
Abstract
Due to their vast roles in human development, differentiation, homeostasis, and disease, bone morphogenetic proteins (BMP) have evolved along with numerous potentiating and inhibitory mechanisms to fine-tune signaling outcomes. As such, this chapter focuses on some of the best-studied and utilized extracellular mechanisms of BMP signal regulation. Due to their inherent binding characteristics, BMP ligands are often found engaged with at least of one of these many interacting partners. From a structural and functional perspective, we discuss our current understanding of how BMP ligands interact with these numerous binding partners, including secreted extracellular antagonists, BMP prodomains, and various co-receptors and noncanonical binding partners. Interestingly, while the BMP ligands themselves exhibit very redundant structural features, the composition and structure of their interacting proteins is quite diverse, lending to different ligand-binding modes and mechanisms, which lead to very different biological outcomes. Collectively, biochemical and structural characterization of these important interactions has provided valuable insight into BMP signal regulation.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Bone morphogenetic proteins (BMPs) were originally identified as osteogenic factors with the ability to induce cartilage and bone formation at ectopic sites [1]. Accumulating evidence thereafter showed that BMPs (of which about 20 members have been identified in mammals) can perform versatile functions in embryonic development and in maintenance of adult tissue homeostasis. BMPs were found to regulate proliferation, survival, migration, differentiation, and lineage commitment of many different cell types [2, 3]. Perturbation in BMP signal transduction processes may lead to disease states, including tumorigenesis [3]. BMPs belong to the transforming growth factor β (TGFβ) superfamily, which are dimeric ligands that signal via specific transmembrane type I and type II serine/threonine kinase receptors and intracellular SMAD transduction factors. Each step of the BMP signaling pathway is carefully regulated, e.g., through ligand-binding proteins that sequester ligand from binding to receptors and coreceptors that present ligand to these receptors [4]. Recent years have seen an increasing interest in the role of BMP signaling in the development and progression of several cancers [5]. Similar as found for TGFβ, BMPs may act as tumor suppressor and/or promoter in a highly contextual manner [5].
BMPs play an important role in the development of embryonic mammary gland [6]. Of interest is also that breast cancer is frequently accompanied by osteolytic metastasis, which accounts for significant morbidity [7]. BMPs are present with high abundance in bone and have the ability to stimulate bone formation [8]. In this review, we aim to overview the recent studies on the relationship between BMPs and breast cancer pathology. After a brief introduction to the key components of BMP signaling pathways and their regulation, we discuss the aberrant expression of canonical BMP/SMAD signaling components and the underlying prognostic value in breast cancer. We then focus on the functions of BMPs in breast cancer initiation, proliferation, apoptosis, tumor microenvironment, as well as the processes of metastasis. The possibilities utilizing these controlling mechanisms of BMPs for therapeutic intervention against breast cancer are also discussed.
2 BMP Signaling and Its Regulation
BMPs are produced as larger dimeric precursor proteins, which are proteolytically processed thereby generating a carboxy-terminal bioactive domain with highly conserved cysteine residues. This mature dimer may undergo further posttranslational modification such as glycosylation [4, 9]. The BMP signaling cascade is initiated by binding of BMPs to two types of transmembrane serine/threonine kinase receptors, i.e., BMP type I and type II receptors (BMPRIs and BMPRIIs, respectively) [10]. Generally, initial binding occurs to BMPRIs, i.e., activin receptor-like kinase (ALK)1, ALK2 (or ACVR1A), ALK3 (or BMPRIA), and ALK6 (or BMPRIB), to which BMPs interact with higher affinity as compared to BMPRIIs. Thereafter, BMPs recruit BMPRII, which is specific for BMPs, or activin type II A receptor (ACVR2A) and activin type IIB receptor (ACVR2B), which are shared type II receptors with the activins (Table 1) [4].
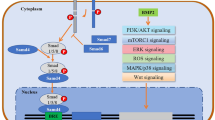
As described in Fig. 1, upon BMP-induced formation of a heteromeric receptor complex, the constitutively active BMPRIIs kinase can phosphorylate BMPRI in the highly conserved glycine-serine-rich (GS) juxtamembrane domain. Then, the activated BMP type I receptor in turn can incur intracellular signaling by phosphorylating specific SMADs (R-SMADs), SMAD1/5/8 [9]. These BMP R-SMADs are distinct from TGFβ and activin receptor-induced R-SMADs, i.e., SMAD2 and SMAD3. Phosphorylated R-SMADs form heteromeric complexes with common-partner SMAD (Co-SMAD), i.e., SMAD4 [11]. Subsequently, these SMAD complexes can translocate into the nucleus where they serve as transcription factors and recognize specific BMP response elements (BRE) (also termed SMAD-binding elements (SBE)) located within the promoters or enhancers of target genes. In collaboration with other transcription factors and transcriptional coactivators/corepressors, they mediate the transcription of BMP target genes, such as inhibitor of differentiation (ID) 1–3, inhibitory SMAD6, and runt-related transcription factor 2 (RUNX2) [12–14]. Besides the canonical SMAD-dependent pathway, BMPs have also been reported to activate non-SMAD pathways, including stress-activated protein kinase/c-Jun NH2-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 mitogen-activated protein kinase (MAPK) pathways, as well as phosphoinositide 3-kinase (PI3K)-AKT, protein kinase C (PKC), TGFβ-activated kinase 1 (TAK1), and small Rho-GTPases pathways [9, 15].

Schematic presentation of the BMP signaling pathway. BMP binds and induces heterotetrameric complex formation of specific single transmembrane-spanning BMP type I and type II receptors. Upon heteromeric complex formation, the extracellular BMP signal is transduced across the membrane by the phosphorylation of BMP type I receptors in the glycine-serine-rich (GS) juxtamembrane domain by the constitutively active type II receptors kinase. The intracellular signal is initiated by the phosphorylation of SMAD1/5/8. These activated R-SMADs can then form heteromeric complexes with SMAD4, which translocate into the nucleus where they collaborate with other DNA-binding transcription factors and transcriptional coactivators/corepressors to regulate the transcription of BMP target genes (SMAD-dependent pathway). The BMP signal can also be transduced via non-SMAD pathways. BMP signaling is subject to multiple regulations, such as extracellular antagonists, coreceptors, membrane promoters/inhibitors, and inhibitory SMAD6/7. There also exists extensive cross talk between BMP signaling pathways and other signaling pathways
The BMP signaling cascade is subject to intricate regulation at multiple levels. Extracellular antagonists prevent binding of BMPs to receptors either by sequestering the BMP ligands or by binding to the BMP receptors themselves [2]. Like the BMP ligands, the BMP antagonists have a cysteine knot structure, which can be divided into several subclasses: twisted gastrulation, Noggin and Chordin family, and differential screening-selected gene aberrative in neuroblastoma (DAN) family (including DAN, Cerberus, Gremlin 1, protein related to Dan or Cerberus (PRDC), Sclerostin, uterine sensitization-associated gene 1 (USAG1), Caronte, and Coco) [9, 16]. Another type of inhibitors involves soluble receptors in the extracellular environment, which also can sequester BMPs from binding to their transmembrane receptors [17]. Regulation at the cell membrane level is mediated by various membrane proteins. The BMP and activin membrane-bound inhibitor (BAMBI) inhibit BMP signaling by interfering with receptor complex formation [18]. In addition, BMP signaling can be potentiated by some membrane proteins, such as members of the repulsive guidance molecule (RGM) family [19], and coreceptors betaglycan [20] and endoglin (CD105) [21, 22].
Within the cell, Endofin acts as an anchor between SMAD1 and activated BMPRIs to facilitate SMAD1 phosphorylation. Meanwhile, Endofin can mediate the dephosphorylation and inactivation of BMPRIs by its motif for protein phosphatase binding [23]. FK506 binding protein 12 (FKBP12) can bind to the GS domain of BMPRIs, thereby shielding the serine and threonine residues from being phosphorylated by BMPRIIs and stabilizing the inactive conformation [24, 25]. The drug FK506 (tacrolimus) that binds FKBP12 was shown to relieve this inhibition and to potentiate BMP signaling [24, 25]. BMP signaling is also restricted intracellularly by the inhibitory SMADs (I-SMADs), i.e., SMAD6 and SMAD7, which compete with SMAD1/5/8 for interaction with BMPRIs and with SMAD4 for complex formation with SMAD1 [26, 27]. Both SMAD1 and SMAD5 can be targeted for proteasomal degradation via addition of ubiquitin chains by SMAD ubiquitin regulatory factors (SMURFs). Additionally, by interacting with I-SMADs that can be recruited to activated BMPRI, SMURFs are also capable of decreasing the stability of BMPRI [28].
Importantly, many of the (negative) regulators of BMP signaling themselves are BMP target genes, creating auto-feedback loops that ensure increased fine-tuning of signaling [2, 28, 29]. Additional facets of BMP signaling include cross talk with other signaling pathways, such as TGFβ, Notch, Janus kinase/signal transducers and activators of transcription (JAK/STAT), Wnt, and Hedgehog, which further broaden the cellular responses to BMP signaling [30]. Thus, the actual outcome of BMP signaling results from levels and activities of all those cellular context-dependent components mentioned above, explaining the diversity of observed effects.
3 Aberrant Expression of BMP Signaling Components in Breast Cancer
In the normal breast, all the necessary components of the canonical BMP signaling pathway (i.e., BMP ligands, BMP receptors, and SMADs) are expressed [31]. Aberrant expression of these components has been observed for breast cancer cell lines with different characteristics and/or has been detected in breast cancer cell lines compared to normal cell lines, in primary tumor tissues compared to normal tissues, and in recurrent tumor tissues compared to primary tumor tissues, however, often with inconsistent and frequent contradictory results. In part, this may be caused by cell lines that were cultured under different conditions and tumors that were not characterized and, for example, not subdivided based upon their genetic alterations and stroma content.
