
Abstract
Alzheimer’s disease is the most common cause of dementia in humans and is rapidly becoming an unmet medical need of epidemic proportions. Although billions of dollars have been allocated to this cause, more than 190 putative Alzheimer’s disease drugs have failed in the clinic, exemplifying the high risk-to-reward ratio of therapeutic development. One factor implicated in this high failure rate is the limitation of animal models to accurately predict clinical outcomes. The fact that some of these models, such as transgenic mice, have a 100% failure rate for predicting clinical outcomes of putative drugs exemplifies their limitations and the need for additional models that better recapitulate the multifactorial nature of Alzheimer’s disease progression. Aged dogs naturally develop Alzheimer’s-like neuropathological changes, as well as cognitive-domain specific impairments consistent with early stages of Alzheimer’s disease progression. Moreover, cross-sectional data in differentially aged dogs suggests that Alzheimer’s relevant biomarker changes are also found in dogs and could be used to monitor early stage Alzheimer’s-like disease progression. The aged dog model accurately predicted the clinical outcome of both the gold standard symptomatic Alzheimer’s disease therapeutic, donepezil, and the failure of active fibrillary amyloid vaccination strategies. The aged dog, therefore, provides a valuable preclinical animal model for assessing Alzheimer’s disease therapeutics with demonstrable translational value for predicting clinical outcomes. Given the translational validity of the model, it is likely that Alzheimer’s disease clinical research could also be used for improving care of senior dogs.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Alzheimer’s disease is the most common cause of dementia in humans and is rapidly becoming an unmet medical need of epidemic proportions. More than 50 million people are expected to be diagnosed with Alzheimer’s disease by 2050 unless efficacious disease-modifying treatments are developed. Although billions of dollars have been allocated to this cause, more than 190 putative Alzheimer’s disease drugs have failed in the clinic, exemplifying the high risk to reward ratio of Alzheimer’s disease therapeutic development. One factor implicated in this high failure rate is the limitation of animal models to accurately predict clinical outcomes.
Animal models are essential for demonstrating potential target relevance and target engagement of novel therapeutics. The fact that some of these models, such as transgenic mice, have a 100% failure rate for predicting clinical outcomes of putative drugs exemplifies their limitations and the need for additional models that better recapitulate the multifactorial nature of Alzheimer’s disease progression.
Aged dogs naturally develop Alzheimer’s-like neuropathological changes, as well as cognitive domain-specific impairments consistent with early stages of Alzheimer’s disease progression. Moreover, cross-sectional data in differentially aged dogs suggests that Alzheimer’s relevant biomarker changes are also found in dogs and could be used to monitor early-stage Alzheimer’s-like disease progression. The naturalistic progression of these changes in canine aging has not been linked to a single target, which suggests aged dogs may be used as a confirmatory animal model for preclinically evaluating putative Alzheimer’s therapeutics. In this respect, the aged dog model accurately predicted the clinical outcome of both the gold standard symptomatic Alzheimer’s disease therapeutic, donepezil, and the failure of active fibrillary amyloid vaccination strategies.
The aged dog, therefore, provides a valuable preclinical animal model for assessing Alzheimer’s disease therapeutics with demonstrable translational value for predicting clinical outcomes of both symptomatic and disease-modifying Alzheimer’s disease interventions. Given the translational validity of the model, it is likely that Alzheimer’s disease clinical research could also be used for improving care of senior dogs.
4.1 Introduction
Alzheimer’s disease (AD) is a progressive and chronic neurodegenerative disease and the major cause of dementia in the elderly, accounting for 50–75% of all dementia cases (Alzheimer 1906; Grossman et al. 2006; Qiu et al. 2009; Selkoe 2001; Uzun et al. 2011). According to diagnostic criteria guidelines (McKhann et al. 1984), AD onset occurs between 40 and 90 years of age, with onset being highest in populations greater than 65 years of age; early-onset cases, associated with dominant familial gene mutations, represent less than 5% of all AD cases (Ott et al. 1995; Selkoe 2001). Current global estimates of AD prevalence range from 15 to 27.7 million cases in the general population, but are anticipated to rise to greater than 115 million cases by 2015, indicating there is unmet medical need of epidemic proportions (WHO 2012).
Longitudinal clinical studies aimed at characterizing the progression of AD in aged human populations suggest that neuropathological changes develop decades prior to clinical diagnosis, which can only be confirmed by postmortem confirmation of both pathological hallmarks of AD—senile plaques and neurofibrillary tangles. Therefore, it is increasingly accepted that there is a substantial prodromal phase of AD, and that disease progression, as well as therapeutic efficacy, could be monitored using pathophysiological biomarkers, which provide a paradigm for targeting patient populations during earlier, and presumably more responsive, stages of the disease.
Although an enormous amount of financial resource has been invested in developing AD therapeutics, more than 190 have failed in human clinical trials, and only four drugs have been approved (Becker and Greig 2012; Cummings et al. 2014). Therefore, there is a significant unmet need for novel, and efficacious, AD-modifying therapeutics that either halt or slow progression to clinical AD (Klafki et al. 2006). The failure to develop efficacious drugs for AD can be attributed in part to translational limitations of preclinical animal models to predict clinical outcomes. The current chapter aims to demonstrate that the aged dog model naturally models several characteristics of AD and can be used to evaluate the potential efficacy of putative therapeutics across varying stages of AD progression. Specifically, the aged dog is a translationally relevant preclinical animal model because the model accurately predicts outcomes of human clinical trials.
4.2 Alzheimer’s Disease
Alzheimer’s disease is a neurodegenerative disease originally described by Alois Alzheimer (Alzheimer 1906) in which patients present with progressive cognitive deficits across multiple cognitive domains that ultimately result in dementia and death. Although the cause of the disease is unknown, there are both distinguishing and associated neuropathological changes present in the disease. More recently, biomarkers of these pathophysiological changes are being investigated in the effort to diagnose disease in early stages of progression, and potentially to monitor disease-modifying interventions. The current section focuses on these three key features–neuropathology, cognitive deficits, and associated biomarkers of pathophysiological progression.
4.2.1 Neuropathological Features
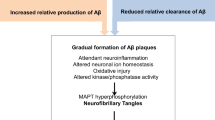
The hallmark neuropathological features of AD are extracellular senile, or neuritic, plaques and intracellular neurofibrillary tangles (NFT) in the neocortex and temporal lobe structures (Hyman and Trojanowski 1997). The senile plaque pathology is a result of extracellular focal deposition of amyloid beta (Aβ) protein, and the NFT pathology is comprised of intracellular paired helical filaments and straight filaments of abnormally, or hyper-, phosphorylated tau protein (Markesbery 2010). These two pathologies can occur decades prior to clinical manifestation of the disease, and are accompanied by extensive neurodegenerative processes that include neuronal and synaptic dysfunction and loss, leading to widespread cortical atrophy and alterations in multiple neurotransmitter systems and neural networks (Jack et al. 2010).
In addition to these hallmark pathological features, several other neuropathological changes are associated with AD, including decline in neurotransmitter systems, particularly the cholinergic system (Bartus 2000; Bartus et al. 1982; Norbury et al. 2005; Picciotto and Zoli 2002; Reinikainen et al. 1987, 1990; Rossor and Iversen 1986), extensive cortical and/or brain region-specific atrophy measured by both postmortem evaluation and in vivo magnetic resonance imaging (MRI) (Double et al. 1996; Fox and Freeborough 1997; Jack et al. 2005), and increased oxidative stress (Markesbery 1997; Smith et al. 2000). Thus, the two neuropathological diagnostic criteria for an AD diagnosis represent only a part of the neuropathological processes occurring in AD progression.
4.2.2 Cognitive Decline
The clinical diagnosis of AD is based on a progressive dementia with cognitive impairment in multiple independent cognitive domains and behavioral (neuropsychiatric) changes (Albert 2011; Marin et al. 1997; McKhann et al. 1984, 2011) that ultimately result in death (Cummings 2000). In the majority of cases, deficits in episodic memory occur in conjunction with changes in at least one other cognitive domain, although other variations of cognitive deficits are described (Albert 2011; McKhann et al. 2011). Moreover, episodic memory deficits are reported in subpopulations with mild cognitive impairment (MCI) who are at risk for development of AD; however, this subgroup does not differ cognitively from normal aged-matched controls on other cognitive measures, which suggests that deficits in episodic memory may precede clinical diagnosis of AD (Sperling et al. 2011).
The most devastating feature of AD is perhaps the progression of cognitive decline leading to overt dementia, in which multiple cognitive domains are affected over several years until individuals are no longer able to live independently (Albert 2011). Cognitive domains most commonly affected in the pathophysiological progression of AD include memory, executive function/attention, visuospatial skills, and language (Albert et al. 2011). The majority of AD patients show a relatively consistent pattern of cognitive decline, which is referred to as progressive amnestic disorder, and is generally characterized by early impairments in episodic memory (Albert 2011; Backman et al. 2000; Grober et al. 1999; Linn et al. 1995; Perry et al. 2000; Petersen 1998; Petersen et al. 1994) consistent with early neuropathological changes in the entorhinal cortex and hippocampal formation (Braak and Braak 1991; Jack et al. 1992, 1997). Executive function deficits precede language and visuospatial deficits in a majority of AD cases (Amieva et al. 2004; Foldi et al. 2002; Grady et al. 1988; Lafleche and Albert 1995; Perry et al. 2000; Reid et al. 1996), although some report that visuospatial deficits also occur early in the course of AD progression (Becker et al. 1988; Johnson et al. 2009). Based on performance across various cognitive tasks, it is suggested that AD progression can be divided into amnestic cases, in which declarative memory deficits are most evident; non-memory or dysexecutive cases, in which deficits on executive function tasks are most evident with sparing of declarative memory; or multi-domain, or mixed, cases in which deficits across multiple cognitive domains are evident, including combined memory and language deficits (Delano-Wood et al. 2009; Eppig et al. 2012; Libon et al. 2010, 2011; Petersen 2004; Petersen and Morris 2005; Ritchie and Touchon 2000).
