
Abstract
The degree of conversion (DC) can be defined as the extent to which monomers react to form polymers or as the ratio of C=C double bonds that are converted into C-C single bonds. In the polymerization of bifunctional methacrylates, the complete conversion is never attainable because diffusional restrictions in later stages of the polymerization reaction prevent a certain amount of monomer molecules from reaching reaction sites. Thus the DC in dental composites usually varies between 50 and 80%. The DC is a fundamental attribute of a cured composite since it affects virtually all other material properties which are important for the clinical success of the restoration. Although the composition of contemporary composites is fine-tuned to attain optimal DC and the related properties if properly handled and light-cured, poor DC due to unfavorable curing conditions or operators’ insufficient understanding of the curing procedure may affect critical material properties and increase the risk of clinical failure. This chapter reviews various factors determining the DC, properties of composite materials which are dependent on the DC, as well as methods used to evaluate the DC.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Composite resins as the most commonly used dental material for a variety of applications have greatly evolved since they were introduced with an aim to fulfill different mechanical, biological, and esthetic requirements [1]. Progress in composite material formulation, such as improved filler morphology and improved existing polymeric matrix, and novel monomer technologies may improve the disadvantages of resin composite materials [2, 3]. Therefore, investigations as well as knowledge and understanding of their properties are the foundations for establishing the clinical use guidelines.
The degree of conversion (DC) can be defined as the extent to which monomers react to form polymers or as the ratio of C=C double bonds that are converted into C–C single bonds [4]. A high degree of composite polymerization is an essential material feature for obtaining optimal physical and mechanical properties and biocompatibility [5,6,7,8,9] and has a crucial impact on the utmost success of a composite restoration. It strongly affects each property of a composite material such as hardness, strength, elastic modulus, water sorption, solubility, color stability, dimensional stability with consequential microleakage, secondary caries, and possible pulpal reactions [3, 6]. Various factors like filler particle size and loading, monomer type and amount, polymerization initiator type and concentration [10, 11], the shade and translucency of the material [7], intensity and wavelength of the light source, and irradiation time [12] can influence the DC of dental composite materials.
It would be ideal to have all of the composite resin monomers converted to polymer during the polymerization reaction. However, the conversion is never complete and reaches a degree of about 50% to 75% for conventional composites [13]; for bulk-fill composite materials, values range from 50% to 81% [14, 15] and for pre-heated conventional and bulk-fill composites from 67% to 84% [16]; 24 h post-cure values vary from 68% to 86% [17], while for the experimental remineralizing composites based on amorphous calcium phosphate, DC values can reach even up to 87% [18].
In this chapter, factors determining DC, composite properties dependent upon DC, and methods for the evaluation of DC will be described.
2 Factors Determining the Degree of Conversion
The characteristics of dental composite resins result from their perplexed composition which means resin and filler type and amount. Factors determining DC can be classified as intrinsic and extrinsic.
2.1 Intrinsic Factors
Intrinsic factors imply composite properties and include resin composition, filler composition, and photoinitiator system [2].
Composite resins consist of the mixtures of various methacrylate monomers which are converted into a cross-linked polymer matrix during the polymerization reaction. Optimal combination of monomers is of exceptional importance in achieving a composite resin with outstanding properties. The DC is primarily determined by the structure and relative ratio of the individual monomers [19, 20]. The most common base monomers in contemporary commercial composites are bisphenol A-glycidyl methacrylate (Bis-GMA, molecular weight: 512 g/mol) and urethane dimethacrylate (UDMA, molecular weight: 471 g/mol). The Bis-GMA monomer is characterized by high molecular weight, rigid structure, and high viscosity. Its main benefits are lower polymerization shrinkage, fast setting, and strong and stiff polymeric networks [19]. However, its high viscosity impairs mobility during polymerization and prevents the polymer from attaining high DC values. For this reason, the Bis-GMA-based resins are admixed with monomers of lower viscosity, e. g., triethyleneglycol dimethacrylate (TEGDMA), urethane dimethacrylate (UDMA), or bisphenol A polyethylene glycol diether dimethacrylate (Bis-EMA) in order to improve the final DC [3]. Comonomers based on Bis-EMA, a monomer of high molecular weight and lower viscosity, usually show higher DC than the Bis-GMA/TEGDMA mixtures [21, 22]. Other monomers with higher molecular weight aimed to reduce shrinkage such as urethane dimethacrylate resin DX511 from Dupont (molecular weight: 895 g/mol) have been developed. On the other hand, dimer acid-based monomers have also relatively high molecular weight, i.e., 673–849 g/mol, and have been shown to have high DC while undergoing lower polymerization shrinkage than Bis-GMA systems [23, 24]. Tricyclodecane (TCD) urethane low viscosity monomers have lower shrinkage and polymerization stress but also high DC compared to others containing conventional dimethacrylates [1, 25].
The DC gradually decreases with increasing thickness of a composite resin layer. Curing light is attenuated by the absorption and scattering as the layer thickness increases and fewer radicals are formed to initiate polymerization [26]. The bulk-fill composite materials have been developed to fulfill challenging request of higher efficiency and enable adequate polymerization up to 4 or 5 mm depth. Optical properties between conventional and bulk-fill composites differ vastly. Bulk-fill composites are generally more translucent in order to ensure sufficient curing of thick increments [27]. Usually, the intensity of the curing light that reaches the bottom of a composite layer is considerably lower than that on the surface. To compensate the polymerization decrease in deeper layers, it is possible to improve the absorption spectrum and the initiator’s reactivity, to increase the translucency of the materials and to optimize the LED light source [28, 29]. This approach combines intrinsic factors dealing with photoinitiator system and translucency of composites with extrinsic ones which include polymerization conditions.
In any case, concerning intrinsic factors, photoinitiator which is almost always used is a combination of camphorquinone (CQ) and various types of tertiary amines [30]. However, other photopolymerization systems such as phenylpropanedione (PPD), mono- or bis-acylphosphine oxides (MAPO and BAPO), benzoyl germanium, or benzil [31, 32] have also been introduced. There is a difference among photoinitiators regarding initiation of polymerization—while each converted CQ molecule generates only one free radical, others are able to generate several, e.g., two for MAPO and four for BAPO [33]. Dibenzoyl germanium derivatives are also more light reactive than CQ [34]. While taking into account advantages of some photoinitiators over the CQ system, one should also consider them in relation to comonomer composition and ratio, filler content and size, as well as some extrinsic factors such as light spectrum, irradiance, and irradiation time.
Unavoidable factor in composite resins are fillers responsible for reducing monomer content and consequently for many crucial properties of the material such as strength, resistance to occlusal loads, shrinkage, handling, translucency, opalescence, radiopacity, etc. To increase depth of cure, it is necessary to increase the translucency of composites by decreasing the filler content and increasing filler size in an effort to ensure that more photons penetrate into deeper layers [28]. The amount of scattered and absorbed light influences the light transmittance through a composite material. Large filler-matrix interface area causes a considerable light scattering because of differences in the refractive indices between filler particles and resin matrix [14], while similar refractive indices of the components of composites have been shown to improve translucency in experimental materials.
2.2 Extrinsic Factors
2.2.1 Light Sources and Curing Conditions
Preferred curing mode of composite resins is photopolymerization, and complete understanding of the photopolymerization process is essential for achieving optimal properties of the material. Light curing has greatly revolutionized dentistry and is required in almost any clinical procedure. Technology behind light sources in dentistry changed from UV curing (wavelength about 365 nm) to a visible light curing (wavelength about 470 nm) that was introduced in 1976.
Quartz-tungsten-halogen lights were the main light source for photopolymerization for decades. During that time, a wide range of adaptations and improvements were made such as bulb power increase, output values increased from average to extreme, the adaptation of curing gun, and introduction of different curing modes [28]. The spectral emission of conventional curing lights includes wavelengths that induce heating of the resin and tooth during curing. Despite their popularity, conventional halogen light-curing units have serious drawbacks, such as a limited effective lifetime (40–100 h), reflector and filter degradation over time due to high operating temperatures, and the significant amount of heat produced during curing. Many halogen units have been shown to not reach the minimum power output specified by the manufacturers [35]. The most frequently used photoinitiator in resin composites, CQ, is sensitive to light in the blue region of the visible spectrum, with an absorption peak at 470 nm. Therefore, every light source with adequate irradiance in this region can be used as a curing unit. To overcome the problems of halogen technology, alternative light sources have been suggested, such as argon laser [36], pulsed laser [37, 38], plasma light [39, 40], and blue superbright light-emitting diodes (LEDs) [8, 9].
Today, LEDs are the light source of choice due to much higher efficiency compared to previous technologies. Their emission in the blue part of the spectrum is attained by using indium gallium nitride and matches the maximal absorption of the most frequently used photoinitiator CQ [28]. The first generation of LEDs (commercial) was introduced in the beginning of the new millennium. Their typical design was a combination of multiple, individual LED from 7 to 64 LEDs [28]. These units have a narrow spectral distribution range and are much more energy efficient (30% compared to 4% of halogen lights is used into light) and can be battery-powered. This LED generation demonstrated an irradiance of 150–350 mW/cm2 [41]. Photoactivated composites can be successfully cured with lower irradiance while maintaining high final DC and minimizing shrinkage stress [8]. Second generation of LEDs implies 1 W chips into curing lights generating 140 mW/cm2 output and 5 W chip generating 600 mW/cm2 output which means great increase in output in the similar wavelength range [28]. Third generations of LEDs were developed in order to enable polymerization of restorative materials using different photoinitiators. They feature high irradiance at multiple wavelengths as well as appropriate shape and dimension of the light guide to closely approach every area in the mouth.
