
Abstract
A long-standing postulate in oncology is that platelets facilitate cancer metastasis (Menter et al. Cancer Metastasis Rev 33:231–269, 2014; Menter et al. Invasion Metastasis 7:109–128, 1987; Gasic et al. Proc Natl Acad Sci USA 61:46–52, 1968; Woods Bulletin der Schweizerischen Akademie der Medizinischen Wissenschaften 20:92–121, 1964). As their most critical biological response, platelets serve as “first responders” during the wounding process and hemostasis. As a part of the metastatic process, platelet receptors recognize complexes of tumor cell receptors and surface-bound matrix proteins or cellular products as they invade blood vessels. This recognition triggers platelet activation and platelet-tumor cell interactions. Once activated by tumor cells, platelets change shape, degranulate, and release proteins, growth factors, bioactive lipids, and other factors that recruit additional platelets and immune cells along with initiating thrombogenesis. Extensive membrane changes occur at bilayer interfaces between platelets and tumor cells. Tumor cells form extensive membrane/cytoskeletal processes that heavily interdigitate with a central platelet aggregate and involves the uptake of platelet fragments and mitochondria. These interactions are thought to result in the suppression of immune recognition/cytotoxicity or the promotion of cell arrest at the endothelium or entrapment in the microvasculature. These responses all support survival and spread of cancer cells and the establishment of secondary lesions. Additional mechanisms of the platelet-metastasis relationship may include the production of platelet exosomes or extravascular migratory behavior of platelets helping to drive cancer progression or preconditioning of secondary metastatic sites. In contrast to the many mechanisms involved in platelet-metastasis relationships, little is known about the role of platelets in precancerous lesion development. This paucity of knowledge exists despite numerous large randomized clinical trials illustrating the cancer preventive effects of nonsteroidal anti-inflammatory drugs (NSAIDs), particularly aspirin in reducing the cancer incidence, mortality, and metastasis. Aspirin covalently acetylates and inactivates platelet cyclooxygenase 1 and thereby eliminates all downstream prostaglandin production from arachidonic acid (AA) by platelets. This includes the key bioactive lipid involved in platelet activation, thromboxane A2 (TxA2). Another prostaglandin, prostacyclin (PGI2), counterbalances and inhibits platelet activation (Honn et al. Science 212:1270–1272, 1981). Metabolically, the genesis of TxA2 and other bioactive lipids are also impacted by ω-3 polyunsaturated fatty acid substrate substitution for AA. Although not well studied, this places platelets not only at the center of the metastasis discussion but also the progression of premalignancies. Since neoangiogenesis produces leaky blood vessels during early cancer progression, it stands to reason that platelets are the “first responders” to extravasate, activate, and release their stroma-stimulating, proangiogenic, chemoattractive, and immunomodulatory contents. These normal platelet functions and products undoubtedly promote precancerous lesion progression as a series of cyclic amplification events. Platelets are suspected to have a key role within the full spectrum of the cancer progression continuum, which makes limiting their first response an important target for both prevention and therapy.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
Platelets in Cancer Metastasis
Platelets as “First Responders”
“First responders” are a perfect moniker for platelets as active participants in the hemostasis, wounding, immune, and metastatic processes (Menter et al. 2014; Gasic et al. 1968; Woods 1964). Platelets are often neglected or overlooked during in vivo experimental or pathologic observations. This is partly due to their small size/volume (mean platelet volumes range is 9.7–12.8 femtoliter or spheres 2.6–2.9 μm in diameter) and the requirement for ultrastructural analysis to effectively observe morphologic or activational structural changes in individual platelets (Fig. 1). Aggregates of activated platelets can be detected by immunohistochemistry at the microscopic level, but this is not routinely done (Qi et al. 2015). As a part of their functional biophysical dynamics, the platelet discoid shape, small size/volume, and physical characteristics cause them to segregate toward the outer fluid shear fields of flowing blood (Fedosov et al. 2014; Kumar and Graham 2012; Tokarev et al. 2011a, b; Lee et al. 2009). Normal human numbers range between 150,000 and 400,000 platelets per microliter (μl), and the concentration of platelets near the vessel wall in vivo is two to three times greater than at the vessel core. Thus, their overall biophysical properties in circulation facilitate platelet distribution toward the endothelial surfaces of vessel walls. Platelet flow patterns, near-wall excess, and proximity enhance their ability to encounter and recognize any lesions in the vascular wall due to a laceration or wound. These platelet recognition properties include the exposure of the subendothelial basement membrane or underlying matrix induced by wounding or endothelial retraction (Menter et al. 1987d; Crissman et al. 1988; Walsh et al. 2015; Kim et al. 2013; Spectre et al. 2012). Platelets can also actively migrate across the inflamed vascular wall in response to stromal cell-derived factor 1 (SDF1 or CXC chemokine ligand 12:CXCL12) and into tumor extravascular spaces (Goubran et al. 2014; Unwith et al. 2015; Schmidt et al. 2012; Kraemer et al. 2010; Brandt et al. 2000; Stone et al. 2012). Platelets can also release SDF1: CXCL12, CXCL1 (GRO-α), CXCL4 (PF4), CXCL5 (ENA-78), CXCL7 (PBP; βTG), and CXCL8 (IL8) among numerous other cytokines and chemokines from storage granules that can potentially initiate the migration and invasion of additional platelets, immune cells (macrophages and leukocytes), endothelial progenitor cells, and tumor cells (Kraemer et al. 2011; Chatterjee et al. 2011; Shenkman et al. 2004; Gleissner et al. 2008). A case for first respondership is further strengthened by the discovery of CXC chemokine receptor 4 and 7 (CXCR4, CXCR7), the cognate receptors for SDF1: CXCL12 on platelets (Rath et al. 2014, 2015; Rafii et al. 2015; Chatterjee et al. 2014b). Expanding upon the concept of migration during potential first responses, platelets are ideally suited for movement because of the highly active cytoskeletal responses linked to activation, adhesion, and aggregation (Menter et al. 1987d, e, 2014; Chopra et al. 1992; Bennett et al. 1999; Jackson et al. 2000; Machlus and Italiano 2013; Qi et al. 2015). Together with an active cytoskeleton, platelets are more streamlined, not only because they are small in size and have minimal displacement volume, but they are also unencumbered by the presence of nuclei, which limits the migration of other immune cells (Breckenridge et al. 2010; Ellingsen et al. 2000; Friedl et al. 2011). The lack of nuclei may also limit the distance that platelets can move into tissue; because of limited protein synthesis capabilities, they cannot indefinitely sustain the replacement of proteins (Bruce and Kerry 1987; Borisova and Markosian 1977). In addition as part of their immune surveillance properties, platelet can also recognize foreign bodies or invading pathogens (Menter et al. 2014; Rossaint and Zarbock 2015; Garraud et al. 2011; von Hundelshausen and Weber 2007). Due to these combined properties, during metastasis, circulating platelets can also elicit a first response to the exposure, sloughing, or active invasion of tumor cells into the blood stream at primary tumor sites. Obstruction of blood flow and angiogenesis associated with primary tumor growth are likely to further enhance the probability of platelet-tumor cell encounters through membrane interactions (Horejsova et al. 1995; Benazzi et al. 2014; Fein and Egeblad 2013). The net result is likely to be platelet activation either to subdue cells at the primary site or generate tumor cell-platelet emboli in circulation (Menter et al. 1987b, 2014; Crissman et al. 1988). Based on such significant numbers in circulation, small size, biophysical shear properties, adhesion, aggregation, and streamline migration properties, platelets are well suited to serve as “first responders” to a variety of pathologic stimuli, including metastasis.

