
Abstract
Objectives
This study investigated light transmittance and polymerization kinetics of experimental remineralizing composite materials based on amorphous calcium phosphate (ACP), reinforced with inert fillers.
Materials and methods
Light-curable composites were composed of Bis-EMA-TEGDMA-HEMA resin and ACP, barium glass, and silica fillers. Additionally, a commercial composite Tetric EvoCeram was used as a reference. Light transmittance was recorded in real-time during curing, and transmittance curves were used to assess polymerization kinetics. To obtain additional information on polymerization kinetics, temperature rise was monitored in real-time during curing and degree of conversion was measured immediately and 24 h post-cure.
Results
Light transmittance values of 2-mm thick samples of uncured ACP composites (2.3–2.9 %) were significantly lower than those of the commercial composite (3.8 %). The ACP composites presented a considerable transmittance rise during curing, resulting in post-cure transmittance values similar to or higher than those of the commercial composite (5.5–7.9 vs. 5.4 %). The initial part of light transmittance curves of experimental composites showed a linear rise that lasted for 7–20 s. Linear fitting was performed to obtain a function whose slope was assessed as a measure of polymerization rate. Comparison of transmittance and temperature curves showed that the linear transmittance rise lasted throughout the most part of the pre-vitrification period.
Conclusions
The linear rise of light transmittance during curing has not been reported in previous studies and may indicate a unique kinetic behavior, characterized by a long period of nearly constant polymerization rate.
Clinical relevance
The observed kinetic behavior may result in slower development of polymerization shrinkage stress but also inferior mechanical properties.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Composites containing amorphous calcium phosphate (ACP) filler are capable of releasing calcium and phosphate ions, thus having the potential to prevent secondary caries [1–3]. Mechanical properties of ACP composites are generally lower than those of their commercial counterparts [1, 4], but can be enhanced by the admixture of reinforcing fillers, such as silanized barium glass and silica [5, 6]. This approach improves the material’s strength, while maintaining its favorable properties, i.e., remineralizing effect and high degree of monomer conversion [5–7]. By fine-tuning the ACP particle size and composition of reinforcing fillers, mechanical properties similar to these of commercial composites may be attained [8]. A more complete understanding and further improvement in mechanical properties of this class of biomaterials may lead to their clinical applicability in the future.
Optical properties of light-curable composites determine not only their esthetic appearance but also their curing depth and polymerization kinetics [9, 10]. Light transmittance of composite materials is determined by absorption and complex light scattering on filler particles of various geometries and refractive indices [11]. During polymerization, light transmittance changes due to a series of events. First, the resin matrix undergoes a change in optical properties due to its rising refractive index and changes in light scattering that occur during gelation and vitrification [12]. Second, the consumption of camphorquinone (CQ) during polymerization [13] reduces light absorption, which in turn increases light transmittance. Third, the exothermic nature of polymerization reaction causes a transient decrease in refractive index of resin due to the density reduction [10]. Fourth, the capability of material to scatter light is positively related to the refractive index mismatch between the polymer and the filler. The refractive index of polymer increases during curing due to the increase in density and reduced polarizability [10] while the refractive index of the filler remains constant, thus their mismatch either increases or decreases, depending on the initial values. If the initial refractive index of the monomer is lower than that of the filler, the refractive index mismatch diminishes during curing, resulting in a gradual increase in light transmittance [14]. However, if the refractive index of monomer is initially higher than that of the filler, curing further increases the refractive index mismatch, progressively decreasing transmittance [15, 16]. It is also possible that the initially lower refractive index of the resin becomes equal to that of the filler at some point of conversion and continues to rise afterwards [17]. Therefore, the change of transmittance during curing is a complex material-dependent phenomenon, influenced by several simultaneously occurring processes.
