < 0.001; Student t test. Scale bar, 100 μm
Further, we observed that MG132 treatment also induced a time-dependent increase of lapidated autophagosome-associated protein LC3-II in both N2a cells and primary neurons (Fig. 2a, b). Consistently, confocal microscopic imaging showed a dramatic increase of LC3 puncta in the cytosols of MG132-treated cells (Fig. 2c, d). The formation of acidic vesicular organelles accompanied with lysosome accumulation, two characteristics of autophagy, were seen through acridine orange (AO) staining [43] and LysoTracker Red staining [44], respectively, in both cells (Fig. 2e, f). Moreover, SQSTM1/p62, an autophagy-selective substrate, was markedly decreased in western blotting analysis (Fig. 2a, b). To exclude the possibility that the LC3-II accumulation was due to a defect in autophagosome turnover, we pre-treated bafilomycin A1 (BAF) 1 h prior to MG132 addition in both cells. Since BAF could inhibit the fusion of autophagosome and lysosome, autophagy flux was supposed to be further blocked, resulting in attenuated protein breakdown. Indeed, dramatic inductions of LC3-II and SQSTM1 were observed (Fig. 2g, h), indicating a bona fide enhancement of autophagic flux in response to UPS impairment. All these results verified the involvement of two different pathways in response to UPS impairment in two neuronal cell models.

MG132 induced autophagy. An increased protein level of LC3-II and a decreased level of SQSTM1 were observed in N2a cells and primary neuronal cells treated with MG132 (a). Quantification of the protein expression was shown in b. LC3 puncta number in every cell (standing for autophagosomes) of both N2a and primary neurons was increased by MG132 treatment (c, d). Acridine orange was used to monitor the acidic vesicle formation as it emits bright red fluorescence in acidic vesicles but dim green in the cytoplasm and nucleus. A transition of green fluorescence toward red was clearly observed in both N2a and primary neurons along with MG132 treatment (e). Similarly, lysosome formation was evaluated with LysoTracker Red staining. Accumulation of lysosomes was seen in both cells (f). The pre-treatment of bafilomycin A1 (BAF) led to the accumulation of LC3-II and SQSTM1 aggregation (g), suggesting a blockade of autophagy flux. Quantification of the protein levels from N2a cells and primary neurons was shown in (h). Data are expressed as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; Student’s t test. Scale bar, 10 μm
Blockade of Autophagy Further Activates p38α and Aggravates Apoptotic Cell Death
To investigate the role of autophagy in proteasome inhibition, autophagy inhibitor 3-methyadenine (3MA) or BAF was recruited to block autophagic flux in different stages of the autophagy process. 3MA is an inhibitor of phosphatidylinositol 3-kinases (PI3K) which plays important roles in many biological processes, including controlling the activation of mammalian target of rapamycin (mTOR), a key regulator of autophagy. Pre-treatment of 3MA diminished the lipidation of LC3 and thus resulted in less LC3-II detection, while BAF blocked the LC3-II turnover and caused its aggregation (Figs. 3a, b and 2g). Nonetheless, both treatments can lead to higher SQSTM1 protein level (Fig. 3a, b), demonstrating that they are able to reduce the autophagy activity. Most notably, p-p38α was dramatically induced in the presence of 3MA or BAF (Fig. 3a, b), suggesting that autophagy is a negative regulator of p38α phosphorylation. As a result, western blot showed that poly-ubiquitinated protein level was further increased by autophagy inhibitor 3MA or BAF (Fig. 3c, d), indicating that autophagy was an aggregated protein clearance mechanism in the first place. Blockade of autophagy eventually activated cell apoptotic signaling (Fig. 3e, f) and significantly worsened the resultant cell viability (Figs. 3g–i and S2c).

Inhibition of autophagy aggravated protein aggregation and apoptotic cell death under UPS-impaired condition. N2a cells and primary neurons were pre-treated with autophagy inhibitor 5 mM 3MA or 50 nM BAF 1 h before MG132 treatment. Representative western blot images and relative quantification of LC3, SQSTM1, p-p38α, and p38α were shown in a, b, respectively. Actin was used as the reference. Representative image and quantification of polyubiquitinated protein aggregations with and without autophagy inhibitor pre-treatment were shown in c, d. Autophagy inhibition activated the apoptotic signaling by increasing the apoptosis-related proteins C-PARP and C-CASP3 (e, f). Autophagy inhibition further increased the proportion of TUNEL-positive cells (g, i) and resulted in greater cell loss (g, h). Data are expressed as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; Student’s t test. Scale bar, 100 μm
Enhancement of Autophagy Antagonizes p38α Activation and Facilitates Cell Survival Against UPS Impairment
We further hypothesized that reinforcement of autophagy could reverse UPS impairment-induced cell death. Pre-treatment of autophagy activator rapamycin resulted in more LC3 puncta observed in cell cytosol (Fig. 4a, b) of both N2a cells and primary neurons. Confocal imaging of increased lysosomal activity and accumulation by AO and LysoTracker Red staining (Fig. 4c, d), together with higher LC3-II protein level and lower SQSTM1 protein level in immunoblotting (Fig. 4e, f), further confirmed the activation of autophagy in the presence of rapamycin. As expected, rapamycin subsequently reduced the MG-132-induced poly-ubiquitinated protein level (Fig. 4g, h) and dramatically decreased p-p38α level (Fig. 4i, j). As a result, cleaved PARP and activated caspase 3 were lower (Fig.4i, j) and the cell viability was increased with the reduction of TUNEL+ cells after rapamycin treatment (Fig. 4k–m). Consistently, fewer cells with apoptotic morphology were observed in rapamycin-treated cultures (Fig. S2d). Taken together, our data strongly suggest a reciprocal correlation between autophagy and p38α activation and between autophagy and apoptosis in UPS impairment cell model.