Platelet first responder properties. (a) The activation of platelets is triggered by tumor cells and microenvironmental factors and constitutes an integral part of their first responder properties. This involves shape change and the release reaction. Multiple components that are released into the microenvironment include dense granules, alpha granules, and platelet microparticles (PMP) or microvesicles. The factors released include (1) adhesion molecules, (2) growth factors, (3) angiogenesis factors, (4) tissue remodeling factors, and (5) immune modulators, among others. (b) Due to their biophysical and biological properties, platelets accumulate near blood vessel walls within the fluid shear parameters of flowing blood. This enables them to elicit first responses to lesions in the vasculature and the exposure of extracellular matrix. This can lead to the migration of nonnucleated platelets into the perivascular space. As tumor cells enter the blood stream, they can interact with platelets to form heterotypic emboli. EC endothelial cell, PC pericyte, BM basement membrane, C collagen, TC tumor cell, PLT platelet
Tumor Cell: Migration, Invasion, and Intravasation
The proliferation and migration of cancer cells within primary tumors drives a number of events that can impact metastasis (Starke et al. 2014; Gritsenko et al. 2012; Friedl et al. 2012, 2014; Haeger et al. 2014). Direct impact can occur by shedding, sloughing, or active entry of tumor cells into the blood vessels. Based on single cell profiling of circulating tumor cells (CTCs), there is a large diversity of cells found in the circulation that reflect tumor heterogeneity (Deng et al. 2014; Powell et al. 2012). Within the diversity spectrum, CTCs also frequently exhibit stem cell properties (Tang et al. 2015). A variety of triggers can initiate entry of CTCs into the circulation. For example, decreased availability of blood vessels can increase the induction of hypoxia as tumors outgrow their blood supply and release of angiogenesis or wounding related factors (Pasula et al. 2012; Hellberg et al. 2010; Carmeliet 2005). These factors stimulate the formation of new blood vessels that are typically abnormal and leaky, enabling entry of tumor cells into the blood stream (Keskin et al. 2015; Nagy et al. 2012; Fukumura and Jain 2008). Although not extensively studied, there is also potential for leakage or migration of platelets into the tumor that may further enhance the angiogenesis/leaky blood vessel genesis cycle (Kisucka et al. 2006; Goubran et al. 2014; Schumacher et al. 2013; O’Byrne and Steward 2001). More aggressive tumor cells that enter the circulation often undergo epithelial-mesenchymal transition (EMT) (Satelli et al. 2015). In fact, direct signaling between platelets and cancer cells induces an EMT and promotes metastasis in vitro and in vivo (Labelle and Hynes 2012; Labelle et al. 2011; van Es et al. 2014). Cells with EMT characteristics are more fibroblastic in morphology and are typically much more motile and invasive as a result (Labelle et al. 2011; Labelle and Hynes 2012). These EMT cells are prone to actively invade blood vessels by using matrix metalloproteinase (MMP) to digest the extracellular matrix and basement membrane of blood vessels (Nistico et al. 2012). As part of the invasion process, interactions between platelets and tumor cells increase the production of MMP-9 (Labelle et al. 2011). This vascular tumor cell invasive process is termed intravasation and is considered an early dissemination step of the hematogenous metastatic cascade (Fidler 1978).
Tumor Cell-Platelet Recognition/Interaction: A Two-Way Street
Surface receptors abound on both platelets and tumor cells that trigger the primary response mechanisms which drive biologic function (Goubran et al. 2014; Menter et al. 2014). In the case of a nucleated platelets, the lack of nuclei and restricted protein synthesis capabilities limit the adaptation dynamics of platelet surface receptor expression, which is generally established during platelet genesis by megakaryocytes in the bone marrow (Menter et al. 2014). The genesis of heterogeneous populations of platelets that contain carbon copy surface receptor subsets from specific megakaryocytes capable of adapting to disease has not been well established but could have a key impact on cancer if this were true (Bakchoul and Sachs 2015; Penington et al. 1976). In contrast to platelets, nucleated tumor cells have both surface and nuclear receptors that respond to a variety of molecules. Tumor cells also retain the capacity to replace or increase receptor expression by upregulating gene expression and new protein synthesis as they adapt to their microenvironment (Menter et al. 2014).
Integrin and Collagen Receptors
Platelet-tumor cell encounters can be initiated by surface contacts. Integrins are one class of surface receptor molecules that interact with certain extracellular matrix proteins or by direct interactions and consist of α-subunit and β-subunit heterodimer complexes (Fig. 2a) (Menter and Dubois 2012). From the platelet side (pink text), integrin heterodimer receptor complexes include those for vitronectin (αv/β3:ITGAV/CD61), fibrinogen (αIIb/β3:CD41/CD61), laminin (α2/β1: CD49b/CD29), and fibronectin (α5/β1:ITGA5/CD29) (Menter et al. 2014; Grossi et al. 1987). On the tumor cell side (purple text), many of the same integrin heterodimer complexes can be involved in binding these matrix proteins along with other classes of integrins (Menter and Dubois 2012).

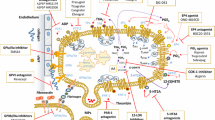
Receptor and factors that mediate platelet-tumor cell interactions. Integrin and collagen receptors mediate cell-cell interactions between platelets and tumor cells along with collagen and extracellular matrix (ECM) proteins. (a) Platelet GPIb complexes recognize vWF tethers, along with thrombospondin, thrombin, αMβ2 integrin, kininogen, and clotting factors XI and XII. The GPIb complex consists of two sets of GPIX, GPIbβ, and GPIbα along with a centrally situated GPV protein. Adhesive contacts with collagen exposed by tumor cells are stabilized by GPVI. The αIIbβ3 integrin has the widest range of interactions and binds to fibrinogen, fibrin, fibronectin, vitronectin, thrombospondin, and vWF. Platelet αIIbβ3 integrin inhibitors include abciximab, tirofiban, and eptifibatide. Additional platelet and tumor cell integrins include: αv/β3(ITGAV/CD61; vitronectin receptors), α2/β1(CD49b/CD29; collagen, laminin receptors), and α5/β1 (ITGA5/CD29; fibronectin, protease receptors). (b) Selectins, cellular adhesion molecules (CAMs), mucins, and lectins can profoundly influence platelet-tumor cell interactions. P-selectin binds to a variety of carbohydrate-rich molecules including: Sialyl Lewisx or Sialyl LewisA as well as tethered (1,3,4,10-18) or soluble (2,5A-C, 6-9,19) mucins and podocalyxin-like protein 1 (PCLP1). Podoplanin on tumor cells can interact with CLEC-2 on platelets. CAMs such as PCAM-1 (CD31) and ICAM-1 (CD54) also regulate platelet tumor cell interactions. (c) G protein-coupled receptors (GPCRs) mediate a wide variety of biological interactions. Tissue factor (thromboplastin or factor III) can form complexes with factor VII and factor X to generate thrombin that in turn binds to and activates protease-activated receptors (PAR) 1 or 4 on platelets, which can be inhibited by PAR-specific inhibitors, E5555, and SCH530348. Bioactive lipids also alter platelet-tumor call interactions. Arachidonic acid (AA) is converted by cyclooxygenase 1 and 2 to prostaglandins (PG) and are inhibited by aspirin, NSAIDs, and COXIBs. COX inhibition inhibits multiple downstream steps in the PG cascade. The key target includes the primary platelet-activating pathway: thromboxane A2 synthase (TXAS) > TxA2 > TxA2 receptor (TP) that can be inhibited by a receptor antagonist, terutroban. This activation is countered by the endogenous prostaglandin I2 synthase (PGIS) > PGI2 > PGI2 receptor (IP) inhibition. Other arms of the PG pathway also influence these heterotypic interactions. Another endogenous inhibitor includes resolvin-E1, a product of eicosapentaenoic acid (EPA). Nucleotide receptors can also activate platelets ADP/purinergic G protein-coupled receptors 1 and 12 (P2Y 1, 2 and 12) that are inhibited by clinically relevant receptor antagonists: clopidogrel, prasugrel, AZD6140, and cangrelor. Serotonin (5HT) receptors are also influenced by the uptake and release of 5-HT by platelets. (d) Immune receptors are involved in responses to pathogens, including FcgammaRIIB and toll-like receptors (TLR). Platelet genesis is influenced by IL-6 receptors CD126 and thrombopoietin (TPO) receptors cMpl/CD110. Platelet chemotaxis and migration are influenced by stromal cell-derived factor 1 (SDF1 or CXC chemokine ligand 12:CXCL12) through CXCR4 and CXCR7 receptors. Neuropeptides that include substance P (SP), calcitonin gene-related peptide (CGRP), and neurokinin A (NPY) stimulate inflammation when released into the microcirculation, where platelets are key responders that react by modulating arachidonate metabolic pathways. In terms of angiogenic response, the platelet-derived form of the sympathetic neurotransmitter neuropeptide Y (NPY) induces vasoconstriction and ischemic angiogenesis. Pink text platelet receptors and factors, purple text tumor cell receptors and factors, maroon text and arrows inhibitors, green arrows stimulation or interaction, red arrows inhibition
Additional matrix protein receptors include those for collagens. On the platelet side, in this case, collagen receptors are relatively unique to platelets that include GPIb complexes (consisting of GPIX, GPIbα, GPIbβ, and GPV subunits) as well as GPVI (Menter et al. 2014). Tumor cells by contrast can express the entire range of collagen receptors including α2/β1 (Menter and Dubois 2012).