The changes in light transmittance during polymerization can be used to gain information on polymerization kinetics. Harrington et al. proposed light transmittance monitoring during light curing as a means of determining the optimal irradiation times, suggesting a cessation of light transmittance change as an indicator of complete cure [14]. Howard et al. described how light transmission profile during curing relates to the initiator concentration, filler load, and irradiance [10]. Changes in light transmittance were used to assess how the reaction kinetics is affected by various photoinitiators [15] and refractive index mismatch and monomer reactivity [17]. Rosentritt et al. compared light transmittance, differential scanning calorimetry, and dielectrical analysis as a means of cure monitoring [18], and Ilie et al. used mathematical modeling to compare the real-time measurements of conversion and light transmittance during polymerization [19]. These studies established that changes in light transmittance allow tracking of the curing progress; however, the quantitative relation between the transmittance changes and the fundamental process of polymerization is poorly understood and recorded changes in transmittance cannot be directly translated into conversion kinetics [17]. Despite that the change of light transmittance during curing is to a certain extent convoluted by other factors (e.g., CQ consumption), the most part of light transmittance change is considered to be due to the changing refractive index of polymerizing resin, which in turn affects light scattering at filler/resin interface [16]. In this regard, transmittance curves reflect the progress of monomer conversion and are considered suitable for assessing polymerization kinetics [19].
Photopolymerization of dental composites is characterized by rapidly increasing viscosity and follows three distinct kinetic regimes: (I) initial phase in which polymeric chains grow without restriction, with kinetics determined by the concentrations of monomer and free radicals; (II) the phase in which rapidly increasing viscosity restricts the termination of free radicals, resulting in an overall increased reaction rate (gel phase); and (III) the phase of extreme viscosity which limits propagation rate and finally ends the reaction (glass phase) [20]. Ilie et al. were able to identify the latter two phases by fitting the real-time conversion data to two exponential functions, each representing one phase [19]. They applied the same approach to the real-time light transmittance data and found transmittance curves less discriminative, able to address only the beginning of the glass phase. Although several other studies used the real-time light transmittance monitoring [10, 14–18], none of them has attempted a mathematical analysis of the transmittance curves as a means of obtaining the polymerization rate.
This paper is one in the series of our investigations on ACP composites reinforced with inert fillers [5–7, 21]. The aims of this study were to (I) assess the light transmittance of ACP composites with reinforcing fillers; (II) use the real-time light transmittance monitoring to obtain a measure of the polymerization rate; (III) compare the light transmittance, conversion, and temperature data to gain information about polymerization kinetics; and (IV) explore the relationship between the polymerization rate and light transmittance.
Materials and methods
Composite materials
Five ACP-based and two control composites were prepared, as described in our previous study [21]. The composition of experimental composites is given in Table 1. For light transmittance and temperature measurements, Tetric EvoCeram (Ivoclar Vivadent, Schaan, Liechtenstein) of shade A2, LOT: S26173, EXP: 06/2017 was used as a reference. Personal communication with the manufacturer indicated that Tetric EvoCeram of shade A2 does not contain any alternative initiators besides CQ.
Light transmittance and temperature
Uncured composite materials were cast into cylindrical Teflon molds (d = 6 mm, h = 2 mm), covered from both sides with a polyethylene terephthalate (PET) film (h = 0.05 mm), and sandwiched between two glass plates (h = 1 mm), as shown in Fig. 1. Five samples were made per material. The curing unit tip was positioned immediately below the glass plate, and the spectra were taken from the opposite side of the sample by a charge-coupled device array fiber spectrometer HR4000 (Ocean Optics, Dunedin, FL, USA). Light transmittance was recorded at the wavelength of 468 nm, corresponding to the CQ absorption peak [22]. The ratio of the light intensity passing through the sample and the light intensity passing through the empty sample compartment was calculated as the light transmittance (%). Temperature was monitored simultaneously with light transmittance, using a T-type thermocouple positioned centrally between the PET film and glass plate at the side opposite to the curing unit.

Cross-sectional scheme of the experimental setup
Curing was performed for 30 s with Bluephase G2 (Ivoclar Vivadent, Schaan, Liechtenstein; wavelength range 380–515 nm, intensity 1185 mW/cm2, as measured with integrating sphere, IS, Gigahertz-Optik GmbH, Puchheim, Germany). After 120 s, an additional illumination (30 s) was performed (Fig. 2). The measurements were performed at the rate of 20 points per second, and five samples were made for each composite. The environmental temperature was 25 ± 1 °C.

An example of simultaneously recorded light transmittance and temperature curves. Transmittance values at the start of curing (T1), end of curing (T2), and during an additional illumination (T3)
Degree of conversion
For each material, five discoid samples (d = 3 mm, h = 2 mm) were made using a stainless steel mold. Uncured composite was cast into the mold, the mold apertures were covered with a PET film, and curing was performed for 30 s through the upper aperture with the Bluephase device. The curing unit tip was positioned at the angle of 90°, immediately adjacent to the mold aperture, contacting the PET film covering the sample. Curing was performed at 25 ± 1 °C. Raman spectra were recorded from the center of the upper sample surface immediately after curing (a single measurement lasted for 15 min) and after 24 h of dark storage in the incubator (Cultura, Ivoclar Vivadent) at 37 ± 1 °C.