In the forthcoming section, we have listed some examples. Significant lower levels of BMP2 transcript and protein were detected in both noninvasive and invasive breast cancer cell lines and/or cancer cells in breast cancer tissues [31–33]. There were no significant differences in the percentage of BMP2-positive tumors found with respect to cancer cell subtype [31] and grades [33]. What is intriguing, BMP2 protein levels were found to be increased significantly in luminal tumor tissues compared to normal tissues [31]. Immunohistochemical (IHC) staining revealed that BMP2 protein was mainly produced by endothelial cells, fibroblasts, and other stromal cells in luminal tumor microenvironment, not by tumor cells themselves [31]. BMP2 is also highly enriched in bone marrow microenvironment during the process of breast cancer bone metastases [34]. These results indicate that breast tumor cells are the target of BMP2, rather than the source of overexpression.
BMP4 is expressed with wide variation in levels among breast cancer cell lines and/or primary cancer tissues [32, 33, 35–39]. While low levels of BMP4 protein were observed only in normal mammary gland tissue, it was strongly stained in 25 % of patients and more frequent in lobular carcinoma compared to the ductal carcinoma, suggesting that strong expression is cancer specific [39]. Breast cancer patients with strong BMP4 staining suffered from increased frequency of local and distant tumor recurrence [39]. Another study showed that a four-marker panel with low methylation in breast cancer (paired-like homeodomain 2 (PITX2), BMP4, fibroblast growth factor (FGF) 4, and family with sequence similarity 110, member A (FAM110A)) is associated with a longer duration to distant metastasis [36]. However, opposite results were reported in a study by Kretschmer and coworkers indicating that BMP4 mRNA and protein are clearly reduced in ductal carcinoma in situ (DCIS) and invasive ductal carcinoma (IDC) compared to nonmalignant human and murine mammary tissues [40]. A negative correlation between BMP4 mRNA level and tumor grade was reported by Ketolainen et al. [37]. Accordingly, lower BMP4 mRNA expression correlated with poor disease-free survival in breast cancer patients [41].
BMP6 mRNA and/or protein expression was consistently found to be significantly downregulated in breast cancer cell lines or primary cancer tissues [33, 42–46]. Downregulation of BMP6 mRNA correlated with the increase in breast tumor histologic grade [46]. Interestingly, compared to estrogen receptor-positive (ER+) breast cancers, BMP6 mRNA level is significantly higher in estrogen receptor-negative (ER−) breast cancers [43, 45, 46].
BMP7 has been described as being amplified at the gene levels [47, 48] and overexpressed at the mRNA and/or protein levels [33, 47, 49–51] frequently in breast cancer cell lines and/or tissues. BMP7 protein expression was also found to be tumor subtype dependent; 57 % of the lobular carcinomas but only 37 % of the ductal carcinomas are BMP7 positive [50]. Increased BMP7 DNA copy number was reported to show significant correlation with a high Ki67 proliferation index and high histological tumor grade [47]. In addition, BMP7 overexpression was regarded as an independent prognostic marker for early bone metastasis development by multivariate analysis, especially in ductal carcinomas [50]. But contradicting results for BMP7 expression in breast cancer to those just mentioned have also been reported. For example, extreme low levels of BMP7 mRNA were detected in aggressive cells [52, 53]. Moreover, BMP7 mRNA levels in primary breast cancers involving bone metastases were found lower when compared with those involving visceral (lung and liver) metastases [52]. In addition, lower BMP7 levels in patients show a moderate and poor clinical outcome [33].
Relatively few studies have appeared on the expression of other BMP ligands in breast cancer. No difference in BMP3 mRNA levels between breast tumors and normal tissues was detected, but lower BMP3 transcript levels correlated with a poorer prognosis [33]. Lower BMP5 mRNA levels were observed in breast tumors compared to normal breast tissues [54] and correlated with cancer recurrence, particularly in patients with ERα-negative cancers [54]. In contrast, another study showed that patients with higher levels of BMP5 transcript were associated with moderate and poor prognosis [33]. Moreover, decreased expression of BMP9 [55], BMP10 [56], growth and differentiation factor (GDF) 9a [57], GDF-9b/BMP15 [57], and BMP12 [58] along with poor prognosis was observed in breast cancer compared with matched normal tissues.
Investigations into the expression profiles of BMP receptors and downstream SMAD signaling components have been conducted rather infrequently for breast cancer. BMPRIs, BMPRIIs, and SMAD4 and inhibitory SMAD6 and 7 were found expressed rather uniformly in breast cancer cells or tissues [35, 38, 59]. DNA homozygous deletion and mRNA downregulation of BMP receptors are rare in breast cancer according to the provisional breast in The Cancer Genome Atlas (TCGA, Provisional) database [60]. BMPRIA [31, 35, 61] and BMPRIB [31, 35, 62] expressions were found overall increased in tumors compared to normal breast tissues. BMPRIB and BMPRII expression is significantly increased in highly metastatic breast cancer cells [51]. Tissue microarrays demonstrated that high expression of BMPRIA [48, 63] and BMPRII [48] correlated with poor relapse-free survival (RFS) or survival. Strong expression of BMPRIB is associated with high proliferation, cytogenetic instability, high grade, and poor prognosis in ER+ breast cancer [62]. However, the results from Bokobza et al. [64] showed that a decreased level of BMPRIB in breast cancer is associated with poor prognosis.
Only a small portion of breast cancer cell lines and clinical samples were identified as homozygous deletion and reduced mRNA and/or protein expression of SMAD4 [48, 65]. But SMAD4 mutations, which are usually found in pancreatic [66] and colorectal [67] cancer, are rare in breast cancer [65]. Secreted BMP antagonists, such as Gremlin 1 [40, 48, 68, 69], Noggin [31, 48], and Chordin [48], are amplified and/or expressed at higher levels in breast cancer tissues compared to nonmalignant tissues. Of which, Gremlin 1 expression was below detection in breast cancer cells [70] but frequently found expressed in stromal cells within the microenvironment of human breast cancers [68]. In addition, a study conducted by Tarragona et al. indicated that higher levels of Noggin were found in breast cancer bone metastatic tissues compared to lung, brain, and liver metastatic tissues [71].
Taken together, the results of the studies above on the expression of BMP signaling components suggest a highly context-dependent and multifunctional role of BMPs in breast cancer.
4 Status of BMP/SMAD Signaling in Breast Cancer
Even though the expression frequencies and levels of BMPs and other BMP signaling components varied considerably among different studies, human breast cancers and their metastases retain BMP/SMAD signaling [48, 61, 72], as well as several mouse models of breast cancer [61].
Strong phospho-SMAD1/5/8 staining, indicative for active BMP receptor signaling, was demonstrated in human breast cancer tissues [48, 61, 72] and not confined to specific cancer cell types within the tumor tissue [48, 61]. This is consistent with the already mentioned finding that the core BMP canonical signaling components were found to be expressed in breast cancer cells. Metastatic breast cancer to the brain, bone, liver, lymph node, and lung was also found to be positive for phospho-SMAD1/5/8 [48, 72]. Lymph node metastasis tissues were demonstrated to be weaker in phospho-SMAD1/5/8 levels than bone metastasis tissues [72]. Moreover, BMP/SMAD signaling is specifically absent in the stroma of human ductal and lobular carcinoma in situ (DCIS and LCIS). Yet after progression to invasion, breast cancers of many distinct subtypes contained a stroma active for BMP signaling [73].
5 Regulation of the Expression of BMP Signaling Components by Other Factors in Breast Cancer
The expression of BMPs and other pathway components has been shown to be regulated by several other factors, such as estrogen [43, 45, 46, 49], epidermal growth factor (EGF) [49], and p53 [74]. Estrogen represents the primary stimulant in the development and progression of breast cancers. ER status is a determinant for selecting endocrine therapies to block estrogen signaling [75]. A possible relationship between BMP signaling and ER is therefore an interesting area of research. Estrogen has been shown to alter BMP signaling by downregulating specific BMPs and their receptors in ER+ MCF-7 cells, including BMP7, BMPRIA, BMPRIB, ACVR2A, and ACVR2B, but no effect was detected on ACVR1 and BMPRII [59, 76]. In addition, estrogen can suppress BMP2-induced activation of the SMAD pathway and BMP-mediated gene expression [77]. This effect probably depends on the direct physical interaction of SMAD4 with ERα/ERβ [78]. The antiestrogen modulator raloxifene can increase the promoter activity of BMP4 in U2OS osteoblast-like cells in the presence of ERα [79]. In contrast, promoter hypermethylation was found to lead to BMP6 downregulation in ER− breast cancer tissues, while lower methylation frequency was detected in ER+ cases [43, 45, 46, 80]. Moreover, BMP6 gene expression can be upregulated by estrogen-mediated demethylation of the BMP6 promoter in ER+ MCF-7 cells in a dose-dependent manner [81].
Apart from upregulation of BMP2 and BMP6, a derivative of vitamin D can reduce inhibitory SMAD6 expression and enhance SMAD1/5 phosphorylation [82, 83]. EGF treatment can also lead to elevated levels of BMP6 mRNA in a dose-dependent manner [42]. FGF8 was found to inhibit BMP receptor-mediated SMAD1/5/8 phosphorylation and mitigate BMP target gene ID1 promoter activity by suppressing BMPRII expression and by increasing I-SMAD expression [84]. Parathyroid hormone-related protein (PTHrP) can function as the upstream regulator of BMP6 through the protein kinase A (PKA) pathway and exert its anti-mitogenic effect through downregulating BMP6 mRNA expression [85]. Furthermore, BMP7 is a target gene of the p53 family [61, 74] and LIM domain only protein 4 (LMO-4) [86], which activate BMP signaling by inducing the expression of BMP7 in breast cancer.
In short, many different signaling pathways regulate BMP signaling; these findings explain in part the contextual functions of BMPs.
6 BMP Signaling in Stem Cell Self-Renewal and Initiation of Breast Cancer
In human breast cancer, a subpopulation of cancer cells with an ALDHhigh/CD44high/CD24low phenotype is highly enriched for cancer stem cells (CSCs), also termed tumor-initiating cells (TICs), which are capable of initiating and sustaining tumorigenesis [87]. CSCs may be generated from the adult somatic stem cell by disturbing the processes of normal self-renewal or from more differentiated cells through certain processes to reacquire stem cell-like characteristics, such as epithelial to mesenchymal transition (EMT) [87, 88]. BMPs are indispensable for tissue homeostasis in adults, regulating somatic stem cells and controlling differentiation. Aberrant regulation of the BMP signaling pathway could therefore be a target in early phases of tumorigenesis [5].