There are many curing factors influencing DC such as the “total energy” concept, meaning that the radiant energy is the product of irradiance and exposure time (which means the higher the irradiance, the shorter the exposure time), temperature buildup during light-curing procedures, maintenance of curing units, as well as measurements of the power density [41].
The photopolymerization method has a profound influence on DC and shrinkage stress [42]. High-intensity lights provide higher DC values, but they also produce higher contraction stress. A slower curing process that allows composite flow may act for stress relaxation during polymerization [43]. The aim of optimal photopolymerization is to enable, for as long as possible, the pre-gel phase and to decrease the rate of polymeric chain growth, allowing more plastic deformation to occur and preventing the buildup of interfacial stresses [13]. For this purpose, modified photo curing protocols with variable intensities throughout the cure cycle were devised [44, 45].
2.2.2 Pre-heating of Composites
Increased curing temperature favors radical and monomer mobility which in turn results with a higher DC [16, 46]. This affects both physical and mechanical polymer properties as well as the biocompatibility of pre-heated composites. Raising the cure temperature reduces viscosity and enhances molecular mobility and collision frequency of reactive species. On the other hand, it postpones diffusion-controlled propagation, which then increases the final limiting conversion [46]. Pre-heating composites before photo curing decreases their viscosity, enhances marginal adaptation, and reduces microleakage [16]. However, increased degree conversion of pre-heated composite resins leads to higher shrinkage and possibly greater polymerization shrinkage stress [47].
The effect of temperature on the DC depends on the photoinitiator system. In the study of Tauböck et al. [16], the only bulk-fill composite material showing higher DC upon pre-heating contains an additional germanium-based photoinitiator besides CQ/amine system [34]. Unlike the aforementioned report [47], this study showed that composite resin pre-heating can reduce polymerization shrinkage forces of bulk-fill and conventional composite resins, whereas the DC remains the same or increases [16].
2.2.3 Post-cure Reaction
Post-cure polymerization is a direct result of the nature of multifunctional methacrylate polymerization reaction. A considerable increase in viscosity and a decrease of reactive species mobility are caused by the initial rapid reaction [18, 48]. Free radicals and unreacted monomers remain trapped within the matrix shortly after the start of illumination. The reaction has the potential to continue at a slow rate [49] and can be continued for as long as there are free radicals available and reactants which are sufficiently mobile [50].
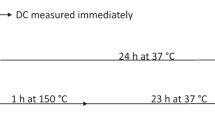
Post-cure reaction was found to be more extensive in materials with an initially lower DC. In cases of highly polymerized materials, reactive sites are immobilized in the polymer network, while in samples with an initially lower DC, a higher amount of unreacted radicals allows increased mobility to make contact with other reactive species. According to Burtscher [49], even a small increase in the extent of DC toward the end of polymerization process can greatly influence the density of cross-linking and optimize the properties of composite resins. From the clinical standpoint, the post-cure DC increase (Fig. 5.1) implies that final mechanical properties and biocompatibility of a composite are not attained immediately after curing but rather developed gradually over the post-cure period. In the study of Par et al. [17], the highest post-cure increase was achieved in materials with patented modified UDMA as well as proprietary monomers analogous to Bis-GMA and Procrylat resin. It is possible that these modified monomers acted to alter polymerization kinetics and retard the conversion of monomers [51].

Degree of conversion of some commercial composites measured immediately after light curing and 24 h post-cure. Error bars denote ±1 standard deviation. Reprinted with permission from [17]
3 Composite Properties Dependent on the Degree of Conversion
The DC is a fundamental attribute of a cured composite since it affects virtually all other material properties [2]. Most of these properties are important for the clinical success of the restoration, either in short or long term [52]. Although the composition of contemporary composites is fine-tuned to attain optimal DC and the related properties if properly handled and light-cured [15, 53, 54], poor DC due to unfavorable curing conditions or operators’ insufficient understanding of the curing procedure may affect critical material properties and increase the risk of clinical failure [55]. The influence of the DC on various composite properties is described below.
3.1 Mechanical Properties
Higher DC implies that more double bonds were consumed for the formation of an infinite polymeric network and that lower amount of unreacted monomer is left to plasticize the network structure [56]. Both of these factors improve mechanical properties, such as strength, elastic modulus, hardness, and glass transition temperature [56,57,58,59,60]. However, the well-known correlation between the DC and mechanical properties is valid only for a given composite formulation since mechanical properties do not depend solely on the DC but also on multiple other factors, most prominently filler load and resin composition [19, 61, 62]. While the filler load and particle geometry are primary determinants of mechanical properties, monomer composition determines the mobility of reactive medium during polymerization, consequently influencing the final DC and polymer structure. In this regard, varying the monomer composition could increase the final DC by virtue of increasing the resin mobility, but this does not necessarily improve material properties [14, 63]. For example, increasing the ratio of the diluent monomer TEGDMA in a Bis-GMA/TEGDMA copolymer improves the DC but renders the composite very brittle [19, 64]. Conversely, increasing the ratio of Bis-GMA may not reduce strength or hardness despite the decline in final DC [56, 57]. This exemplifies that the molecular architecture of monomers is an important determinant of mechanical properties—excessively increasing DC at the cost of the ratio of high molecular weight monomers impairs final mechanical properties of the cured resin. Thus, tailoring a composite with optimal DC and mechanical properties implies attaining a delicate balance between the stiffer “base” monomers and the DC-improving diluent monomers [19].
The kinetics of polymerization reaction, which is for a given composite determined by light-curing conditions, governs the final polymeric structure and the relative amount of cycles, linear chains, and cross-links [65]. Additionally, the polymeric network is substantially heterogeneous, containing high-DC microgel regions, regions of lower DC and unreacted monomer pools [66]. The heterogeneity is DC dependent and increases at higher conversions [67]. These complex structural features are not described by the DC [68, 69] and can only be assessed indirectly through measurements of material softening, glass transition temperature, or dielectric properties [58, 70, 71]. Thus, although the mechanical properties on macro- and microscale might appear constant and correlate well with the DC, a considerable heterogeneity always exists on the fundamental scale. The DC is simply the average measure of consumed double bonds which provides information on neither structural heterogeneity nor cross-linking density [72, 73]. Even within a given material, the final polymeric structure is highly dependent on light-curing conditions [74, 75], and the same DC values may not necessarily suggest identical polymer architecture and mechanical properties [45, 76]. Besides, small changes in the DC at late stages of polymerization can have a considerable effect on mechanical properties, as they mostly contribute to cross-linking [71], resulting in an exponential improvement in mechanical properties [77]. All of these facts indicate that although the DC correlates well with mechanical properties on the clinically relevant scale, it does not provide a complete description of subtle differences in network structure on the molecular level.
3.2 Polymerization Shrinkage and Shrinkage Stress
The polymerization reaction of methacrylates is inseparably linked to volumetric shrinkage, due to the conversion of intermolecular distances among separate monomer molecules of 0.3–0.4 nm into the distance of covalent bonds of about 0.15 nm [19]. As described previously, the amount of shrinkage is linearly related to the DC [78]. While a high DC is desirable to ensure optimal composite properties, it is accompanied with high volumetric shrinkage, contributing to the interfacial stress [79]. Attaining high DC and preserving marginal integrity are therefore conflicted aims, and this issue persists throughout the history of dental composites [80].
Unlike volumetric shrinkage, shrinkage stress developed under constrained conditions shows a highly nonlinear dependence on the DC. Shrinkage stress is a function of volumetric shrinkage and elastic modulus, both of which rise simultaneously during polymerization and are directly dependent on the DC [81]. Whereas low levels of the DC either allow viscous flow (prior to gelation) or high compliance due to low elastic modulus (prior to vitrification), at later stages of polymerization, the stress buildup increases rapidly [82]. After the vitrification point, the material is characterized by high elastic modulus, and small increases DC can have a huge impact on the shrinkage stress [83].
A popular approach for mitigating shrinkage stress in a clinical setting is by using modulated curing protocols, so-called soft start or pulse delay curing [44, 45]. The former combines a period of low irradiance followed by a high irradiance, whereas the latter introduces a time delay between two expositions. Both approaches are intended to allow more viscous flow to diminish the shrinkage stress buildup as well as to change the relative amount of linear chains and cross-links [44, 75]. Some authors argued that the observed benefits in terms of reduced shrinkage stress are not due to delayed gelation or vitrification but due to inferior DC or network microstructural differences resulting from the modulated curing protocols [60, 84]. It is important to note that some of the modulated protocols may impair material properties without being reflected on the DC [45]. The previously discussed subtle structural differences arising from different curing conditions may be the reason for the conflicting literature reports on the stress-decreasing efficiency of modulated curing protocols [2, 60, 83, 85, 86]. The material dependence of the effect of different curing protocols is another possible explanation for the inconsistent literature evidence about their benefits [87].