Activation of autophagy decreased protein aggregation and apoptotic cell death under UPS-impaired condition. N2a cells and primary neurons derived from cortical NPCs were pre-treated with autophagy agonist 100 nM rapamycin (Rapa) 1 h before MG132 treatment. LC3 puncta formation was more obvious with Rapa treatment, presented by representative immunostaining images and LC3 puncta per cell counting (a, b). Representative images of AO and LysoTracker Red staining of both cells with or without Rapa pre-treatment were shown (c, d) to confirm the detection of autophagy. Western blotting showed an increased level of LC3II and a decreased level of SQSTM1 in Rapa-treated cells (e, f). Poly-ubiquitinated protein aggregation was also reduced by Rapa treatment (g, h). A decreased level of p-p38α was induced by Rapa, with decreased levels of apoptosis markers C- PARP and C-CASP3 (i, j). The eventual outcomes of Rapa treatment were the increased cell viability and rescued cell number as shown in k–m. Data are expressed as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; Student’s t test
Inhibition of p38α Phosphorylation Enhances Autophagy and Rescues Cell Loss
As previously shown in Fig. 1h–j, pre-treatment of p38α inhibitor BIRB796 caused a reduction of p38α phosphorylation and increased cell viability by inhibiting apoptosis. This survival outcome might result from clearance of cellular stress by autophagy. We noticed that BIRB796 markedly increased the cytosol puncta of LC3 (Fig. 5a, b), formation of acidic vesicles (Fig. 5c), accumulation of lysosomes (Fig. 5d), and the immunoblots of LC3-II (Fig. 5e, f). In addition, we found that the protein level of SQSTM1was reduced (Fig. 5e, f), demonstrating that de-activation of p38α enhances autophagy activity under proteasome inhibition. Meanwhile, the level of poly-ubiquitinated proteins was decreased (Fig. 5g, h), supporting the protective role of autophagy in this case.

De-activation of p38α enhanced autophagy and alleviated apoptotic cell death under UPS-impaired condition. Pre-treatment of a p38α-specific inhibitor BIRB796 1 h before MG132 increased the number of LC3 puncta in every single cell cytosol, presented by representative confocal imaging and puncta counting (a, b). AO and LysoTracker Red staining was performed to support the detection of autophagy as a result of BIRB796 (c, d). BIRB796 efficacy to reduce p-p38α was confirmed in e, f. Immunoblotting level of LC3-II was increased accompanied by a decrease in the level of SQSTM1 by BIRB796 treatment (e, f). BIRB796 treatment also reduced the polyubiquitinated protein aggregation (g, h). Data are expressed as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; Student’s t test
P-p38α Negatively Regulates Autophagy Through the mTOR-Dependent Pathway
Rapamycin is an autophagy activator through the mTOR-dependent pathway. We found that treatment of rapamycin decreased the phosphorylation levels of several key components of the mTOR pathway including AKT, mTOR, and RPS6KB in both N2a and primary neuronal cells (Fig. 6a, b). As inhibition of p-p38α displayed the same effects as rapamycin in autophagy activation and cell protection, we asked whether p-p38α also regulates autophagy through the mTOR-dependent pathway. We measured the phosphorylation levels of AKT, MTOR, and RPS6KB by western blotting in MG132-treated N2a and primary neuronal cells with or without BIRB796 pre-treatment. We found that all of the measured phosphorylation levels were down-regulated by p38α inhibitor BIRB796 (Fig. 6c, d), indicating that the mTOR-dependent pathway may mediate the p-p38α regulation of autophagy. To exclude the possibility that BIRB796 regulates autophagy through inhibition of mTOR without involvement of p-p38α, we employed a shRNA-mediated lentivirus to specifically knock down p38α in N2a cells. Similar to BIRB796, shp38α resulted in a reduced phosphorylation of p38α and an inhibition of mTOR signaling (Fig. 6e, f). Moreover, BIRB796 failed to affect autophagy and the mTOR pathway in the absence of MG132 (Fig. 6g, h), demonstrating that BIRB796 itself was not able to regulate autophagy without involvement of p-p38α. Taken together, we conclude that de-activation of p38α by BIRB796 regulates autophagy through the mTOR-dependent pathway. On the basis of our data, we propose a model that illustrates a role for p-p38α in the regulation of autophagy and apoptosis when there is dysfunction in the UPS (Fig. 7).