Cell Adhesion Molecules, Selectins, and Mucins
Cell adhesion molecules (CAMs) are transmembrane proteins that bind to extracellular matrix and cell-cell adhesion (Fig. 2b). The primary CAM expressed on platelets that regulates binding to tumor cells is relatively uniformly expressed (PECAM-1:CD31) (Pang et al. 2015a; Coupland and Parish 2014; Aceto et al. 2014). On the tumor cell side, the expression of surface receptors is not as uniform and can vary extensively with tumor heterogeneity and adaptability (Gunning et al. 2008; Moins-Teisserenc et al. 2015; Raeisossadati et al. 2011). These tumor cell receptors include PCAM-1:CD31, intercellular (ICAM-1:CD54), and epithelial (EpCAM: CD326). Selectins are another family of CAMs consisting of single-chain transmembrane glycoproteins with lectin and cell adhesion properties that bind carbohydrate. Platelets express P-selectin (CD62), which interacts with a variety of carbohydrate-rich molecules produced by tumor cells that include numerically identified mucins that are either tethered (1, 3, 4, 10–18) or free (2, 5A–C, 6–9, 19). Some reports also show evidence of platelets actively recruiting ovarian cancer cells and upregulating tissue factor (Orellana et al. 2015). P-selectin also interacts with Sialyl Lewisx or Sialyl LewisA as well as podocalyxin-like protein 1 (PCLP1) expressed by tumor cells. Platelets can also express C-type lectin receptor 2 (CLEC-2), which interacts with podoplanin on tumor cells (Menter et al. 2014; Goubran et al. 2015).
G Protein-Coupled Receptors (GPCR)
A series of cell-signaling G protein-coupled receptors (GPCRs) can help mediate interactions between platelets and tumor cells (Fig. 2c). Once again on the platelet side, the receptor/signaling networks are very complex (Boyanova et al. 2012; Menter et al. 2014). Tissue factor (thromboplastin or factor III) can form complexes with factor VII and factor X to generate thrombin, which binds to and activates protease-activated receptors (PAR) 1 or 4 on platelets (Menter et al. 2014; Goubran et al. 2014). Tissue factor-dependent thrombin is also enhanced by phospholipid-esterified eicosanoids in agonist-activated platelets (Thomas et al. 2010). The 12-lipoxygenase arachidonate metabolite, 12(S)-HETE, is a bioactive lipid that contributes to endothelial cell retraction as a free acid (Honn et al. 1994). However in its esterified form, after platelet activation, it appears to contribute to coagulation as a result of its translocation to the platelet outer surface. This is supported by the observation that in Scott syndrome, platelets that are defective in cell surface coagulation, the esterified HETE is not presented to the cell surface despite being made to the same degree as in thrombin-stimulated platelets from normal patients. Both the PAR1 and PAR4 agonists (TFLLR-NH2 and AY-NH2, respectively) stimulate esterification of 12(S)-HETE with PC and PE to a similar degree (Thomas et al. 2010).
Prostaglandin (PG) receptors are prominent GPCRs expressed by platelets that respond to PGs, which are also important bioactive lipids that profoundly alter platelet function. The PG receptor for thromboxane A2 (TxA2), for example, is among the strongest agonists to stimulate platelet activation via thromboxane receptors (TP) (Fontana et al. 2014; Gleim et al. 2013). A differently structured PG is prostaglandin I2 (prostacyclin) that binds to a separate prostaglandin I2 receptor (IP1) and exhibits the strongest inhibition of platelets (Honn et al. 1981; Hubertus et al. 2014). Platelets also express PGE2 receptors (EP 2, 3 and 4) and PGD2 receptors that can activate or inhibit platelet function (Petrucci et al. 2011; Cooper and Ahern 1979; Hubertus et al. 2014). Platelets also express additional bioactive lipid receptors. One orphan receptor, chemerin receptor 23 (ChemR23; chemokine-like receptor1: CMKLR1) binds resolvin E1, a bioactive lipid synthesized from ω3-polyunsaturated fatty acid eicosapentaenoic acid (EPA) found in fish oil that inhibits ADP-induced platelet activation (Fredman et al. 2010; Petrucci et al. 2011). This receptor also serves as a chemokine (C-C motif) receptor-like 2 (CCRL2) that binds chemerin contained in platelets, an adipokine/chemoattractant that has reported roles in inflammation, adipogenesis, and insulin resistance (Du et al. 2009). Tumor cells by contrast express a wide variety of PG receptors depending on tissue origin. Both cyclooxygenase 2 (COX-2) and microsomal PGE2 synthase1 (mPGES1) are commonly upregulated in cancer, and the production of PGE2 activates EP receptors (Menter et al. 2010).
Platelets express a number of additional GPCRs. These include thrombin/protease-activated receptors 1 and 4 (PAR 1 and 4) along with ADP/purinergic G protein-coupled receptors 1, 2, and 12 (P2Y 1, 2, and 12) (Franchi and Angiolillo 2015). Both sets of receptors typically trigger platelet activation, and a variety of clinically relevant inhibitors exist for target intervention (Raju et al. 2008). PAR1 and PAR4 are activated following thrombin cleavage that exposes a tethered ligand, which binds to PAR and causes a conformational change and interactions with G proteins (Fu et al. 2015; Wallace and Smyth 2013). Platelet PAR activation can lead to platelet aggregation and thrombosis as well as neutrophil recruitment (Fu et al. 2015; Wallace and Smyth 2013). In contrast, the binding of ADP to P2Y receptors mobilizes intracellular calcium ions via activation of phospholipase C that activates platelets (Cattaneo 2015; Liverani et al. 2014) along with stimulating tumor-cell transendothelial migration and metastasis (Schumacher et al. 2013).
Neurogenic Inflammation and Platelets
The connection between receptor-mediated neurogenic inflammation and cancer is being actively investigated for its role both in potentiating local tumor growth and perineural cancer invasion (Stopczynski et al. 2014; Liebig et al. 2009; Black 2002). Substance P (SP), calcitonin gene-related peptide (CGRP), and neurokinin A (TAC1) are neuropeptides that stimulate inflammation when released into the microcirculation, where platelets are key responders that react by modulating arachidonate metabolic pathways (Gecse et al. 1999). In terms of angiogenic response, the platelet-derived form of the sympathetic neurotransmitter neuropeptide Y (NPY) is required for capillary angiogenesis during late-phase recovery from ischemic injury (Tilan et al. 2013) and is also produced in animals with hypertension (Ogawa et al. 1992), a condition whose association with cancer has long been suspected (Hamet 1996; Stocks et al. 2012). As a number of cells upregulate NPY to promote metastasis and NPY receptor has been described in the context of its role in cancer, it is conceivable that platelet-derived NPY provided through platelet-tumor cell interactions may also contribute to metastasis (Hong et al. 2015; Medeiros et al. 2012; Magni and Motta 2001).
Serotonin Receptors
Platelet dense granules contain serotonin (5-HT) that is taken up through serotonin transporter (SERT) activity to regulate 5-HT circulating levels (Mauler et al. 2015; Jedlitschky et al. 2012; Linder et al. 2007). More than 95 % of total body 5-HT is synthesized in the enterochromaffin cells of the intestine. Once released from platelets following stimulation, 5-HT binds to cognate receptors and induces vasoconstriction (Mauler et al. 2015; Jedlitschky et al. 2012; Linder et al. 2007). The role of platelet serotonin in malignancy has been pondered for decades (Crawford et al. 1967). Platelet-derived serotonin has been linked to tissue fibrosis (Dees et al. 2011) [considered a form of EMT, reviewed in Tucker and Honn (2013), Crooks et al. (2014), Lopez-Novoa and Nieto (2009)], where platelet aggregation has also been described (Kahaleh et al. 1982). Furthermore, serotonin may be a marker for hepatocellular carcinoma (Pang et al. 2015b; Pai et al. 2009) and has been proposed as a contributing factor to breast cancer (Pai et al. 2009). Platelet serotonergic mechanisms have also been invoked in bladder cancer (Pawlak et al. 2000).