3.3 Biocompatibility
Various components are released in an aqueous environment from a cured composite material: monomers, components of the photoinitiator system, and various degradation products [88, 89]. As the most abundant mobile species present within the cured composite, residual monomer has traditionally been attributed the majority of the toxic effect, but other compounds may elute in comparable or even higher amounts [89] and exert a toxic effect [90]. The final DC of contemporary dental composites ranges from about 50% to 80%, suggesting that 20–50% of double bonds remained unreacted [21]. However, not all of these bonds can be eluted, since most of them are embedded in the polymeric network as pendant double bonds [91]. It is estimated that only 5–10% of the total amount of unreacted double bonds is capable of being eluted from the cured composite, amounting to approximately 2 wt% of the resin component [88]. The inverse relationship between the DC and the amount of eluted monomers is well documented in the literature [91,92,93]. Higher DC not only leaves less free monomer available for leaching out of the cured composite but also implies a denser network which reduces water diffusivity [94], thus hindering the release of monomer and other leachable species. The inverse relationship between the DC and biocompatibility is particularly relevant for the class of “bulk-fill” composites, whose application in thick layers may lead to the higher release of potentially toxic compounds [92]. An additional concern regarding bulk-fill composites is due to manufacturer’s recommendations of very short curing times which may be insufficient to attain optimal DC throughout thick layers [15, 95]. It should be noted that despite an established correlation, the DC is not a single determinant of the amount of available leachable species, since different architectures of polymer network as a function of curing conditions may lead to different amounts of leachable molecules within a single material at similar DC values [75, 91].
In addition to the release of potentially toxic components, biocompatibility issues related to low DC arise from the propensity of unreacted monomers to promote bacterial colonization [96]. Furthermore, some products of composite degradation can influence biofilm formation and survival of S. mutans on restoration surfaces [97]. This is particularly relevant to the development of secondary caries at the proximal cavity floor [98], which has the highest risk of remaining undercured in a clinical situation, especially when the “bulk-fill” technique is used. The risk of secondary caries at this site is additionally heightened by faster degradation of poorly cured composite [99].
3.4 Water Sorption, Solubility, Degradation, and Color Stability
The amount of water absorbed by a composite is mainly determined by the filler ratio and hydrophilicity of the resin [94, 100]. Although the polymeric matrix is insoluble, it can be penetrated and expanded by water molecules which show affinity to certain functional groups or bonds contained within the network, e.g., hydroxyl groups and ester or ether bonds [101]. By affecting the network density, the DC determines the amount and diffusivity of absorbed water [94]. Additionally, DC also determines the amount of unreacted monomer, which can leach out of the polymerized material. Thus, both water sorption and solubility show a good correlation with the DC for a given composite [102].
All composite components undergo a degradation process when exposed to oral environment; polymeric network is cleaved by hydrolysis and enzymatic breakdown, siloxane bonds at the filler/matrix interface are hydrolyzed, and components of filler particles are solubilized and eroded [88, 103, 104]. Since all of these reactions occur in an aqueous medium, the DC-dependent mobility and amount of water within the polymeric network determine the extent and rate of degradation [94]. Additionally, DC affects the amount of unreacted monomer which by its elution forms porosities within the material, allowing more water sorption and facilitating degradation [101]. Color stability is jeopardized by material degradation and uptake of pigment particles from oral environment, both of which depend on porosity and water sorption [105, 106]. Exposition to water diminishes mechanical properties by plasticization and swelling of the matrix [107]. These effects are more pronounced in the case of low DC and lead to increased abrasive wear [108].
As is the case with other DC-dependent properties, the correlation of water sorption, solubility, and degradation with the DC does not hold for comparison across different composites due to the influence of multiple other factors. Additionally, the same material with similar DC values attained under various curing conditions can show different water sorption and susceptibility to degradation [45], once again highlighting the fact that DC by itself is insufficient for a complete characterization of the network structure, as it is considerably influenced by structural complexities that cannot be described with a simple proportion of converted double bonds [65, 76, 109].
4 Methods for the DC Evaluation
4.1 Vibrational Spectroscopies: Infrared and Raman
4.1.1 Theoretical Background
Every molecule is characterized by a specific set of energetic states that are determined by its atomic arrangement. The collection of energetic transitions between these states gives rise to the pattern that is unique for a certain chemical compound. Probing the sample by means of vibrational spectroscopies gives a spectrum in which the transitions between vibrational states are represented by spectral bands. The position of the band within the spectrum is determined by transition energy between vibrational states and is commonly expressed in wavenumbers (cm−1). By assigning the specific vibrational band to a certain functional group or bond within the molecule, vibrational spectroscopies can provide both qualitative and quantitative information [110]. The translation of spectroscopic data into absolute concentration values requires the usage of calibration curves, which relate the concentration of a particular species with the intensity of its respective vibrational band. Alternatively, the ratio of concentrations can be calculated without using calibration curves; instead the spectra can be normalized by using the internal standard, i.e., the vibrational band whose intensity remains constant [111]. In the case of dental composites, this means using a band which undergoes no change throughout the polymerization. Thus, the DC calculations do not require calibration curves, as only the relative amount of the double bonds consumed during polymerization is evaluated [112, 113]. Since the double C=C bonds are contained within different monomers, the underlying assumption is that their infrared absorptivity and Raman scattering intensity are independent on the molecular structure of the monomer. This assumption holds for infrared absorptivity, while the Raman scattering intensity of the C=C bonds may differ among different monomers [114]. However, this difference is neglected when evaluating the DC by means of Raman spectroscopy, since the spectra of polymerized and unpolymerized material used for the calculation have qualitatively the same monomer composition [114].
The physical phenomena behind the infrared and Raman spectroscopy are different; however, both methods are equally used for the DC measurements and give similar results [29, 115]. The choice of the method probably depends on the convenience for a particular experimental design and availability of the instrumentation. Overall, infrared spectroscopy is considered more traditional method for DC measurements and is more frequently used [15, 16, 53, 68, 116].
4.1.2 Infrared Spectroscopy
Infrared spectroscopy is based on absorption of photons with energy equal to the energy difference between molecular vibrational energy states. The infrared spectrometer operates by illuminating the sample with infrared beam and recording the radiation that is transmitted through or reflected from the sample. The remaining radiation carries information on how much of the incident radiation was absorbed for a particular wavelength and is used to produce the vibrational infrared spectrum. Most of the contemporary infrared spectrometers are based on an interferometer and use Fourier transform to convert an interferogram into a spectrum, thus the term Fourier-transform infrared (FTIR) spectroscopy [117].
Infrared spectroscopy can be performed in different regions of the infrared spectrum, which are named after their relative position to the visible light as near-infrared (14,000–4000 cm−1), mid-infrared (4000–400 cm−1), and far-infrared (400–4 cm−1) [117]. The DC of dental composites is commonly measured in the mid-infrared region by quantifying the change in the intensity of the vibrational band at 1640 cm−1 that corresponds to the stretching vibration of aliphatic C=C bonds in methacrylate molecules [112]. Assuming the proportionality of the aliphatic C=C bonds concentration to the corresponding vibrational band intensity, information on the amount of converted bonds can be extracted by comparing the band intensity in polymerized and unpolymerized material [114]. As an internal standard, the band at 1610 cm−1 representing the aromatic C=C absorption is commonly used. The availability of this vibrational band depends on the presence of methacrylate monomers containing an aromatic core, such as Bis-GMA and Bis-EMA. In composites based on aliphatic monomers, e.g., UDMA, the vibrational band at 1610 cm−1 is absent, and other alternative reference bands are used [53, 118].
For spectrum processing, various methods of baseline subtraction and band deconvolution can be performed [119,120,121]. The linear relationship between the aliphatic C=C concentration and the corresponding band intensity is highly dependent on the baseline selection. Different baselines may cause considerable variations in the final DC results, reaching up to 20% [120]. Further issue related to the baseline position is the change in intensity of the nearby C=O band at 1715 cm−1 due to the conjugation of C=O group with C=C bonds consumed during polymerization [64, 122]. Thus, some authors recommended using band height instead of the band area, since the former is less affected by the baseline change [115]. Another procedure that may affect the DC values is the decomposition procedure (peak fitting) that is performed to resolve partially overlapping bands at 1610 and 1640 cm−1 [119]. However, it was shown that accurate DC calculations can also be performed using original, unfitted spectra [120]; thus, some authors find the fitting procedure unnecessary [115]. In any case, the variations in spectra processing do affect the final results to a various degree [115, 120, 123], and this fact should be considered when comparing DC values among different studies. Unfortunately, the detailed information about spectra processing is rarely reported in research papers.
Vibrational band intensities are used to calculate the DC by the equation [124]:
where I C=C and I reference represent the band intensity of aliphatic C=C stretching (1640 cm−1) and the intensity of the reference band (Fig. 5.2). In this equation, the expression contained within the fraction essentially represents the ratio of the aliphatic C=C bonds remaining after polymerization, while subtracting this value from unity gives the percentage of the consumed double bonds, which is the definition of DC [112].

The part of the infrared spectrum used for the DC evaluation. The spectra of uncured and cured sample are normalized to the reference band at 1610 cm−1, and the change in intensity of the band at 1640 cm−1 is used to calculate the DC
Sample preparation for infrared measurements varies according to the mode of collecting the spectra. The most classical technique for recording in transmission mode involves grinding the tested material, mixing it with potassium bromide (KBr) powder, and pressing the mixture into a thin pellet [16]. The pellet contains only a small concentration of the sample (0.3–1% by weight), dispersed in the KBr matrix which is transparent for infrared radiation. The pellet is then illuminated with an infrared beam, and the transmitted radiation is registered by a detector (Fig. 5.3). Main shortcomings of this approach are its destructiveness and elaborate sample preparation. Alternatively, infrared spectrum can be recorded in transmittance mode by pressing a composite into a sufficiently thin film [124] or in reflectance modes—attenuated total reflectance (ATR) and diffuse and specular reflectance. ATR mode is gaining popularity due to its convenience [125], whereas other reflectance modes are practically possible but not widely employed. In the ATR mode, the sample is positioned in a tight contact with the crystal of high refractive index, e.g., zinc selenide, diamond, or germanium. By directing the infrared beam at a certain angle onto the crystal, light is totally reflected from the border between the crystal and the sample. During the process of total reflection, infrared light penetrates the sample, while the absorption occurs at the crystal surface in contact with the sample. After multiple internal reflections, attenuated beam exiting the crystal is guided to the detector [117]. The major advantage of ATR is that it requires no additional sample preparation besides applying the uncured composite paste or clamping the polymerized composite sample onto the crystal (Fig. 5.4). ATR mode also allows evaluation of polymerization kinetics through the real-time DC measurements during the light curing with high data acquisition rate [126, 127].