MTOR-dependent pathway mediated the interaction of p-p38α and autophagy. Representative western blot images and quantification of the phosphorylated forms of mTOR-related proteins AKT1, mTOR, and RPS6KB with and without Rapa pre-treatment were shown in a, b. Representative western blot images and quantification of the phosphorylated forms of mTOR-related proteins with and without pre-treatment of a p38α-specific inhibitor BIRB796 were shown in c, d. To further verify a direct relationship between p-p38α and the mTOR-dependent pathway, levels of p38α and p-p38α were knocked down by shRNA-mediated lentivirus in N2a cells, and it showed a similar pattern of mTOR as BIRB796 (e, f). mTOR-related proteins were also evaluated in N2a cells with and without BIRB796 in the absence of MG132 (g, h). Data are expressed as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; Student’s t test

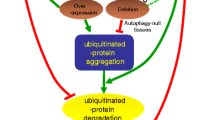
Hypothetical model for regulation of autophagy and apoptosis through p-p38α. When UPS is impaired, both autophagy and apoptosis are activated. In this process, the key modulator is p-p38α that positively regulates apoptosis through caspase cascades and negatively regulates autophagy through the mTOR pathway. Pharmacological inhibition of p-p38α will have dual effects on neuronal cell survival
Discussion
Increasing evidence has converged to indicate that failure of the UPS to degrade misfolded proteins plays a critical role in the pathogenesis of neurodegenerative diseases. These diseases are generally accompanied with enhanced autophagy and apoptotic cell death in affected regions of the central nervous system [20, 45, 46]. However, the mechanisms underlying the relationship and the key mediator among UPS, autophagy, and apoptosis still remain largely unknown. In this study, we showed that UPS impairment can activate both apoptosis and the autophagy pathway in neuronal cells. Furthermore, we identified p-p38α as a key mediator involved in both processes and cells utilized it to seesaw the cell death and survival in response to disturbed proteostasis. In addition, we demonstrated that the direct de-activation of p38α resulted in both anti-apoptotic and pro-autophagic functions, thus minimizing the UPS impairment-induced neuronal cell stress.
Our focus on the neuronal cell response to this pathological insult is extremely important for the understanding of the pathogenesis of a wide variety of neurodegenerative diseases. Progressive decline of UPS activity and the resultant accumulation of misfolded proteins are the common pathologies associated with aging that is the most significant risk factor for these diseases [17, 47, 48]. How to restore the proteostasis, alleviate the cytotoxicity, and rescue the neuronal cell loss remains largely unclear. MG132-treated N2a and primary cortical neuron cell cultures serve as preferable in vitro UPS impairment cell models that mimic such pathological conditions as the imbalanced proteostasis, increased cytotoxicity, and loss of neuronal cells in vivo. Under MG132-induced cellular stress of polyubiquitinated protein accumulation, p38α is activated to execute apoptotic cell death probably due to upstream endoplasmic reticulum (ER) stress or mitochondrial dysfunction as previously suggested [32, 36, 49, 50]. Meanwhile, cells also recruited autophagy as an alternative cell defense mechanism to degrade protein aggregates and relieve cellular stress. However, the autophagic protein clearance mechanisms surrendered to the overwhelming increase of protein aggregates due to continuous blockade of UPS, leading to the progressive cell loss.
The p38 MAPK in mammals has four subtypes, p38α, p38β, p38γ, and p38δ. P38α and P38β are widely expressed in various cell types, while p38γ is mainly expressed in skeletal muscle and p38δ mainly in the pancreas, kidney, and endocrine glands [51]. Each subtype of p38 MAPK has its own activators and substrates. Compared with other subtypes, phosphorylation of p38α is suggested as the main p38 signaling participating in multiple biological functions and physiological responses of neuronal cells in the brain [52]. Moreover, p-p38α is also implicated in the pathological brains. In line with our results of the beneficial effects of de-activating p38α in UPS-impaired neuronal cells, a recent finding has shown that deficiency of neuronal p38α-MAPK attenuated amyloid pathology in Alzheimer’s mouse and cell models through facilitating lysosomal degradation of BACE1 [53].