FT-Raman spectroscopy measurements were performed using a Spectrum GX spectrometer (PerkinElmer, Waltham, USA). The excitation was an Nd:YaG laser at 1064 nm wavelength, with laser power of 400 mW, resolution of 4 cm−1, and the exposed sample surface of 0.5 mm in diameter. For each spectrum, 100 scans were recorded. Spectra of the uncured composites (n = 5) were recorded in the same manner. The spectra were processed with the Kinetics add-on for MATLAB (MathWorks, Natick, Massachusetts, USA).
Degree of conversion (DC) calculation was performed by comparing the relative change of the band at 1640 cm−1, representing the aliphatic C=C stretching mode to the aromatic C=C band at 1610 cm−1, before and after polymerization. Peak heights of aliphatic C=C and aromatic C=C bands were used for DC calculation by the following equation: DC = 1 − Rpolymerized / Runpolymerized, where R = (aliphatic C=C peak height) / (aromatic C=C peak height). A previous study by our group [21] showed that the DC values measured at sample surfaces were statistically similar to those measured at depths up to 2 mm, thus the surface DC values were considered representative for the samples used in transmittance measurements, despite the different geometry.
Scanning electron microscopy
The light-cured (30 s), disk-shaped samples of 1 mm thickness, unpolished and unsputtered, were examined with FE-SEM JSM 7000 (JEOL, Peabody, MA, USA).
Statistical analysis
Normality of distribution and homogeneity of variances were confirmed using Shapiro-Wilk’s and Levene’s test, respectively. Light transmittance values at the start of curing (T1), end of curing (T2), and during an additional illumination (T3) were compared among composites by one-way ANOVA and Tukey post hoc test. Within a given material, T1, T2, and T3 values were compared using repeated measurements ANOVA and Bonferroni post hoc test.
Mean values of the variables used to describe polymerization kinetics (duration of the linear transmittance rise, time of the temperature peak, slope of the linear transmittance rise, and DC) were compared among materials by one-way ANOVA and Tukey post hoc test. Additionally, paired t test was used to compare the duration of the linear transmittance rise and time of the temperature peak within each composite. Paired t test was also used to compare the DC values immediately after curing and 24 h post-cure.
Pearson’s correlation analysis was used to correlate the slope of the linear part of transmittance rise and transmittance values at three time points (T1, T2, and T3). Statistical analysis was performed in SPSS 20 (IBM, Armonk, NY, USA) with α = 0.05.
Results
The light transmittance and temperature curves are exemplified in Fig. 2. A linear transmittance rise can be observed in the initial part of the curve, followed by an exponential-like behavior towards the end of illumination. The temperature curve during the first illumination features a distinct peak that was used to approximate the onset of vitrification [23]. Light transmittance values used in calculations were taken from transmittance curves at three time points: start of curing (T1), end of curing (T2), and during an additional illumination (T3). These values and results of statistical analysis are presented in Fig. 3.

Light transmittance (%) at three time points. Same uppercase letters denote statistically homogeneous groups within a single time point. Error bars represent ± one standard deviation. Horizontal line above the bars denotes statistical homogeneity between different time points, which was found only between T2 and T3 for Tetric EvoCeram
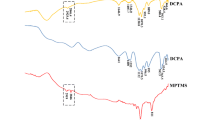
Figure 4 shows light transmittance curves representative for (a) the ACP composites, (b) control composites, and (c) Tetric EvoCeram. To describe polymerization kinetics, an initial linear part of light transmittance curves was fitted to a linear function in the range for which R 2 >0.98 could be obtained and its slope (Table 2) was assessed as a measure of the polymerization rate. Each curve was normalized to its transmittance change during curing (T2 − T1), so that the slope represents the rate at which transmittance increases in terms of percentage of the total transmittance change during 30 s of curing. The slope values were plotted against transmittance values for each sample (total n = 35) and analyzed by Pearson correlation. A significant correlation and high r values were observed (Fig. 5). Since the linear transmittance rise was not detectable for Tetric EvoCeram (Fig. 4c), it was excluded from further kinetic analysis.