Sample preparation for infrared spectroscopy in transmission mode. A small amount of composite sample (a) is ground with potassium bromide into powder (b), pressed into a pellet (c, d) which is mounted into a holder (e), and positioned inside the sample compartment of (f) an infrared spectrometer

Attenuated total reflectance (ATR) accessory of the infrared spectrometer and experimental setup for real-time measurements. Uncured composite is applied in the direct contact with zinc selenide crystal of the ATR accessory (a). The light-curing unit tip is positioned immediately above the composite sample, allowing spectra recording and light curing to be performed simultaneously (b)
The caution must be exercised when recording the spectra in the mid-infrared region, since the vibrational bands of interest (1640 and 1610 cm−1) are located within the absorption spectrum of water [117]. Therefore, the samples must be dry, and the air humidity in the sample compartment must be kept constant; otherwise, water vibrational bands may interfere with the DC measurement. The effect of water can be eliminated by purging the sample compartment with inert gas (e.g., nitrogen) or by switching to the near-infrared region in which the vibrational bands used for DC evaluation are not affected by the presence of water [111]. Measuring the DC in near-infrared region provides some additional advantages. Due to lower absorptivity in the near-infrared region, much higher path-lengths are allowed (up to 4 mm), enabling measurements through the bulk of the samples without any previous preparation [128]. If the sample geometry is precisely determined, using near-infrared eliminates the need for the internal standard, and only the intensity of the =C-H2 vibration band at 6165 cm−1 is assessed for the DC calculation [128, 129]. Despite these benefits, the near-infrared region is much less often used for DC measurements than the mid-infrared.
4.1.3 Raman Spectroscopy
Raman spectroscopy probes molecular vibrations by means of inelastic Raman scattering. Unlike the elastic (Rayleigh) scattering, inelastic scattering is characterized by different energies of incident and scattered photons. Raman scattering is induced by exciting a molecule to a virtual energy state and its subsequent relaxation to an energy state that is either higher or lower than the original state. If the molecule undergoes an excitation from ground energy state and then relaxes to one of the higher energy states, the energy of scattered photon is lower than that of the incident photon (Stokes Raman scattering). Inversely, the excitation from one of the excited vibrational energy states and then relaxation to the ground energy state result with the scattered photon having higher energy than the incident photon (anti-Stokes Raman scattering, Fig. 5.5). In both cases, the energy difference represents the Raman shift and corresponds to the characteristic vibrational energy transition of a molecule [110].

A schematic representation of elastic (Rayleigh) and inelastic (Raman) scattering. Inelastically scattered photons have either lower (Stokes) or higher (anti-Stokes) energy than the incident photons. This energy difference (ΔE) represents Raman shift
In a Raman spectrometer, the scattering effect is induced by illuminating the sample with monochromatic laser radiation in the ultraviolet, visible, or near-infrared region. The light scattered on the sample is collected with a lens, and the elastically scattered photons (Rayleigh) are filtered out. These photons have the same energy as the incident photons emitted by a laser and carry no information on vibrational spectrum of the molecule. Only the small portion of photons which were inelastically scattered (about 1 in 10 million) is led to the detector for the reconstruction of the Raman spectrum [130]. Various Raman spectrometers offer a wide range of excitation laser wavelengths. Since the Raman scattering intensity is proportional to the negative fourth power of the excitation wavelength, shorter wavelengths are beneficial due to higher signal intensity. However, shorter wavelengths induce more unwanted fluorescence which interferes with the signal in the spectral region of interest. This can be overcome by using the excitation laser of longer wavelength (near-infrared), since its energy is insufficient to excite the energetic transitions responsible for fluorescence [114]. Such infrared lasers are usually a part of the hybrid FTIR/FT-Raman instruments that share the same optics and hardware for both techniques. The drawback of near-infrared excitation is lower intensity of Raman scattering and the need to use higher laser power or longer acquisition times in order to improve the signal [18]. Another important consideration regarding the selection of excitation wavelength is to avoid its overlapping with the absorption spectrum of the photoinitiator system [131], in order to avoid any additional photocuring during the measurement.
Raman and infrared spectroscopy are complementary techniques, as each vibrational transition may be active in infrared, Raman, or both. Generally, infrared spectroscopy is more sensitive for polar bonds, yielding strong vibrational bands with high signal-to-noise ratio, whereas Raman is more appropriate for examining nonpolar bonds. The vibrational bands pertaining to aliphatic C=C and aromatic C=C used for the DC assessment are well detectable in both Raman and FTIR; thus, both methods can be used interchangeably. The Raman spectrum is processed by baseline subtraction and deconvolution similarly to the infrared spectrum, and the same equation (Eq. 5.1) is used for the DC calculation. As in the case of the infrared spectroscopy, the parameters of spectra processing that influence the final DC results [115] are often reported with insufficient parameters, affecting the inter-study comparability of DC values.
Raman can be advantageous over the infrared spectroscopy due to much simpler sample preparation. The uncured or cured composite is mounted on the sample holder “as is” and aligned with the laser beam (Fig. 5.6), eliminating the need for laborious preparation of KBr pellets or using the accessories for various reflectance modes in the case of infrared spectroscopy [17]. The measurements can be done in situ, on samples of any geometry and without damaging the samples. Another advantage of Raman spectroscopy is its insensitivity to water, which eliminates the need to control the environmental humidity and allows measurements on wet samples. Raman spectrometer can be coupled with a microscope to enable focusing of the laser beam onto small areas of the sample (micro-Raman) for assessing the local DC (Fig. 5.7). A set of such local measurements can be used to create a DC map and investigate the DC as a function of the position within the sample [132].

Sample compartment of a FT-Raman spectrometer. The sample in its original state is mounted on the universal holder and aligned with the excitation laser beam. The lens collects scattered photons, which are subsequently filtered and led to the detector

Raman spectrum of the uncured composite (black) and spectra collected from various depths of the cured sample. The sample of a conventional composite was light cured from one side, and measurements were done at increasing distance (“depth”) from the cured surface. All spectra are normalized to the reference band at 1610 cm−1 so that intensities of the band at 1640 cm−1 can be directly compared. Lower intensities of the band at 1640 cm−1 indicate higher consumption of aliphatic C=C double bonds at the surface and nearby depths (1 and 2 mm), whereas DC considerably declines as the measurement depth increases further, approaching that of the uncured material
There are several drawbacks regarding the use of Raman for DC measurements. Due to the low intensity of Raman scattering, longer acquisition times are needed to obtain a good signal-to-noise ratio compared to the infrared measurements. This prevents real-time DC monitoring and may also present a problem if measurements are performed before the slowly developing DC has stabilized [133]. Thus, Raman is more suitable for assessing the DC in what can be considered a static system, which usually means 24 h after light curing [134]. Another concern regarding Raman spectroscopy may be localized temperature increase at the spot excited with laser beam, which may increase the mobility of unreacted monomers and free radicals, thus furthering the polymerization. The concerns about detrimental effect of heating by excitation laser were expressed for biological samples [135], but there are no reports of the laser heating effect on the DC of dental composites.
4.1.4 Limitations of the Spectroscopic Techniques
The inherent shortcoming of infrared and Raman spectroscopy is their inability to differentiate between the aliphatic C=C bonds that originate from methacrylate monomers from these contained within the silane molecules. The double bonds in silane molecules undergo conversion in the same way as the monomeric double bonds, but they show lower reactivity, especially if located in deeper parts of the silane layer [136]. The amount of silane varies greatly among composites due to differences in the filler load and particle size [54]. Both these factors determine the effective particle surface area that needs to be covered with silane. For instance, in experimental composites with low filler load (40–50 wt%) and a small amount of nano-sized particles, up to 4.4% of the total number of C=C bonds were contained within silane molecules [18]. Commercial composites contain much higher filler loads, and the contribution of the silane to the total filler load ranging from 2.8% to 9.0% [137] is not negligible. In light of these facts, the net conversion of C=C bonds from methacrylate monomers is generally somewhat higher than the spectroscopically determined DC, which takes into account both monomeric and silane C=C bonds.
A major issue regarding laboratory investigations of DC is its sensitivity to experimental conditions that are often not sufficiently controlled. The DC is commonly investigated as a function of material, layer thickness, and curing protocol [15]; however, some important factors are either ignored or beyond the control of investigators. For example, mold size and reflectivity play an important role in the reflection of curing light from mold borders [138, 139]. Furthermore, the mold material determines its thermal capacitance and conductivity, thus affecting the heat dissipation from the specimen. The amount of the material contained in the specimen influences the amount of released heat and temperature rise, which has an impact on polymerization kinetics and final DC values [140]. Additionally, environmental temperature and storage conditions affect the polymerization kinetics both during and after light curing [133]. The aforementioned differences in spectra processing further contribute to the variability of the DC results. All of these facts must be considered when comparing DC results obtained from different experimental setups. Currently no standardized method for the DC evaluation exists, but developing one in the future would highly improve the interlaboratory comparability of results [141].