Enhancement of autophagy is a rational option to regain the proteostasis and increase the cell survival. Interestingly, we found that this strategy could be reached by de-activation of p38α. We demonstrated that p-p38α is a negative regulator of autophagy and suppression of p-p38α can result in a pro-survival effect of autophagy. Autophagy can be a pro-apoptosis or pro-survival process depending on cell types and conditions [54, 55]. P-P38α has been reported to be involved in both processes, while its function and relationship with autophagy are controversial. It has been reported that pharmacological p38α blockade in colorectal cancer cells promoted autophagic cell death and further triggered the cancer cell type-specific apoptosis [25]. However, treatment of a natural product Piperlongumine was shown to induce autophagic cell death through activation of the p38 pathway in human osteosarcoma cells [56]. In senescent T cells, it was suggested that the mitochondrial dysfunction was due to autophagy inhibition resulted from higher level of p-p38 [57]. These seemingly conflicting results suggest that the interaction of p-p38α and autophagy is also highly cell type- and condition-specific. Nevertheless, in our in vitro UPS impairment cell models, inhibition of p-p38α served as an appropriate way to enhance autophagy for the cell proteostasis.
Reversely, we also demonstrated that activation of autophagy by rapamycin further down-regulated p38α activation, suggesting p-p38α as a downstream target of autophagy in autophagic cell survival mechanism. Consistent with our results, autophagy has been reported to suppress p38 activation and enhance human skin cell survival under UVB radiation [23, 39], whereas Atg7 knockdown dramatically increases p38 phosphorylation [58].
The interactions between p-p38α and autophagy appear to be reciprocal in this study. We showed evidence of p-p38α regulation on autophagy through the mTOR pathway, whereas the autophagy regulation on p-p38α might depend on overall cellular conditions. Mechanistically, a direct outcome of autophagy in our study is the elimination of polyubiquitinated proteins, the accumulation of which was known to further result in ER stress and mitochondrial dysfunction [59, 60]. Chronic ER stress and mitochondrial dysfunction lead to heavy cellular stress and greatly impair the intracellular environment of neuronal cells. ER stress and mitochondrial dysfunction have been shown to be main inducers for p38α activation and its downstream apoptotic cascades [61]. Furthermore, phosphorylation of p38α has been shown to be positively regulated by autophagy receptor p62 and work together with p62 in response to inflammation stimuli [60]. P62 protein has been shown to directly bind to p38α and affect its phosphorylation [62]. In our study, the elimination of p62 in neuronal cells was a hallmark of autophagy and concurred with the reduced phosphorylation of p38α. Thus, it is likely that the observed negative regulation of p-p38α could be an overall outcome of the pro-survival effect of autophagy through multiple cellular biological functions, including the elimination of polyubiquitinated proteins, reduced ER stress, repaired mitochondrial dysfunction, and reduced p62. Key molecules that mediate the autophagy regulation on p-p38α need to be further explored in future studies.
De-activation of p38α showed similar effects to rapamycin treatment in promoting autophagy and subsequent outcomes. Further molecular signaling study demonstrated that they both work through the inhibition of the mTOR-dependent pathway. In the neurodegenerative diseases, inhibition of mTOR with activation of autophagy has been reported as neuroprotective [19]. In AD mouse models, autophagic clearance of cortical β-amyloid provided protection against memory impairment through mTOR signaling inhibition [55]. In PD models, induction of autophagy and inhibition of mTOR eliminated α-synuclein toxicity and protected dopaminergic neurons [19]. Defects in autophagic pathways resulted in neurodegeneration in patients with Lewy body disease and in models of α-synucleinopathy [63]. Inhibition of the mTOR pathway was shown to prevent the cell death of nigral neurons overexpressing α-synuclein by reducing α-synuclein and promoting the generation of autophagic vacuoles [64, 65]. The growth factor erythropoietin has also been shown to protect against dopaminergic neurotoxicity through autophagy induction and mTOR inhibition [66]. As for HD, mTOR protein could interact with Htt protein, whose mutation is the direct cause of HD pathogenesis [67]. Induction of autophagy could help the clearance of mutant Htt [68]. All these results including ours suggest that manipulation of p38α activity might offer a potential therapeutical strategy to treat neurodegenerative diseases through regulation of mTOR-dependent autophagy.
Our previous study has found that p53 mediated the proteasome inhibition-induced autophagy activation in human dopaminergic neuroblastoma cell SH-SY5Y and UPS-impaired mouse model of PD [23]. And the enhancement of autophagy in turn could partially block p53 and its downstream mitochondria-dependent apoptotic pathway. The major concern with utilizing p53 as a potential regulatory target arises from the worry that activation of p53 will also activate the apoptotic pathway and counteract the anti-apoptotic effect from autophagy. The strategy of utilizing de-activation of p38α to enhance autophagy will exclude the apoptosis risk as in p53, but rather include additional cell survival benefits because de-activation of p38α will also reduce the apoptosis cascade signaling.