Immune Response Receptors
Additional critical receptors that can impact interactions between platelets and tumor cells include those influencing immunomodulation (Fig. 2d). Platelets express a wide range of receptors that interact with pathogens (Hamzeh-Cognasse et al. 2015) including complement. These receptors include: FcγRII, toll-like receptors (TLR), and also integrins conventionally described in the hemostatic response, such as αIIbβ3 or GPIb (Hamzeh-Cognasse et al. 2015). Some of these receptors such as Fcγ receptor IIa can mediate platelet-tumor cell cross talk and initiate tumor cell-induced platelet secretion (TCIPS) (Mitrugno et al. 2014). Interactions with histocompatibility antigen also influence platelet behavior in cancer patients, but this has not been extensively studied (Messerschmidt et al. 1988). Also as previously mentioned, chemokine/cytokine release and subsequent binding to immune receptors on platelets or other cells in the local or systemic environment can stimulate the migration and growth of a variety of cells (Kraemer et al. 2011; Chatterjee et al. 2011, 2014a, b; Shenkman et al. 2004; Gleissner et al. 2008; Rath et al. 2014, 2015; Rafii et al. 2015).
Cancer-Induced Thrombocytosis
Finally, cancer-induced thrombocytosis significantly increases the number of circulating platelets and is linked to tumor cell proliferation (Bouvenot et al. 1977; Honn et al. 1992; Levin and Conley 1964; Cho et al. 2012). This constitutes an important link to Trousseau’s syndrome, but the mechanisms responsible remained poorly understood until recently (Stone et al. 2012) when a multicenter study involving 619 ovarian cancer patients examined the associations between platelet counts and disease outcome (Stone et al. 2012; Davis et al. 2014). During this study, thrombocytosis was elevated in association with circulating levels of thrombopoietin (TPO) and interleukin-6 (IL-6). This elevation was associated with advanced disease and shortened survival in ovarian cancer patients and was verified in orthotopic mouse models (Stone et al. 2012). In pancreatic cancer studies as well, paraneoplastic thrombocytosis predicts poor prognosis in patients with locally advanced disease (Chadha et al. 2015). These tumor-liver-bone marrow-platelet feedback mechanisms are thought to occur through IL-6 receptors: CD126 (Marino et al. 2013) and TPO receptors: cMpl/CD110 (Hitchcock and Kaushansky 2014).
Anticoagulant Therapy for VTE Prevention in Cancer Patients
It is estimated that individuals with cancer have a fivefold higher incidence of venous thromboembolic events (VTEs) than the general population (Chao et al. 2011). Chemotherapy can also contribute to elevated levels of thrombosis. In healthy individuals, venous thrombosis can be the first manifestation of malignancy. Since the 1970s when the Veterans Administration Cooperative Study Program first tested warfarin in cancer patients, many other studies have examined agents that interrupt the coagulation pathway as a means to attenuate VTEs and impede cancer progression (Chao et al. 2011). Unfortunately, the doses of warfarin required to achieve therapeutic results were not without side effects (Zacharski et al. 2005), and heparin required continuous intravenous administration (Hull et al. 1986). As a result of numerous clinical trials, low molecular weight heparin (LMWH or nadroparin) replaced unfractionated heparin (UFH) nearly 20 years ago as the preferred treatment and prophylaxis agent for several reasons (Chao et al. 2011; Salzman et al. 1980). Its chemical structural properties allow it to specifically inhibit factor Xa without all of the off-target serine protease inhibition seen with UFH. Compared to UFH, LMWH is easier to administer and significantly lowers recurrent thrombosis, major bleeding, and mortality (van Dongen et al. 2004). There is some suggestion that the beneficial effects of LMWH in treating cancer patients may be direct against the tumor cells in addition to the anticoagulant effect (Barni et al. 2014). Even with these advances and new reagents, a major concern to consider is the potential to induce heparin-induced thrombocytopenia (Wharin and Tagalakis 2014; Greinacher 2015).
To Decorate, Cloak, or Hold Tumor Cells: That Is the Question
As part of their function in immune surveillance, platelets interact with viral particles, bacteria, and parasites as well as virally transformed tumor cells (Chabert et al. 2015; Hamzeh-Cognasse et al. 2015; Kofman et al. 2013; Ahmed et al. 2014). Many of these interactions essentially help to decorate pathogens and trigger platelet aggregation and the release of factors that stimulate leukocyte activation and neutrophil extracellular trap (NET) formation (Hamzeh-Cognasse et al. 2015). In contrast, tumor cell-platelet interactions are postulated to suppress immune recognition/cytotoxicity and promote arrest at the endothelium or entrapment in the microvasculature. These responses support survival and spread of cancer cells and the establishment of secondary lesions. The current dogma suggests that once tumor cells enter the circulation, they can become enveloped or decorated by a coating of platelets that shields them from recognition by cytotoxic immune cells. While this notion has received support from numerous investigators, it may not be exactly what occurs. When examining heterotypic aggregates that formed during tumor cell-induced platelet aggregation (TCIPA), the platelets tend to cluster at the center, not the external surfaces (Menter et al. 1987a, c) (Fig. 3). In most cases the tumor cells are not completely enveloped enough following TCIPA to fully encase and hide the tumor cells from immune cells (Menter et al. 1987a, c, e). Furthermore, from a logistical standpoint, the feasibility of completely covering the large tumor cell surface area with much smaller platelets is in doubt.

Early events in platelet-tumor cell interactions. (a) Elutriated W256 tumor cells are shown interacting with platelets in an aggregometer cuvette at the plateau phase (10 min). A reduction in tumor cell surface microvilli occurs by this time (asterisks, scanning electron micrograph; magnification x3900). (b) Elutriated W256 tumor cells project small processes (arrows) into platelet aggregates by midphase of aggregation that become more (c) prominent and protrude deeper into forming platelet emboli as these interactions plateau (arrows) that associates with tumor cell engulfment of platelet fragments (open arrows) (transmission electron micrographs; magnifications—(b) x3000, (c) x4300). (d) Elutriated Lewis lung tumor cells form intravascular aggregates in the mouse lung that involves process penetration (arrows) into syngeneic C57Bl/6 platelet emboli, which occurs 2 min after tail vein injection and (e) engulfing platelet fragments (arrows; transmission electron micrographs; magnifications—(d) x6800, (e) x12,000, scale bars 1 μm). (a–c) Reprinted from Honn et al. (1983) with permission from S. Karger AG, Basel; (d, e) courtesy of Menter
Platelet Changes in Cancer Patients
Recent studies have demonstrated that there are profound ultrastructural changes in platelets from cancer patients as compared to normal controls or individuals with benign disease. Electron cryotomography (cryo-ET) was used to examine platelets from patients with invasive ovarian cancer and controls (either benign adnexal mass or free from disease). Significant morphological differences were detected between the cancer and control platelets including disruption of microtubules as well as altered numbers of mitochondria (Wang et al. 2015). Furthermore, RNA-seq studies have revealed that molecular pathway changes in tumor-educated blood platelets (TEPs), thus connecting them to systemic and local responses to tumor growth resulting in altered RNA profiles (Best et al. 2015).
Heterotypic Emboli, Platelet Interactions, and Extensive Membrane/Structural Changes
The heterotypic contacts between platelets and tumor cell lead to dynamic focal interactions during TCIPA (Menter et al. 1987a, c, e). As platelets cluster centrally during heterotypic aggregate formation, they change shape and degranulate and release proteins, growth factors, bioactive lipids, and other factors that recruit additional platelets toward the central core of the interaction (Menter et al. 1987a, c, e). Extensive membrane changes occur at the membrane interfaces between platelets and tumor cells. There is very little data on whether the tumor cells or platelets drive these membrane interactions, but they appear to be bidirectional. Extensive membrane/cytoskeletal tumor cell processes heavily interdigitate with a central platelet aggregate, demonstrating a very active process. As these interactions progress, extensive uptake of platelet fragments and mitochondria occurs into the tumor cells in vitro (Menter et al. 1987a, c, e). This interdigitation and uptake of platelet fragments by tumor cells are also observed in vivo following tail vein injections of syngeneic tumor cells into mice (Crissman et al. 1985, 1988; Menter et al. 1987b).