4.2.3 Pathophysiological Biomarkers of Progression
It is increasingly accepted that pathophysiological biomarkers can provide diagnostic and prognostic information for improved clinical management of patients in early stages of AD progression (Buerger et al. 2006; Hampel et al. 2008; Jack et al. 2010; Shaw et al. 2007). To a large extent, this has been facilitated by large longitudinal clinical trials such as the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer’s Network (DIAN), which collect longitudinal data in patients with AD, mild cognitive impairment, and elderly controls with the aim of understanding disease progression (Jack et al. 2008; Morris et al. 2012; Moulder et al. 2013; Weiner et al. 2013). The emerging data indicate that AD processes begin decades before clinical manifestation, and to this end, Jack et al. (2010) propose a trajectory of neuropathological events progressing through cerebral amyloidosis, neuronal injury and synaptic dysfunction, and neurodegeneration, with memory impairment occurring prior to clinically diagnosed dementia. Specifically, markers of amyloidosis including increased amyloid positron emission tomography (PET) tracer retention and reduced cerebrospinal fluid (CSF) Aβ42 are detected early in the disease process in patients that are clinically normal (de Leon et al. 2001; Jagust et al. 2006; Petrie et al. 2009). Neuronal and synaptic dysfunction can be monitored by increased CSF tau and decreased fluorodeoxyglucose uptake on PET imaging, respectively. Neurodegeneration can be identified by magnetic resonance imaging (MRI) measures of cerebral atrophy (Bobinski et al. 1999; Frisoni et al. 2010; Ramani et al. 2006; Scheltens et al. 2002). Moreover, combinations of biomarkers demonstrate improved ability to predict conversion to AD better than one marker alone (Vemuri et al. 2009a, b, 2010). Ultimately, biomarkers may serve as a method to track the in vivo efficacy of AD-modifying treatments in patient populations that are presymptomatic (Jack et al. 2010).
In addition to the abovementioned biomarkers, there are several other in vivo imaging, CSF, and blood-based biomarkers currently being explored. For example, metabolic markers of neuronal health can be measured by proton magnetic resonance spectroscopy (H-MRS). In patients with MCI or AD, levels of n-acetylaspartate (NAA) and myoinositol (INS) are of particular interest. NAA is an amino acid made in the mitochondria, which are located in neuronal cell bodies, axons, and dendrites (Birken and Oldendorf 1989; Simmons et al. 1991). Thus, NAA is thought to be a biomarker for neuronal loss. H-MRS studies have shown decreased NAA in MCI and AD patients (Kantarci et al. 2000; Rose et al. 1999). By contrast, INS is a glial metabolite. Kantarci et al. (2000) reported that INS levels are increased in MCI and AD patients. Given the large-scale longitudinal trials and the intensity of interest in novel biomarker of AD progression, it is likely that key biomarkers will be identified and validated in the near future.
4.3 The Aged Dog Model of Alzheimer’s Disease Progression
Canine aging is associated with behavioral and neuropathological changes that parallel the progressive nature of human AD. Recent data examining in vivo imaging and CSF pathophysiological biomarkers in canine aging also demonstrate parallels to the biomarker changes observed in AD progression. The current section compares these three features of canine aging to AD progression to demonstrate the parallels of this naturally occurring disease process in dogs and humans.
4.3.1 Neuropathological Changes
Like AD patients, aged dogs also demonstrate amyloid plaques, and numerous studies have examined the composition and accumulation pattern of senile plaques in the aged canine brain (Cummings et al. 1996a, c; Mirra et al. 1991). This research is driven largely by the popularity of the amyloid hypothesis of AD that proposes that Aβ is a causative factor in disease progression (Hardy 2006). Mirra et al. (1991) found that senile plaques are composed of Aβ protein, which is identical in sequence between dogs and humans (Johnstone et al. 1991; Selkoe et al. 1987). The Aβ peptide is produced by the sequential cleavage of amyloid precursor protein (APP) by beta-secretase and gamma-secretase in both humans and dogs (Borghys et al. 2012; Murphy et al. 2010; Selkoe 1996), resulting in similar isoforms in both species (Portelius et al. 2010). In both dog and human, the 42-amino acid isoform (Aβ42) is the predominant isoform found in the insoluble plaque deposits (Selkoe 2001; Wisniewski et al. 1990). The regional and temporal pattern of Aβ deposition in the human brain is well characterized (Braak and Braak 1991; Braak et al. 1993; Wisniewski et al. 1970). Similar to humans, Aβ deposition occurs earliest in the prefrontal cortex, and later in the temporal cortex, hippocampus, and occipital cortex of beagles (Giaccone et al. 1990; Head et al. 2000; Ishihara et al. 1991; Thal et al. 2002) and other breeds of dogs (Rofina et al. 2003, 2004, 2006; Smolek et al. 2016). In contrast to human plaque pathology, which includes both diffuse and dense core plaque, canine amyloid plaques are mainly of the diffuse subtype (Cummings et al. 1993; Giaccone et al. 1990; Morys et al. 1994; Okuda et al. 1994; Russell et al. 1992; Uchida et al. 1992). In the cerebrovasculature, the deposition of the 40-amino acid isoform of Aβ occurs in both dogs and humans, resulting in cerebrovascular amyloid angiopathy (Attems 2005; Attems et al. 2005; Herzig et al. 2006). Vascular Aβ deposits can result in disruption of the blood-brain barrier and vessel wall viability (Prior et al. 1996), as well as microhemorrhages (Uchida et al. 1991).
Several studies in beagle dogs indicate a positive correlation between cognitive impairments and increasing brain Aβ plaque load in addition to increasing age (Colle et al. 2000; Cummings et al. 1996a, c; Head et al. 1998; Rofina et al. 2006). In this respect, standardized neuropsychological tests (described in the subsequent section) have been useful for determining the impact of brain region-specific amyloid burden on cognitive domain-specific function independently of age. High levels of Aβ in the prefrontal cortex, for instance, are associated with reversal learning and complex working memory deficits, whereas high levels of Aβ in the entorhinal cortex are associated with impairments in size discrimination and reward- and object-approach learning (Cummings et al. 1996a; Head et al. 1998; Tapp et al. 2004); the prefrontal pathology-linked impairment is consistent with the known dependency of executive processes to prefrontal integrity. In pet dogs, the extent of Aβ plaque load also correlates significantly with behavioral changes independently of age (Colle et al. 2000; Rofina et al. 2006). On the other hand, impairments in complex working memory (measured by the delayed non-matching to position task (DNMP)) occur early in canine aging and likely precede amyloid plaque deposition (Studzinski et al. 2006). This pre-plaque impairment may be attributable to soluble amyloid oligomeric species, which are highly toxic, impair synaptic function, and correlate with cognitive dysfunction in humans (Lacor et al. 2004; Selkoe 2008; Tomic et al. 2009). Oligomeric amyloid also is increased with age in beagle dogs (Pop et al. 2012) and may serve as a target for AD-modifying therapeutics early in the disease process.
The second neuropathological hallmark of AD is NFT neuropathology, which generally is absent in aged animals, including dogs (Morys et al. 1994; Russell et al. 1992; Selkoe et al. 1987; Uchida et al. 1992); however, others report the presence of NFTs in aged dogs, albeit to a lesser extent than humans (Papaioannou et al. 2001; Schmidt et al. 2015). Regardless, several phosphorylated tau epitopes, as well as neuropil threads that are consistent with AD pathology in humans are found in the aged canine brain and may be linked to behavioral changes associated with cognitive dysfunction syndrome (Pugliese et al. 2006, Schmidt et al. 2015; Smolek et al. 2016; Yu et al. 2011). Therefore, it is unclear if tauopathy in dogs leads to synaptic and cell loss. Aged dogs often also show marked ventriculomegaly, thinning of the cerebral cortex, and reduced white matter (Gonzalez-Soriano et al. 2001; Kimotsuki et al. 2005; Su et al. 1998; Vandevelde et al. 2012). Moreover, brain atrophy varies by region; for example, prefrontal cortical volume decreases prior to hippocampal volume (Tapp et al. 2004). Significantly fewer neurons are found in the hilus of the dentate gyrus of aged dogs, and fewer Purkinje cells are found in the cerebellum of impaired dogs (Pugliese et al. 2007; Siwak-Tapp et al. 2008), which is consistent with the extensive neuronal loss in AD (Bobinski et al. 1997; West 1993). These reductions may reflect decreased neurogenesis in aged dogs (Pekcec et al. 2008; Siwak-Tapp et al. 2007). Another potential source of neurodegeneration is the increase in oxidative stress and reduced antioxidant capacity in canine aging, which is linked to cognitive deficits (Head et al. 2002; Hwang et al. 2008; Kiatipattanasakul et al. 1997; Opii et al. 2008; Papaioannou et al. 2001; Rofina et al. 2004, 2006; Skoumalova et al. 2003) and may be a result of age-related mitochondrial dysfunction (Head et al. 2009).
Although age-related modification in neurotransmitter systems has not been extensively investigated in the dog, there is growing evidence that alterations in neurotransmitter systems may be a cause of cognitive dysfunction and may be a result or cause of other AD-like neuropathological changes. Like humans, aged dogs are more sensitive to cognitive disruption caused by scopolamine than young dogs, which may be due in part to reduced muscarinic receptor numbers across multiple brain regions except the cerebellum (Araujo et al. 2011b; Reinikainen et al. 1987, 1990). Complex working memory performance is particularly susceptible to cholinergic disruption and is improved using cholinesterase inhibitors in both dogs and humans (Araujo et al. 2004, 2011a; Drachman and Leavitt 1974). Cognitively impaired dogs also show a significant reduction in the noradrenergic neurons in the locus coeruleus, which are also implicated in AD (Dringenberg 2000; Grudzien et al. 2007; Insua et al. 2010).