Neuroblastoma N2a cells and post-mitotic primary neurons share large similarity in embryonic origin, environmental responses, intracellular machinery, and neuronal vulnerability. Our study shows that regardless of their differential mitosis status, a conventional molecular pathway exists in both cancer and normal cells for the determination of survival and death. Hence, it further raises the question about how to differentially utilize our knowledge of p-p38α in service of leading the death of cancer cells and guarding the survival of neurons. It also remains unknown whether the p-p38α-mediated balance between autophagy and apoptosis is neuron-specific or more universal. Further studies targeting these questions will enhance the potential practicability of strategy of p-p38α de-activation. We believe that an optimal manipulation of p-p38α offers great promise and exciting avenues for future pre-clinical and pharmaceutical applications in neurodegenerative disorders.
Materials and Methods
Reagents, Chemicals, and Cells
The following antibodies and chemicals were used in this study: mouse anti monoclonal β-actin (Santa Cruz, SC47778), rabbit polyclonal anti-p38α (Cell Signaling Technology, 9218), rabbit polyclonal anti-p-p38α (Merck Millipore, 09-272), rabbit monoclonal anti-LC3B (Sigma, 7543), rabbit monoclonal anti-SQSTM1/p62 (Abcam, 109012), rabbit polyclonal anti-active caspase 3 (Abcam, 2302), mouse anti-cleaved PARP (Cell Signaling Technology, 9544), MG132 (Sigma, M8699), 3MA (Sigma, M9281), bafilomycin A1 (Cayman, 11038), rapamycin (Sigma, V900930), and BIRB796 (Selleck, S1574).
The mouse neuroblastoma cell line N2a was maintained in DMEM (Gibco, 11995-065) medium containing 10% fetal bovine serum (Millipore, AYK172765) and 0.1% penicillin/streptomycin (Invitrogen, 15070-063) at 37 °C in a humidified atmosphere incubator with 5% CO2.
Primary NPCs were isolated and cultured from embryonic day 12 mouse cortices (ICR) as previously described [69]. NPCs were expanded by adding basic fibroblast growth factor (bFGF, 20 ng/ml; R&D Systems) and epithelial growth factor (EGF, 20 ng/ml; R&D Systems) in serum-free RHB-A medium (Cellartis, Y40001). NPCs were then passaged once followed by further cell proliferation. Differentiation of NPCs into neurons was conducted through withdrawal of bFGF and EGF. Four days after differentiation, neuronal cells were applied for various assays.
Immunocytochemistry and Confocal Microscopy Imaging
For immunocytochemical analysis, cultured cells were fixed with cold methanol at −20 °C for 30 min. After washing with PBS, the fixed cells were incubated with blocking buffer (0.2% saponin (PBS) + 2% BSA) for 30 min at room temperature, followed by an overnight incubation at 4 °C with the following primary antibody: LC3 (Cell Signaling Technology, 2775). After being washed with 0.2% saponin for three times, cells were incubated with secondary antibody conjugated with Alexa Fluor 594 (Invitrogen, A11037) at room temperature for 1 h then washed with PBS, and the nuclei were stained with DAPI (Beyotime, C1002) for 10 min. Cells were observed under a fluorescence microscope (Zeiss, AX10) or confocal microscope (Zeiss, LSM 710).
TUNEL Staining
TUNEL assays were performed with the one-step TUNEL kit (Beyotime, C1089) according to the manufacturer’s instructions. In detail, cells were fixed with 4% paraformaldehyde for 30 min at room temperature, followed by permeabilization with 0.1% Triton X-100 for 2 min on ice. The cells were applied with the TUNEL reaction mixture and incubated for 1 h at 37 °C in the dark. Cells were imaged under a fluorescent microscope (Zeiss, AX10).
AO and LysoTracker Red Staining
For AO staining, 1 μg/mL AO (Sigma, A8097) was added into cells within the culture for 5 min. After a brief wash with PBS, cells were fixed with paraformaldehyde for 30 min and applied for Hoechst staining. For LysoTracker Red staining, a similar protocol as AO staining was performed with LysoTracker Red probe (Beyotime, C1046).
Cell Counting and Statistical Analysis
DAPI-stained cells were counted in at least 20 randomly chosen areas of each coverslip. Data are expressed as mean ± SEM. Statistical comparisons were made by Student’s t test.
Immunoblotting
Cells were harvested at desired time points and lysed for 30 min in freshly prepared western lysis buffer (Beyond, P0013) with PMSF (Beyond, ST506-2). Lysates were centrifuged at 12,000 rpm, 30 min, and 4 °C; protein concentration was measured with Pierce BCA Assay Kit (Pierce, 23225); and 5 μg of protein was separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membrane was blocked with 5% (w/v) skim milk (Cell Signaling Technology, 9999S) for 1 h at room temperature. The membrane was incubated with primary antibodies against LC3B (Sigma, 7543) and SQSTM1 (Abcam, 109012) overnight at 4 °C. The membranes were washed three times with Tris-buffered saline and Tween-20 followed by incubation with the peroxidase-conjugated anti-mouse (Millipore, AP124P) or anti-rabbit (Millipore, AP132P) for 1 h at room temperature.