The evidence points activation of BMP signaling as an early event during primary breast cancer initiation from malignant transformation [31, 48, 61]. Clinically defined samples demonstrate increased BMP signaling in premalignant luminal epithelial cells within the area of DCIS lesions [61]. BMP signaling is also hyperactivated in both epithelium and surrounding stroma in the premalignant mammary gland of transgenic mice model with mouse mammary tumor virus (MMTV)-derived oncogene expression [48, 61]. Chapellier et al. [31] showed that stimulation with BMP2 rapidly induced sustained upregulation of a well-known luminal differentiation regulator, GATA3, and progressive switch of the forkhead box (FOX)A1/FOXC1 balance in favor of FOXA1 through BMPRIB-dependent signaling, thereby leading to differentiation of normal mammary epithelial cell to luminal and expansion of luminal immature progenitors. In addition, abnormal high levels of BMP2 are produced in the mammary microenvironment upon exposure to common carcinogens. Chronic exposure of MCF10A breast epithelial cells to high levels of BMP2 thus initiates transformation of luminal immature progenitor cells toward a luminal tumorlike phenotype in vitro [31].
The small-molecule BMPRIs kinase inhibitor Dorsomorphin and its more selective analogs LDN193189 and DMH1 provide the chance to evaluate the effects of BMP type I receptor signaling on tumorigenesis. In vitro analysis revealed that suppressing BMP signaling in premalignant murine mammary cells or immortalized mammary epithelial cells (IMECs) repressed mammosphere formation [89] and clonogenic capacity and diminishes the CSC-enriched ALDH1high population [61]. Accordingly, the expression of stem markers, spinocerebellar ataxia type 1(SCA1) and NOTCH1, are markedly reduced [89]. Consistently, BMP4 stimulation increased the number and size of primary mammospheres [89]. Thus, BMP signaling is essential for maintenance of CSCs in breast cancer. Importantly, the BMP receptor kinase inhibitor blocks the ability of ALDH1high fraction to resubstitute the mixed ALDH1high/ALDH1low parental culture, implicating that BMP signaling may control the aspects of cellular plasticity within tumor hierarchies [61]. Furthermore, LDN193189 restricts the tumorigenic capacity of allografts and increases tumor latency in vivo [61]. Therefore, these data implicate that BMP signaling is central to regulating mammary epithelial cell stemness, plasticity, and potentially supports maintenance and progression of tumorigenesis.
Interestingly, BMPs also seem to pose a substantial barrier to tumor stemness, when it comes to aggressive and metastatic breast cancers, or rather metastasis-initiating cells. Besides reduced BMP7 expression, an aggressive clone from MCF-7 cell line shows CD44 upregulation and CD24 downregulation, indicative of a CSC phenotype [90]. BMP4 inhibits mammosphere-forming and tumor-initiating ability in IMEC-transformed derivatives with high motility and high percentage of CD44high/CD24low subpopulation [91]. Multiple BMPs (BMP2, BMP7, BMP2/7) decrease the size of ALDHhigh/CD44high/CD24low stem/progenitor subpopulation in MDA-MB-231 [92]. Elevated expression of BMP6 in MDA-MB-231 cells results in decreased tumorigenesis in vivo [93]. Furthermore, colonization of metastatic cancer cells in the target organs is thought of as another type of tumor initiation, while CSCs are commonly considered as the culprits [94]. High-metastatic cells expressing high levels of the BMP antagonist Noggin [71] or Coco [95] are associated with CSCs traits, with the ability to form more tumor spheres and a higher CD44high/CD24low population that display a higher capacity for metastatic colonization. Mechanistically, Coco induces CSC traits of metastatic cells by sustaining the expression of stem cell transcription factors, NANOG, SRY-related HMG-box (SOX) 2, octamer-binding transcription factor (OCT) 4, and transcriptional coactivator TAFAZZIN (TAZ). BMP4 suppresses their expression [95].
Taken together, with respect to CSCs development and tumorigenesis, it can be concluded that BMP signaling can act as promoter of premalignant mammary cells and as suppressor of aggressive mammary cancer cells.
7 Effects of BMPs on Breast Cancer Proliferation and Apoptosis
BMPs have been reported to regulate breast cancer cell growth with context pleiotropy. For the same BMP ligand, the responses can vary within different tumor types. For example, BMP7 was reported to promote cell proliferation of BT-474 and MDA-MB-231 breast cancer cells but to decrease cell proliferation of other breast cancer cell lines (including MDA-MB-361, HCC1954, ZR-75-30, and T-47D) [53]. Even for the same BMP ligand and cell line, different conditions may cause a different response. BMP4 does not have any inhibitory effects on the proliferation of MDA-MB-231 cells in two-dimensional (2D) cell culture but inhibits proliferation in 3D [96]. BMP2 was found to inhibit the hormone-independent growth of MCF-7 in vitro [97–99], but the contrary was reported in vivo [100]. BMP4 and BMP7 have also been shown to promote anchorage-independent MCF-7 cell proliferation [51, 89].
In most of the studies, BMP2 [31, 97–103], BMP4 [31, 37, 96], BMP6 [46, 93, 104], BMP9 [105, 106], and BMP10 [56] were found to trigger cytostatic effects on multiple breast cancer cells. The underlying mechanism could be that BMP signaling has evident effects on the expression of mitotic checkpoint proteins. Chemical inhibition of BMP signaling by BMPRIs kinase inhibitor Dorsomorphin abrogates Nocodazole-mediated mitotic arrest [107]. Simultaneously, levels of mitotic checkpoint proteins, budding uninhibited by benzimidazoles 3 (BUB3), highly expressed protein in cancer (HEC1), monopolar spindle 1 (MPS1), and mitotic arrest deficient 2 (MAD2), which ensures proper chromosome segregation during mitosis, were dramatically downregulated. Overexpressing these proteins significantly recovers the defect in mitotic arrest caused by BMP inhibition [107]. Some of BMPs are demonstrated to delay cell cycle reentry in breast cancer cells. BMP2 [99, 102, 108, 109], BMP4 [37, 96], and BMP6 [46, 93, 104] induce G1 cell cycle arrest caused by increased expression of the cell cycle inhibitor p21 [96, 99, 102, 108, 109]. p21 promoter activity in turn inactivates cyclin D1 and cyclin E and results in retinoblastoma protein (pRb) hypophosphorylation [101]. The process of cell cycle arrest requires active BMPRIs, and the cytoplasmic signal transducers SMAD1/5 and SMAD4 are indispensable [102]. Upregulation of protein tyrosine phosphatases (PTPs), such as protein tyrosine phosphatase gamma (PTPRG), MAPK phosphatase (MKP), and phosphatase and tensin homolog (PTEN), may also contribute to increased levels of p21 in cells where BMP induced antiproliferative effects [110, 111]. In addition, BMP7 [84] and BMP9 [105] can lead to an accumulation of the G2/M phase in breast cancer cells.
BMPs can also influence the effect of other factors on breast cancer cell proliferation. BMP4 itself cannot significantly stimulate the proliferation but potently enhances the mitogenic activity of EGF, FGF, and hepatocyte growth factor (HGF) on murine mammary epithelial cells [112]. BMP2, in contrast to BMP4, prevents EGF-induced proliferation of MDA-MB-231 cells [108]. The estrogen-induced mitotic effects can be suppressed by BMP2 [59, 101], BMP4 [59], BMP6 [59], and BMP7 [59, 84], with the effects of BMP6 and BMP7 being more potent than those of BMP2 and BMP4 [59]. AB215, an activin A/BMP2 chimera, has increased BMP2-like signaling potency via the SMAD1/5/8 pathway and exerts stronger inhibitory effects on estradiol-induced proliferation in ER+ breast cancer cells than BMP2 [113]. Estradiol rapidly activates MAPK phosphorylation including ERK1/2, p38, and JNK pathways [59, 84]. BMP6 and 7 can preferentially inhibit estradiol-induced p38 phosphorylation [59]. BMP6 is also believed to decrease the chemoresistance of MCF-7 breast cancer cells to doxorubicin through inactivation of ERK signaling and upregulation of P-glycoprotein (P-GP) [46]. Furthermore, BMP9 can inhibit expression of HER2, phosphorylation of ERK1/2 (without effect on p38 and JNK), and PI3K/AKT in SK-BR-3 cells, thereby suppressing the growth of HER2-positive SK-BR-3 cells in vitro and in vivo [106].
Obviously, the distinct BMP receptors present also explain the diversity of effects of BMP signaling on breast cancer proliferation. BMPRIA was identified as a positive regulator of breast cancer at primary and secondary sites through activation of the SMAD pathway [72]. In contrast, another type I receptor, BMPRIB, plays a negative role in the proliferation of breast cancer cells. Downregulation of BMPRIB in MDA-MB-231 cells leads to promotion of cell growth in vitro [64]. Overexpression of a BMPRII-dominant negative (DN) mutant interferes with the phosphorylation of SMAD1, resulting in G1 phase cell cycle arrest of T-47D cells [109]. However, in the MMTV polyoma middle T antigen mice model of spontaneous mammary tumor formation, BMPRII-DN-expressing tumor cells have higher proliferation rates [114].
A few studies have pointed out pro-apoptotic roles for BMPs in breast cancer cells [86, 99, 105, 115]. BMP2 regulates the expression of apoptosis-related genes, especially protein kinase R (PKR) and activates its substrate α-subunit of eukaryotic initiation factor 2, thereby showing a pro-apoptotic effect in MCF-7 cells under normal culture conditions [115]. However, when these cells are deprived of serum, BMPs display a contrasting function by exerting an anti-apoptotic effect. BMP2 increases the resistance to hypoxia-induced apoptosis in MCF-7 cells via activation of the MAPK and ID1 pathways and suppression of caspase-3 [116, 117]. In parallel, BMP6, which can inhibit the proliferation of MDA-MB-231 cells, inhibits serum starvation-induced apoptosis through SMAD-dependent upregulation of Survivin and non-SMAD-dependent activation of p38 MAPK [104].