4.3.2 Cognitive Decline
Consistent with AD-like cognitive impairments, cognitive function tasks can be used to demonstrate that domain-specific cognitive decline which also occurs in canine aging (Araujo et al. 2008; Chan et al. 2002; Landsberg et al. 2012; Rofina et al. 2006; Siwak et al. 2001; Snigdha et al. 2012; Studzinski et al. 2006; Tapp et al. 2003). For example, memory and executive functions are impaired relatively early in the aging process (Studzinski et al. 2006; Tapp et al. 2001), and these impairments are positively correlated with brain region-specific amyloid burden and atrophy (Cummings et al. 1996c; Head et al. 1998; Rofina et al. 2006; Tapp et al. 2004). To objectively assess cognitive function across multiple cognitive domains in dogs, a number of validated laboratory-based neuropsychological tasks have been developed (Landsberg et al. 2012). These tasks are tester administered and utilize food rewards to reinforce correct responses.
Although the behavioral changes associated with canine cognitive dysfunction (CDS) are presumably due to brain neuropathology and impaired cognitive function (Landsberg et al. 2012, see Chaps. 1 and 5), early and subtle cognitive deficits may be difficult to identify in the home or clinical environment. Moreover, the behavioral changes associated with CDS generally occur later in life than the cognitive impairments detected using neuropsychological testing. In fact, impaired spatial working memory performance predicts behavioral changes that include altered sleep-wake cycles and decreased exploration and interaction (Siwak et al. 2001, 2003).
The test apparatus and neuropsychological tasks have been described extensively elsewhere (Landsberg et al. 2012), but here we describe a subset of tasks that are sensitive to both aging and therapeutic intervention. Importantly, these tasks can be used to examine therapeutic interventions both as a model for AD progression and as a preclinical model for CDS.
Delayed Non-matching to Position Task (DNMP): The DNMP task can be used to assess both complex visuospatial learning and short-term visuospatial working memory (Chan et al. 2002; Studzinski et al. 2006), although the former can only truly be tested once, limiting its use in assessing interventions. On each trial of the task, the dog is presented initially with a single object (e.g., white block) over one of the three possible locations and then is required to displace the object to obtain a food reward (Fig. 4.1a). The tray is then removed from the dog’s sight, and a delay is initiated. Following the delay, the animal is then presented with two objects identical to that used in the original presentation. The dog is required to displace the object in the new location and receives a reward for responding correctly (see Fig. 4.1b).

(a) Sample phase of the DNMP task. The dog is required to displace a single block in one of the three positions to obtain a food reward. (b) Choice phase of the DNMP task. Following sample presentation and a predetermined delay, the dog must select the block in the new (non-match) position to obtain a food reward. (c) Simple discrimination learning and reversal task. During learning, the dog is required to continually select one of the two objects until learning criteria are reached. Subsequently, the reward contingency is reversed during reversal learning discrimination. (d) Selective attention task. The animal is required to select a single object and not a distractor; however, the number of distractors (i.e., negative stimuli) can vary from 0 to 3 during each trial
The delay between the first and second presentations may be varied to modify the memory demands of the task. Old dogs can be separated into three groups based on DNMP performance—unimpaired, impaired, and severely impaired—which may be analogous to the various cognitive stages of AD progression (Adams et al. 2000). Also, learning and memory impairments on the DNMP can be detected as early as middle age (Studzinski et al. 2006). In terms of assessing learning, this is a particularly useful task because performance varies directly with age: young dogs learn the task rapidly, older dogs learn more slowly, and very old dogs may be unable to learn even with extensive training (Studzinski et al. 2006); specifically, age predicted 48.2% of the variability in learning the DNMP task. Beagle dogs ranging from 1 to 11.99 years generally made more errors with increasing age, and mild visuospatial deficits were detected by 6 years, which precedes the typical onset of Aβ accumulation in the dog brain by approximately 2 years. This suggests the DNMP task can serve as an early marker for cognitive decline in the dog, and that age-related changes in visuospatial function in the dog models are seen in humans (Studzinski et al. 2006), possibly due to the influence of the cholinergic system on its performance (Araujo et al. 2004, 2011a, b).
Object Discrimination and Reversal Learning Tasks: Discrimination learning requires the animal to select one of the two different objects to obtain a food reward (Milgram et al. 1994). Objects may vary in color, shape, size, or a combination of the three (see Fig. 4.1c). Discrimination learning performance typically remains intact with age, possibly because an associative learning strategy can be used (Cotman et al. 2002). However, increasing difficulty of the task (by using more similar objects) exacerbates age-related deficits (Tapp et al. 2003). Once the task is learned, reversal learning can be tested by modifying the reward contingency such that the previously non-rewarded object is rewarded. Reversal learning is highly age sensitive in dogs, as the task predominately relies on executive function, requiring the subject to inhibit previously rewarded actions and shift responses to a new stimulus (Mongillo et al. 2013; Tapp et al. 2003). Specifically, both delayed learning and perseverative responding increase with increasing age.
Selective Attention Task: The selective attention task, or variable object discrimination task, is similar to discrimination learning in that the dog is required to select a single object for a reward; however, in this task, rather than one negative stimulus, there is a variable number of distractor objects (0 to 3) on any given trial (see Fig. 4.1d). With increasing distractor number, accuracy decreases significantly, and latency to respond increases across dogs of all ages (Snigdha et al. 2014), which is consistent with human selective attention impairments on tasks such as the visual search task (Parasuraman et al. 1995; Snigdha et al. 2012). Senior dogs are significantly impaired compared to both young and old dogs, and old dogs are intermediate in performance between young and senior age groups (Snigdha et al. 2014). These results suggest that aging impairs the ability of canines to discriminate between task-relevant and task-irrelevant stimuli, likely due to impairments in attentional processes related to inhibitory control and engagement.
The selective attention task employs a previously learned object to permit repeated testing in longitudinal aging or therapeutic intervention studies. A similar paradigm, oddity discrimination learning, examines learning when the incorrect object is present in duplicate and in which difficulty can be increased by using objects that are more similar (Milgram et al. 2002b; Tapp et al. 2003). On the oddity discrimination task, performance declines with both increasing age and task difficulty (Milgram et al. 2002b), presumably due to the increased attentional demands associated with increased distractor number (Araujo et al. 2008).
4.3.3 Pathophysiological Biomarkers
The longitudinal pattern of AD-relevant biomarker changes has not been well studied in the dog, but there is increasing evidence from cross-sectional studies suggesting biomarkers of brain amyloidosis, neuronal and synaptic dysfunction, and neurodegeneration, which parallel those seen in AD progression, are also evident in canine aging. Head et al. (2008) demonstrated that percent Aβ42 CSF levels decline from middle to senior age in dogs, and that this decline is inversely correlated with increasing brain amyloid burden. This parallels the findings in humans that decreasing levels of Aβ42 in CSF correlate with increased brain amyloid burden measured using amyloid-binding ligands with PET imaging (Fagan et al. 2006; Grimmer et al. 2009; Jagust et al. 2009; Sperling et al. 2011; Tolboom et al. 2009). However, the amyloid-binding ligands currently used for clinical or research purposes have not demonstrated specific amyloid binding of canine plaques, which may be due to the diffuse nature of the canine amyloid plaques (Cummings et al. 1996b; Czasch et al. 2006; Rofina et al. 2004; Torp et al. 2000a, b; Yoshino et al. 1996; Verhoeff et al. 2003).
More recently, we investigated changes in CSF levels of amyloid in 130 beagle dogs divided into four age groups (young, n = 17, 2.00–2.58 years; middle age, n = 21, 6.33–7.25 years; old, n = 57, 8.00–11.92 years; and senior, n = 35, 12.00–17.33 years) (Araujo et al. 2013a). A significant effect of age on percent Aβ42 was found, which reflected significantly higher levels in middle-aged dogs compared to all other age groups. This finding supports the hypothesis that a threshold concentration of Aβ42 is achieved during middle age prior to brain amyloid deposition (Sutphen et al. 2015). Percent Aβ42 was significantly lower in senior dogs compared to old dogs, which is consistent with the findings of Head et al. (2008). We also examined CSF levels of total tau in a subset of these dogs and found a significant age-dependent effect on CSF total tau levels, which increased from middle age to old and from old to senior, analogously to findings in human AD (Buerger et al. 2006). The mean within-subject coefficient of variance was 9.9% for Aβ42, 10.8% for Aβ40, and 9.7% for total tau, suggesting CSF Aβ42 and total tau levels may serve as reliable and translational biomarkers for preclinical aged canine studies investigating AD progression and possibly for predicting CDS.
Many in vivo imaging studies have been conducted in dogs, and several results parallel human AD findings. A pilot study examining fluorodeoxyglucose uptake on PET imaging in six adult and six senior dogs revealed significantly decreased fluorodeoxyglucose uptake across multiple brain regions consistent with the hypometabolism reported in AD patients (Araujo et al. 2013c). When cognitive tasks of varying difficulty were administered prior to imaging, brain region-specific engagement and disengagement were seen in adult, but not senior dogs, which suggests impairments in synaptic function and cortical processing (Horwitz et al. 1999). Magnetic resonance imaging studies demonstrate age-related ventriculomegaly as well as tissue volume loss of the frontal lobe, which occurs prior to atrophy of the hippocampus (Dimakopoulos and Mayer 2002; Su et al. 1998, 2005; Tapp et al. 2004). Moreover, a longitudinal MRI study in 47 dogs ranging from 8 to 11 years of age at the start of the 4-year study indicated that ventricular volume increased significantly over the last 2 years of the study and that the increase in ventricular volume was greater in older dogs than younger dogs; a 2.8% compared to 1.1% increase in ventricular volume was seen in dogs that were 11 compared to 8 years old at the start of the study, respectively (Su et al. 2005). Imaging studies using H-MRS also indicate an age-dependent reduction in NAA levels in dogs, which also is reported in AD patients (Adalsteinsson et al. 2000; de Rivera et al. 2007).