Abbreviations
- ALP:
-
Autophagy-lysosome pathway
- p-p38α:
-
Phosphorylated p38α
- TUNEL:
-
Transferase-mediated deoxyuridine triphosphate-biotin nick end-labeling
- 3-MA:
-
3-Methyladenine
- BAF:
-
Bafilomycin A1
- PI3K:
-
Phosphatidylinositol 3-kinases
- Rapa:
-
Rapamycin
- PARP:
-
Poly ADP-ribose polymerase
- C-CASP3:
-
Cleaved caspase 3
- mTOR:
-
Mammalian target of rapamycin
- AD:
-
Alzheimer’s disease
- PD:
-
Parkinson’s disease
- HD:
-
Huntington’s disease
- ALS:
-
Amyotrophic lateral sclerosis
References
Lecker SH (2006) Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17:1807–1819. doi:10.1681/ASN.2006010083
Wang J, Maldonado MA (2006) The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol Immunol 3:255–261
Saxena S, Caroni P (2011) Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71:35–48. doi:10.1016/j.neuron.2011.06.031
Mattson MP, Magnus T (2006) Ageing and neuronal vulnerability. Nat Rev Neurosci 7:278–294. doi:10.1038/nrn1886
Ross C, Poirier M (2004) Protein aggregation and neurodegenerative disease. Nat Med 10(Suppl):S10–S17. doi:10.1038/nm1066
McKinnon C, Tabrizi SJ (2014) The ubiquitin-proteasome system in neurodegeneration. Antioxid Redox Signal 0:1–20. doi:10.1089/ars.2013.5802
Dantuma NP, Bott LC (2014) The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci 7:70. doi:10.3389/fnmol.2014.00070
Cecconi F, Levine B (2008) The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell 15:344–357. doi:10.1016/j.devcel.2008.08.012.The
Fitzwalter BE, Thorburn A (2015) Recent insights into cell death and autophagy. FEBS J 282:n/a-n/a. doi: 10.1111/febs.13515
Behrends C, Fulda S (2012) Receptor proteins in selective autophagy. Int J Cell Biol. doi:10.1155/2012/673290
Wang S, Xia P, Rehm M, Fan Z (2015) Autophagy and cell reprogramming. Cell Mol Life Sci 1699–1713. doi: 10.1007/s00018-014-1829-3
Loos B, du Toit A, Hofmeyr J-HS (2014) Defining and measuring autophagosome flux-concept and reality. Autophagy 8627:37–41. doi:10.4161/15548627.2014.973338
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147:728–741. doi:10.1016/j.cell.2011.10.026
Fuchs Y, Steller H (2011) Programmed cell death in animal development and disease. Cell 147:742–758. doi:10.1016/j.cell.2011.10.033
Wang XJ, Yu J, Wong SH et al (2013) A novel crosstalk between two major protein degradation systems. Autophagy 9:1500–1508. doi:10.4161/auto.25573
Korolchuk VI, Menzies FM, Rubinsztein DC (2010) Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584:1393–1398. doi:10.1016/j.febslet.2009.12.047
Lamark T, Johansen T (2010) Autophagy: links with the proteasome. Curr Opin Cell Biol 22:192–198. doi:10.1016/j.ceb.2009.11.002
Laussmann MA, Passante E, Düssmann H et al (2011) Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ 18:1584–1597. doi:10.1038/cdd.2011.27
Pan T, Kondo S, Zhu W et al (2008) Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol Dis 32:16–25. doi:10.1016/j.nbd.2008.06.003
Shen Y, Tang Y, Zhang X et al (2013) Adaptive changes in autophagy after UPS impairment in Parkinson’s disease. Acta Pharmacol Sin 34:667–673. doi:10.1038/aps.2012.203
Khaminets A, Behl C, Dikic I (2016) Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol 26:6–16. doi:10.1016/j.tcb.2015.08.010
Zhang X, Chen S, Song L et al (2014) MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 10:588–602. doi:10.4161/auto.27710
Du Y, Yang D, Li A et al (2009) An insight into the mechanistic role of p53-mediated autophagy induction in response to proteasomal inhibition-induced neurotoxicity. Autophagy 5:663–675. doi:10.4161/auto.5.5.8377
Pandey UB, Nie Z, Batlevi Y et al (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447:859–863. doi:10.1038/nature05853
Comes F, Matrone A, Lastella P et al (2007) A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ 14:693–702. doi:10.1038/sj.cdd.4402076
Zarubin T, Han J (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res 15:11–18. doi:10.1038/sj.cr.7290257
Cao J, Semenova MM, Solovyan VT et al (2004) Distinct requirements for p38α and c-Jun N-terminal kinase stress-activated protein kinases in different forms of apoptotic neuronal death. J Biol Chem 279:35903–35913. doi:10.1074/jbc.M402353200