4.2 Indirect Methods for the DC Evaluation
In addition to the spectroscopic techniques which are considered “direct” methods for the DC evaluation, the extent of conversion can be assessed indirectly by several methods that measure various other properties which change along with the DC. Due to the complex influence of multiple factors that are either unknown or difficult to assess, some of these properties are useful only as approximate indicators of the polymerization progress, and most of them are impossible to translate into absolute DC values. Among the indirect methods, microhardness testing is by far the most frequently used but also often criticized. Together with another classical (and disputed) method, the ISO 4049 depth of cure, microhardness is classically used to evaluate the efficiency of curing at depth. These and other less conventional methods for indirect DC assessment are briefly described below.
4.2.1 Microhardness
Microhardness measurements are performed by impressing a diamond indenter of a specific geometry into the composite surface. The surface of indentation is measured and together with a known force of indentation used to calculate the microhardness value [142]. In practical terms, microhardness represents the resistance of a material to plastic deformation and wear by abrasion. Due to a good correlation between microhardness and DC [69, 77], this method has a long history of use for indirectly measuring the efficiency of cure [73] (Fig. 5.8). The correlation is valid only when the DC and microhardness values are compared for samples of various conversions within a single composite formulation and does not hold for different composites as both properties are independently influenced by other factors [143]. Microhardness shows an exponential dependence on DC [77, 144] since it is considerably influenced by cross-linking which is scarce in the early stage of the polymerization and increases as the polymerization progresses [48]. Thus, the sensitivity of microhardness measurements increases at higher conversions, allowing the detection of small changes in the DC [77].

Micrograph of indentations made on the top (a) and bottom (b) of a 6-mm-thick specimen of a bulk-fill composite. Greater surface area of the bottom indentation (b) reflects DC decline due to the curing light attenuation at depth
4.2.2 The Depth of Cure (DoC) Concept
Since the curing light is attenuated while passing through a composite material due to light scattering and absorption, there is a gradual decline in the DC through the composite layer (Fig. 5.1) [17]. The DoC concept is intended to detect the distance from the composite surface at which the DC heterogeneity becomes clinically unacceptable. Thus, the DoC is defined as the maximum thickness of a composite material that can be adequately cured in a single layer [69]. The term “adequately” has raised much dispute rendering the DoC concept itself rather controversial. Basically, the cure is defined as adequate if microhardness values are higher than 80% of the maximum value which is often, but not always, obtained at the surface [15]. Although originally devised for microhardness, the similar principle of identifying the depth with 80% of the maximum value has also been applied to the DC [145]. Alternatively, the 90% of the maximum DC value is sometimes considered as the “adequate cure” [132], since it was estimated that 80% reduction in hardness corresponds to 90% reduction in DC [146]. A major issue with the DoC concept is that the postulated cutoff values (whether microhardness or DC) are selected completely arbitrarily with neither physical meaning nor any measurable change in composite properties occurring at the threshold value [147]. The gradual decline in all composite properties with depth is inevitable, but there is no clear rationale for setting either the 80% or 90% cutoff value for any of the measured properties. Despite the controversy and lack of physical foundation, the DoC concept continues to be used to assess curing efficiency, and 80% of the maximum microhardness value is routinely quoted as adequate [15, 145, 146, 148]. An alternative definition of the DoC, as the depth of transition from glassy to rubbery state, has been proposed [69]. Despite being more physically justified, this approach is not commonly used as a criterion for the DoC.
4.2.3 ISO 4049 Depth of Cure
The simplest method to evaluate light-curing efficiency as a function of depth is devised by the International Organization for Standardization (ISO) and known as the ISO 4049 DoC. Briefly, a cylindrical composite specimen is prepared in a stainless-steel mold with the diameter of 4 mm. Both apertures of the mold are covered with Mylar strips, and the composite is cured through the upper aperture. Immediately after light curing, the specimen is removed from the mold, and the uncured material is removed with a plastic spatula. The height of the remaining cylinder is measured with a micrometer and divided by two. The obtained value is regarded as the maximum thickness of the composite increment that can be cured in a single exposition, i.e., the DoC [149].
Unlike other DoC testing methods, the ISO 4049 DoC requires no sophisticated instrumentation. It is intended for a quick and cost-effective screening of the relative performance of various combinations of composite materials and curing conditions, without claiming any correlation with mechanical properties or clinical performance [138]. There is much criticism of the ISO 4049 DoC, arising from the lack of either physical or clinical rationale for the procedure and calculation performed, poor correlation with values obtained by other DoC methods [69], sensitivity on the operator technique, and dependence on the mold characteristics [138, 139]. Despite criticism, the ISO 4049 DoC test fulfills the basic requirement of a standard testing method, which is to be as simple and as reproducible as possible [150] and is currently mandatory for determining maximum layer thickness and recommended curing times for all commercial composites [139].
4.2.4 Other Indirect Indicators of Conversion
Differential scanning calorimetry [151] and differential thermal analysis [152] measure the enthalpy of the polymerization reaction and make use of the known amount of heat released for a mole of the methacrylate functional group converted (approximately 55 kJ/mol) to calculate the DC [153]. By measuring the heat flow in real time, information on polymerization kinetics can be obtained [154].
Polymerization shrinkage can be used to indirectly evaluate the DC in a similar manner. Since each mole of converted double bonds contributes to a particular amount of polymerization shrinkage of approximately 20 cm3/mol [155], shrinkage data can be translated into DC [156].
Softening in ethanol and glass transition temperature are indirect measures of cross-linking density, since more cross-linked polymeric networks are less prone to hardness deterioration in a solvent and undergo glass transition at higher temperatures [58, 70]. Although the cross-linking density increases along with the DC, it must not be considered a simple function of the DC since cross-linking is independently affected by curing conditions which influence the rates of initiation and termination and thus control the number of active growth centers [2].
Light transmittance undergoes a gradual change as the DC increases, due to the changes in resin refractive index and consumption of photoinitiator [157]. Although the clear relationship with DC cannot be determined due to the convolution of many simultaneously occurring processes, monitoring the light transmittance in real time is a simple method that can provide some insight into polymerization kinetics [126, 158].
Impedance spectroscopy operates by measuring electrical properties of the sample over the range of alternating current frequencies. Since the formation of polymeric network occurring during polymerization is reflected as the change in electrical conductivity, the latter can be used for real-time monitoring of the polymerization kinetics [159, 160].
References
Ilie N, Hickel R. Resin composite restorative materials. Aust Dent J. 2011;56(Suppl 1):59–66.
Leprince JG, Palin WM, Hadis MA, Devaux J, Leloup G. Progress in dimethacrylate-based dental composite technology and curing efficiency. Dent Mater. 2013;29(2):139–56.
Ferracane JL. Resin composite—state of the art. Dent Mater. 2011;27(1):29–38.
Yoshida K, Greener EH. Effects of two amine reducing agents on the degree of conversion and physical properties of an unfilled light-cured resin. Dent Mater. 1993;9(4):246–51.
Calheiros FC, Daronch M, Rueggeberg FA, Braga RR. Degree of conversion and mechanical properties of a BisGMA:TEGDMA composite as a function of the applied radiant exposure. J Biomed Mater Res B Appl Biomater. 2008;84(2):503–9.
Krifka S, Seidenader C, Hiller KA, Schmalz G, Schweikl H. Oxidative stress and cytotoxicity generated by dental composites in human pulp cells. Clin Oral Investig. 2012;16(1):215–24.
Tarle Z, Meniga A, Ristic M, Sutalo J, Pichler G, Davidson CL. The effect of the photopolymerization method on the quality of composite resin samples. J Oral Rehabil. 1998;25(6):436–42.
Knezević A, Tarle Z, Meniga A, Sutalo J, Pichler G, Ristić M. Degree of conversion and temperature rise during polymerization of composite resin samples with blue diodes. J Oral Rehabil. 2001;28(6):586–91.
Tarle Z, Meniga A, Knezević A, Sutalo J, Ristić M, Pichler G. Composite conversion and temperature rise using a conventional, plasma arc, and an experimental blue LED curing unit. J Oral Rehabil. 2002;29(7):662–7.
Miletic V, Santini A. Optimizing the concentration of 2,4,6-trimethylbenzoyldiphenylphosphine oxide initiator in composite resins in relation to monomer conversion. Dent Mater J. 2012;31(5):717–23.
Turssi CP, Ferracane JL, Vogel K. Filler features and their effects on wear and degree of conversion of particulate dental resin composites. Biomaterials. 2005;26(24):4932–7.
Uctasli S, Tezvergil A, Lassila LV, Vallittu PK. The degree of conversion of fiber-reinforced composites polymerized using different light-curing sources. Dent Mater. 2005;21(5):469–75.
Tarle Z, Knezevic A, Demoli N, Meniga A, Sutaloa J, Unterbrink G, Ristic M, Pichler G. Comparison of composite curing parameters: effects of light source and curing mode on conversion, temperature rise and polymerization shrinkage. Oper Dent. 2006;31(2):219–26.
Czasch P, Ilie N. In vitro comparison of mechanical properties and degree of cure of bulk fill composites. Clin Oral Investig. 2012;17(1):227–35.