Therefore, biomarker patterns seen in AD progression are also found in canine aging, albeit the temporal sequence of these events has not been well studied in canine longitudinal studies. The same biomarkers may also prove valuable for predicting likely progression to CDS in pet dogs; however, the cost and invasiveness currently limit their use in the clinical setting. Regardless, biomarkers can be incorporated into laboratory canine studies designed to understand the progression of neuropathological events, their impact on cognitive function, and evaluation of novel therapeutics for AD-modifying drugs.
4.4 Predictive Validity of the Aged Dog Model
The validity of an animal model or test can be evaluated using various criteria which are beyond the scope of the current chapter. Rather, the ability of animal model to accurately predict clinical outcomes, or predictive validity, is the focus of the current section, which is pertinent given the absence of available AD-modifying therapeutic agents. In this respect, (pharmacological) predictive validity can be evaluated by determining how accurately an animal model predicts the results of human therapeutic clinical trials. While several animal models accurately predicted the symptomatic benefits of cholinesterase inhibitors on AD-like memory deficits, more than 190 compounds that were positive in animal models of AD have failed in clinical trials. This high number of false positives demonstrates a general lack of pharmacological validity in the most commonly used animal models, and transgenic mouse models, for example, have failed to accurately predict a positive result (Zahs and Ashe 2010). Although the absence of a gold standard AD-modifying therapeutics limits the ability to evaluate predictive validity for disease-modifying therapeutics, the dog has demonstrated ability to detect both true positives and false negatives (Studzinski et al. 2005) and also shows predictive value in non-pharmacological studies.
4.4.1 Symptomatic Treatment
The current gold standard for treatment of AD is donepezil hydrochloride (Aricept®), a cholinesterase inhibitor, which works by inhibiting the breakdown of acetylcholine in the brain. The rationale for this drug class is to compensate for the cholinergic deficits that are consistently reported in AD by increasing cholinergic neurotransmission (Bartus 2000; Bartus et al. 1982). Two cholinesterase inhibitors have been evaluated in the canine model: the first was the gold standard, donepezil, and the second was phenserine tartrate (Araujo et al. 2011a). Donepezil (1.5 mg/kg, PO) improved memory performance on the DNMP at the longer delays during treatment compared to both baseline and washout. By contrast, phenserine (0.5 mg/kg, PO) significantly improved DNMP performance at the longest delay compared to washout, and improved learning on a difficult version of the oddity discrimination task compared to placebo. In a study replicating the effects of donepezil in which the pharmacokinetic profile was also examined, DNMP improvement corresponded with plasma levels of donepezil consistent with those reported in humans (Araujo et al. 2013b; Matsui et al. 1999). Similarly, the canine model has accurately predicted symptomatic treatments that failed human clinical trials (i.e., false positives), such as the ampakines (Studzinski et al. 2005). Collectively, these data support the predictive validity of the aged dog model for future screening of symptomatic therapeutics for AD, as well as for investigating the links among cholinergic function, Aβ pathology, and cognitive decline in both AD and CDS.
4.4.2 Disease-Modifying Treatments
The prevailing theory in AD is that Aβ is toxic and that its accumulation in the brain is one of the leading causes of cognitive dysfunction (Hardy 2006). Consequently, eliminating the accumulation of Aβ in the brain has been a major research and drug development focus since the development of the amyloid cascade hypothesis. For example, amyloid vaccines resulted in plaque clearance as well as cognitive benefits in several transgenic mouse lines (Janus and Westaway 2001; Schenk et al. 1999; Sigurdsson et al. 2001); however, the vaccination approach has repeatedly failed in phase 3 human clinical trials (Alves et al. 2014; Holmes et al. 2008).
In contrast to transgenic mouse models, the aged dog predicted the clinical outcome of active vaccine trials, thereby identifying a false-positive finding in transgenic mice (Head et al. 2008). In both old and young dogs, immunization with fibrillar Aβ42 and a Th1 adjuvant (TiterMax Gold) resulted in primarily IgG2 and IgM antibody responses, and also caused a nonsignificant increase in CSF Aβ40 and decrease in cortical Aβ40/42, which was consistent with findings in transgenic models and humans (Head et al. 2006). In a subsequent 2.4-year vaccination study investigating both Aβ pathology and effect on cognitive dysfunction in aged dogs, no improvement on measures of learning, spatial attention, or spatial memory was found; however, after extended treatment, maintenance of prefrontal-dependent reversal learning ability was found (Head et al. 2008). Moreover, levels of soluble and insoluble Aβ40 and Aβ42 and the extent of diffuse plaque accumulation in the brain were significantly decreased in several cortical regions, with largest reductions in the prefrontal cortex. This predicted the outcome of similar human clinical vaccination trials and also suggests that reducing total Aβ may be of limited therapeutic benefit, particularly late in AD progression (Head et al. 2008; Holmes et al. 2008).
Current research extending beyond vaccination strategies includes beta- and gamma-secretase inhibitors and modulators, as well as numerous approaches targeting tau pathology. While the dog has been utilized to evaluate biomarker changes (i.e., CSF amyloid markers) following administration of secretase modulators (Borghys et al. 2012), the absence of canine studies aimed at predicting clinical trials, as well as the lack of a gold standard, limits the ability to further evaluate predictive validity of the canine model for accurately identifying successful (true-positive) disease-modifying drugs.
4.4.3 Non-pharmacological Studies
Non-pharmacological intervention studies in humans have focused primarily on lifestyle changes to see if these may improve or ameliorate the clinical signs associated with the early stages of Alzheimer’s disease. Two lines of research have shown efficacious results in humans: moderate exercise and dietary modification. In humans, moderate exercise has been shown to promote brain health via changes in specific brain mechanisms (Dao et al. 2013; Radák et al. 2001; van Praag et al. 1999; Liu et al. 2011). These changes have been shown to delay the onset of cognitive delay associated with AD (e.g., Rolland et al. 2007). Aged dogs trained to run on a treadmill for 10 min daily were tested on three different memory tasks: a concurrent discrimination task, a novel object recognition task, and a novel object location task (Snigdha et al. 2014). Acute exercise was able to improve performance on both the concurrent discrimination task and the novel object location task on a 24-h retest, and chronic exercise improved performance on the object location memory task. Interestingly, one of the brain changes reported after exercise in humans is a reduction of reactive oxygen species (ROS). Alternate methods to reduce ROS are via consumption of a diet rich with antioxidants. In a large prospective study held in the Netherlands called the Rotterdam Study, it was reported that a diet high in antioxidants vitamins C and E may lower the risk for Alzheimer’s disease (Engelhart et al. 2002). By comparison, a diet enriched with antioxidants alone and when combined with environmental enrichment improves cognitive function and prevents cognitive decline in aged dogs (Cotman et al. 2002; Milgram et al. 2002a, b; 2004). It is important to note that the general absence of phase 3 clinical trial data for non-pharmacological interventions impacts the ability to robustly evaluate predictive validity.
4.5 Summary
Aging dogs show AD-like cognitive domain-specific deficits and associated neuropathology that result in biomarker patterns consistent with those seen in AD progression. Therefore, the dog provides a natural model of AD progression that can be used to evaluate novel therapeutics or potentially be used to learn more about human disease progression and the sequential pathological events associated with disease progress. Moreover, aged dogs demonstrate the ability to accurately predict clinical outcomes and have successfully predicted symptomatic drugs that were approved and disease-modifying drugs that failed, such as active vaccine approaches. Consequently, the aged dog is a unique model of sporadic AD in which the progression of disease can be monitored using CSF and in vivo imaging biomarkers currently used in AD clinical research and drug development. On the other hand, developments in the AD field may also prove valuable for improved diagnosis, monitoring, and treatment of CDS.