8 BMPs and the Tumor Microenvironment
Accumulating evidence indicates that the tumor microenvironment is a pathologically active niche that shapes tumor evolution. Hypoxia, low pH, immune evasion, chronic inflammation, and neovasculature can be considered as enabling characteristics [118]. Disruption of BMP signaling brings about alterations in the breast tumor microenvironment and accelerates tumor progression [41, 114, 119]. Deletion of BMPRII in mammary tumors [114] or in fibroblasts within the tumor stroma [119] can result in increased expression of chemokines, such as chemokine (C-C motif) ligand 5 and 9 (CCL5, 9), interferon gamma-induced protein 10 (IP-10), and granulocyte colony-stimulating factor (G-CSF), which facilitate inflammation by a sustained increase of myeloid cells infiltration, especially myeloid-derived suppressor cells (MDSCs) [114, 119]. Accordingly, the T-cell population is reduced due to a main function of MDSCs in the inhibition of T-cell proliferation [114]. As a classical stress response pathway, nuclear factor-κB (NF-κB) activation can be detected in a majority of cancers [120]. BMP4 has been shown to attenuate NF-κB activity in breast cancer [41]. Thereby lower levels of chemokines result from the attenuation of its known regulator NF-κB, leading to reduced numbers and immunosuppressive activity of MDSCs [41, 114]. Meanwhile, increased T-cell populations are observed within stromal tissues, and many immune-related genes are significantly upregulated by BMP4, indicating BMP4 triggers an enhanced antitumor immune response [41]. Therefore, it can be concluded that BMP signaling could inhibit inflammatory infiltrates and tumor progression through suppressing an inflammatory chemokine profile in tumor microenvironment.
Intriguingly, BMP signaling could also induce a series of cytokines which trigger CAF-mediated pro-tumorigenic stimulation on epithelial cells directly. BMP4 treatment of normal mammary fibroblasts or carcinoma-associated mammary fibroblasts (CAFs) induces an increase in secreted matrix metalloproteases (MMPs) and pro-inflammatory cytokines, which enhance mammary carcinoma cell invasion [73, 121]. Furthermore, inhibition of BMP signaling alters fibroblasts, macrophages, and lymphatic vessels to be less tumor promoting in vivo [48].
It has been reported that BMPs can promote endothelial cell (EC) proliferation and migration [122]. Consistent with this notion, BMP signaling is required for appropriate angiogenesis [123]. BMP2 promotes vascularization by stimulating the ID1 and p38 MAPK pathways. Overexpression of BMP2 in MCF-7 cells induces vascularized tumors eventually upon injection in vivo [124]. The signaling mediated by BMP type I receptor ALK1 has a critical role in regulation of both developmental and pathologic blood vessel formation [125]. ALK1 is mainly expressed at the sites of angiogenesis during embryogenesis and is expressed at lower levels in adult vasculature. Yet its expression increases in neoangiogenic vessels of wounds and cancer [125]. BMP9 binds to ALK1 in ECs with high affinities [126]. There have been divergent results with respect to the effects of BMP9/ALK1 signaling on ECs. Some reports demonstrate that high-dose BMP9/ALK1 signaling exhibits antiangiogenic effects, by inhibiting FGF-induced angiogenesis [127, 128], while other reports have shown induction of proliferation by low dose of BMP9 in several types of ECs and proangiogenic effects of BMP9 in Matrigel plug assays [129, 130]. The apparent discrepancy between these reports might reflect the contextual function of BMPs, in which the concentration plays an important role. In addition, common proangiogenic factors (VEGF-A and bFGF) can stimulate ALK1-mediated BMP/SMAD-like signaling, leading to cell spreading, and tubulogenesis of ECs [131]. Inhibition of ALK1 signaling by gene silencing, ligand traps, or antibodies can significantly suppress the growth and progression of tumors, including breast cancer, with substantial reduction of angiogenesis, supporting the notion that ALK1 is an important target for antiangiogenic treatment [131, 132].
9 Roles of BMPs in the Migration, Invasion, and Metastasis of Breast Cancer
It is clear that BMPs and their receptors modulate key pathways mediating breast cancer cell invasion and migration, critical parameters of metastatic dissemination. But the conclusions also seem paradoxical, indicating dependence on particular cell types and contexts.
9.1 BMPs and EMT
The development of metastasis involves the replacement with new phenotypes in cancer cells to facilitate detachment from the primary site [133]. Many epithelial cancer cells can acquire sufficient phenotypic plasticity by EMT, which implies the conversion of a proliferative epithelial state into nonproliferative mesenchymal state with the ability to migrate and invade adjacent tissue [134]. Restriction in BMP signaling level is frequently needed for efficient EMT [54, 91, 135]. Significant downregulation of some BMPs and upregulation of two secreted BMP antagonists, Chordin-like (CHRDL) 2 and Gremlin, were observed when human mammary epithelial cells pass through an EMT [91]. A subsequent study showed that the transcription factor zinc finger E-box-binding homeobox 1 (ZEB1) which mediates EMT can directly upregulate the expression of the BMPs antagonists Noggin, Follistatin, and CHRDL1 [135]. Likewise, a newly identified EMT pathway mediated by the transcriptional repressor Blimp-1 (PRDM1) leads to SNAIL induction via repression of BMP5 [54]. Of note, during acquisition of metastatic ability, EMT in mammary cells is strongly correlated with a CD44high/CD24low stem cell phenotype [90, 91, 136]. These studies thus support a mechanistic link between BMP downregulation, EMT, and stem cell signature in cancer.
In addition, some BMPs are capable of reversing EMT or EMT markers in breast cancer cells [52, 80, 137]. E-cadherin-mediated cell-to-cell adhesion can be restored through inhibition of ZEB1 by BMP6 in breast cancer cells [44, 137, 138]. Stimulation with exogenous BMP7, which can decrease vimentin and increase cytokeratin expression in vitro and in vivo, gives rise to an epithelial-like phenotype [52]. BMPs can also oppose EMT inducers, e.g., TGFβ, in normal mammary epithelial cells or IMECs [54, 91, 139–142] and in breast cancer cells [52, 92, 140]. For example, the loss of E-cadherin expression on the surface of NMuMG cells in response to TGFβ1 is largely overridden by BMP5, and the fibroblastoid phenotype is also substantially reversed [54]. BMP7 has also been shown to reverse TGFβ-induced EMT [139–141], which increases E-cadherin expression through upregulation of ID2 and ID3. Interestingly, when knocking down ID2 or ID3, BMP7 actually induces the expression of α-smooth muscle actin (αSMA) and stimulates EMT [140, 141]. Thus, BMP signaling impedes the progression of breast cancer to an invasive state and prevents metastasis in the aforementioned studies. However, the BMP pathway was found to maintain a mesenchymal stem cell phenotype of breast cancer cells and render cells more migratory, invasive in other in vitro [89, 143, 144] and in vivo [61, 143] studies. BMP2 transforms MCF-7 cells from a round-like shape into a spindle-like shape with some specialized structures, such as filopodia, lamellipodia, and membrane protrusions, which are essential for cell migration and spreading [100, 144]. BMP4 blocks the capacity of mammary epithelial cells to form polarized lumen-containing structures and renders them invasive properties [145]. Of note, in 4T1.2 cells expressing BMP4, genes associated with EMT are upregulated but no change was observed in their migratory capacity [41].
9.2 BMPs and Components of the Extracellular Matrix (ECM)
EMT is not an “all-or-nothing” event; it’s highly dynamic. Studies have shown that BMPs induce MMP-dependent migration and invasion of breast cancer [48, 96, 121]. MMPs are known for degrading surrounding ECM components during cancer invasion and metastasis [146]. Treatment of primary tumors with BMPRI kinase inhibitor DMH1 reduced MMP2 and CCL9 in CAFs [48]. BMP4 induces the expression of multiple MMPs in mouse mammary fibroblasts and in cancer-associated human mammary fibroblasts [121] and dramatically increases MMP3 and MMP4 expression in 3D-cultured MDA-MB-231 cells [96]. However, another study showed that BMP4 suppresses the activity of MMP9 in 2D culture, rather than MMP1 and MMP3 [147]. Moreover, BMP6 was found to inhibit MMP9 activation via SMAD-dependent induction of heme oxygenase 1 (HO1) in MCF-7 cells [148]. BMP9 can inhibit MMP9 by inhibiting the AKT signaling pathway [106, 149].
ECM-associated protein Wnt1-inducible secreted protein 3 (WISP-3/CCN6) binds directly to BMP4 to antagonize BMP4-induced SMAD-independent activation of TAK1/p38 kinases, decreases the invasiveness of breast cancer cells in 3D, and also reduces distant metastasis in xenografts [143]. In contrast, the expression of ECM proteins tenascin-W, which can promote the motility of breast cancer cells expressing α8 integrin, is induced by BMP2-mediated p38 MAPK and JNK signaling pathways [150].
9.3 Interplay Between BMPs and TGFβ
Apart from EMT as previously mentioned, other features of cancer cells such as migration and invasion are also affected by a mutual antagonism between BMPs and TGFβ. Overexpression of type III TGFβ receptor inhibited BMP-mediated SMAD1/5/8 phosphorylation and BMP-induced migration [151]. BMP7 treatment significantly increases migration and invasion in MDA-MB-231 cells [53, 152]. This effect is substantially inhibited by costimulation with TGFβ by inducing the formation of complexes involving phosphorylated SMAD1/5 and SMAD3 [152]. Moreover, BMP2-mediated upregulation of ID1 may be a contributing factor in BMP2-related aggressiveness of breast cancer cells. Aberrant activation of SRC kinase resulting in increased SMAD1/5 signaling can change ID1 expression, which is positively controlled via SMAD1/5 by BMP2 and negatively via SMAD2/3 by TGFβ [153]. Conversely, BMP7 inhibits TGFβ-induced expression of αvβ3 integrin and invasion of the metastatic breast cancer cell line MCF-10CA1a in a spheroid model [154].