Averaged light transmittance curves for materials ACP40 (a), Ba40 (b), and Tetric EvoCeram (c). The insets show a magnified part of curves during the light curing, range of linear fitting (R 2 >0.98), and fit result (dotted line)

Linear correlation between the slope of the linear part of light transmittance rise and transmittance values at the start of curing (a), end of curing (b), and during an additional illumination (c)
Table 2 shows variables used for describing the polymerization kinetics (duration of the linear transmittance rise, time of the temperature peak, and slope of the linear transmittance rise) and DC values immediately after curing and 24 h post-cure. The linear transmittance rise was observed for all of the experimental materials, with notable differences in its duration between the ACP-based and control materials. The ACP composites presented high immediate DC with insignificant post-cure DC increase, while the controls showed the opposite behavior: low immediate DC and high post-cure DC increase.
Figure 6 is a SEM micrograph of a cured sample of ACP40, showing irregularly shaped ACP agglomerates of approximately 2–5 μm.

SEM micrograph of ACP40 demonstrating a homogeneous distribution of ACP particles with sizes of 2–5 μm
Discussion
The effect of composition on light transmittance
Initial light transmittance values (T1) of the ACP composites (2.3–2.9 %) were lower than those of the commercial reference Tetric EvoCeram (3.8 %). However, the ACP composites presented a considerable transmittance rise during curing, which led to T2 and T3 values similar to or higher than those of Tetric EvoCeram (Fig. 3). This indicates that the initially lower potential of the ACP composites to transmit light improves during the curing to a level comparable to that of the commercial composite. The effect of the addition of inert fillers was the most prominent for material ACP40-Si10 which showed significantly lower light transmittance than in the other ACP composites. Silica fillers exist as agglomerates of different sizes, some of which have the size that maximizes the scattering of curing light (about 0.2 μm for a wavelength of 468 nm), and are responsible for lowering the light transmittance [24]. Other combinations of inert fillers slightly decreased the light transmittance, though not statistically significant. It is interesting to note that ACP40, ACP40-Ba10, and ACP40-Ba9Si1 had statistically similar transmittance values within any measurement time (T1, T2, and T3), suggesting that the addition of up to 10 wt% barium glass produced a negligible effect on transmittance.
For all of the experimental composites, T3 was significantly higher than T2, evidencing that post-cure polymerization continued after the initial curing of 30 s (Fig. 3). An additional feature that gives information on post-cure behavior is the shape of the curve during an additional illumination—a plateau observed for the ACP composites (Fig. 4a) suggests that the post-cure reaction was mostly finished before the additional illumination, while the curves of the control composites showed a slight slope (Fig. 4b) evidencing the continuation of the post-cure reaction. Whether this was caused by the additional illumination or the reaction would continue by itself “in dark” [25] was outside the scope of this work. Nevertheless, real-time light transmittance monitoring appears as a simple means for assessment of the polymerization “completeness” [14].
Polymerization kinetics
Real-time light transmittance measurements enable monitoring of the curing progress. An interesting kinetic feature observed for experimental composites is the linear transmittance rise that lasted for 6.6–20.2 s (Fig. 4, Table 2). Such kinetic behavior has not been reported in previous studies [10, 14–19], which presented light transmittance curves resembling the function f(x) = 1 − exp (−x) and was shown to be well represented by a sum of two exponential functions [19]. The exponential transmittance rise was also demonstrated for the commercial reference Tetric EvoCeram in our study, whose transmittance curves could be described by the function f(t) = 0.0612 + 0.0394 × (1 − exp (−0.2048 t)) + 0.0536 × (1 − exp (−24.2659 t)), where f(t) = transmittance (unitless) and t = time (s). Despite that the polymerization progress observed through light transmittance curves cannot be directly translated into conversion, the existence of a linear part in transmittance curves of experimental composites suggests a constant reaction rate during a certain period of polymerization. This is unusual, since the diffusional restrictions that occur very early during methacrylate polymerization are expected to considerably affect the reaction rate through processes known as autoacceleration and autodeceleration [20]. However, Dickens et al. have demonstrated that low-viscosity, TEGDMA-rich resins may present a nearly constant reaction rate throughout a wide range of conversion [26]. In their study, Bis-EMA/TEGDMA comonomers with 0–50 mol% of Bis-EMA showed a nearly constant reaction rate and delayed diffusional restrictions, possibly due to the better network mobility and dominance of cyclization over crosslinking. Our composites had the Bis-EMA content similar to that of Dickens’ study (46 mol%) and featured an additional 26 mol% of HEMA. The propensity of monofunctional HEMA to promote linear chain growth instead of crosslinking can even more postpone diffusional restrictions [20] and contribute to the distinct polymerization behavior seen in our composites.