Park G, Tan J, Garcia G et al (2015) Regulation of histone acetylation by autophagy in Parkinson disease. J Biol Chem. doi:10.1074/jbc.M115.675488
Tian L, Chen J, Chen M et al (2014) The p38 pathway regulates oxidative stress tolerance by phosphorylation of mitochondrial protein IscU. J Biol Chem 289:31856–31865. doi:10.1074/jbc.M114.589093
Ge B (2002) MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science (80- ) 295:1291–1294. doi:10.1126/science.1067289
Ashraf MI, Ebner M, Wallner C et al (2014) A p38MAPK/MK2 signaling pathway leading to redox stress, cell death and ischemia/reperfusion injury. Cell Commun Signal 12:6. doi:10.1186/1478-811X-12-6
Han YH, Park WH (2010) Treatment with p38 inhibitor partially prevents Calu-6 lung cancer cell death by a proteasome inhibitor, MG132. Cancer Genet Cytogenet 199:81–88. doi:10.1016/j.cancergencyto.2010.02.004
Choi CH, Lee BH, Ahn SG, Oh SH (2012) Proteasome inhibition-induced p38 MAPK/ERK signaling regulates autophagy and apoptosis through the dual phosphorylation of glycogen synthase kinase 3?? Biochem Biophys Res Commun 418:759–764. doi:10.1016/j.bbrc.2012.01.095
Navas TA, Nguyen AN, Hideshima T et al (2006) Inhibition of p38alpha MAPK enhances proteasome inhibitor-induced apoptosis of myeloma cells by modulating Hsp27, Bcl-X(L), Mcl-1 and p53 levels in vitro and inhibits tumor growth in vivo. Leuk Off J Leuk Soc Am Leuk Res Fund UK 20:1017–1027. doi:10.1038/sj.leu.2404200
Du Y, Li X, Yang D et al (2008) Multiple molecular pathways are involved in the neuroprotection of GDNF against proteasome inhibitor induced dopamine neuron degeneration in vivo. Exp Biol Med 233:881–890. doi:10.3181/0712-RM-329
N’Diaye E-N, Kajihara KK, Hsieh I et al (2009) PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 10:173–179. doi:10.1038/embor.2008.238
Webber JL, Tooze SA (2010) Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J 29:27–40. doi:10.1038/emboj.2009.321
Moruno-Manchón JF, Pérez-Jiménez E, Knecht E (2012) Glucose induces autophagy under starvation conditions by a p38 MAPK-dependent pathway. Biochem J 506:497–506. doi:10.1042/BJ20121122
Qiang L, Wu C, Ming M et al (2013) Autophagy controls p38 activation to promote cell survival under genotoxic stress. J Biol Chem 288:1603–1611. doi:10.1074/jbc.M112.415224
Li L, Liu Y, Chen H et al (2015) Impeding the interaction between Nur77 and p38 reduces LPS-induced inflammation. Nat Chem Biol 11:339–346. doi:10.1038/nchembio.1788
Zhang X, Chen S, Li L et al (2008) Folic acid protects motor neurons against the increased homocysteine, inflammation and apoptosis in SOD1G93A transgenic mice. Neuropharmacology 54:1112–1119. doi:10.1016/j.neuropharm.2008.02.020
Zhu W, Downey JS, Gu J et al (2000) Regulation of TNF expression by multiple mitogen-activated protein kinase pathways. J Immunol 164:6349–6358. doi:10.4049/jimmunol.164.12.6349
Paglin S, Hollister T, Delohery T et al (2001) A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 61:439–444
Gan Q, Lu X, Dong W et al (2012) Endosomal pH-activatable magnetic nanoparticle-capped mesoporous silica for intracellular controlled release. J Mater Chem 22:15960. doi:10.1039/c2jm32020g
Ghavami S, Shojaei S, Yeganeh B et al (2014) Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol 112:24–49. doi:10.1016/j.pneurobio.2013.10.004
Agarwal S, Tiwari SK, Seth B et al (2015) Activation of autophagic flux against xenoestrogen bisphenol-A-induced hippocampal neurodegeneration via AMP kinase (AMPK)/mammalian target of rapamycin (mTOR) pathways. J Biol Chem 290:21163–21184. doi:10.1074/jbc.M115.648998
Pan T, Kondo S, Le W, Jankovic J (2008) The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 131:1969–1978. doi:10.1093/brain/awm318
Johansen T, Lamark T (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy 7:279–296. doi:10.4161/auto.7.3.14487
Park HS, Jun DY, Han CR et al (2011) Proteasome inhibitor MG132-induced apoptosis via ER stress-mediated apoptotic pathway and its potentiation by protein tyrosine kinase p56 lck in human Jurkat T cells. Biochem Pharmacol 82:1110–1125. doi:10.1016/j.bcp.2011.07.085