10 BMPs and Metastasis
Common sites of metastatic dissemination, such as the bone and lung, are the main targets of metastatic breast cancer [7]. In the process of bone metastasis, breast cancer triggers predominantly an osteoclast-mediated osteolytic lesion [155]. BMP signaling is shown to shift the osteoblast/osteoclast differentiation balance in favor of stimulating osteoblast differentiation [70, 71, 156]. By inactivating BMP signaling, BMP antagonists, such as Noggin, Follistatin, and CHRDL1, have been linked to the induction of osteoclast differentiation, as well as the formation of osteolytic bone metastases [71, 135, 156]. Lack of Noggin expression by breast cancer cells is a determinant of osteoblastic activities [70]. In an intracardiac xenograft model, evidence was found that Noggin is expressed in metastatic breast cancer cells during the late events of metastasis. In particular, it facilitates the metastatic capabilities of breast cancer cells to the bone by promoting osteoclast differentiation and bone degradation [71].
In contrast, when MCF-7 or MDA-MB-231 cells are cocultured with osteoblast-like cells, Noggin effectively inhibits migration and invasion of breast cancer cells by downregulating MMP1 and CXCR4 and improves bone remodeling by increasing the ratio of osteoprotegerin (OPG)/nuclear factor kappa B ligand (RANKL) [38]. The BMP target gene and cofactor RUNX2 are required for breast cancer osteolytic metastases [157, 158]. miR-135 impairs the BMP-RUNX2 axis by directly targeting SMAD5 and subsequently reduces the osteolytic properties of breast cancer cells [158]. Likewise, expression of dominant-negative receptors (DN-ALK3) for BMPs reduces interleukin-11 (IL-11) expression and inhibits bone metastasis in xenograft model [72].
As for individual BMP, BMP9, which is one of the most effective BMPs in osteogenesis, can inhibit osteolytic injury and bone metastasis caused by MDA-MB-231 cells by downregulating PTHrP, IL6, RANKL, and connective tissue growth factor (CTGF) [55, 149]. BMP2, 7, and 2/7 heterodimer inhibits bone metastases formation in MDA-MB-231 cells [52, 92]. Contradicting results showed that BMP7 overexpression could lead to accelerated bone metastasis formation of breast cancer cells [50, 51, 53].
BMP signaling can also prevent the colonization of metastatic cells in the lung by repressing key CSCs traits and enforcing cancer cells into dormancy. Overexpression of the BMP antagonist Coco permits a few dormant cancer cells to break through the barrier imposed by BMP signaling and to establish clinically meaningful metastases [95].
11 Conclusions and Perspectives
As discussed above, there are conflicting views regarding the significance of BMPs in breast cancer, based both on in vitro and in vivo studies. This has been attributed to multiple factors, including the (dose- and context-dependent) differential effects of different BMP ligands and differences in the genetic patterns of breast cancer subtypes, as well as differences in the research models that were used. Most results are obtained using only a few types of cancer cell lines or single and different animal models and are therefore difficult to compare to each other. What is clear is that BMPs are emerging as key factors in many aspects of breast cancer. Aberrant changes in BMP signaling/components have been detected in breast cancer and metastatic recurrence and have deepened our understanding of the pathogenesis of breast cancer. The majority of studies indicate that BMP signaling is a critical negative regulator in multiple breast cancer cell lines both in vitro and in vivo. Restoration or amplification of specific aspects of BMP signaling may be potentially exploited for therapeutic intervention strategies.
To this point, context is critical. For instance, even an agonist or coactivator with precisely delivered BMP signaling input will not make any contribution to overcome the shortages that derive from functional deficiency of BMP receptors or any critical downstream components. It is therefore necessary to identify more potential targets or markers of the specific signaling defect(s). This might be pursued by using the latest types of high-throughput (epi)genetic, proteomic, and metabolomic analysis to systematically investigate the BMP responses to multiple cell types of the different breast cancer subclasses and/or patient-derived (organoid) (co)cultures grown in 3D and investigating the effect of misexpression of BMP receptor components or pharmacological inhibition of BMP receptor signaling in relevant transgenic mouse models and patient-derived xenografts with clear classification of histological pathology. This may provide effective principles to better illuminate the context-dependent roles of BMP family signaling in breast cancer. Via these approaches the opportunities for pharmacological intervention to rectify aberrant BMP family signaling in specific contexts are likely to be increased.
References
Urist MR (1965) Bone: formation by autoinduction. Science 150:893–899
Brazil DP, Church RH, Surae S, Godson C, Martin F (2015) BMP signalling: agony and antagony in the family. Trends Cell Biol 25:249–264
Wang RN, Green J, Wang Z, Deng Y, Qiao M, Peabody M, Zhang Q, Ye J, Yan Z, Denduluri S (2014) Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis 1:87–105
Miyazono K, Kamiya Y, Morikawa M (2010) Bone morphogenetic protein receptors and signal transduction. J Biol Chem 147:35–51
Ehata S, Yokoyama Y, Takahashi K, Miyazono K (2013) Bi-directional roles of bone morphogenetic proteins in cancer: another molecular Jekyll and Hyde? Pathol Int 63:287–296
Robinson GW (2008) Cooperation of signalling pathways in embryonic mammary gland development. Nat Rev Genet 9:566–566
Lorusso G, Rüegg C (2012) New insights into the mechanisms of organ-specific breast cancer metastasis. Semin Cancer Biol 22:226–233
Long F (2012) Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol 13:27–38
Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, Nohe A (2011) Bone morphogenetic proteins: a critical review. Cell Signal 23:609–620
Wakefield LM, Hill CS (2013) Beyond TGFβ: roles of other TGFβ superfamily members in cancer. Nat Rev Cancer 13:328–341
Heldin CH, Miyazono K, Ten Dijke P (1997) TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 390:465–471
López-Rovira T, Chalaux E, Massagué J, Rosa JL, Ventura F (2002) Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. J Biol Chem 277:3176–3185
Ishida W, Hamamoto T, Kusanagi K, Yagi K, Kawabata M, Takehara K, Sampath TK, Kato M, Miyazono K (2000) Smad6 is a Smad1/5-induced Smad inhibitor characterization of bone morphogenetic protein-responsive element in the mouse Smad6 promoter. Trends Cell Biol 20:244–256
Lee KS, Kim HJ, Li QL, Chi XZ, Ueta C, Komori T, Wozney JM, Kim EG, Choi JY, Ryoo HM (2000) Runx2 is a common target of transforming growth factor β1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol Cell Biol 20:8783–8792
Zhang YE (2009) Non-Smad pathways in TGF-β signaling. Cell Res 19:128–139
Walsh DW, Godson C, Brazil DP, Martin F (2010) Extracellular BMP-antagonist regulation in development and disease: tied up in knots. Trends Cell Biol 20(5):244–256
Singhatanadgit W, Salih V, Olsen I (2006) Shedding of a soluble form of BMP receptor-IB controls bone cell responses to BMP. Bone 39:1008–1017
Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massague J, Niehrs C (1999) Silencing of TGF-β signalling by the pseudoreceptor BAMBI. Nature 401:480–485
Halbrooks PJ, Ding R, Wozney JM, Bain G (2007) Role of RGM coreceptors in bone morphogenetic protein signaling. J Mol Signal 2:4
Kirkbride KC, Townsend TA, Bruinsma MW, Barnett JV, Blobe GC (2008) Bone morphogenetic proteins signal through the transforming growth factor-β type III receptor. J Biol Chem 283:7628–7637
Scherner O, Meurer SK, Tihaa L, Gressner AM, Weiskirchen R (2007) Endoglin differentially modulates antagonistic transforming growth factor-β1 and BMP-7 signaling. J Biol Chem 282:13934–13943
Alt A, Miguel-Romero L, Donderis J, Aristorena M, Blanco FJ, Round A, Rubio V, Bernabeu C, Marina A (2012) Structural and functional insights into endoglin ligand recognition and binding. PLoS One 7:e29948
Shi W, Chang C, Nie S, Xie S, Wan M, Cao X (2007) Endofin acts as a Smad anchor for receptor activation in BMP signaling. J Cell Sci 120:1216–1224
Kugimiya F, Yano F, Ohba S, Igawa K, Nakamura K, Kawaguchi H, Chung UI (2005) Mechanism of osteogenic induction by FK506 via BMP/Smad pathways. Biochem Biophys Res Commun 338:872–879
Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El-Bizri N, Sawada H, Haghighat R, Chan R (2013) FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 123:3600–3613
Ten Dijke P, Goumans MJ, Itoh F, Itoh S (2002) Regulation of cell proliferation by Smad proteins. J Cell Physiol 191:1–16
Massagué J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19:2783–2810
Itoh S, ten Dijke P (2007) Negative regulation of TGF-β receptor/Smad signal transduction. Curr Opin Cell Biol 19:176–184