ACP composites showed longer durations of linear transmittance rise than the controls Ba40 and Ba40Si10 (Table 2), suggesting the later onset of diffusional restrictions in case of the former. Similar to the way in which TEGDMA delays gelation by favoring cyclization over crosslinking [26], the unsilanized ACP particles may be hypothesized to favor high local conversion while hindering the formation of infinite network. Early polymerization of resins containing TEGDMA and HEMA is dominated by cyclization reactions, resulting in poorly crosslinked microgel regions. As polymerization progresses, the microgels become interconnected by crosslinks, leading to macroscopic gelation [20]. Unsilanized ACP particles may present obstacles for joining of the microgels, thus hindering crosslinking and delaying gelation. In contrast, silanized particles are capable of bonding to the growing polymeric network and may allow earlier crosslinking. The earlier onset of diffusional restrictions caused by reactive fillers was demonstrated in a study of composites filled with reactive and nonreactive nanogels [27]. Another contributing factor for the observed differences in kinetic behavior may have been comparatively larger particles of ACP filler (Fig. 6), which had lower surface area in contact with the resin thus allowing better mobility [28, 29]. In any case, all of the ACP composites showed similar duration of the linear transmittance rise (Table 2), demonstrating that the addition of 10 % of silanized fillers did not considerably affect their kinetic behavior.
The differences in polymerization kinetics between ACP composites and controls are furthermore evidenced by the DC results obtained immediately and 24 h after cure. For the ACP composites, very high DC immediately after curing and a statistically insignificant post-cure DC increase were observed (Table 2), suggesting high mobility of reactive species during curing. In contrast, the controls showed lower initial DC and a significant post-cure DC increase, indicating that severe mobility restrictions occurred much before the composites attained their final conversion. The explanatory hypothesis that this difference may be due to presence of unsilanized particles which enabled better mobility during curing is supported by the studies of rubbers which demonstrated that local mobility and glass transition temperature depends upon the ability of filler particles to form chemical bonds with the matrix [30, 31]. For dental composites, the effect of reactive particle surfaces on network mobility was suggested by lower conversions of composites containing silanized particles [32, 33]. Additionally, the fact that commercial composites encompassing a wide range of resin compositions, filler loads, and geometries [34] commonly show conversions in the range of 50–75 %, contrasting the ACP composites which attain conversions of 80 % or higher [1, 21, 35–37], supports a probable role of unsilanized particles on polymerization kinetics.
To obtain additional information on polymerization progress, temperature rise was recorded simultaneously with light transmittance (Fig. 2). The peak in temperature curve occurs when the temperature buildup ceases due to the sudden decrease in reaction rate that occurs at the onset of vitrification [23]. Thus, the time of the temperature peak (Table 2) can be used to approximate the glass point. The glass point of the ACP composites was observed at 19.9–24.0 s, while for Ba40 and Ba40Si10 the glass point occurred at 22.6 and 16.6 s, respectively. Among the ACP composites, ACP40-Si10 had the latest occurrence of peak, possibly due to its comparatively lowest transmittance that reduced the initiation rate, thus delaying vitrification. The influence of other inert filler additions on the time of temperature peak for ACP composites was statistically insignificant. The early onset of peak for Ba40Si10 suggests the fastest vitrification among all of the materials. A possible explanation is high silanized surface area that may have contributed to more rapid immobilization of the polymerizing matrix [32, 33]. Much earlier vitrification seen in Ba40Si10 than in Ba40 can be attributed to increased viscosity due to the addition of small silica particles [28]. On the other hand, the addition of 10 wt% of silica to ACP composites did not cause such a pronounced shift in vitrification point. This may have been compensated by large ACP particles with unsilanized surfaces that allowed better mobility, as discussed previously. Comparison of the times of the temperature peak and the duration of linear transmittance rise showed that the largest part of the pre-glass polymerization of ACP composites was characterized by a linear transmittance rise, which lasted for approximately 17–20 s and ended 0–3 s before vitrification (Table 2). This suggests that the polymerization proceeded with a nearly constant rate until very close to vitrification. In the case of ACP40-Ba10, the nearly constant reaction rate lasted throughout the whole pre-glass period. In contrast, the linear part of transmittance rise for the control composites Ba40 and Ba40Si10 ended approximately 10–13 s before the temperature peak, indicating that mobility restrictions occurred at the earlier phase of the polymerization.