The engulfment of platelets or platelet particles such as mitochondria may be a form of emperipolesis or entosis. These are processes whereby cells take up other cells without degradation with varied outcomes (Xia et al. 2008; Rastogi et al. 2014). This is reminiscent of frustrated phagocytosis where organisms ride along the plasma membrane or become engulfed only to be subsequently released. Some metastatic tumor cells even take on behaviors of M2 polarized macrophages (Caruso et al. 2012; Djaldetti and Strauss 1982).
While genomic and epigenetic elements are fundamental to the cancer continuum, the contribution of exosomes/microvesicles, free nucleic acid, and cytoplasmic elements is just as intriguing. It has been suggested that the reaction of the mitochondrial genetic system to genomic (nuclear DNA) stress response genes leads to mitochondrial dysfunction and could account for the marked increase in metabolic diseases and cancer (Wallace 2005; Brahimi-Horn et al. 2015). Studies have demonstrated that tumorigenic phenotypes can be down-modulated when mitochondrial DNA is ablated and can be complemented by reintroduction of mitochondria (Cavalli et al. 1997). Therefore, hyperpolarization of mitochondria in activated platelets and the observation that platelets release mitochondria to potentiate immune response (Zharikov and Shiva 2013; Boudreau et al. 2014) raise the specter that platelet mitochondria potentially make a direct contribution to malignancy by regulating tumor cells. Whether platelet mitochondria compensate for tumor cell mitochondrial deficits or augment some tumor cell function through genetic cross programming remains to be determined. With respect to the latter idea, mitochondrial transcription factor binding sites are seen throughout the nuclear genome. Coordinated regulation of nuclear genes in response to mitochondrial stress or mitochondrial uptake has not yet been established, but it is a topic being scrutinized (Haynes et al. 2013).
Secondary Sites, Exosomes, and Extravasation: Differences or Similarities?
Tumor cell exosomes play key roles in cancer progression and metastasis (Melo et al. 2014, 2015; Kahlert and Kalluri 2013; Milane et al. 2015; An et al. 2015; Robbins and Morelli 2014; Colombo et al. 2014). Functionally, these exosomes can influence the biology of both the local tumoral and systemic environments (Kahlert and Kalluri 2013). Tumor cell exosome biogenesis often begins with endocytosis, then endosome genesis, followed by the formation of multivesicular bodies (MVBs) or by microvesicle formation (Huber and Holvoet 2015; Zhang et al. 2015). Exosomes formed within MVBs are released following membrane fusion into the surrounding microenvironment or circulation. Locally, release of tumor cell exosomes into the microenvironment can influence proliferation and chemoresistance and initiate fibroblast or immune cell activation and recruitment along with stimulating angiogenesis. Systemically, tumor cell exosomes can stimulate a hypercoagulable state, initiate preneoplastic niches at secondary metastatic sites, recruit bone marrow-derived hematopoietic and immune cells, or elicit immunosuppression (Lima et al. 2013). Classically by contrast, platelets contain numerous electron-dense subcellular organelles called alpha granules, electron-dense and very electron-dense granules, lysosomes, and dense bodies (Fukami and Salganicoff 1977). The many factors contained in these granules constitute what is commonly found in serum used to grow cancer cells in tissue culture and in regenerative medicine (Dhillon et al. 2014; Burnouf et al. 2013; Ross et al. 1978). These organelles are sometimes called secretory granules, since the appropriate stimuli causes degranulation that deposits granule contents in the extracellular surroundings. These organelles were identified and visualized by electron microscopy, microprobe analysis, secretion experiments, and subcellular fractionation studies from patients with platelet storage pool deficiency diseases. In many respects, this degranulation process can be equated with the formation of platelet microparticles (PMP), which are essentially very exosome-like (Goubran et al. 2015; Mezouar et al. 2014). Platelet extravascular migratory behavior and exosome deposition is likely to drive cancer progression or preconditioning of secondary metastatic sites (Goubran et al. 2015). Preventing platelet activation inhibits the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo (Mezouar et al. 2015).
Blood-borne CTCs are commonly found in the blood stream of patients with metastases (Aceto et al. 2014; Li et al. 2015; Joosse et al. 2015). In a patient study of CTCs isolated from breast and prostate cancer patients, their presence correlated with poor prognosis (Aceto et al. 2014). This study focused on comparing single CTCs to CTC clusters and found that markers for platelets were present in both populations (Aceto et al. 2014). The presence of these clusters was also associated with high plakoglobin expression and poor prognosis (Aceto et al. 2014). Since plakoglobin plays an important role in the regulation of CAM interactions (Ilan et al. 2001; Rival et al. 1996; Reynolds et al. 1992, 1994), its upregulation may influence platelet/tumor cell heterotypic CTC cluster formation. In a separate randomized phase II trial, therapeutically targeting platelet function did not significantly change the number of CTCs in circulation in patients with metastatic breast cancer. Unfortunately, the number of circulating cells was lower than expected, so a more definitive study is needed (Roop et al. 2013). In early mouse studies, although large numbers of CTCs may be present in circulation, only those with specific characteristics survive to establish metastasis (Fidler and Nicolson 1978; Fidler 1975; Hart and Fidler 1980). In other mouse hematogenous metastasis studies, cell arrest at secondary sites precedes the establishment of lesions. This may occur by a variety of mechanisms. Tumor cell interactions directly with the vascular endothelium can initiate the process. In this case, the engagement of platelets can occur during or after tumor cell arrest has occurred, and platelets can fill in around arrested tumor cells. Tumor cell-platelet heterotypic interactions can also occur in suspension in flowing blood to initiate TCIPA. The resulting heterotypic platelet-tumor cell emboli can then undergo attachment or entrapment in secondary organ sites. Platelet-tumor cell emboli initially interact with vascular surfaces at cellular junctions or gaps in the endothelial cell monolayer (Menter et al. 1987e). Platelets are also capable of forming chains that tether tumor cells to these attachment sites (Menter et al. 1987e). As these cellular interactions progress, platelets engage both endothelial cell surfaces and gaps in the endothelial cell monolayer facilitating the linkage to tumor cell membranes (Menter et al. 1987e). In other studies, agent-based, computational, theoretical modeling simulated the basic dynamics of circulating tumor cell adhesion involving endothelial, neutrophil, and platelet interactions and proposed multiple therapeutic targets involving platelets (Uppal et al. 2014). Other studies have utilized intravital microscopy to visualize human red fluorescent protein (RFP) expressing HT29 (RFP-HT29) colon cancer cells injected into the spleens of GFP expressing nude mice (Tanaka et al. 2014). These studies revealed extensive interactions between GFP platelets and RFP-HT29 in liver sinusoids (Tanaka et al. 2012, 2014). Intravital imaging has also been done using a syngeneic C57/Bl6 mouse model, and interactions were shown to involve αvβ3 integrins on B16 melanoma cells to αIIbβ3 on platelets using blocking antibodies (Lonsdorf et al. 2012).
Tumor cells can exit the vasculature at secondary sites (Crissman et al. 1985), and platelets can facilitate this process known as extravasation (Coupland et al. 2012; Crissman et al. 1985; Fein and Egeblad 2013; Pang et al. 2015a; Schumacher et al. 2013). Thrombin activation involving platelet membranes can facilitate the activation of ECM degradation by tumor cells in a P-selectin and αIIbβ3-dependent manner (Pang et al. 2015a). Taken together these studies support the notion of platelet involvement in the establishment of hematogenous metastasis.
New Thoughts: Platelets in Premalignancy
So What About Aspirin, Arachidonic Acid, and Omega-3 PUFA?
Meta-analyses of large randomized clinical trials (Bousser et al. 2011; Chan et al. 2012; Chan and Cook 2012; Cuzick et al. 2015; Goldstein and Rothwell 2012; Langley and Rothwell 2013, 2014; Rothwell 2013a) show that aspirin reduces the incidence (Algra and Rothwell 2012), metastasis (Rothwell et al. 2012), and mortality (Rothwell et al. 2012) of gastrointestinal and other cancers, especially adenocarcinomas (Algra and Rothwell 2012; Chan et al. 2012; Cuzick et al. 2015; Langley and Rothwell 2013; Rothwell 2013a, b; Rothwell et al. 2012). Despite substantial cumulative knowledge regarding cancer prevention by aspirin (Umar et al. 2016), little is known about the role of the primary target found in platelets during precancerous lesion development. Aspirin covalently acetylates Ser-530 and irreversibly inactivates platelet cyclooxygenase 1 (DeWitt et al. 1990; Rowlinson et al. 2000), thereby eliminating all downstream prostaglandin production from arachidonic acid by platelets. This includes the key bioactive lipid involved in platelet activation, thromboxane A2 (TxA2) (Fontana et al. 2014; Fitzpatrick and Gorman 1977; Hammarstrom and Falardeau 1977). Pronounced elevation of circulating TxA2 levels accompanies CRC progression in familial adenomatous polyposis (FAP) patients (Li et al. 2015). As a strong platelet TP receptor agonist, TxA2 produced by TxA2 synthase in platelets is counterbalanced by an equally strong platelet inhibitory response by IP1 receptors. IP1 receptors are activated by PGI2 that is produced by PGI2 synthase in endothelial cells lining the vasculature. Both TxA2 and PGI2 have epoxide bonds that contain considerable molecular strain, which limits their half-life in circulation. Activation by TCIPA shifts this delicate eicosanoid balance triggering multiple pro-carcinogenic responses centered on platelets.