Massagué J, Chen YG (2000) Controlling TGF-β signaling. Genes Dev 14:627–644
Guo X, Wang XF (2009) Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res 19:71–88
Chapellier M, Bachelard-Cascales E, Schmidt X, Clément F, Treilleux I, Delay E, Jammot A, Ménétrier-Caux C, Pochon G, Besançon R (2015) Disequilibrium of BMP2 levels in the breast stem cell niche launches epithelial transformation by overamplifying BMPR1B cell response. Stem Cell Rep 4:239–254
Reinholz MM, Iturria SJ, Ingle JN, Roche PC (2002) Differential gene expression of TGF-β family members and osteopontin in breast tumor tissue: analysis by real-time quantitative PCR. Breast Cancer Res Treat 74:255–269
Davies SR, Watkins G, Douglas-Jones A, Mansel RE, Jiang WG (2007) Bone morphogenetic proteins 1 to 7 in human breast cancer, expression pattern and clinical/prognostic relevance. J Exp Ther Oncol 7:327–338
Zhang XHF, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, Foekens JA, Massagué J (2009) Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 16:67–78
Alarmo EL, Kuukasjärvi T, Karhu R, Kallioniemi A (2007) A comprehensive expression survey of bone morphogenetic proteins in breast cancer highlights the importance of BMP4 and BMP7. Breast Cancer Res Treat 103:239–246
Hartmann O, Spyratos F, Harbeck N, Dietrich D, Fassbender A, Schmitt M, Eppenberger-Castori S, Vuaroqueaux V, Lerebours F, Welzel K (2009) DNA methylation markers predict outcome in node-positive, estrogen receptor-positive breast cancer with adjuvant anthracycline-based chemotherapy. Clin Cancer Res 15:315–323
Ketolainen JM, Alarmo EL, Tuominen VJ, Kallioniemi A (2010) Parallel inhibition of cell growth and induction of cell migration and invasion in breast cancer cells by bone morphogenetic protein 4. Breast Cancer Res Treat 124:377–386
Guo D, Huang J, Gong J (2012) Bone morphogenetic protein 4 (BMP4) is required for migration and invasion of breast cancer. Mol Cell Biochem 363:179–190
Alarmo EL, Huhtala H, Korhonen T, Pylkkänen L, Holli K, Kuukasjärvi T, Parkkila S, Kallioniemi A (2013) Bone morphogenetic protein 4 expression in multiple normal and tumor tissues reveals its importance beyond development. Mod Pathol 26:10–21
Kretschmer C, Conradi A, Kemmner W, Sterner-Kock A (2011) Latent transforming growth factor binding protein 4 (LTBP4) is downregulated in mouse and human DCIS and mammary carcinomas. Cell Oncol 34:419–434
Cao Y, Slaney CY, Bidwell BN, Parker BS, Johnstone CN, Rautela J, Eckhardt BL, Anderson RL (2014) BMP4 inhibits breast cancer metastasis by blocking myeloid-derived suppressor cell activity. Cancer Res 74:5091–5102
Clement JH, Sänger J, Höffken K (1999) Expression of bone morphogenetic protein 6 in normal mammary tissue and breast cancer cell lines and its regulation by epidermal growth factor. Int J Cancer 80(2):250–256
Zhang M, Wang Q, Yuan W, Yang S, Wang X, Yan JD, Du J, Yin J, Gao SY, Sun BC (2007) Epigenetic regulation of bone morphogenetic protein-6 gene expression in breast cancer cells. J Steroid Biochem Mol Biol 105(1):91–97
Du J, Yang S, An D, Hu F, Yuan W, Zhai C, Zhu T (2009) BMP-6 inhibits microRNA-21 expression in breast cancer through repressing δEF1 and AP-1. Cell Res 19(4):487–496
Barekati Z, Radpour R, Lu Q, Bitzer J, Zheng H, Toniolo P, Lenner P, Zhong XY (2012) Methylation signature of lymph node metastases in breast cancer patients. BMC Cancer 12:244
Lian WJ, Liu G, Liu YJ, Zhao ZW, Yi T, Zhou HY (2013) Downregulation of BMP6 enhances cell proliferation and chemoresistance via activation of the ERK signaling pathway in breast cancer. Oncol Rep 30:193–200
Alarmo EL, Rauta J, Kauraniemi P, Karhu R, Kuukasjärvi T, Kallioniemi A (2006) Bone morphogenetic protein 7 is widely overexpressed in primary breast cancer. Genes Chromosomes Cancer 45:411–419
Owens P, Pickup MW, Novitskiy SV, Giltnane JM, Gorska AE, Hopkins CR, Hong CC, Moses HL (2014) Inhibition of bmp signaling suppresses metastasis in mammary cancer. Oncogene 34:2437–2449
Schwalbe M, Sänger J, Eggers R, Naumann A, Schmidt A, Höffken K, Clement JH (2003) Differential expression and regulation of bone morphogenetic protein 7 in breast cancer. Int J Oncol 23:89–95
Alarmo EL, Korhonen T, Kuukasjärvi T, Huhtala H, Holli K, Kallioniemi A (2007) Bone morphogenetic protein 7 expression associates with bone metastasis in breast carcinomas. Ann Oncol 19:308–314
Sakai H, Furihata M, Matsuda C, Takahashi M, Miyazaki H, Konakahara T, Imamura T, Okada T (2012) Augmented autocrine bone morphogenic protein (BMP) 7 signaling increases the metastatic potential of mouse breast cancer cells. Clin Exp Metastasis 29:327–338
Buijs JT, Henriquez NV, Van Overveld PG, Van der Horst G, Que I, Schwaninger R, Rentsch C, Ten Dijke P, Cleton-Jansen AM, Driouch K (2007) Bone morphogenetic protein 7 in the development and treatment of bone metastases from breast cancer. Cancer Res 67:8742–8751
Alarmo EL, Pärssinen J, Ketolainen JM, Savinainen K, Karhu R, Kallioniemi A (2009) BMP7 influences proliferation, migration, and invasion of breast cancer cells. Cancer Lett 275:35–43
Romagnoli M, Belguise K, Yu Z, Wang X, Landesman-Bollag E, Seldin DC, Chalbos D, Barillé-Nion S, Jézéquel P, Seldin ML (2012) Epithelial-to-mesenchymal transition induced by TGF-β1 is mediated by Blimp-1–dependent repression of BMP-5. Cancer Res 72:6268–6278
Ren W, Sun X, Wang K, Feng H, Liu Y, Fei C, Wan S, Wang W, Luo J, Shi Q (2014) BMP9 inhibits the bone metastasis of breast cancer cells by downregulating CCN2 (connective tissue growth factor, CTGF) expression. Mol Biol Rep 41:1373–1383
Ye L, Bokobza S, Li J, Moazzam M, Chen J, Mansel RE, Jiang WG (2010) Bone morphogenetic protein-10 (BMP-10) inhibits aggressiveness of breast cancer cells and correlates with poor prognosis in breast cancer. Cancer Sci 101:2137–2144
Hanavadi S, Martin T, Watkins G, Mansel R, Jiang W (2007) The role of growth differentiation factor-9 (GDF-9) and its analog, GDF-9b/BMP-15, in human breast cancer. Ann Surg Oncol 14:2159–2166
Li J, Ye L, Parr C, Douglas-Jones A, Kyanaston H, Mansel RE, Jiang WG (2009) The aberrant expression of bone morphogenetic protein 12 (BMP-12) in human breast cancer and its potential prognostic value. Gene Ther Mol Biol 13:186–193
Takahashi M, Otsuka F, Miyoshi T, Otani H, Goto J, Yamashita M, Ogura T, Makino H, Doihara H (2008) Bone morphogenetic protein 6 (BMP6) and BMP7 inhibit estrogen-induced proliferation of breast cancer cells by suppressing p38 mitogen-activated protein kinase activation. J Endocrinol 199(3):445–455
Network CGA (2012) Comprehensive molecular portraits of human breast tumours. Nature 490:61–70
Balboni AL, Hutchinson JA, DeCastro AJ, Cherukuri P, Liby K, Sporn MB, Schwartz GN, Wells WA, Sempere LF, Paul BY (2013) ΔNp63α-mediated activation of bone morphogenetic protein signaling governs stem cell activity and plasticity in normal and malignant mammary epithelial cells. Cancer Res 73:1020–1030
Helms MW, Packeisen J, August C, Schittek B, Boecker W, Brandt BH, Buerger H (2005) First evidence supporting a potential role for the BMP/SMAD pathway in the progression of oestrogen receptor-positive breast cancer. J Pathol 206:366–376
Hover LD, Pickup MW, Gorska AE, Chytil A, Guo Y, Novitskiy SV, Moses HL, Owens P (2015) Deletion of the BMP receptor BMPR1a results in EMT and impairs mammary gland tumor formation and metastasis. Cancer Res 75:4083–4083
Bokobza SM, Ye L, Kynaston HE, Mansel RE, Jiang WG (2009) Reduced expression of BMPR-IB correlates with poor prognosis and increased proliferation of breast cancer cells. Cancer Genom Proteom 6:101–108
Zhong D, Morikawa A, Guo L, Colpaert C, Xiong L, Nassar A, Chen C, Lamb N, Dong J-T, Zhou W (2006) Homozygous deletion of SMAD4 in breast cancer cell lines and invasive ductal carcinomas. Cancer Biol Ther 5:601–607
Valero V III, Saunders TJ, He J, Weiss MJ, Cameron JL, Dholakia A, Wild AT, Shin EJ, Khashab MA, O’Broin-Lennon AM (2015) Reliable detection of somatic mutations in fine needle aspirates of pancreatic cancer with next-generation sequencing. Ann Surg 263:153–161
Voorneveld PW, Kodach LL, Jacobs RJ, Liv N, Zonnevylle AC, Hoogenboom JP, Biemond I, Verspaget HW, Hommes DW, de Rooij K (2014) Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology 147:196–208
Sneddon JB, Zhen HH, Montgomery K, van de Rijn M, Tward AD, West R, Gladstone H, Chang HY, Morganroth GS, Oro AE (2006) Bone morphogenetic protein antagonist gremlin 1 is widely expressed by cancer-associated stromal cells and can promote tumor cell proliferation. Proc Natl Acad Sci 103:14842–14847
Ma X-J, Dahiya S, Richardson E, Erlander M, Sgroi DC (2009) Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res 11:R7
Schwaninger R, Rentsch CA, Wetterwald A, van der Horst G, van Bezooijen RL, van der Pluijm G, Löwik CW, Ackermann K, Pyerin W, Hamdy FC (2007) Lack of noggin expression by cancer cells is a determinant of the osteoblast response in bone metastases. Am J Pathol 170:160–175