The linear part of transmittance rise was fitted to a linear function whose slope was assessed as a measure of the polymerization rate. Theoretically, before the onset of diffusional limitations, the polymerization rate is a function of the concentrations of monomer and free radicals [38]. For early phase of the polymerization, two assumptions can be made: that the monomer concentration remains constant and that all of the present free radicals are in fact the radicalized initiator. Such simplification yields a constant polymerization rate, which is linearly dependent on the concentration of the radicalized initiator [39]. Although this approximation is not valid for long-time ranges through which we performed linear fitting (7–20 s, Table 2), a good fit throughout the whole range means that the slope remained the same as in the earliest stage of polymerization where the approximation holds. Assuming that the slope of the linear part of transmittance rise is a valid measure of the polymerization rate, a relationship between slope and transmittance should exist, as transmittance determines the effective irradiance in the bulk of the sample, which in turn dictates the concentration of the radicalized initiator. To examine this relationship, transmittance values of seven experimental composites at the start of curing (T1), end of curing (T2), and during an additional illumination (T3) were plotted against the slope values and Pearson correlation coefficient was calculated (Fig. 5). In a linear function written as y = ax + b, where y = normalized slope and x = transmittance, a values (s−1) were 0.0082, 0.0053, and 0.0047 respectively for T1, T2, and T3. Respective values of Pearson’s r were 0.962, 0.918, and 0.906, statistically significant at p < 0.01. The trend of decreasing a values in the order T1 > T2 > T3 shows that the linear dependence of reaction rate is steeper for earlier transmittance values, while the trend of decreasing r values in the same order shows that earlier transmittance values were more predictive of the reaction rate. Higher correlation for earlier transmittance values is expected, since these values determined the concentration of the radicalized initiator.
Despite that the kinetic inferences drawn from light transmittance curves were not confirmed by real-time conversion measurements, we observed a kinetic behavior that has not been reported previously and is suggestive of a rather long period of a nearly constant reaction rate for composites composed of Bis-EMA/TEGDMA/HEMA resin and unsilanized ACP particles. Since remineralizing composites usually contain unsilanized particles of various calcium phosphates [8], there are possible consequences relevant to this group of materials. A kinetic behavior with no evidence of autoacceleration suggests slower development of polymerization shrinkage stress and possibly a prolonged pre-gel phase, which is favorable from a clinical standpoint. On the other hand, the final polymeric network may be highly heterogeneous and consist of many poorly interconnected microgel regions [20], resulting in impaired mechanical properties. This indicates the need to further investigate the kinetic behavior of remineralizing composites.
References
Skrtic D, Antonucci JM, Eanes ED (2003) Amorphous calcium phosphate-based bioactive polymeric composites for mineralized tissue regeneration. J Res Natl Inst Stand Technol 108(3):167–182
Li F, Wang P, Weir MD, Fouad AF, Xu HH (2014) Evaluation of antibacterial and remineralizing nanocomposite and adhesive in rat tooth cavity model. Acta Biomater 10(6):2804–2813. doi:10.1016/j.actbio.2014.02.033
Weir MD, Chow LC, Xu HH (2012) Remineralization of demineralized enamel via calcium phosphate nanocomposite. J Dent Res 91(10):979–984. doi:10.1177/0022034512458288
Skrtic D, Antonucci JM (2007) Dental composites based on amorphous calcium phosphate—resin composition/physicochemical properties study. J Biomater Appl 21(4):375–393. doi:10.1177/0885328206064823