References
Adalsteinsson E, Sullivan EV, Kleinhans N, Spielman DM, Pfefferbaum A (2000) Longitudinal decline of the neuronal marker N-acetyl aspartate in Alzheimer’s disease. Lancet. 355(9216):1696–1697
Adams B, Chan A, Callahan H, Siwak C, Tapp D, Ikeda-Douglas C, Atkinson P, Head E, Cotman CW, Milgram NW (2000) Use of a delayed non-matching to position task to model age-dependent cognitive decline in the dog. Behav Brain Res 108(1):47–56
Albert MS (2011) Changes in cognition. Neurobiol Aging 32(1):S58–S63
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH (2011) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute of Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia 7(3):270–279
Alves RPS, Yang MJ, Batista MT, Ferreira LCS (2014) Alzheimer’s disease: is a vaccine possible? Brazil J Med Biol Res 47(6):438–444
Alzheimer A (1906) Über einen eigenartigen schweren Erkrankungsprozeß der Hirnrinde. Neurologisches Centralblatt 23:1129–1136
Amieva H, Phillips LH, Della Sala S, Henry JD (2004) Inhibitory functioning in Alzheimer’s disease. Brain 127(5):949–964
Araujo JA, Chan ADF, Winka LL, Seymour PA, Milgram NW (2004) Dose-specific effects of scopolamine on canine cognition: impairment of visuospatial memory, but not visuospatial discrimination. Psychopharmacology 175(1):92–98
Araujo JA, de Rivera C, Milgram NW (2008) Visual attention is decreased in aged dogs. Alzheimer’s Dementia 4(4):T205–T206
Araujo JA, Greig NH, Ingram DK, Sandin J, de Rivera C, Milgram NW (2011a) Cholinesterase inhibitors improve both memory and complex learning in aged beagle dogs. J Alzheimer’s Dis 26(1):143–155
Araujo JA, Nobrega JN, Raymond R, Milgram NW (2011b) Aged dogs demonstrate both increased sensitivity to scopolamine impairment and decreased muscarinic receptor density. Pharmacol Biochem Behav 98(2):203–209
Araujo J, de Rivera C, Baulk J, Kelly S, Chakravarthy B (2013a) The reliability of and age effects on CSF measures of beta-amyloid 42 in beagle dogs: implications for a natural animal model of Alzheimer’s disease progression. Alzheimer’s Dementia 9(4):P851
Araujo JA, de Rivera C, Milgram NW, Sandin J (2013b) Pharmacological validation of the canine model of Alzheimer’s disease: donepezil improves memory in cognitively impaired aged beagle dogs. J Alzheimer’s Dis 5(4):e23
Araujo J, Sokolnicki K, Hesterman J, Hoppin J, Araujo D, Dobson H (2013c) Aged beagle dogs demonstrate reduced brain metabolism measured using PET-MR. Alzheimer’s Dementia 9(4):P851
Attems J (2005) Sporadic cerebral amyloid angiopathy; pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol 110(4):345–359
Attems J, Jellinger KA, Lintner F (2005) Alzheimer’s disease pathology influences severity and topographical distribution of cerebral amyloid angiopathy. Acta Neuropathol 110(3):222–231
Backman L, Ginovart N, Dixon RA, Robins Wahlin TB, Wahlin A, Halldin C, Farde L (2000) Age-related cognitive deficits mediated by changes in the striatal dopamine system. Am J Psychiatry 157(4):635–637
Bartus RT (2000) On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp Neurol 163(2):495–529
Bartus RT, Dean R, Beer B, Lippa AS (1982) The cholinergic hypothesis of geriatric memory dysfunction. Science 217(4558):408–414
Becker RE, Greig NH (2012) Increasing the success rate of Alzheimer’s disease drug discovery and development. Expert Opin Drug Discov 7(4):367–370
Becker JT, Huff J, Nebes RD, Holland A, Boller F (1988) Neuropsychological function in Alzheimer’s disease: pattern of impairment and rates of progression. Archiv Neurol 45(3):263–268
Birken DL, Oldendorf WH (1989) N-acetyl-L-aspartic acid: a literature review of a compound prominent in 1H-NMR spectroscopic studies of brain. Neurosci Biobehav Rev 13(1):23–31
Bobinski M, Wegiel J, Tarnawski M, Bobinski M, Reisberg B, De Leon MJ, Miller DC, Wisniewski HM (1997) Relationships between regional neuronal loss and neurofibrillary changes in the hippocampal formation and duration and severity of Alzheimer disease. J Neuropathol Exp Neurol 56(4):414–420
Bobinski M, De Leon MJ, Wegiel J, Desanti S, Convit A, Saint Louis LA, Rusinek H, Wisniewski HM (1999) The histological validation of post mortem magnetic resonance imaging-determined hippocampal volume in Alzheimer’s disease. Neuroscience 95(3):721–725
Borghys H, Tuefferd M, Van Broeck B, Clessens E, Dillen L, Cools W, Vinken P, Straetemans R, De Ridder F, Gijsen H, Mercken M (2012) A canine model to evaluate efficacy and safety of γ-secretase inhibitors and modulators. J Alzheimer’s Dis 28(4):809–822
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259
Braak H, Braak E, Bohl J (1993) Staging of Alzheimer-related cortical destruction. Eur Neurol 33(6):403–408
Buerger K, Ewers M, Pirttilä T, Zinkowski R, Alafuzoff I, Teipel SJ, DeBernardis J, Kerkman D, McCulloch C, Soininen H, Hampel H (2006) CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain 129(11):3035–3041
Chan AD, Nippak P, Murphey H, Ikeda-Douglas CJ, Muggenburg B, Head E, Cotman CW, Milgram NW (2002) Visuospatial impairments in aged canines (Canis familiaris): the role of cognitive-behavioral flexibility. Behav Neurosci 116(3):443
Colle MA, Hauw JJ, Crespeau F, Uchihara T, Akiyama H, Checler F, Pageat P, Duykaerts C (2000) Vascular and parenchymal Aβ deposition in the aging dog: correlation with behavior. Neurobiol Aging 21(5):695–704
Cotman CW, Head E, Muggenburg BA, Zicker S, Milgram NW (2002) Brain aging in the canine: a diet enriched in antioxidants reduces cognitive dysfunction. Neurobiol Aging 23(5):809–818
Cummings JL (2000) Cognitive and behavioral heterogeneity in Alzheimer’s disease: seeking the neurobiological basis. Neurobiol Aging. 21(6):845–861
Cummings BJ, Su JH, Cotman CW, White R, Russell MJ (1993) β-Amyloid accumulation in aged canine brain: a model of early plaque formation in Alzheimer’s disease. Neurobiol Aging 14(6):547–560
Cummings BJ, Head E, Afagh AJ, Milgram NW, Cotman CW (1996a) β-amyloid accumulation correlates with cognitive dysfunction in the aged canine. Neurobiol Learning Memory 66(1):11–23
Cummings BJ, Head E, Ruehl W, Milgram NW, Cotman CW (1996b) The canine as an animal model of human aging and dementia. Neurobiol Aging 17(2):259–268
Cummings BJ, Pike CJ, Shankle R, Cotman CW (1996c) Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol Aging 17(6):921–933
Cummings JL, Morstorf T, Zhong K (2014) Alzheimer’s disease drug development pipeline: few candidates, frequent failures. Alzheimer’s Res Therapy. 6(4):37
Czasch S, Paul S, Baumgartner W (2006) A comparison of immunohistochemical and silver staining methods for the detection of diffuse plaques in the aged canine brain. Neurobiol Aging 27(2):293–305
Dao AT, Zagaar MA, Levine AT, Salim S, Eriksen JL, Alkadhi KA (2013) Treadmill exercise prevents learning and memory impairment in Alzheimer’s disease-like pathology. Curr Alzheimer Res 10(5):507–515
De Leon MJ, Convit A, Wolf OT, Tarshish CY, DeSanti S, Rusinek H, Tsui W, Kandil E, Scherer AJ, Roche A, Imossi A (2001) Prediction of cognitive decline in normal elderly subjects with 2-[18F] fluoro-2-deoxy-D-glucose/positron-emission tomography (FDG/PET). Proc Natl Acad Sci 98(19):10966–10971
de Rivera C, Konyer B, Dobson H, Araujo JA, Milgram NW (2007) Magnetic resonance spectroscopy reveals similarities in brain metabolites between puppies and kittens. Society Neurosci 868.18/B12
Delano-Wood L, Bondi MW, Sacco J, Abeles N, Jak AJ, Libon DJ, Bozoki A (2009) Heterogeneity in mild cognitive impairment: differences in neuropsychological profile and associated white matter lesion pathology. J Int Neuropsychol Soc 15(6):906–914
Dimakopoulous AC, Mayer RJ (2002) Aspects of neurodegeneration in the canine brain. J Nutrition 132(6):1579S–1582S
Double KL, Halliday GM, Krill JJ, Harasty JA, Cullen K, Brooks WS, Creasey H, Broe GA (1996) Topography of brain atrophy during normal aging and Alzheimer’s disease. Neurobiol Aging 17(4):513–521
Drachman DA, Leavitt J (1974) Human memory and the cholinergic system: a relationship to aging? Archives Neurol 30(2):113–121
Dringenberg HC (2000) Alzheimer’s disease: more than a ‘cholinergic disorder’—evidence that cholinergic–monoaminergic interactions contribute to EEG slowing and dementia. Behav Brain Res 115(2):235–249
Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JC, Breteler MM (2002) Dietary intake of antioxidants and risk of Alzheimer disease. J Am Med Assoc 287(24):3223–3229
Eppig J, Wambach D, Nieves C, Price CC, Lamar M, Delano-Wood L, Giovannetti T, Bettcher BM, Penney DL, Swenson R, Lippa C, Kabasakalian A, Bondi MW, Libon DJ (2012) Dysexecutive functioning in mild cognitive impairment: derailment in temporal gradients. J Int Neuropsychol Soc 18(1):20–28
Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST (2006) Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann Neurol 59(3):512–519
Foldi NS, Lobosco JJ, Schaefer LA (2002) The effect of attentional dysfunction in Alzheimer’s disease: theoretical and practical implications. Semin Speech Lang 23(2):139–150. Copyright © 2002 by Thieme Medical Publishers, Inc., 333 Seventh Avenue, New York, NY 10001, USA