Tarragona M, Pavlovic M, Arnal-Estapé A, Urosevic J, Morales M, Guiu M, Planet E, González-Suárez E, Gomis RR (2012) Identification of NOG as a specific breast cancer bone metastasis-supporting gene. J Biol Chem 287:21346–21355
Katsuno Y, Hanyu A, Kanda H, Ishikawa Y, Akiyama F, Iwase T, Ogata E, Ehata S, Miyazono K, Imamura T (2008) Bone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through Smad pathway. Oncogene 27:6322–6333
Owens P, Polikowsky H, Pickup MW, Matise LA, Gorska AE, Shaw AK, Novitskiy SV, Aakre ME, Hong CC, Moses HL (2012) Bone morphogenetic proteins stimulate mammary fibroblasts to promote mammary tumorigenesis. Cancer Res 72:1500–1500
Yan W, Chen X (2007) Targeted repression of bone morphogenetic protein 7, a novel target of the p53 family, triggers proliferative defect in p53-deficient breast cancer cells. Cancer Res 67:9117–9124
Huang B, Warner M (2015) Gustafsson J-Å (2014) Estrogen receptors in breast carcinogenesis and endocrine therapy. Mol Cell Endocrinol 418:240–244
Kusumegi T, Tanaka J, Kawano M, Yonemoto J, Tohyama C, Sone H (2004) BMP7/ActRIIB regulates estrogen-dependent apoptosis: New biomarkers for environmental estrogens. J Biochem Mol Toxicol 18:1–11
Yamamoto T, Saatcioglu F, Matsuda T (2002) Cross-talk between bone morphogenic proteins and estrogen receptor signaling. Endocrinology 143:2635–2642
Páez-Pereda M, Giacomini D, Refojo D, Nagashima AC, Hopfner U, Grübler Y, Chervin A, Goldberg V, Goya R, Hentges ST (2003) Involvement of bone morphogenetic protein 4 (BMP-4) in pituitary prolactinoma pathogenesis through a Smad/estrogen receptor crosstalk Proc Natl Acad Sci 100: 1034–1039
Van den Wijngaard A, Mulder W, Dijkema R, Boersma C, Mosselman S, van Zoelen E, Olijve W (2000) Antiestrogens specifically up-regulate bone morphogenetic protein-4 promoter activity in human osteoblastic cells. Mol Endocrinol 14:623–633
Liu G, Liu YJ, Lian WJ, Zhao ZW, Yi T, Zhou HY (2014) Reduced BMP6 expression by DNA methylation contributes to EMT and drug resistance in breast cancer cells. Oncol Rep 32:581–588
Zhang M, Yan JD, Zhang L, Wang Q, Lü SJ, Zhang J, Zhu TH (2005) Activation of bone morphogenetic protein-6 gene transcription in MCF-7 cells by estrogen. Chin Med J (Engl) 118:1629–1636
Lee HJ, Liu H, Goodman C, Ji Y, Maehr H, Uskokovic M, Notterman D, Reiss M, Suh N (2006) Gene expression profiling changes induced by a novel Gemini Vitamin D derivative during the progression of breast cancer. Biochem Pharmacol 72:332–343
Lee HJ, Wislocki A, Goodman C, Ji Y, Ge R, Maehr H, Uskokovic M, Reiss M, Suh N (2006) A novel vitamin D derivative activates bone morphogenetic protein signaling in MCF10 breast epithelial cells. Mol Pharmacol 69:1840–1848
Masuda H, Otsuka F, Matsumoto Y, Takano M, Miyoshi T, Inagaki K, Shien T, Taira N, Makino H, Doihara H (2011) Functional interaction of fibroblast growth factor-8, bone morphogenetic protein and estrogen receptor in breast cancer cell proliferation. Mol Cell Endocrinol 343:7–17
Mi D, Zhang M, Yan JD, Zhang J, Wang X, Wang Q, Yang S, Zhu TH (2011) PTHrP inhibits BMP-6 expression through the PKA signaling pathway in breast cancer cells. J Cancer Res Clin Oncol 137:295–303
Wang N, Lin K, Lu Z, Lam K, Newton R, Xu X, Yu Z, Gill G, Andersen B (2007) The LIM-only factor LMO4 regulates expression of the BMP7 gene through an HDAC2-dependent mechanism, and controls cell proliferation and apoptosis of mammary epithelial cells. Oncogene 26:6431–6441
Wei W, Lewis MT (2015) Identifying and targeting tumor-initiating cells in the treatment of breast cancer. Endocr Relat Cancer 22:R135–R155
Oshimori N, Fuchs E (2012) The harmonies played by TGF-β in stem cell biology. Cell Stem Cell 11:751–764
Garulli C, Kalogris C, Pietrella L, Bartolacci C, Andreani C, Falconi M, Marchini C, Amici A (2014) Dorsomorphin reverses the mesenchymal phenotype of breast cancer initiating cells by inhibition of bone morphogenetic protein signaling. Cell Signal 26:352–362
Uchino M, Kojima H, Wada K, Imada M, Onoda F, Satofuka H, Utsugi T, Murakami Y (2010) Nuclear β-catenin and CD44 upregulation characterize invasive cell populations in non-aggressive MCF-7 breast cancer cells. BMC Cancer 10:414
Scheel C, Eaton EN, Li SH-J, Chaffer CL, Reinhardt F, Kah K-J, Bell G, Guo W, Rubin J, Richardson AL (2011) Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145:926–940
Buijs J, Van Der Horst G, Van Den Hoogen C, Cheung H, De Rooij B, Kroon J, Petersen M, Van Overveld P, Pelger R, Van Der Pluijm G (2012) The BMP2/7 heterodimer inhibits the human breast cancer stem cell subpopulation and bone metastases formation. Oncogene 31:2164–2174
Hu F, Meng X, Tong Q, Liang L, Xiang R, Zhu T, Yang S (2013) BMP-6 inhibits cell proliferation by targeting microRNA-192 in breast cancer. Biochim Biophys Acta 1832:2379–2390
Clevers H (2011) The cancer stem cell: premises, promises and challenges. Nat Med 17:313–319
Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, Shen R, Brogi E, Brivanlou AH, Giancotti FG (2012) The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 150:764–779
Ampuja M, Jokimäki R, Juuti-Uusitalo K, Rodriguez-Martinez A, Alarmo EL, Kallioniemi A (2013) BMP4 inhibits the proliferation of breast cancer cells and induces an MMP-dependent migratory phenotype in MDA-MB-231 cells in 3D environment. BMC Cancer 13:429
Arnold S, Tims E, McGrath B (1999) Identification of bone morphogenetic proteins and their receptors in human breast cancer cell lines: importance of BMP2. Cytokine 11:1031–1037
Wang D, Huang P, Zhu B, Sun L, Huang Q, Wang J (2012) Induction of estrogen receptor α-36 expression by bone morphogenetic protein 2 in breast cancer cell lines. Mol Med Rep 6:591–559
Chen A, Wang D, Liu X, He S, Yu Z, Wang J (2012) Inhibitory effect of BMP-2 on the proliferation of breast cancer cells. Mol Med Rep 6:615–620
Clement JH, Raida M, Sänger J, Bicknell R, Liu J, Naumann A, Geyer A, Waldau A, Hortschansky P, Schmidt A (2005) Bone morphogenetic protein 2 (BMP-2) induces in vitro invasion and in vivo hormone independent growth of breast carcinoma cells. Int J Oncol 27:401–407
Ghosh-Choudhury N, Ghosh-Choudhury G, Celeste A, Ghosh PM, Moyer M, Abboud SL, Kreisberg J (2000) Bone morphogenetic protein-2 induces cyclin kinase inhibitor p21 and hypophosphorylation of retinoblastoma protein in estradiol-treated MCF-7 human breast cancer cells. Biochim Biophys Acta 1497:186–196
Pouliot F, Labrie C (2002) Role of Smad1 and Smad4 proteins in the induction of p21WAF1, Cip1 during bone morphogenetic protein-induced growth arrest in human breast cancer cells. J Endocrinol 172:187–198
Dumont N, Arteaga CL (2003) A kinase-inactive type II TGFβ receptor impairs BMP signaling in human breast cancer cells. Biochem Biophys Res Commun 301:108–112
Du J, Yang S, Wang Z, Zhai C, Yuan W, Lei R, Zhang J, Zhu T (2008) Bone morphogenetic protein 6 inhibit stress-induced breast cancer cells apoptosis via both smad and P38 pathways. J Cell Biochem 103:1584–1597
Wang K, Feng H, Ren W, Sun X, Luo J, Tang M, Zhou L, Weng Y, He TC, Zhang Y (2011) BMP9 inhibits the proliferation and invasiveness of breast cancer cells MDA-MB-231. J Cancer Res Clin Oncol 137:1687–1696
Ren W, Liu Y, Wan S, Fei C, Wang W, Chen Y, Zhang Z, Wang T, Wang J, Zhou L (2014) BMP9 Inhibits proliferation and metastasis of HER2-positive SK-BR-3 breast cancer cells through ERK1/2 and PI3K/AKT pathways. PLoS One 9:e96816
Yan H, Zhu S, Song C, Liu N, Kang J (2012) Bone morphogenetic protein (BMP) signaling regulates mitotic checkpoint protein levels in human breast cancer cells. Cell Signal 24:961–968
Ghosh-Choudhury N, Woodruff K, Qi W, Celeste A, Abboud SL, Choudhury GG (2000) Bone morphogenetic protein-2 blocks MDA MB 231 human breast cancer cell proliferation by inhibiting cyclin-dependent kinase-mediated retinoblastoma protein phosphorylation. Biochem Biophys Res Commun 272:705–711
Pouliot F, Blais A, Labrie C (2003) Overexpression of a dominant negative type II bone morphogenetic protein receptor inhibits the growth of human breast cancer cells. Cancer Res 63:277–281
Waite KA, Eng C (2003) BMP2 exposure results in decreased PTEN protein degradation and increased PTEN levels. Hum Mol Genet 12:679–684
Rodriguez-Martinez A, Alarmo E-L, Saarinen L, Ketolainen J, Nousiainen K, Hautaniemi S, Kallioniemi A (2011) Analysis of BMP4 and BMP7 signaling in breast cancer cells unveils time-dependent transcription patterns and highlights a common synexpression group of genes. BMC Med Genomics 4:80
Montesano R, Sarközi R, Schramek H (2008) Bone morphogenetic protein-4 strongly potentiates growth factor-induced proliferation of mammary epithelial cells. Biochem Biophys Res Commun 374:164–168
Jung JW, Shim SY, Lee DK, Kwiatkowski W, Choe S (2014) An Activin A/BMP2 chimera, AB215, blocks estrogen signaling via induction of ID proteins in breast cancer cells. BMC Cancer 14:549
Owens P, Pickup MW, Novitskiy SV, Chytil A, Gorska AE, Aakre ME, West J, Moses HL (2012) Disruption of bone morphogenetic protein receptor 2 (BMPR2) in mammary tumors promotes metastases through cell autonomous and paracrine mediators. Proc Natl Acad Sci 109:2814–2819