Marovic D, Tarle Z, Hiller KA, Muller R, Ristic M, Rosentritt M, Skrtic D, Schmalz G (2014) Effect of silanized nanosilica addition on remineralizing and mechanical properties of experimental composite materials with amorphous calcium phosphate. Clin Oral Investig 18(3):783–792. doi:10.1007/s00784-013-1044-x
Marovic D, Tarle Z, Hiller KA, Muller R, Rosentritt M, Skrtic D, Schmalz G (2014) Reinforcement of experimental composite materials based on amorphous calcium phosphate with inert fillers. Dent Mater 30(9):1052–1060. doi:10.1016/j.dental.2014.06.001
Marovic D, Tarle Z, Ristic M, Music S, Skrtic D, Hiller KA, Schmalz G (2011) Influence of different types of fillers on the degree of conversion of ACP composite resins. Acta Stomatol Croat 45:231–238
Xu HH, Weir MD, Sun L, Moreau JL, Takagi S, Chow LC, Antonucci JM (2010) Strong nanocomposites with Ca, PO4, and F release for caries inhibition. J Dent Res 89(1):19–28. doi:10.1177/0022034509351969
Kawaguchi M, Fukushima T, Miyazaki K (1994) The relationship between cure depth and transmission coefficient of visible-light-activated resin composites. J Dent Res 73(2):516–521. doi:10.1177/00220345940730020601
Howard B, Wilson ND, Newman SM, Pfeifer CS, Stansbury JW (2010) Relationships between conversion, temperature and optical properties during composite photopolymerization. Acta Biomater 6(6):2053–2059. doi:10.1016/j.actbio.2009.11.006
Musanje L, Darvell BW (2006) Curing-light attenuation in filled-resin restorative materials. Dent Mater 22(9):804–817. doi:10.1016/j.dental.2005.11.009
Shibayama M, Ozeki S, Norisuye T (2005) Real-time dynamic light scattering on gelation and vitrification. Polymer 46(7):2381–2388. doi:10.1016/j.polymer.2005.01.018
Seghi RR, Gritz MD, Kim J (1990) Colorimetric changes in composites resulting from visible-light-initiated polymerization. Dent Mater 6(2):133–137. doi:10.1016/S0109-5641(05)80044-2
Harrington E, Wilson HJ, Shortall AC (1996) Light-activated restorative materials: a method of determining effective radiation times. J Oral Rehabil 23(3):210–218. doi:10.1111/j.1365-2842.1996.tb01235.x
Ogunyinka A, Palin WM, Shortall AC, Marquis PM (2007) Photoinitiation chemistry affects light transmission and degree of conversion of curing experimental dental resin composites. Dent Mater 23(7):807–813. doi:10.1016/j.dental.2006.06.016
Fujita K, Ikemi T, Nishiyama N (2011) Effects of particle size of silica filler on polymerization conversion in a light-curing resin composite. Dent Mater 27(11):1079–1085. doi:10.1016/j.dental.2011.07.010
Shortall AC, Palin WM, Burtscher P (2008) Refractive index mismatch and monomer reactivity influence composite curing depth. J Dent Res 87(1):84–88. doi:10.1177/154405910808700115
Rosentritt M, Shortall AC, Palin WM (2010) Dynamic monitoring of curing photoactive resins: a methods comparison. Dent Mater 26(6):565–570. doi:10.1016/j.dental.2010.02.006
Ilie N, Durner J (2014) Polymerization kinetic calculations in dental composites: a method comparison analysis. Clin Oral Investig 18(6):1587–1596. doi:10.1007/s00784-013-1128-7
Andrzejewska E (2001) Photopolymerization kinetics of multifunctional monomers. Prog Polym Sci 26(4):605–665. doi:10.1016/S0079-6700(01)00004-1
Par M, Gamulin O, Marovic D, Skenderovic H, Klaric E, Tarle Z (2016) Conversion and temperature rise of remineralizing composites reinforced with inert fillers. J Dent. doi:10.1016/j.jdent.2016.03.008
Emami N, Sjodahl M, Soderholm KJ (2005) How filler properties, filler fraction, sample thickness and light source affect light attenuation in particulate filled resin composites. Dent Mater 21(8):721–730. doi:10.1016/j.dental.2005.01.002
Atai M, Motevasselian F (2009) Temperature rise and degree of photopolymerization conversion of nanocomposites and conventional dental composites. Clin Oral Investig 13(3):309–316. doi:10.1007/s00784-008-0236-2
Clewell DH (1941) Scattering of light by pigment particles. J Opt Soc Am 31:512–517. doi:10.1364/JOSA.31.000521