Tarle Z, Attin T, Marovic D, Andermatt L, Ristic M, Taubock TT. Influence of irradiation time on subsurface degree of conversion and microhardness of high-viscosity bulk-fill resin composites. Clin Oral Investig. 2015;19(4):831–40.
Taubock TT, Tarle Z, Marovic D, Attin T. Pre-heating of high-viscosity bulk-fill resin composites: effects on shrinkage force and monomer conversion. J Dent. 2015;43(11):1358–64.
Par M, Gamulin O, Marovic D, Klaric E, Tarle Z. Raman spectroscopic assessment of degree of conversion of bulk-fill resin composites—changes at 24 hours post cure. Oper Dent. 2015;40(3):E92–101.
Par M, Gamulin O, Marovic D, Skenderovic H, Klaric E, Tarle Z. Conversion and temperature rise of remineralizing composites reinforced with inert fillers. J Dent. 2016;48:26–33.
Peutzfeldt A. Resin composites in dentistry: the monomer systems. Eur J Oral Sci. 1997;105(2):97–116.
Amirouche-Korichi A, Mouzali M, Watts DC. Effects of monomer ratios and highly radiopaque fillers on degree of conversion and shrinkage-strain of dental resin composites. Dent Mater. 2009;25(11):1411–8.
Stansbury JW. Dimethacrylate network formation and polymer property evolution as determined by the selection of monomers and curing conditions. Dent Mater. 2012;28(1):13–22.
Skrtic D, Antonucci JM. Effect of chemical structure and composition of the resin phase on vinyl conversion of amorphous calcium phosphate-filled composites. Polym Int. 2007;56(4):497–505.
Lu H, Trujillo-Lemon M, Ge J, Stansbury JW. Dental resins based on dimer acid dimethacrylates: a route to high conversion with low polymerization shrinkage. Compend Contin Educ Dent. 2010;31(Special Issue 2):1–4.
Bracho-Troconis C, Trujillo-Lemon M, Boulden J, Wong N, Wall K, Esquibel K. Characterization of N’Durance: a nanohybrid composite based on new nano-dimer technology. Compend Contin Educ Dent. 2010;31(Special Issue 2):5–9.
Kurokawa R, Finger WJ, Hoffmann M, Endo T, Kanehira M, Komatsu M, Manabe A. Interactions of self-etch adhesives with resin composites. J Dent. 2007;35(12):923–9.
Lee SK, Kim TW, Son SA, Park JK, Kim JH, Kim HI, Kwon YH. Influence of light-curing units on the polymerization of low-shrinkage composite resins. Dent Mater J. 2013;32(5):688–94.
Bucuta S, Ilie N. Light transmittance and micro-mechanical properties of bulk fill vs. conventional resin based composites. Clin Oral Investig. 2014;18(8):1991–2000.
Rueggeberg FA. State-of-the-art: dental photocuring—a review. Dent Mater. 2011;27(1):39–52.
Leloup G, Holvoet PE, Bebelman S, Devaux J. Raman scattering determination of the depth of cure of light-activated composites: influence of different clinically relevant parameters. J Oral Rehabil. 2002;29(6):510–5.
Jakubiak J, Allonas X, Fouassier JP, Sionkowska A, Andrzejewska E, Linden LÅ, Rabek JF. Camphorquinone–amines photoinitiating systems for the initiation of free radical polymerization. Polymer. 2003;44(18):5219–26.
Neshchadin D, Rosspeintner A, Griesser M, Lang B, Mosquera-Vazquez S, Vauthey E, Gorelik V, Liska R, Hametner C, Ganster B, Saf R, Moszner N, Gescheidt G. Acylgermanes: photoinitiators and sources for Ge-centered radicals. Insights into their reactivity. J Am Chem Soc. 2013;135(46):17314–21.
Leprince JG, Hadis M, Shortall AC, Ferracane JL, Devaux J, Leloup G, Palin WM. Photoinitiator type and applicability of exposure reciprocity law in filled and unfilled photoactive resins. Dent Mater. 2011;27(2):157–64.
Neumann MG, Schmitt CC, Ferreira GC, Correa IC. The initiating radical yields and the efficiency of polymerization for various dental photoinitiators excited by different light curing units. Dent Mater. 2006;22(6):576–84.
Moszner N, Fischer UK, Ganster B, Liska R, Rheinberger V. Benzoyl germanium derivatives as novel visible light photoinitiators for dental materials. Dent Mater. 2008;24(7):901–7.
Jandt KD, Mills RW, Blackwell GB, Ashworth SH. Depth of cure and compressive strength of dental composites cured with blue light emitting diodes (LEDs). Dent Mater. 2000;16(1):41–7.
Pradhan RD, Melikechi N, Eichmiller F. The effect of irradiation wavelength bandwidth and spot size on the scraping depth and temperature rise in composite exposed to an argon laser or a conventional quartz–tungsten–halogen source. Dent Mater. 2002;18(3):221–6.
Tarle Z, Meniga A, Ristic M, Sutalo J, Pichler G. Polymerization of composites using pulsed laser. Eur J Oral Sci. 1995;103(6):394–8.
Meniga A, Tarle Z, Ristic M, Sutalo J, Pichler G. Pulsed blue laser curing of hybrid composite resins. Biomaterials. 1997;18(20):1349–54.
Park SH, Krejci I, Lutz F. Microhardness of resin composites polymerized by plasma arc or conventional visible light curing. Oper Dent. 2002;27(1):30–7.
Knezević A, Tarle Z, Meniga A, Sutalo J, Pichler G, Ristić M. Photopolymerization of composite resins with plasma light. J Oral Rehabil. 2002;29(8):782–6.
Burtscher P. Visible light curing of composite resins—report. Research and development Ivoclar Vivadent AG, Schaan, Liechtenstein 18; 2007. p. 29–32.
Silikas N, Eliades G, Watts DC. Light intensity effects on resin-composite degree of conversion and shrinkage strain. Dent Mater. 2000;16(4):292–6.
Yap AU, Seneviratne C. Influence of light energy density on effectiveness of composite cure. Oper Dent. 2001;26(5):460–6.
Ilie N, Jelen E, Hickel R. Is the soft-start polymerisation concept still relevant for modern curing units? Clin Oral Investig. 2011;15(1):21–9.
Asmussen E, Peutzfeldt A. Influence of pulse-delay curing on softening of polymer structures. J Dent Res. 2001;80(6):1570–3.
Daronch M, Rueggeberg FA, De Goes MF, Giudici R. Polymerization kinetics of pre-heated composite. J Dent Res. 2006;85(1):38–43.
Lohbauer U, Zinelis S, Rahiotis C, Petschelt A, Eliades G. The effect of resin composite pre-heating on monomer conversion and polymerization shrinkage. Dent Mater. 2009;25(4):514–9.
Andrzejewska E. Photopolymerization kinetics of multifunctional monomers. Prog Polym Sci. 2001;26:605–65.
Burtscher P. Stability of radicals in cured composite materials. Dent Mater. 1993;9(4):218–21.
Leprince J, Lamblin G, Truffier-Boutry D, Demoustier-Champagne S, Devaux J, Mestdagh M, Leloup G. Kinetic study of free radicals trapped in dental resins stored in different environments. Acta Biomater. 2009;5(7):2518–24.
Par M, Lapas-Barisic M, Gamulin O, Panduric V, Spanovic N, Tarle Z. Long term degree of conversion of two bulk-fill composites. Acta Stomatol Croat. 2016;50(4):292–300.
Opdam NJ, van de Sande FH, Bronkhorst E, Cenci MS, Bottenberg P, Pallesen U, Gaengler P, Lindberg A, Huysmans MC, van Dijken JW. Longevity of posterior composite restorations: a systematic review and meta-analysis. J Dent Res. 2014;93(10):943–9.
Alshali RZ, Silikas N, Satterthwaite JD. Degree of conversion of bulk-fill compared to conventional resin-composites at two time intervals. Dent Mater. 2013;29(9):e213–7.
Ilie N, Hickel R. Investigations on mechanical behaviour of dental composites. Clin Oral Investig. 2009;13(4):427–38.
Price RB, Ferracane JL, Shortall AC. Light-curing units: a review of what we need to know. J Dent Res. 2015;94(9):1179–86.
Ferracane JL, Greener EH. The effect of resin formulation on the degree of conversion and mechanical properties of dental restorative resins. J Biomed Mater Res. 1986;20(1):121–31.
Asmussen E. Restorative resins: hardness and strength vs. quantity of remaining double bonds. Scand J Dent Res. 1982;90(6):484–9.
Lovell LG, Lu H, Elliott JE, Stansbury JW, Bowman CN. The effect of cure rate on the mechanical properties of dental resins. Dent Mater. 2001;17(6):504–11.
Lovell LG, Newman SM, Donaldson MM, Bowman CN. The effect of light intensity on double bond conversion and flexural strength of a model, unfilled dental resin. Dent Mater. 2003;19(6):458–65.
Dewaele M, Asmussen E, Peutzfeldt A, Munksgaard EC, Benetti AR, Finne G, Leloup G, Devaux J. Influence of curing protocol on selected properties of light-curing polymers: degree of conversion, volume contraction, elastic modulus, and glass transition temperature. Dent Mater. 2009;25(12):1576–84.
Chung KH, Greener EH. Correlation between degree of conversion, filler concentration and mechanical properties of posterior composite resins. J Oral Rehabil. 1990;17(5):487–94.