Steinert S, Kroll TC, Taubert I, Pusch L, Hortschansky P, Höffken K, Wölfl S, Clement JH (2008) Differential expression of cancer-related genes by single and permanent exposure to bone morphogenetic protein 2. J Cancer Res Clin Oncol 134:1237–1245
Clement JH, Marr N, Meissner A, Schwalbe M, Sebald W, Kliche K-O, Höffken K, Wölfl S (2000) Bone morphogenetic protein 2 (BMP-2) induces sequential changes of Id gene expression in the breast cancer cell line MCF-7. J Cancer Res Clin Oncol 126:271–279
Raida M, Clement JH, Ameri K, Han C, Leek RD, Harris A (2005) Expression of bone morphogenetic protein 2 in breast cancer cells inhibits hypoxic cell death. Int J Oncol 26:1465–1470
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Pickup MW, Hover LD, Polikowsky ER, Chytil A, Gorska AE, Novitskiy SV, Moses HL, Owens P (2015) BMPR2 loss in fibroblasts promotes mammary carcinoma metastasis via increased inflammation. Mol Oncol 9:179–191
Perkins ND (2012) The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer 12:121–132
Owens P, Polikowsky H, Pickup MW, Gorska AE, Jovanovic B, Shaw AK, Novitskiy SV, Hong CC, Moses HL (2013) Bone morphogenetic proteins stimulate mammary fibroblasts to promote mammary carcinoma cell invasion. PLoS One 8:e67533
Cai J, Pardali E, Sánchez-Duffhues G, ten Dijke P (2012) BMP signaling in vascular diseases. FEBS Lett 586:1993–2002
David L, Feige JJ, Bailly S (2009) Emerging role of bone morphogenetic proteins in angiogenesis. Cytokine Growth Factor Rev 20:203–212
Raida M, Clement JH, Leek RD, Ameri K, Bicknell R, Niederwieser D, Harris AL (2005) Bone morphogenetic protein 2 (BMP-2) and induction of tumor angiogenesis. J Cancer Res Clin Oncol 131:741–750
Cunha SI, Pietras K (2011) ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood 117:6999–7006
Brown MA, Zhao Q, Baker KA, Naik C, Chen C, Pukac L, Singh M, Tsareva T, Parice Y, Mahoney A (2005) Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J Biol Chem 280:25111–25118
Scharpfenecker M, van Dinther M, Liu Z, van Bezooijen RL, Zhao Q, Pukac L, Löwik CW, ten Dijke P (2007) BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J Cell Sci 120:964–972
David L, Mallet C, Keramidas M, Lamandé N, Gasc JM, Dupuis-Girod S, Plauchu H, Feige JJ, Bailly S (2008) Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ Res 102:914–922
Suzuki Y, Montagne K, Nishihara A, Watabe T, Miyazono K (2008) BMPs promote proliferation and migration of endothelial cells via stimulation of VEGF-A/VEGFR2 and angiopoietin-1/Tie2 signalling. J Biochem 143:199–206
Suzuki Y, Ohga N, Morishita Y, Hida K, Miyazono K, Watabe T (2010) BMP-9 induces proliferation of multiple types of endothelial cells in vitro and in vivo. J Cell Sci 123:1684–1692
Hu-Lowe DD, Chen E, Zhang L, Watson KD, Mancuso P, Lappin P, Wickman G, Chen JH, Wang J, Jiang X (2011) Targeting activin receptor-like kinase 1 inhibits angiogenesis and tumorigenesis through a mechanism of action complementary to anti-VEGF therapies. J Biochem 143:199–206
Cunha SI, Pardali E, Thorikay M, Anderberg C, Hawinkels L, Goumans MJ, Seehra J, Heldin CH, ten Dijke P, Pietras K (2010) Genetic and pharmacological targeting of activin receptor-like kinase 1 impairs tumor growth and angiogenesis. J Exp Med 207:85–100
Chaffer CL, Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331:1559–1564
Polyak K, Weinberg RA (2009) Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9:265–273
Mock K, Preca B, Brummer T, Brabletz S, Stemmler M, Brabletz T (2015) The EMT-activator ZEB1 induces bone metastasis associated genes including BMP-inhibitors. Oncotarget 6:14399–14412
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–715
Yang S, Du J, Wang Z, Yuan W, Qiao Y, Zhang M, Zhang J, Gao S, Yin J, Sun B (2007) BMP-6 promotes E-cadherin expression through repressing δEF1 in breast cancer cells. BMC Cancer 7:211
Yang S, Du J, Wang Z, Yan J, Yuan W, Zhang J, Zhu T (2009) Dual mechanism of δEF1 expression regulated by bone morphogenetic protein-6 in breast cancer. Int J Biochem Cell Biol 41:853–861
Zeisberg M, J-i H, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R (2003) BMP-7 counteracts TGF-β1–induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 9:964–968
Kowanetz M, Valcourt U, Bergström R, Heldin C-H, Moustakas A (2004) Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor β and bone morphogenetic protein. Mol Cell Biol 24:4241–4254
Valcourt U, Kowanetz M, Niimi H, Heldin C-H, Moustakas A (2005) TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell 16:1987–2002
Buijs JT, Henriquez NV, van Overveld PG, van der Horst G, ten Dijke P, van der Pluijm G (2007) TGF-β and BMP7 interactions in tumour progression and bone metastasis. Clin Exp Metastasis 24:609–617
Pal A, Huang W, Li X, Toy KA, Nikolovska-Coleska Z, Kleer CG (2012) CCN6 modulates BMP signaling via the Smad-independent TAK1/p38 pathway, acting to suppress metastasis of breast cancer. Cancer Res 72:4818–4828
Jin H, Pi J, Huang X, Huang F, Shao W, Li S, Chen Y, Cai J (2012) BMP2 promotes migration and invasion of breast cancer cells via cytoskeletal reorganization and adhesion decrease: an AFM investigation. Appl Microbiol Biotechnol 93:1715–1723
Montesano R (2007) Bone morphogenetic protein-4 abrogates lumen formation by mammary epithelial cells and promotes invasive growth. Biochem Biophys Res Commun 353:817–822
Friedl P, Alexander S (2011) Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147:992–1009
Shon SK, Kim A, Kim JY, Kim KI, Yang Y, Lim JS (2009) Bone morphogenetic protein-4 induced by NDRG2 expression inhibits MMP-9 activity in breast cancer cells. Biochem Biophys Res Commun 385:198–203
Wang C, Hu F, Guo S, Mi D, Shen W, Zhang J, Qiao Y, Zhu T, Yang S (2011) BMP-6 inhibits MMP-9 expression by regulating heme oxygenase-1 in MCF-7 breast cancer cells. J Cancer Res Clin Oncol 137:985–995
Wan S, Liu Y, Weng Y, Wang W, Ren W, Fei C, Chen Y, Zhang Z, Wang T, Wang J (2014) BMP9 regulates cross-talk between breast cancer cells and bone marrow-derived mesenchymal stem cells. Cell Oncol 37:363–375
Scherberich A, Tucker RP, Degen M, Brown-Luedi M, Andres A-C, Chiquet-Ehrismann R (2005) Tenascin-W is found in malignant mammary tumors, promotes alpha8 integrin-dependent motility and requires p38MAPK activity for BMP-2 and TNF-alpha induced expression in vitro. Oncogene 24:1525–1532
Gatza CE, Elderbroom JL, Oh SY, Starr MD, Nixon AB, Blobe GC (2014) The balance of cell surface and soluble type III TGF-β receptor regulates BMP signaling in normal and cancerous mammary epithelial cells. Neoplasia 16:489–500
Grönroos E, Kingston IJ, Ramachandran A, Randall RA, Vizán P, Hill CS (2012) Transforming growth factor β inhibits bone morphogenetic protein-induced transcription through novel phosphorylated Smad1/5-Smad3 complexes. Mol Cell Biol 32:2904–2916
Gautschi O, Tepper CG, Purnell PR, Izumiya Y, Evans CP, Green TP, Desprez PY, Lara PN, Gandara DR, Mack PC (2008) Regulation of Id1 expression by SRC: implications for targeting of the bone morphogenetic protein pathway in cancer. Cancer Res 68:2250–2258
Naber HP, Wiercinska E, Pardali E, van Laar T, Nirmala E, Sundqvist A, van Dam H, van der Horst G, van der Pluijm G, Heckmann B (2012) BMP-7 inhibits TGF-β-induced invasion of breast cancer cells through inhibition of integrin β3 expression. Cell Oncol 35:19–28
Roodman GD (2004) Mechanisms of bone metastasis. N Engl J Med 350(16):1655–1664
Bunyaratavej P, Hullinger TG, Somerman MJ (2000) Bone morphogenetic proteins secreted by breast cancer cells upregulate bone sialoprotein expression in preosteoblast cells. Exp Cell Res 260:324–333
Javed A, Barnes GL, Pratap J, Antkowiak T, Gerstenfeld LC, Van Wijnen AJ, Stein JL, Lian JB, Stein GS (2005) Impaired intranuclear trafficking of Runx2 (AML3/CBFA1) transcription factors in breast cancer cells inhibits osteolysis in vivo. Proc Natl Acad Sci 102:1454–1459
Taipaleenmäki H, Browne G, Akech J, Zustin J, van Wijnen AJ, Stein JL, Hesse E, Stein GS, Lian JB (2015) Targeting of Runx2 by miR-135 and miR-203 impairs progression of breast cancer and metastatic bone disease. Cancer Res 75:1433–1444
Acknowledgments
We are grateful to Philip Owens, Miriam de Boeck, and Hans van Dam for critical reading and comments. Our studies on BMP in cancer and vascular diseases are supported by the Cancer Genomics Centre, Netherlands, and Swedish Cancerfonden (090773).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Ren, J., ten Dijke, P. (2017). Bone Morphogenetic Proteins in the Initiation and Progression of Breast Cancer. In: Vukicevic, S., Sampath, K. (eds) Bone Morphogenetic Proteins: Systems Biology Regulators. Progress in Inflammation Research. Springer, Cham. https://doi.org/10.1007/978-3-319-47507-3_18
Download citation
DOI: https://doi.org/10.1007/978-3-319-47507-3_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-47505-9
Online ISBN: 978-3-319-47507-3
eBook Packages: MedicineMedicine (R0)