Diets rich in ω-3 PUFA, EPA, and docosahexaenoic acid (DHA) inhibit carcinogenesis (Holla et al. 2008; Schroeder et al. 2007; Yang et al. 2006, 2014a, b). EPA feeds into the COX-1 and COX-2 pathways to produce TXA3 and PGE3 lipid metabolites that are ineffective at PG receptor activation and protect against CVD and lung or colon tumorigenesis (Algra and Rothwell 2012; Chan et al. 2012; Chan and Cook 2012; Cuzick et al. 2015; Goldstein and Rothwell 2012; Holla et al. 2008; Langley and Rothwell 2013, 2014; Liyasova et al. 2010; Macdonald et al. 1999; Pinckard et al. 1968; Rothwell 2013a; Rothwell et al. 2012; Schroeder et al. 2007; Yang et al. 2006, 2014a). Taking aspirin in combination with EPA and DHA leads to the formation of resolvins, maresins, and protectins (Serhan et al. 2006, 2009; Spite et al. 2014; Spite and Serhan 2010; Krishnamoorthy et al. 2015; Serhan 2014). Resolvins were originally named for their ability to resolve inflammation. More specifically, aspirin-inactivated COX-2 generates a longer half-life R-epimer of resolvin-D1 (Arita et al. 2005; Serhan et al. 2004; Sun et al. 2007) from DHA and resolvin-E1 from EPA (Dona et al. 2008; Pirman et al. 2013). Although not well studied, this places platelets not only at the center of the metastasis discussion but also the progression of premalignancies.
Leaky Vessels, Extravasation, and “First Response”
The role of angiogenesis in premalignancy remains an important question, particularly how early this change occurs (Menakuru et al. 2008) (Fig. 4). Biomarkers of this process include factor VIII, CD31, and CD34. Increased microvascular density can occur in ductal carcinoma in situ (DCIS) (Menakuru et al. 2008; Sapino et al. 2001). These premalignant lesions range in size from 1 to 1.5 cm (Allegra et al. 2009). Newly formed vessels can be identified by CD105 (Fox and Harris 2004; Smith et al. 2012; Ratajczak et al. 2012; Dubinski et al. 2012; Marioni et al. 2011; Sapino et al. 2001). Platelet-derived thrombospondin 1 helps regulate angiogenesis and serves as a potential biomarker (Zaslavsky et al. 2010). The presence of highly permeable, disorganized, poorly formed, and leaky blood vessels contribute to progression in advanced cancers (Fang et al. 2011). These leaky vessels provide ample opportunity for platelet first responders to extravasate and enter the precancerous microenvironment. Once in the perivascular space, platelet activation triggers the release of growth-promoting factors found in serum. These serum factors are highly likely to provide a permissive and growth-promoting microenvironment for transformation.

Platelet influences on angiogenic and immune switches. Angiogenic and immune switches can turn on at different stages of carcinogenesis. (a) Biological maintenance of normal tissue involves a delicate balance between cell growth and apoptosis and homeostasis that is regulated by immune surveillance, tissue repair, and immune clearance. (b) Premalignant disease may involve the earliest stages of angiogenic and inflammatory switch activation. Premalignancy-associated inflammation triggers immune cell infiltration in response to cytokines and prostaglandins that promotes vessel dilation and the detachment of pericytes from blood vessels. During these early stages of premalignancy, blood factors accumulate in the perivascular space. This provides the opportunity for platelets to migrate into the perivascular space along with activation and the release of platelet granules containing vascular endothelial cell growth factor (VGEF), basic fibroblast growth factor (bFGF), platelet-derived growth factor (PDGF), epithelial growth factor (EGF), and angiopoietin-2 (Ang2) that enhances both angiogenic and immune switch activation. (c) The onset of malignancy, heightened hypoxia, and apoptosis further increases vessel leakiness, platelet activation, platelet migration, and granule release. The cycle accelerates as angiogenic sprouting and tip cell-mediated migration into the growing tumor lead to further recruitment of additional platelets, immune cells, and tumor cell migration and proliferation as cancer progression continues
Angiogenic and immune stimulation often switch on during premalignancy, before cells achieve the invasive state (Ferrati et al. 2012; Folkman et al. 1989). Many factors influence this switching, including microRNAs (Bergers and Benjamin 2003; Grandis and Argiris 2009). Switching has been elegantly demonstrated in the adenoma-adenocarcinoma transition of colorectal cancer and the transition from DCIS to invasive ductal carcinoma of the breast. Specifically, the initiation of angiogenesis accompanies dysplasia during adenoma formation in colorectal cancer progression (Staton et al. 2007) and hyperplasia during DCIS progression to invasive breast cancer (Bluff et al. 2009). Mechanistically, mice that lack histidine-rich glycoprotein (HRG) exhibit an accelerated angiogenic switch and enhanced platelet activation, inducing molecular changes in the pre-tumorigenic environment accompanied by increased survival, angiogenesis, and EMT (Cedervall et al. 2013). Although more studies are needed, these effects suggest an early, vital role for platelets early in the premalignancy to malignancy transition. Abnormal or leaky blood vessels that arise during tumor-initiated angiogenesis provide adequate opportunity for the leakage or migration of platelets into the precancerous microenvironment. Subsequent activation and release of their stroma-stimulating, proangiogenic, chemoattractive, and immunomodulatory contents can then promote precancerous lesion progression as a series of cyclic amplification events. In support of this hypothesis, release of VEGF and other platelet factors occurs during early breast cancer (McDowell et al. 2005), and platelet GPIbα also increases in invasive intraductal breast carcinoma tissues (Oleksowicz et al. 1997). These early angiogenic and immunogenic switching events during premalignancy suggest that platelets are likely contributors to early cancer progression along with potential targets for early diagnosis (Han et al. 2014).
Immunotherapy?
Platelets are derived from megakaryocytes (Menter et al. 2014; Machlus and Italiano 2013), arising during the process of megakaryopoiesis from hematopoietic stem cell lineage that share commonalities with both lymphoid and granulocytic cells prior to differentiating toward hematopoietic progenitors. Specifically, in the bone marrow, hematopoiesis produces hemangioblasts that give rise to hematopoietic stem cells (HSCs) and multipotent progenitor cells (MPP) (Deutsch and Tomer 2013; Meng et al. 2012; Thon and Italiano 2010; Akbar et al. 2011). Genesis from MPP forms common myeloid progenitor cells (CMP) (Deutsch and Tomer 2013; Akbar et al. 2011; Thon and Italiano 2010; Meng et al. 2012) that give rise to megakaryocyte-erythroid progenitor (MEP) cells followed by megakaryopoietic progenitors (MKP) (Kanz et al. 1987). MKP then generate immature—followed by mature—megakaryocytes before finally generating platelets (Deutsch and Tomer 2013; Geddis 2010; Machlus and Italiano 2013; Tijssen and Ghevaert 2013; Meng et al. 2012). Immune thrombocytopenia (ITP) involves cellular checkpoints during the normal regulation of immunological self-reactivity and the development of B and T cells (Cines et al. 2009). This may occur through cell deletion, gene editing, induction of anergy, and extrinsic cellular suppression. When immune checkpoints fail, tolerance to self-antigens may be downregulated (Toltl et al. 2011; Cines et al. 2009). Checkpoint failure is a key target of immune checkpoint blockers that are being used in combination with interferon to overcome immune tolerance (Rafique et al. 2015). Importantly, platelet genesis and lineage development are also influenced by interferon (Rivadeneyra et al. 2015; D’Atri et al. 2015). With the advent of immune checkpoint cancer therapy, the influence of these agents may cross over to impact platelet biology and function (Topalian et al. 2015; Miller and Sadelain 2015; Gubin et al. 2014), which may also influence immune-related adverse events that are autoinflammatory in nature (Weber et al. 2015). Most notably, preclinical data demonstrate that pharmacologic inhibition or genetic deletion of COX-1 and COX-2 synergizes with anti-PD-1 blockade to induce the eradication of tumors more effectively than PD-1 treatment alone (Zelenay et al. 2015). Aspirin treatment was highly effective in synergizing with anti-PD1, but platelet function was not followed in these studies and deserves additional study (Zelenay et al. 2015). In view of their “first responder” properties, it is critical to develop a better understanding of this role or potentially harness platelets in some fashion to alter checkpoint blockade. One could envision taking bone marrow aspirates from cancer patients and engineering in vitro cultures of megakaryocytes (Nishikii et al. 2015; Hatami et al. 2015; Meinders et al. 2015; Panuganti et al. 2013; Thiele et al. 2012; Emond et al. 2012; Pallotta et al. 2009) to alter tumor recognition and inflammatory responses and reinfuse these reengineered platelets though adoptive bone marrow transfer.