Leprince JG, Palin WM, Vanacker J, Sabbagh J, Devaux J, Leloup G. Physico-mechanical characteristics of commercially available bulk-fill composites. J Dent. 2014;42(8):993–1000.
Musanje L, Ferracane JL. Effects of resin formulation and nanofiller surface treatment on the properties of experimental hybrid resin composite. Biomaterials. 2004;25(18):4065–71.
Sideridou I, Tserki V, Papanastasiou G. Effect of chemical structure on degree of conversion in light-cured dimethacrylate-based dental resins. Biomaterials. 2002;23(8):1819–29.
Hadis M, Leprince JG, Shortall AC, Devaux J, Leloup G, Palin WM. High irradiance curing and anomalies of exposure reciprocity law in resin-based materials. J Dent. 2011;39(8):549–57.
Bowman CN, Kloxin CJ. Toward an enhanced understanding and implementation of photopolymerization reactions. AIChE J. 2008;54(11):2775–95.
Kannurpatti AR, Anseth JW, Bowman CN. A study of the evolution of mechanical properties and structural heterogeneity of polymer networks formed by photopolymerizations of multifunctional (meth)acrylates. Polymer. 1998;39(12):2507–13.
Frauscher KE, Ilie N. Degree of conversion of nano-hybrid resin-based composites with novel and conventional matrix formulation. Clin Oral Investig. 2013;17(2):635–42.
Leprince JG, Leveque P, Nysten B, Gallez B, Devaux J, Leloup G. New insight into the “depth of cure” of dimethacrylate-based dental composites. Dent Mater. 2012;28(5):512–20.
Schneider LF, Moraes RR, Cavalcante LM, Sinhoreti MA, Correr-Sobrinho L, Consani S. Cross-link density evaluation through softening tests: effect of ethanol concentration. Dent Mater. 2008;24(2):199–203.
Lovell LG, Berchtold KA, Elliott JE, Lu H, Bowman CN. Understanding the kinetics and network formation of dimethacrylate dental resins. Polym Adv Technol. 2001;12:335–45.
Soh MS, Yap AU. Influence of curing modes on crosslink density in polymer structures. J Dent. 2004;32(4):321–6.
Ferracane JL. Correlation between hardness and degree of conversion during the setting reaction of unfilled dental restorative resins. Dent Mater. 1985;1(1):11–4.
Peutzfeldt A, Asmussen E. Resin composite properties and energy density of light cure. J Dent Res. 2005;84(7):659–62.
Ilie N, Obermaier J, Durner J. Effect of modulated irradiation time on the degree of conversion and the amount of elutable substances from nano-hybrid resin-based composites. Clin Oral Investig. 2014;18(1):97–106.
Barszczewska-Rybarek IM. Structure-property relationships in dimethacrylate networks based on Bis-GMA, UDMA and TEGDMA. Dent Mater. 2009;25(9):1082–9.
Rueggeberg FA, Craig RG. Correlation of parameters used to estimate monomer conversion in a light-cured composite. J Dent Res. 1988;67(6):932–7.
Rueggeberg F, Tamareselvy K. Resin cure determination by polymerization shrinkage. Dent Mater. 1995;11(4):265–8.
Braga RR, Ferracane JL. Contraction stress related to degree of conversion and reaction kinetics. J Dent Res. 2002;81(2):114–8.
Ferracane JL. Buonocore Lecture. Placing dental composites—a stressful experience. Oper Dent. 2008;33(3):247–57.
Schneider LF, Cavalcante LM, Silikas N. Shrinkage stresses generated during resin-composite applications: a review. J Dent Biomech. 2010;2010:131630.
Braga RR, Ballester RY, Ferracane JL. Factors involved in the development of polymerization shrinkage stress in resin-composites: a systematic review. Dent Mater. 2005;21(10):962–70.
Stansbury JW, Trujillo-Lemon M, Lu H, Ding X, Lin Y, Ge J. Conversion-dependent shrinkage stress and strain in dental resins and composites. Dent Mater. 2005;21(1):56–67.
Asmussen E, Peutzfeldt A. Two-step curing: influence on conversion and softening of a dental polymer. Dent Mater. 2003;19(6):466–70.
Feng L, Suh BI. A mechanism on why slower polymerization of a dental composite produces lower contraction stress. J Biomed Mater Res B Appl Biomater. 2006;78(1):63–9.
Cunha LG, Alonso RC, Pfeifer CS, Correr-Sobrinho L, Ferracane JL, Sinhoreti MA. Modulated photoactivation methods: influence on contraction stress, degree of conversion and push-out bond strength of composite restoratives. J Dent. 2007;35(4):318–24.
Hofmann N, Denner W, Hugo B, Klaiber B. The influence of plasma arc vs. halogen standard or soft-start irradiation on polymerization shrinkage kinetics of polymer matrix composites. J Dent. 2003;31(6):383–93.
Ferracane JL. Elution of leachable components from composites. J Oral Rehabil. 1994;21(4):441–52.
Van Landuyt KL, Nawrot T, Geebelen B, De Munck J, Snauwaert J, Yoshihara K, Scheers H, Godderis L, Hoet P, Van Meerbeek B. How much do resin-based dental materials release? A meta-analytical approach. Dent Mater. 2011;27(8):723–47.
Nomura Y, Teshima W, Kawahara T, Tanaka N, Ishibashi H, Okazaki M, Arizono K. Genotoxicity of dental resin polymerization initiators in vitro. J Mater Sci Mater Med. 2006;17(1):29–32.
Durner J, Obermaier J, Draenert M, Ilie N. Correlation of the degree of conversion with the amount of elutable substances in nano-hybrid dental composites. Dent Mater. 2012;28(11):1146–53.
Pongprueksa P, De Munck J, Duca RC, Poels K, Covaci A, Hoet P, Godderis L, Van Meerbeek B, Van Landuyt KL. Monomer elution in relation to degree of conversion for different types of composite. J Dent. 2015;43(12):1448–55.
Randolph LD, Palin WM, Bebelman S, Devaux J, Gallez B, Leloup G, Leprince JG. Ultra-fast light-curing resin composite with increased conversion and reduced monomer elution. Dent Mater. 2014;30(5):594–604.
Martin N, Jedynakiewicz N. Measurement of water sorption in dental composites. Biomaterials. 1998;19(1–3):77–83.
Rueggeberg FA, Cole MA, Looney SW, Vickers A, Swift EJ. Comparison of manufacturer-recommended exposure durations with those determined using biaxial flexure strength and scraped composite thickness among a variety of light-curing units. J Esthet Restor Dent. 2009;21(1):43–61.
Brambilla E, Gagliani M, Ionescu A, Fadini L, Garcia-Godoy F. The influence of light-curing time on the bacterial colonization of resin composite surfaces. Dent Mater. 2009;25(9):1067–72.
Khalichi P, Singh J, Cvitkovitch DG, Santerre JP. The influence of triethylene glycol derived from dental composite resins on the regulation of Streptococcus mutans gene expression. Biomaterials. 2009;30(4):452–9.
Mjor IA. Clinical diagnosis of recurrent caries. J Am Dent Assoc. 2005;136(10):1426–33.
da Silva EM, Almeida GS, Poskus LT, Guimarães JG. Relationship between the degree of conversion, solubility and salivary sorption of a hybrid and a nanofilled resin composite. J Appl Oral Sci. 2008;16(2):161–6.
Yiu CK, King NM, Carrilho MR, Sauro S, Rueggeberg FA, Prati C, Carvalho RM, Pashley DH, Tay FR. Effect of resin hydrophilicity and temperature on water sorption of dental adhesive resins. Biomaterials. 2006;27(9):1695–703.
Ferracane JL. Hygroscopic and hydrolytic effects in dental polymer networks. Dent Mater. 2006;22(3):211–22.
Pearson GJ, Longman CM. Water sorption and solubility of resin-based materials following inadequate polymerization by a visible-light curing system. J Oral Rehabil. 1989;16(1):57–61.
Drummond JL. Degradation, fatigue, and failure of resin dental composite materials. J Dent Res. 2008;87(8):710–9.
Soares LE, Nahorny S, Martin AA. FT-Raman spectroscopy study of organic matrix degradation in nanofilled resin composite. Microsc Microanal. 2013;19(2):327–34.
Dietschi D, Campanile G, Holz J, Meyer JM. Comparison of the color stability of ten new-generation composites: an in vitro study. Dent Mater. 1994;10(6):353–62.
Asmussen E. Factors affecting the color stability of restorative resins. Acta Odontol Scand. 1983;41(1):11–8.
Ferracane JL, Berge HX, Condon JR. In vitro aging of dental composites in water—effect of degree of conversion, filler volume, and filler/matrix coupling. J Biomed Mater Res. 1998;42(3):465–72.
Ruyter IE. Methacrylate-based polymeric dental materials: conversion and related properties. Summary and review. Acta Odontol Scand. 1982;40(5):359–76.
Sideridou ID, Achilias DS. Elution study of unreacted Bis-GMA, TEGDMA, UDMA, and Bis-EMA from light-cured dental resins and resin composites using HPLC. J Biomed Mater Res B Appl Biomater. 2005;74(1):617–26.
Chalmers JM, Griffiths PR, editors. Handbook of vibrational spectroscopy. Chichester: Wiley; 2001.
Moraes LG, Rocha RS, Menegazzo LM, de Araújo EB, Yukimito K, Moraes JC. Infrared spectroscopy: a tool for determination of the degree of conversion in dental composites. J Appl Oral Sci. 2008;16(2):145–9.