Fox NC, Freeborough PA (1997) Brain atrophy progression measured from registered serial MRI: validation and application to Alzheimer’s disease. J Magnet Reson Imag 7(6):1069–1075
Frisoni GB, Fox NC, Jack CR, Scheltens P, Thompson PM (2010) The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol 6(2):67–77
Giaccone G, Verga L, Finazzi M, Pollo B, Tagliavini F, Frangione B, Bugiani O (1990) Cerebral preamyloid deposits and congophilic angiopathy in aged dogs. Neurosci Lett 114(2):178–183
Gonzalez-Soriano J, Garcia PM, Contreras-Rodriguez J, Martinez-Sainz P, Rodriguez-Veiga E (2001) Age-related changes in the ventricular system of the dog brain. Ann Anatomy-Anatomischer Anzeiger 183(3):283–291
Grady CL, Haxby JV, Horwitz B, Sundaram M, Berg G, Schapiro M, Friedland RP, Rapoport SI (1988) Longitudinal study of the early neuropsychological and cerebral metabolic changes in dementia of the Alzheimer type. J Clin Exp Neuropsychol 10(5):576–596
Greig NH, Sambamurti K, Yu QS, Brossi A, Bruinsma GB, Lahiri DK (2005) An overview of phenserine tartrate, a novel acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Curr Alzheimer Res 2(3):281–290
Grimmer T, Riemenschneider M, Förstl H, Henriksen G, Klunk WE, Mathis CA, Shiga T, Wester HJ, Kurz A, Drzezga A (2009) Beta amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol Psychiatry 65(11):927–934
Grober E, Dickson D, Sliwinski MJ, Buschke H, Katz M, Crystal H, Lipton RB (1999) Memory and mental status correlates of modified Braak staging. Neurobiol Aging. 20(6):573–579
Grossman H, Bergmann C, Parker S (2006) Dementia: a brief review. Mount Sinai J Med 73(7):985–992
Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM (2007) Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging. 28(3):327–335
Hampel H, Bürger K, Teipel SJ, Bokde AL, Zetterberg H, Blennow K (2008) Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimer’s Dementia 4(1):38–48
Hardy J (2006) Alzheimer’s disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimer’s Dis 9(3):151–153
Head E, Callahan H, Muggenburg BA, Cotman CW, Milgram NW (1998) Visual-discrimination learning ability and β-amyloid accumulation in the dog. Neurobiol Aging 19(5):415–425
Head E, McCleary R, Hahn FF, Milgram NW, Cotman CW (2000) Region-specific age at onset of β-amyloid in dogs. Neurobiol Aging. 21(1):89–96
Head E, Liu J, Hagen TM, Muggenburg BA, Milgram NW, Ames BN, Cotman CW (2002) Oxidative damage increases with age in a canine model of human brain aging. J Neurochem 82(2):375–381
Head E, Barrett EG, Murphy MP, Das P, Nistor M, Sarsoza F, Glabe CC, Kayed R, Milton S, Vasilevko V, Milgram NW (2006) Immunization with fibrillar Aβ 1–42 in young and aged canines: Antibody generation and characteristics, and effects on CSF and brain Aβ. Vaccine 24(15):2824–2834
Head E, Pop V, Vasilevko V, Hill M, Saing T, Sarsoza F, Nistor M, Christie LA, Milton S, Glabe C, Barrett E (2008) A two-year study with fibrillar β-amyloid (Aβ) immunization in aged canines: effects on cognitive function and brain Aβ. J Neurosci 28(14):3555–3566
Head E, Nukala VN, Fenoglio KA, Muggenburg BA, Cotman CW, Sullivan PG (2009) Effects of age, dietary, and behavioral enrichment on brain mitochondria in a canine model of human aging. Exp Neurol 220(1):171–176
Herzig MC, Van Nostrand WE, Jucker M (2006) Mechanism of cerebral beta-amyloid angiopathy: murine and cellular models. Brain Pathol 16(1):40–54
Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E (2008) Long-term effects of Aβ 42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372(9634):216–223
Horwitz B, Tagamets MA, McIntosh AR (1999) Neural modeling, functional brain imaging, and cognition. Trends Cognit Sci 3(3):91–98
Hwang IK, Yoon YS, Yoo KY, Li H, Choi JH, Kim DW, Yi SS, Seong JK, Lee IS, Won MH (2008) Differences in lipid peroxidation and Cu, Zn-superoxide dismutase in the hippocampal CA1 region between adult and aged dogs. J Veterinary Med Sci 70(3):273–277
Hyman BT, Trojanowski JQ (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathy Exp Neurol 56(10):1095–1097
Insua D, Suárez ML, Santamarina G, Sarasa M, Pesini P (2010) Dogs with canine counterpart of Alzheimer’s disease lose noradrenergic neurons. Neurobiol Aging 31(4):625–635
Ishihara T, Gondo T, Takahashi M, Uchino F, Ikeda SI, Allsop D, Imai K (1991) Immunohistochemical and immunoelectron microscopical characterization of cerebrovascular and senile plaque amyloid in aged dogs’ brains. Brain Res 548(1):196–205
Jack CR, Petersen RC, O'Brien PC, Tangalos EG (1992) MR-based hippocampal volumetry in the diagnosis of Alzheimer’s disease. Neurology 42(1):183–183
Jack CR, Petersen RC, Xu YC, Waring SC, O'Brien PC, Tangalos EG, Smith GE, Ivnik RJ, Kokmen E (1997) Medial temporal atrophy on MRI in normal aging and very mild Alzheimer’s disease. Neurology 49(3):786–794
Jack CR, Shiung MM, Weigand SD, O’Brien PC, Gunter JL, Boeve BF, Knopman DS, Smith GE, Ivnik RJ, Tangalos EG, Petersen RC (2005) Brain atrophy rates predict subsequent clinical conversion in normal elderly and amnestic MCI. Neurology 65(8):1227–1231
Jack CR, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, Borowski B, Britson PJ, Whitwell JL, Ward C, Dale AM (2008) The Alzheimer’s disease neuroimaging initiative (ADNI): MRI methods. J Magnet Res Imag 27(4):685–691
Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Trojanowski JQ (2010) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9(1):119–128
Jagust W, Gitcho A, Sun F, Kuczynski B, Mungas D, Haan M (2006) Brain imaging evidence of preclinical Alzheimer’s disease in normal aging. Ann Neurol 59(4):673–681
Jagust WJ, Landau SM, Shaw LM, Trojanowski JQ, Koeppe RA, Reiman EM, Foster NL, Petersen RC, Weiner MW, Price JC, Mathis CA (2009) Relationships between biomarkers in aging and dementia. Neurology 73(15):1193–1199
Janus C, Westaway D (2001) Transgenic mouse models of Alzheimer’s disease. Physiol Behav 73(5):873–886
Johnson DK, Storandt M, Morris JC, Galvin JE (2009) Longitudinal study of the transition from healthy aging to Alzheimer’s disease. Archiv Neurol 66(10):1254–1259
Johnstone EM, Chaney MO, Norris FH, Pascual R, Little SP (1991) Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Mol Brain Res 10(4):299–305
Kantarci KJCJ, Jack CR, Xu YC, Campeau NG, O’Brien PC, Smith GE, Ivnik RJ, Boeve BF, Kokmen E, Tangalos EG, Petersen RC (2000) Regional metabolic patterns in mild cognitive impairment and Alzheimer’s disease a 1h mrs study. Neurology 55(2):210–217
Kiatipattanasakul W, Nakamura SI, Kuroki K, Nakayama H (1997) Immunohistochemical detection of anti-oxidative stress enzymes in the dog brain. Neuropathology 17(4):307–312
Kimotsuki T, Nagaoka T, Yasuda M, Tamahara S, Matsuki N, Ono K (2005) Changes of magnetic resonance imaging on the brain in beagle dogs with aging. J Veterinary Med Sci 67(10):961–967
Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J (2006) Therapeutic approaches to Alzheimer’s disease. Brain 129(11):2840–2855
Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA (2004) Synaptic targeting by Alzheimer’s-related amyloid β oligomers. J Neurosci 24(45):10191–10200
Lafleche G, Albert MS (1995) Executive function deficits in mild Alzheimer’s disease. Neuropsychology 9(3):313–320
Landsberg GM, Nichol J, Araujo JA (2012) Cognitive dysfunction syndrome: a disease of canine and feline brain aging. Veterinary Clin N Am: Small Animal Pract 42(4):749–768
Libon DJ, Xie SX, Eppig J, Wicas G, Lamar M, Lippa C, Bettcher BM, Price CC, Giovannetti T, Swenson R, Wambach DM (2010) The heterogeneity of mild cognitive impairment: a neuropsychological analysis. J Int Neuropsychol Soc 16(1):84–93
Libon DJ, Bondi MW, Price CC, Lamar M, Eppig J, Wambach DM, Nieves C, Delano-Wood L, Giovannetti T, Lippa C, Kabasakalian A, Cosentino S, Swenson R, Penney DL (2011) Verbal serial list learning in mild cognitive impairment: a profile analysis of interference, forgetting, and errors. J Int Neuropsychol Soc 17(5):905–914
Linn RT, Wolf PA, Bachman DL, Knoefel JE, Cobb JL, Belanger AJ, Kaplan EF, D'Agostino RB (1995) The ‘preclinical phase’ of probable Alzheimer’s disease: a 13-year prospective study of the Framingham cohort. Archiv Neurol 52(5):485–490
Liu HL, Zhao G, Cai K, Zhao HH (2011) Treadmill exercise prevents decline in spatial learning and memory in APP/PS1 transgenic mice through improvement of hippocampal long-term potentiation. Behav Brain Res 218(2):308–314
Marin DB, Green CR, Schmeidler J, Harvey PD, Lawlor BA, Ryan TM, Aryan M, Davis KL, Mohs RC (1997) Noncognitive disturbances in Alzheimer’s disease: frequency, longitudinal course, and relationship to cognitive symptoms. J Am Geriatrics Soc. 45(11):1331–1338
Markesbery WR (1997) Oxidative stress hypothesis in Alzheimer’s disease. Free Radical Biol Med 23(1):134–147
Markesbery WR (2010) Neuropathologic alterations in mild cognitive impairment: a review. J Alzheimer’s Dis 19(1):221–228
Matsui K, Taniguchi S, Yoshimura T (1999) Correlation of the intrinsic clearance of donepezil (Aricept®) between in vivo and in vitro studies in rat, dog and human. Xenobiotica 29(11):1059–1072