Platelets: “First Responders” in the Complete Cancer Continuum
Platelets as vital contributors to the early stages of cancer formation are a new concept. Support for this notion comes from a variety of sources. One line of evidence is the long-standing link between chronic inflammation and the risk of developing colon cancer. Patients with inflammatory bowel disease or Crohn’s disease, for example, are at high risk for developing colon cancer (Herszenyi et al. 2015; Burisch and Munkholm 2015; Sanduleanu and Rutter 2014). A potential association with platelet biology may be found in mean platelet volume changes that accompany Crohn’s inflammatory disease states, among others (Kilincalp et al. 2015; Tang et al. 2015; Huang et al. 2013; Harris et al. 2011). Similarly, changes in mean platelet volume are found during adenoma formation (Kilincalp et al. 2015) along with colorectal cancer (Li et al. 2014; Kemal et al. 2014).
Additional support for this notion comes from Apc min\+ mouse crossbreeding with P-selectin −\− (CD62P) knockout mice, which lack expression on activated platelets and reduced VEGF expression (Qi et al. 2015). These crosses resulted in reduced formation of low- and high-grade adenomas accompanied by decreased vessel density (Qi et al. 2015). This study also examined human adenomas and stages I–IV of CRC progression by IHC for CD41 (integrin GPIIb subunit) positivity. Similar levels of platelet staining were observed in the polyp and stage I CRC tissues (Qi et al. 2015), suggesting that the platelets were already involved at the polyp stage. This CD41 staining increased in intensity with each progressive stage II–IV of CRC tumor development. Our group has also recently observed extravascular platelet structures in Apc min\+ polyps (Fig. 5). These data, along with our previous studies illustrating the dynamic activation of platelets in heterotypic platelet-tumor cell aggregates and syngeneic intravascular metastasis, highlight a role for platelets throughout the cancer continuum. Collectively, these data strongly suggest that altering the “first responder” capabilities of platelets is an important target for cancer prevention and therapy.

Platelet migration into adenoma perivascular spaces. Ten-week-old adenomatous polyposis coli Apc min\+ mice were euthanized and necropsied, and the intestinal tract was excised and fixed. Adenoma polyps were dissected and processed for transmission electron microscopy. The examination of polyp tissue revealed platelet structures in the perivascular space of blood vessels (arrows; insets). These platelets were underlying endothelial cells (EC) and had exited the vascular lumen (x4000, scale bars 2 μm)
Take Home Messages
-
Platelets are “First Responders” as active participants in the hemostasis, wounding, immune, and metastatic processes.
-
Platelets are often neglected or overlooked during in vivo experimental or pathologic observations due to their small size and lack of defining nuclei.
-
Surface receptors abound on both platelets and tumor cells trigger the primary response mechanisms that drive biologic function.
-
When examining heterotypic aggregates that form during tumor cell-induced platelet aggregation (TCIPA), the platelets tend to cluster at the center.
-
Platelets can also actively migrate across the inflamed vascular wall in response to a variety of microenvironmental and tumor factors.
-
Additional critical receptors that can impact interactions between platelets and tumor include those influencing immunomodulation.
-
Platelets serve as “First Responders” in the complete cancer continuum.
References
Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, Brannigan BW, Kapur R, Stott SL, Shioda T, Ramaswamy S, Ting DT, Lin CP, Toner M, Haber DA, Maheswaran S (2014) Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158:1110–1122
Ahmed AM, Pinheiro MM, Divis PC, Siner A, Zainudin R, Wong IT, Lu CW, Singh-Khaira SK, Millar SB, Lynch S, Willmann M, Singh B, Krishna S, Cox-Singh J (2014) Disease progression in Plasmodium knowlesi malaria is linked to variation in invasion gene family members. PLoS Neglect Trop Dis 8:e3086
Akbar H, Shang X, Perveen R, Berryman M, Funk K, Johnson JF, Tandon NN, Zheng Y (2011) Gene targeting implicates Cdc42 GTPase in GPVI and non-GPVI mediated platelet filopodia formation, secretion and aggregation. PLoS One 6:e22117
Algra AM, Rothwell PM (2012) Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol 13:518–527
Allegra CJ, Aberle DR, Ganschow P, Hahn SM, Lee CN, Millon-Underwood S, Pike MC, Reed SD, Saftlas AF, Scarvalone SA, Schwartz AM, Slomski C, Yothers G, Zon R (2009) NIH state-of-the-science conference statement: diagnosis and management of ductal carcinoma in situ (DCIS). NIH consensus and state-of-the-science statements 26:1–27
An T, Qin S, Xu Y, Tang Y, Huang Y, Situ B, Inal JM, Zheng L (2015) Exosomes serve as tumour markers for personalized diagnostics owing to their important role in cancer metastasis. J Extracell Vesic 4:27522
Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, Blumberg RS, Serhan CN (2005) Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci USA 102:7671–7676
Bakchoul T, Sachs UJ (2015) Platelet destruction in immune thrombocytopenia. Understanding the mechanisms. Hamostaseologie 35
Barni S, Bonizzoni E, Verso M, Gussoni G, Petrelli F, Perrone T, Agnelli G (2014) The effect of low-molecular-weight heparin in cancer patients: the mirror image of survival? Blood 124:155–156
Benazzi C, Al-Dissi A, Chau CH, Figg WD, Sarli G, de Oliveira JT, Gartner F (2014) Angiogenesis in spontaneous tumors and implications for comparative tumor biology. ScientificWorldJournal 2014:919570
Bennett JS, Zigmond S, Vilaire G, Cunningham ME, Bednar B (1999) The platelet cytoskeleton regulates the affinity of the integrin alpha(IIb)beta(3) for fibrinogen. J Biol Chem 274:25301–25307
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3:401–410
Best MG, Sol N, Kooi I, Tannous J, Westerman BA, Rustenburg F, Schellen P, Verschueren H, Post E, Koster J, Ylstra B, Ameziane N, Dorsman J, Smit EF, Verheul HM, Noske DP, Reijneveld JC, Nilsson RJ, Tannous BA, Wesseling P, Wurdinger T (2015) RNA-Seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer cell 28:666–676
Black PH (2002) Stress and the inflammatory response: a review of neurogenic inflammation. Brain Behav Immun 16:622–653
Bluff JE, Menakuru SR, Cross SS, Higham SE, Balasubramanian SP, Brown NJ, Reed MW, Staton CA (2009) Angiogenesis is associated with the onset of hyperplasia in human ductal breast disease. Br J Cancer 101:666–672
Borisova TA, Markosian RA (1977) Age and biosynthesis and breakdown of thrombocyte proteins. Biulleten’ eksperimental’noi biologii i meditsiny 83:20–21
Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, Pare A, Rousseau M, Naika GS, Levesque T, Laflamme C, Marcoux G, Lambeau G, Farndale RW, Pouliot M, Hamzeh-Cognasse H, Cognasse F, Garraud O, Nigrovic PA, Guderley H, Lacroix S, Thibault L, Semple JW, Gelb MH, Boilard E (2014) Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 124:2173–2183