Ruyter IE, Svendsen SA. Remaining methacrylate groups in composite restorative materials. Acta Odontol Scand. 1978;36(2):75–82.
Ruyter IE, Oysaed H. Conversion in different depths of ultraviolet and visible light activated composite materials. Acta Odontol Scand. 1982;40(3):179–92.
Shin WS, Li XF, Schwartz B, Wunder SL, Baran GR. Determination of the degree of cure of dental resins using Raman and FT-Raman spectroscopy. Dent Mater. 1993;9(5):317–24.
Pianelli C, Devaux J, Bebelman S, Leloup G. The micro-Raman spectroscopy, a useful tool to determine the degree of conversion of light-activated composite resins. J Biomed Mater Res. 1999;48(5):675–81.
Al-Ahdal K, Ilie N, Silikas N, Watts DC. Polymerization kinetics and impact of post polymerization on the degree of conversion of bulk-fill resin-composite at clinically relevant depth. Dent Mater. 2015;31(10):1207–13.
Smith BC. Fundamentals of Fourier transform infrared spectroscopy. 2nd ed. Boca Raton, FL: CRC Press, Taylor & Francis Group; 2011.
Marovic D, Taubock TT, Attin T, Panduric V, Tarle Z. Monomer conversion and shrinkage force kinetics of low-viscosity bulk-fill resin composites. Acta Odontol Scand. 2015;73(6):474–80.
Acquaviva PA, Cerutti F, Adami G, Gagliani M, Ferrari M, Gherlone E, Cerutti A. Degree of conversion of three composite materials employed in the adhesive cementation of indirect restorations: a micro-Raman analysis. J Dent. 2009;37(8):610–5.
Rueggeberg FA, Hashinger DT, Fairhurst CW. Calibration of FTIR conversion analysis of contemporary dental resin composites. Dent Mater. 1990;6(4):241–9.
Nomoto R, Asada M, McCabe JF, Hirano S. Light exposure required for optimum conversion of light activated resin systems. Dent Mater. 2006;22(12):1135–42.
De Santis A. Photo-polymerisation effects on the carbonyl CO bands of composite resins measured by micro-Raman spectroscopy. Polymer. 2005;46(14):5001–4.
Ilie N, Hickel R. Can CQ be completely replaced by alternative initiators in dental adhesives? Dent Mater J. 2008;27(2):221–8.
Ferracane JL, Greener EH. Fourier transform infrared analysis of degree of polymerization in unfilled resins—methods comparison. J Dent Res. 1984;63(8):1093–5.
Ilie N, Fleming GJ. In vitro comparison of polymerisation kinetics and the micro-mechanical properties of low and high viscosity giomers and RBC materials. J Dent. 2015;43(7):814–22.
Ilie N, Durner J. Polymerization kinetic calculations in dental composites: a method comparison analysis. Clin Oral Investig. 2014;18(6):1587–96.
Scherzer T, Decker U. Real-time FTIR–ATR spectroscopy to study the kinetics of ultrafast photopolymerization reactions induced by monochromatic UV light. Vib Spectrosc. 1999;19(2):385–98.
Stansbury JW, Dickens SH. Determination of double bond conversion in dental resins by near infrared spectroscopy. Dent Mater. 2001;17(1):71–9.
Trujillo M, Newman SM, Stansbury JW. Use of near-IR to monitor the influence of external heating on dental composite photopolymerization. Dent Mater. 2004;20(8):766–77.
Lewis IR, Edwards HGM, editors. Handbook of Raman spectroscopy. New York: CRC Press, Taylor & Francis Group; 2001.
Stansbury JW. Curing dental resins and composites by photopolymerization. J Esthet Dent. 2000;12(6):300–8.
Li X, Pongprueksa P, Van Meerbeek B, De Munck J. Curing profile of bulk-fill resin-based composites. J Dent. 2015;43(6):664–72.
Par M, Gamulin O, Marovic D, Klaric E, Tarle Z. Effect of temperature on post-cure polymerization of bulk-fill composites. J Dent. 2014;42(10):1255–60.
Pilo R, Cardash HS. Post-irradiation polymerization of different anterior and posterior visible light-activated resin composites. Dent Mater. 1992;8(5):299–304.
Marigheto NA, Kemsley EK, Potter J, Belton PS, Wilson RH. Effects of sample heating in FT-Raman spectra of biological materials. Spectrochim Acta A Mol Biomol Spectrosc. 1996;52(12):1571–9.
Halvorson RH, Erickson RL, Davidson CL. The effect of filler and silane content on conversion of resin-based composite. Dent Mater. 2003;19(4):327–33.
Sabbagh J, Ryelandt L, Bachérius L, Biebuyck JJ, Vreven J, Lambrechts P, Leloup G. Characterization of the inorganic fraction of resin composites. J Oral Rehabil. 2004;31(11):1090–101.
Price RB, Rueggeberg FA, Harlow J, Sullivan B. Effect of mold type, diameter, and uncured composite removal method on depth of cure. Clin Oral Investig. 2016;20(7):1699–707.
Erickson RL, Barkmeier WW. Curing characteristics of a composite. Part 2: the effect of curing configuration on depth and distribution of cure. Dent Mater. 2014;30(6):e134–45.
Price RB, Whalen JM, Price TB, Felix CM, Fahey J. The effect of specimen temperature on the polymerization of a resin-composite. Dent Mater. 2011;27(10):983–9.
Sakaguchi RL. Review of the current status and challenges for dental posterior restorative composites: clinical, chemistry, and physical behavior considerations. Summary of discussion from the Portland Composites Symposium (POCOS) June 17–19, 2004, Oregon Health and Science University, Portland, Oregon. Dent Mater. 2005;21(1):3–6.
Watts DC, Amer OM, Combe EC. Surface hardness development in light-cured composites. Dent Mater. 1987;3(5):265–9.
Marovic D, Panduric V, Tarle Z, Ristic M, Sariri K, Demoli N, Klaric E, Jankovic B, Prskalo K. Degree of conversion and microhardness of dental composite resin materials. J Mol Struct. 2013;1044:299–302.
Erickson RL, Barkmeier WW, Halvorson RH. Curing characteristics of a composite – part 1: cure depth relationship to conversion, hardness and radiant exposure. Dent Mater. 2014;30(6):e125–33.
Bouschlicher MR, Rueggeberg FA, Wilson BM. Correlation of bottom-to-top surface microhardness and conversion ratios for a variety of resin composite compositions. Oper Dent. 2004;29(6):698–704.
Miletic V, Pongprueksa P, De Munck J, Brooks NR, Van Meerbeek B. Curing characteristics of flowable and sculptable bulk-fill composites. Clin Oral Investig. 2016;21:1201–12.
Musanje L, Darvell BW. Curing-light attenuation in filled-resin restorative materials. Dent Mater. 2006;22(9):804–17.
Ilie N, Stark K. Effect of different curing protocols on the mechanical properties of low-viscosity bulk-fill composites. Clin Oral Investig. 2015;19(2):271–9.
International Standard 4049. Polymer-based restorative materials. Geneva: ISO; 2009.
Della Bona A, Wozniak WT, Watts DC. International dental standards—order out of chaos? Dent Mater. 2011;27(7):619–21.
Antonucci JM, Toth EE. Extent of polymerization of dental resins by differential scanning calorimetry. J Dent Res. 1983;62(2):121–5.
Imazato S, McCabe JF, Tarumi H, Ehara A, Ebisu S. Degree of conversion of composites measured by DTA and FTIR. Dent Mater. 2001;17(2):178–83.
Miyazaki K, Horibe T. Polymerization of multifunctional methacrylates and acrylates. J Biomed Mater Res. 1988;22(11):1011–22.
Rosentritt M, Shortall AC, Palin WM. Dynamic monitoring of curing photoactive resins: a methods comparison. Dent Mater. 2010;26(6):565–70.
Dewaele M, Truffier-Boutry D, Devaux J, Leloup G. Volume contraction in photocured dental resins: the shrinkage-conversion relationship revisited. Dent Mater. 2006;22(4):359–65.
Venhoven BA, de Gee AJ, Davidson CL. Polymerization contraction and conversion of light-curing BisGMA-based methacrylate resins. Biomaterials. 1993;14(11):871–5.
Howard B, Wilson ND, Newman SM, Pfeifer CS, Stansbury JW. Relationships between conversion, temperature and optical properties during composite photopolymerization. Acta Biomater. 2010;6(6):2053–9.
Par M, Marovic D, Skenderovic H, Gamulin O, Klaric E, Tarle Z. Light transmittance and polymerization kinetics of amorphous calcium phosphate composites. Clin Oral Investig. 2016;21:1173–82.
Steinhaus J, Hausnerova B, Haenel T, Grossgarten M, Moginger B. Curing kinetics of visible light curing dental resin composites investigated by dielectric analysis (DEA). Dent Mater. 2014;30(3):372–80.
Par M, Santic A, Gamulin O, Marovic D, Mogus-Milankovic A, Tarle Z. Impedance changes during setting of amorphous calcium phosphate composites. Dent Mater. 2016;32(11):1312–21.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Tarle, Z., Par, M. (2018). Degree of Conversion. In: Miletic, V. (eds) Dental Composite Materials for Direct Restorations. Springer, Cham. https://doi.org/10.1007/978-3-319-60961-4_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-60961-4_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-60960-7
Online ISBN: 978-3-319-60961-4
eBook Packages: MedicineMedicine (R0)