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease. Neurology 34(7):939–944
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia 7(3):263–269
Milgram NW, Head E, Muggenburg B, Holowachuk D, Murphey H, Estrada J, Ikeda-Douglas CJ, Zicker SC, Cotman CW (2002a) Landmark discrimination learning in the dog: effects of age, an antioxidant fortified food, and cognitive strategy. Neurosci Biobehav Rev 26(6):679–695
Milgram NW, Zicker SC, Head E, Muggenburg BA, Murphey H, Ikeda-Douglas CJ, Cotman CW (2002b) Dietary enrichment counteracts age-associated cognitive dysfunction in canines. Neurobiol Aging 23(5):737–745
Milgram NW, Head E, Zicker SC, Ikeda-Douglas C, Murphey H, Muggenberg BA, Siwak CT, Tapp PD, Lowry SR, Cotman CW (2004) Long-term treatment with antioxidants and a program of behavioral enrichment reduces age-dependent impairment in discrimination and reversal learning in beagle dogs. Exp Gerontol 39(5):753–765
Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L (1991) The consortium to establish a registry for Alzheimer’s disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41(4):479–486
Mongillo P, Araujo JA, Pitteri E, Carnier P, Adamelli S, Regolin L, Marinelli L (2013) Spatial reversal learning is impaired by age in pet dogs. Age 35(6):2273–2282
Morris JC, Aisen PS, Bateman RJ, Benzinger TL, Cairns NJ, Fagan AM, Ghetti B, Goate AM, Holtzman DM, Klunk WE, McDade E (2012) Developing an international network for Alzheimer’s research: the Dominantly Inherited Alzheimer Network. Clin Investig 2(10):975–984
Morys J, Narkiewicz O, Maciejewska B, Wegiel J, Wisniewski HM (1994) Amyloid deposits and loss of neurones in the claustrum of the aged dog. Neuroreport 5(14):1825–1828
Moulder KL, Snider BJ, Mills SL, Buckles VD, Santacruz AM, Bateman RJ, Morris JC (2013) Dominantly Inherited Alzheimer Network: facilitating research and clinical trials. Alzheimer’s Res Therapy 5(5):1
Murphy MP, Morales J, Beckett TL, Astarita G, Piomelli D, Weidner A, Studzinski CM, Dowling AL, Wang X, LeVine H III, Kryscio RJ (2010) Changes in cognition and amyloid-β processing with long term cholesterol reduction using atorvastatin in aged dogs. J Alzheimer’s Dis 22(1):135–150
Norbury R, Travis MJ, Erlandsson K, Waddington W, Owens J, Pimlott S, Ell PJ, Murphy DGM (2005) In vivo imaging of muscarinic receptors in the aging female brain with (R, R)[123 I]-I-QNB and single photon emission tomography. Exp Gerontol 40(3):137–145
Okuda R, Uchida K, Tateyama S, Yamaguchi R, Nakayama H, Goto N (1994) The distribution of amyloid beta precursor protein in canine brain. Acta Neuropathol 87(2):161–167
Opii WO, Joshi G, Head E, Milgram NW, Muggenburg BA, Klein JB, Pierce WM, Cotman CW, Butterfield DA (2008) Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer’s disease. Neurobiol Aging 29(1):51–70
Ott A, Breteler MM, Van Harskamp F, Claus JJ, Van Der Cammen TJ, Grobbee DE, Hofman A (1995) Prevalence of Alzheimer’s disease and vascular dementia: association with education. The Rotterdam study. BMJ 310(6985):970–973
Papaioannou N, Tooten PC, van Ederen AM, Bohl JR, Rofina J, Tsangaris T, Gruys E (2001) Immunohistochemical investigation of the brain of aged dogs. I. Detection of neurofibrillary tangles and of 4-hydroxynonenal protein, an oxidative damage product, in senile plaques. Amyloid 8(1):11–21
Parasuraman R, Greenwood PM, Alexander GE (1995) Selective impairment of spatial attention during visual search in Alzheimer’s disease. Neuroreport 6(14):1861–1864
Pekcec A, Baumgärtner W, Bankstahl JP, Stein VM, Potschka H (2008) Effect of aging on neurogenesis in the canine brain. Aging Cell 7(3):368–374
Perry E, Martin-Ruiz C, Lee M, Griffiths M, Johnson M, Piggott M, Haroutunian V, Buxbaum JD, Nasland J, Davis K, Gotti C, Clementi F, Tzartos S, Cohen O, Soreq H, Jaros E, Perry R, Ballard C, McKeith I, Court J (2000) Nicotinic receptor subtypes in human brain ageing, Alzheimer and Lewy body diseases. Eur J Pharmacol 393(1):215–222
Petersen RC (1998) Clinical subtypes of Alzheimer’s disease. Dementia Geriatr Cognit Disorders 9(3):16–24
Petersen RC (2004) Mild cognitive impairment as a diagnostic entity. J Int Med 256(3):183–194
Petersen RC, Morris JC (2005) Mild cognitive impairment as a clinical entity and treatment target. Archiv Neurol 62(7):1160–1163
Petersen RC, Smith GE, Ivnik RJ, Kokmen E, Tangalos EG (1994) Memory function in very early Alzheimer’s disease. Neurology 44(5):867–867
Petrie EC, Cross DJ, Galasko D, Schellenberg GD, Raskind MA, Peskind ER, Minoshima S (2009) Preclinical evidence of Alzheimer changes: convergent cerebrospinal fluid biomarker and fluorodeoxyglucose positron emission tomography findings. Archiv Neurol 66(5):632–637
Picciotto MR, Zoli M (2002) Nicotinic receptors in aging and dementia. J Neurobiol 53(4):641–655
Pop V, Head E, Berchtold NC, Glabe CG, Studzinski CM, Weidner AM, Murphy MP, Cotman CW (2012) Aβ aggregation profiles and shifts in APP processing favor amyloidogenesis in canines. Neurobiol Aging. 33(1):108–120
Portelius E, Van Broeck B, Andreasson U, Gustavsson MK, Mercken M, Zetterberg H, Borghys H, Blennow K (2010) Acute effect on the Aβ isoform pattern in CSF in response to γ-secretase modulator and inhibitor treatment in dogs. J Alzheimer’s Dis 21(3):1005–1012
Prior R, D'Urso D, Frank R, Prikulis I, Pavlakovic G (1996) Loss of vessel wall viability in cerebral amyloid angiopathy. Neuroreport 7(2):562–564
Pugliese M, Mascort J, Mahy N, Ferrer I (2006) Diffuse beta-amyloid plaques and hyperphosphorylated tau are unrelated processes in aged dogs with behavioral deficits. Acta Neuropathol 112(2):175–183
Pugliese M, Gangitano C, Ceccariglia S, Carrasco JL, Del Fà A, Rodríguez MJ, Michetti F, Mascort J, Mahy N (2007) Canine cognitive dysfunction and the cerebellum: acetylcholinesterase reduction, neuronal and glial changes. Brain Res 1139:85–94
Qiu C, Kivipelto M, von Strauss E (2009) Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci 11(2):111–128
Radák Z, Kaneko T, Tahara S, Nakamoto H, Pucsok J, Sasvári M, Nyakas C, Goto S (2001) Regular exercise improves cognitive function and decreases oxidative damage in rat brain. Neurochem Int 38(1):17–23
Ramani A, Jensen JH, Helpern JA (2006) Quantitative MR Imaging in Alzheimer Disease 1. Radiology 241(1):26–44
Reid W, Broe G, Creasey H, Grayson D, McCusker E, Bennett H, Longley W, Sulway MR (1996) Age at onset and pattern of neuropsychological impairment in mild early-stage Alzheimer disease: a study of a community-based population. Archiv Neurol 53(10):1056–1061
Reinikainen KJ, Riekkinen PJ, Halonen T, Laakso M (1987) Decreased muscarinic receptor binding in cerebral cortex and hippocampus in Alzheimer’s disease. Life Sci 41(4):453–461
Reinikainen KJ, Soininen H, Riekkinen PJ (1990) Neurotransmitter changes in Alzheimer’s disease: implications to diagnostics and therapy. J Neurosci Res 27(4):576–586
Ritchie K, Touchon J (2000) Mild cognitive impairment: conceptual basis and current nosological status. Lancet 355(9199):225–228
Rofina J, Andel IV, Van Ederen AM, Papaioannou N, Yamaguchi H, Gruys E (2003) Canine counterpart of senile dementia of the Alzheimer type: amyloid plaques near capillaries but lack of spatial relationship with activated microglia and macrophages. Amyloid 10(2):86–96
Rofina JE, Singh K, Skoumalova-Vesela A, van Ederen AM, van Asten AJ, Wilhelm J, Gruys E (2004) Histochemical accumulation of oxidative damage products is associated with Alzheimer-like pathology in the canine. Amyloid 11(2):90–100
Rofina JE, Van Ederen AM, Toussaint MJM, Secreve M, Van Der Spek A, Van Der Meer I, Van Eerdenburg FJCM, Gruys E (2006) Cognitive disturbances in old dogs suffering from the canine counterpart of Alzheimer’s disease. Brain Res 1069(1):216–226
Rolland Y, Pillard F, Klapouszczak A, Reynish E, Thomas D, Andrieu S, Rivière D, Vellas B (2007) Exercise program for nursing home residents with Alzheimer’s disease: a 1-year randomized, controlled trial. J Am Geriatr Soc 55(2):158–165
Rose S, de Zubicaray G, Wang D, Galloway G, Chalk J, Eagle S, Semple J, Doddrell D (1999) A 1H MRS study of probable Alzheimer’s disease and normal aging: implications for longitudinal monitoring of dementia progression. Magn Res Imag 17(2):291–299
Rossor M, Iversen LL (1986) Non-cholinergic neurotransmitter abnormalities in Alzheimer’s disease. Br Med Bullet 42(1):70–74
Russell MJ, White R, Patel E, Markesbery WR, Watson CR, Geddes JW (1992) Familial influence on plaque formation in the beagle brain. Neuroreport 3(12):1093–1096
Scheltens P, Fox N, Barkhof F, De Carli C (2002) Structural magnetic resonance imaging in the practical assessment of dementia: beyond exclusion. Lancet Neurol 1(1):13–21
Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D (1999) Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400(6740):173–177
Schmidt F, Boltze J, Jäger C, Hofmann S, Willems N, Seeger J, Härtig W, Stolzing A (2015) Detection and Quantification of β-Amyloid, Pyroglutamyl Aβ, and Tau in Aged Canines. J Neuropathol Exp Neurol 74(9):912–923
Selkoe DJ (1996) Amyloid β-protein and the genetics of Alzheimer’s disease. J Biol Chem 271(31):18295–18298