Zanotto-Filho A, Braganhol E, Oliveira Battastini AM, Fonseca Moreira JC (2012) Proteasome inhibitor MG132 induces selective apoptosis in glioblastoma cells through inhibition of PI3K/Akt and NFkappaB pathways, mitochondrial dysfunction, and activation of p38-JNK1/2 signaling. Investig New Drugs 30:2252–2262. doi:10.1007/s10637-012-9804-z
Tanoue T, Nishida E (2003) Molecular recognitions in the MAP kinase cascades. Cell Signal 15:455–462. doi:10.1016/S0898-6568(02)00112-2
Corrêa SAL, Eales KL (2012) The role of p38 MAPK and its substrates in neuronal plasticity and neurodegenerative disease. J Signal Transduct 2012:649079. doi:10.1155/2012/649079
Schnöeder L, Hao W, Qin Y, et al. (2015) Deficiency of neuronal p38α-MAPK attenuates amyloid pathology in Alzheimer’s mouse and cell models through facilitating lysosomal degradation of BACE1. J Biol Chem 1:jbc.M115.695916. doi: 10.1074/jbc.M115.695916
Zhang X, Li L, Chen S et al (2011) Rapamycin treatment augments motor neuron degeneration in SOD1G93A mouse model of amyotrophic lateral sclerosis. Autophagy 7:412–425. doi:10.4161/auto.7.4.14541
Wang Y, Mandelkow E (2012) Degradation of tau protein by autophagy and proteasomal pathways. Biochem Soc Trans 40:644–652. doi:10.1042/BST20120071
Wang Y, Wang J-W, Xiao X et al (2013) Piperlongumine induces autophagy by targeting p38 signaling. Cell Death Dis 4:e824. doi:10.1038/cddis.2013.358
Henson SM, Lanna A, Riddel NE et al (2014) P38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J Clin Invest 124:4004–4016. doi:10.1172/JCI75051
Zhuang Y, Li Y, Li X et al (2016) Atg7 knockdown augments concanavalin A-induced acute hepatitis through an ROS-mediated p38/MAPK pathway. PLoS One 11:1–15. doi:10.1371/journal.pone.0149754
Xu C, Bailly-Maitre B, Reed J (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115:2656–2664. doi:10.1172/JCI26373.2656
Hashimoto M, Rockenstein E, Crews L, Masliah E (2003) Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neruomol Med 4:21–36. doi:10.1385/NMM:4:1-2:21
Kawakami T, Gomez IG, Ren S, et al. (2014) Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol 1–13. doi: 10.1681/ASN.2013111202
Sudo T, Maruyama M, Osada H (2000) p62 functions as a p38 MAP kinase regulator. Biochem Biophys Res Commun 269:521–525. doi:10.1006/bbrc.2000.2333
Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19:983–997. doi:10.1038/nm.3232
Pino E, Amamoto R, Zheng L et al (2014) FOXO3 determines the accumulation of α-synuclein and controls the fate of dopaminergic neurons in the substantia nigra. Hum Mol Genet 23:1435–1452. doi:10.1093/hmg/ddt530
Maiese K (2015) FoxO proteins in the nervous system. Anal Cell Pathol. doi:10.1155/2015/569392
Maiese K (2015) Targeting molecules to medicine with mTOR, autophagy and neurodegenerative disorders. Br J Clin Pharmacol. doi:10.1111/bcp.12804
Berger Z, Ravikumar B, Menzies FM et al (2006) Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet 15:433–442. doi:10.1093/hmg/ddi458
Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC (2009) Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 16:46–56. doi:10.1038/cdd.2008.110
Park CH, Kang JS, Yoon EH et al (2008) Proneural bHLH neurogenin 2 differentially regulates Nurr1-induced dopamine neuron differentiation in rat and mouse neural precursor cells in vitro. FEBS Lett 582:537–42. doi:10.1016/j.febslet.2008.01.018
Acknowledgements
This work was supported by funding from the National Natural Science Foundation of China (NSFC 81430021 and 81370470), the Program for Liaoning Innovative Research Team in University (LT2015009), and the Liaoning Science and Technology Project (2015225008).
Author information
Authors and Affiliations
Corresponding author
Additional information
Fang Guo and Xi-Biao He contributed equally to this work.
Rights and permissions
About this article

Cite this article
Guo, F., He, XB., Li, S. et al. A Central Role for Phosphorylated p38α in Linking Proteasome Inhibition-Induced Apoptosis and Autophagy. Mol Neurobiol 54, 7597–7609 (2017). https://doi.org/10.1007/s12035-016-0260-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0260-1