Pilo R, Cardash HS (1992) Post-irradiation polymerization of different anterior and posterior visible light-activated resin composites. Dent Mater 8(5):299–304. doi:10.1016/0109-5641(92)90104-K
Dickens SH, Stansbury JW, Choi KM, Floyd CJE (2003) Photopolymerization kinetics of methacrylate dental resins. Macromolecules 36(16):6043–6053. doi:10.1021/ma021675k
Moraes RR, Garcia JW, Barros MD, Lewis SH, Pfeifer CS, Liu J, Stansbury JW (2011) Control of polymerization shrinkage and stress in nanogel-modified monomer and composite materials. Dent Mater 27(6):509–519. doi:10.1016/j.dental.2011.01.006
Beun S, Bailly C, Dabin A, Vreven J, Devaux J, Leloup G (2009) Rheological properties of experimental Bis-GMA/TEGDMA flowable resin composites with various macrofiller/microfiller ratio. Dent Mater 25(2):198–205. doi:10.1016/j.dental.2008.06.001
Hadis M, Leprince JG, Shortall AC, Devaux J, Leloup G, Palin WM (2011) High irradiance curing and anomalies of exposure reciprocity law in resin-based materials. J Dent 39(8):549–557. doi:10.1016/j.jdent.2011.05.007
Berriot J, Lequeux F, Monnerie L, Montes H, Long D, Sotta P (2002) Filler–elastomer interaction in model filled rubbers, a 1H NMR study. J Non-Cryst Solids 307-310:719–724. doi:10.1016/S0022-3093(02)01552-1
Ou YC, Yu ZZ, Vidal A, Donnet JB (1996) Effects of alkylation of silicas on interfacial interaction and molecular motions between silicas and rubbers. J Appl Polym Sci 59(8):1321–1328. doi:10.1002/(SICI)1097-4628(19960222)59:8<1321::AID-APP16>3.0.CO;2-8
Wilson KS, Zhang K, Antonucci JM (2005) Systematic variation of interfacial phase reactivity in dental nanocomposites. Biomaterials 26(25):5095–5103. doi:10.1016/j.biomaterials.2005.01.008
Ferracane JL, Berge HX, Condon JR (1998) In vitro aging of dental composites in water—effect of degree of conversion, filler volume, and filler/matrix coupling. J Biomed Mater Res 42(3):465–472. doi:10.1002/(SICI)1097-4636(19981205)42:3<465::AID-JBM17>3.0.CO;2-F
Ilie N, Hickel R (2009) Investigations on mechanical behaviour of dental composites. Clin Oral Investig 13(4):427–438. doi:10.1007/s00784-009-0258-4
Skrtic D, Antonucci JM (2011) Bioactive polymeric composites for tooth mineral regeneration: physicochemical and cellular aspects. J Funct Biomater 2(3):271–307. doi:10.3390/jfb2030271
Skrtic D, Antonucci JM, Liu DW (2006) Ethoxylated bisphenol dimethacrylate-based amorphous calcium phosphate composites. Acta Biomater 2(1):85–94. doi:10.1016/j.actbio.2005.10.004
Tarle Z, Knežević A, Matošević D, Škrtić D, Ristić M, Prskalo K, Musić S (2009) Degree of vinyl conversion in experimental amorphous calcium phosphate composites. J Mol Struct 924-926:161–165. doi:10.1016/j.molstruc.2008.11.024
Watts DC (2005) Reaction kinetics and mechanics in photo-polymerised networks. Dent Mater 21(1):27–35. doi:10.1016/j.dental.2004.10.003
Steinhaus J, Hausnerova B, Haenel T, Grossgarten M, Moginger B (2014) Curing kinetics of visible light curing dental resin composites investigated by dielectric analysis (DEA). Dent Mater 30(3):372–380. doi:10.1016/j.dental.2013.12.013
Acknowledgments
We thank Mira Ristić and Marijan Marciuš from the Division of Materials Chemistry, Ruđer Bošković Institute, for the SEM micrographs. We also gratefully acknowledge Drago Skrtic for providing us with the zirconia-hybridized ACP.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
This investigation was supported by Croatian Science Foundation (Project 08/31 Evaluation of new bioactive materials and procedures in restorative dental medicine).
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study, formal consent is not required.
Rights and permissions
About this article

Cite this article
Par, M., Marovic, D., Skenderovic, H. et al. Light transmittance and polymerization kinetics of amorphous calcium phosphate composites. Clin Oral Invest 21, 1173–1182 (2017). https://doi.org/10.1007/s00784-016-1880-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00784-016-1880-6