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81(2):741–766
Selkoe DJ (2008) Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav Brain Res 192(1):106–113
Selkoe DJ, Bell DS, Podlisny MB, Price DL, Cork LC (1987) Conservation of brain amyloid proteins in aged mammals and humans with Alzheimer’s disease. Science 235(4791):873–877
Shaw LM, Korecka M, Clark CM, Lee VMY, Trojanowski JQ (2007) Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat Rev Drug Discov 6(4):295–303
Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T (2001) Immunization with a nontoxic/nonfibrillar amyloid-β homologous peptide reduces Alzheimer’s disease-associated pathology in transgenic mice. Am J Pathol 159(2):439–447
Simmons ML, Frondoza CG, Coyle JT (1991) Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies. Neuroscience 45(1):37–45
Siwak CT, Tapp PD, Milgram NW (2001) Effect of age and level of cognitive function on spontaneous and exploratory behaviors in the beagle dog. Learning Memory 8(6):317–325
Siwak CT, Tapp PD, Zicker SC, Murphey HL, Muggenburg BA, Head E, Cotman CW, Milgram NW (2003) Locomotor activity rhythms in dogs vary with age and cognitive status. Behav Neurosci 117(4):813
Siwak-Tapp CT, Head E, Muggenburg BA, Milgram NW, Cotman CW (2007) Neurogenesis decreases with age in the canine hippocampus and correlates with cognitive function. Neurobiol Learn Memory 88(2):249–259
Siwak-Tapp CT, Head E, Muggenburg BA, Milgram NW, Cotman CW (2008) Region specific neuron loss in the aged canine hippocampus is reduced by enrichment. Neurobiol Aging 29(1):39–50
Skoumalova A, Rofina J, Schwippelova Z, Gruys E, Wilhelm J (2003) The role of free radicals in canine counterpart of senile dementia of the Alzheimer type. Exp Gerontol 38(6):711–719
Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G (2000) Oxidative stress in Alzheimer’s disease. Biochem Biophys Acta Mol Basis Dis 1502(1):139–144
Smolek T, Madari A, Farbakova J, Kandrac O, Jadhav S, Cente M, Brezovakova V, Zilka N (2016) Tau hyperphosphorylation in synaptosomes and neuroinflammation are associated with canine cognitive impairment. J Comparative Neurol 524(4):874–895
Snigdha S, Christie LA, De Rivera C, Araujo JA, Milgram NW, Cotman CW (2012) Age and distraction are determinants of performance on a novel visual search task in aged beagle dogs. Age 34(1):67–73
Snigdha S, de Rivera C, Milgram NW, Cotman C (2014) Exercise enhances memory consolidation in the aging brain. Front Aging Neurosci 6:3
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ, Park DC (2011) Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia 7(3):280–292
Studzinski CM, Araujo JA, Milgram NW (2005) The canine model of human cognitive aging and dementia: pharmacological validity of the model for assessment of human cognitive-enhancing drugs. Progr Neuro-Psychopharmacol Biol Psychiatr 29(3):489–498
Studzinski CM, Christie LA, Araujo JA, Burnham WM, Head E, Cotman CW, Milgram NW (2006) Visuospatial function in the beagle dog: an early marker of cognitive decline in a model of human aging and dementia. Neurobiol Learning Memory 86(2):197–204
Su MY, Head E, Brooks WM, Wang Z, Muggenburg BA, Adam GE, Sutherland R, Cotman CW, Nalcioglu O (1998) Magnetic resonance imaging of anatomic and vascular characteristics in a canine model of human aging. Neurobiol Aging 19(5):479–485
Su MY, Tapp PD, Vu L, Chen YF, Chu Y, Muggenburg B, Chiou JY, Chen C, Wang J, Bracco C, Head E (2005) A longitudinal study of brain morphometrics using serial magnetic resonance imaging analysis in a canine model of aging. Progr Neuro-Psychopharmacol Biol Psychiatry 29(3):389–397
Sutphen CL, Jasielec MS, Shah AR, Macy EM, Xiong C, Vlassenko AG, Benzinger TL, Stoops EE, Vanderstichele HM, Brix B, Darby HD (2015) Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol 72(9):1029–1042
Tapp PD, Siwak CT, Head E, Cotman CW, Milgram NW (2001) Sex differences in the effect of oestrogen on size discrimination learning and spatial memory. In: Proceedings of the Third International Congress on Veterinary Behavioral Medicine. Universities Federation for Animal Welfare, Wheathamstead, pp 136–138
Tapp PD, Siwak CT, Estrada J, Head E, Muggenburg BA, Cotman CW, Milgram NW (2003) Size and reversal learning in the beagle dog as a measure of executive function and inhibitory control in aging. Learning Memory. 10(1):64–73
Tapp PD, Siwak CT, Gao FQ, Chiou JY, Black SE, Head E, Muggenburg BA, Cotman CW, Milgram NW, Su MY (2004) Frontal lobe volume, function, and β-amyloid pathology in a canine model of aging. J Neurosci 24(38):8205–8213
Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58(12):1791–1800
Tolboom N, van der Flier WM, Yaqub M, Boellaard R, Verwey NA, Blankenstein MA, Windhorst AD, Scheltens P, Lammertsma AA, van Berckel BN (2009) Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J Nucl Med 50(9):1464–1470
Tomic JL, Pensalfini A, Head E, Glabe CG (2009) Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiol Dis 35(3):352–358
Torp R, Head E, Cotman CW (2000a) Ultrastructural analyses of β-amyloid in the aged dog brain: Neuronal β-amyloid is localized to the plasma membrane. Progr Neuro-Psychopharmacol Biol Psychiatry 24(5):801–810
Torp R, Head E, Milgram NW, Hahn F, Ottersen OP, Cotman CW (2000b) Ultrastructural evidence of fibrillar β-amyloid associated with neuronal membranes in behaviorally characterized aged dog brains. Neuroscience 96(3):495–506
Uchida K, Nakayama H, Goto N (1991) Pathological studies on cerebral amyloid angiopathy senile plaques and amyloid deposition in visceral organs in aged dogs. J Veterinary Med Sci 53(6):1037–1042
Uchida K, Tani Y, Uetsuka K, Nakayama H, Goto N (1992) Immunohistochemical studies on canine cerebral amyloid angiopathy and senile plaques. J Veterinary Med Sci 54(4):659–667
Uzun S, Kozumplik O, Folnegović-Šmalc V (2011) Alzheimer’s dementia: current data review. Colleg Antropol 35(4):1333–1337
Van Praag H, Christie BR, Sejnowski TJ, Gage FH (1999) Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci 96(23):13427–13431
Vandevelde M, Higgins R, Oevermann A (2012) Veterinary neuropathology: essentials of theory and practice. Wiley, New York
Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CR, Alzheimer’s Disease Neuroimaging Initiative (2009a) MRI and CSF biomarkers in normal, MCI, and AD subjects: diagnostic discrimination and cognitive correlations. Neurology 73(4):287–293
Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CR, Alzheimer’s Disease Neuroimaging Initiative (2009b) MRI and CSF biomarkers in normal, MCI, and AD subjects: predicting future clinical change. Neurology 73(4):294–301
Vemuri P, Wiste HJ, Weigand SD, Knopman DS, Trojanowski JQ, Shaw LM, Bernstein MA, Aisen PS, Weiner M, Petersen RC, Jack CR (2010) Serial MRI and CSF biomarkers in normal aging, MCI, and AD. Neurology 75(2):143–151
Verhoeff NPLG, Wilson AA, Nobrega J, Milgram NW, Westaway D, Head E, Hussey D, Tapp D, Trop L, Giuliano F, Araujo J, Ginovart N, Richardson L, Singh K, Houle S (2003) Development of positron emission tomography (PET) tracers for beta-amyloid imaging in vivo. J Cerebral Blood Flow Metab 23(1):685
Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC, Harvey D, Jack CR, Jagust W, Liu E, Morris JC (2013) The Alzheimer’s Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimer’s Dementia 9(5):111–194
West MJ (1993) Regionally specific loss of neurons in the aging human hippocampus. Neurobiol Aging 14(4):287–293
Wisniewski H, Johnson AB, Raine CS, Kay WJ, Terry RD (1970) Senile plaques and cerebral amyloidosis in aged dogs. A histochemical and ultrastructural study. Lab Investig 23:287–296
Wisniewski HM, Wegiel J, Morys J, Bancher C, Soltysiak Z, Kim KS (1990) Aged dogs: an animal model to study beta-protein amyloidogenesis. In: Alzheimer’s disease. Epidemiology, neuropathology, neurochemistry, and clinics. Springer, Vienna, pp 151–168
World Health Organization (2012) Dementia: a public health priority. World Health Organization, Geneva
Yoshino T, Uchida K, Tateyama S, Yamaguchi R, Nakayama H, Goto N (1996) A retrospective study of canine senile plaques and cerebral amyloid angiopathy. Veterinary Pathol Online 33(2):230–234
Yu CH, Song GS, Yhee JY, Kim JH, Im KS, Nho WG, Lee JH, Sur JH (2011) Histopathological and immunohistochemical comparison of the brain of human patients with Alzheimer’s disease and the brain of aged dogs with cognitive dysfunction. J Comparative Pathol 145(1):45–58
Zahs KR, Ashe KH (2010) ‘Too much good news’–are Alzheimer mouse models trying to tell us how to prevent, not cure, Alzheimer's disease? Trends Neurosci 33(8):381–389
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Araujo, J.A., Baulk, J., de Rivera, C. (2017). The Aged Dog as a Natural Model of Alzheimer’s Disease Progression. In: Landsberg, G., Maďari, A., Žilka, N. (eds) Canine and Feline Dementia. Springer, Cham. https://doi.org/10.1007/978-3-319-53219-6_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-53219-6_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-53218-9
Online ISBN: 978-3-319-53219-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)