Bousser MG, Amarenco P, Chamorro A, Fisher M, Ford I, Fox KM, Hennerici MG, Mattle HP, Rothwell PM, de Cordoue A, Fratacci MD, Investigators PS (2011) Terutroban versus aspirin in patients with cerebral ischaemic events (PERFORM): a randomised, double-blind, parallel-group trial. Lancet 377:2013–2022
Bouvenot G, Escande M, Xeridat B, Simonin G, Boucoiran J, Delboy C (1977) Thrombocytosis and cancer. Apropos of a chronological series of 100 patients. La semaine des hopitaux : organe fonde par l’Association d’enseignement medical des hopitaux de Paris 53:1921–1925
Boyanova D, Nilla S, Birschmann I, Dandekar T, Dittrich M (2012) PlateletWeb: a systems biologic analysis of signaling networks in human platelets. Blood 119:e22–e34
Brahimi-Horn MC, Giuliano S, Saland E, Lacas-Gervais S, Sheiko T, Pelletier J, Bourget I, Bost F, Feral C, Boulter E, Tauc M, Ivan M, Garmy-Susini B, Popa A, Mari B, Sarry JE, Craigen WJ, Pouyssegur J, Mazure NM (2015) Knockout of Vdac1 activates hypoxia-inducible factor through reactive oxygen species generation and induces tumor growth by promoting metabolic reprogramming and inflammation. Cancer Metab 3:8
Brandt E, Ludwig A, Petersen F, Flad HD (2000) Platelet-derived CXC chemokines: old players in new games. Immunol Rev 177:204–216
Breckenridge MT, Egelhoff TT, Baskaran H (2010) A microfluidic imaging chamber for the direct observation of chemotactic transmigration. Biomed Microdevices 12:543–553
Bruce IJ, Kerry R (1987) The effect of chloramphenicol and cycloheximide on platelet aggregation and protein synthesis. Biochem Pharmacol 36:1769–1773
Burisch J, Munkholm P (2015) The epidemiology of inflammatory bowel disease. Scand J Gastroenterol 50:942–951
Burnouf T, Goubran HA, Chen TM, Ou KL, El-Ekiaby M, Radosevic M (2013) Blood-derived biomaterials and platelet growth factors in regenerative medicine. Blood Rev 27:77–89
Carmeliet P (2005) VEGF as a key mediator of angiogenesis in cancer. Oncology 69(Suppl 3):4–10
Caruso RA, Fedele F, Finocchiaro G, Arena G, Venuti A (2012) Neutrophil-tumor cell phagocytosis (cannibalism) in human tumors: an update and literature review. Exp Oncol 34:306–311
Cattaneo M (2015) P2Y12 receptors: structure and function. J Thromb Haemost 13(Suppl 1):S10–S16
Cavalli LR, Varella-Garcia M, Liang BC (1997) Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ 8:1189–1198
Cedervall J, Zhang Y, Ringvall M, Thulin A, Moustakas A, Jahnen-Dechent W, Siegbahn A, Olsson AK (2013) HRG regulates tumor progression, epithelial to mesenchymal transition and metastasis via platelet-induced signaling in the pre-tumorigenic microenvironment. Angiogenesis 16:889–902
Chabert A, Hamzeh-Cognasse H, Pozzetto B, Cognasse F, Schattner M, Gomez RM, Garraud O (2015) Human platelets and their capacity of binding viruses: meaning and challenges? BMC Immunol 16:26
Chadha AS, Kocak-Uzel E, Das P, Minsky BD, Delclos ME, Mahmood U, Guha S, Ahmad M, Varadhachary GR, Javle M, Katz MH, Fleming JB, Wolff RA, Crane CH, Krishnan S (2015) Paraneoplastic thrombocytosis independently predicts poor prognosis in patients with locally advanced pancreatic cancer. Acta Oncol 54:971–978
Chan AT, Cook NR (2012) Are we ready to recommend aspirin for cancer prevention? Lancet 379:1569–1571
Chan AT, Arber N, Burn J, Chia WK, Elwood P, Hull MA, Logan RF, Rothwell PM, Schror K, Baron JA (2012) Aspirin in the chemoprevention of colorectal neoplasia: an overview. Cancer Prev Res 5:164–178
Chao BH, Lepeak L, Leal T, Robins HI (2011) Clinical use of the low-molecular-weight heparins in cancer patients: focus on the improved patient outcomes. Thrombosis 2011:530183
Chatterjee M, Huang Z, Zhang W, Jiang L, Hultenby K, Zhu L, Hu H, Nilsson GP, Li N (2011) Distinct platelet packaging, release, and surface expression of proangiogenic and antiangiogenic factors on different platelet stimuli. Blood 117:3907–3911
Chatterjee M, Borst O, Walker B, Fotinos A, Vogel S, Seizer P, Mack A, Alampour-Rajabi S, Rath D, Geisler T, Lang F, Langer HF, Bernhagen J, Gawaz M (2014a) Macrophage migration inhibitory factor limits activation-induced apoptosis of platelets via CXCR7-dependent Akt signaling. Circ Res 115:939–949
Chatterjee M, Seizer P, Borst O, Schonberger T, Mack A, Geisler T, Langer HF, May AE, Vogel S, Lang F, Gawaz M (2014b) SDF-1alpha induces differential trafficking of CXCR4-CXCR7 involving cyclophilin A, CXCR7 ubiquitination and promotes platelet survival. FASEB J 28:2864–2878
Cho MS, Bottsford-Miller J, Vasquez HG, Stone R, Zand B, Kroll MH, Sood AK, Afshar-Kharghan V (2012) Platelets increase the proliferation of ovarian cancer cells. Blood 120:4869–4872
Chopra H, Timar J, Rong X, Grossi IM, Hatfield JS, Fligiel SE, Finch CA, Taylor JD, Honn KV (1992) Is there a role for the tumor cell integrin alpha IIb beta 3 and cytoskeleton in tumor cell-platelet interaction? Clin Exp Metastasis 10:125–137
Cines DB, Bussel JB, Liebman HA, Luning Prak ET (2009) The ITP syndrome: pathogenic and clinical diversity. Blood 113:6511–6521
Colombo M, Raposo G, Thery C (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30:255–289
Cooper B, Ahern D (1979) Characterization of the platelet prostaglandin D2 receptor. Loss of prostaglandin D2 receptors in platelets of patients with myeloproliferative disorders. J Clin Invest 64:586–590
Coupland LA, Parish CR (2014) Platelets, selectins, and the control of tumor metastasis. Semin Oncol 41:422–434
Coupland LA, Chong BH, Parish CR (2012) Platelets and P-selectin control tumor cell metastasis in an organ-specific manner and independently of NK cells. Cancer Res 72:4662–4671
Crawford N, Sutton M, Horsfield GI (1967) Platelets in the carcinoid syndrome: a chemical and ultrastructural investigation. Br J Haematol 13:181–188
Crissman JD, Hatfield J, Schaldenbrand M, Sloane BF, Honn KV (1985) Arrest and extravasation of B16 amelanotic melanoma in murine lungs. A light and electron microscopic study. Lab Invest 53:470–478
Crissman JD, Hatfield JS, Menter DG, Sloane B, Honn KV (1988) Morphological study of the interaction of intravascular tumor cells with endothelial cells and subendothelial matrix. Cancer Res 48:4065–4072
Crooks MG, Fahim A, Naseem KM, Morice AH, Hart SP (2014) Increased platelet reactivity in idiopathic pulmonary fibrosis is mediated by a plasma factor. PLoS One 9:e111347
Cuzick J, Thorat MA, Bosetti C, Brown PH, Burn J, Cook NR, Ford LG, Jacobs EJ, Jankowski JA, La Vecchia C, Law M, Meyskens F, Rothwell PM, Senn HJ, Umar A (2015) Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann Oncol 26:47–57
D’Atri LP, Etulain J, Rivadeneyra L, Lapponi MJ, Centurion M, Cheng K, Yin H, Schattner M (2015) Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J Thromb Haemost 13:839–850
Davis AN, Afshar-Kharghan V, Sood AK (2014) Platelet effects on ovarian cancer. Semin Oncol 41:378–384
Dees C, Akhmetshina A, Zerr P, Reich N, Palumbo K, Horn A, Jungel A, Beyer C, Kronke G, Zwerina J, Reiter R, Alenina N, Maroteaux L, Gay S, Schett G, Distler O, Distler JH (2011) Platelet-derived serotonin links vascular disease and tissue fibrosis. J Exp Med 208:961–972
Deng G, Krishnakumar S, Powell AA, Zhang H, Mindrinos MN, Telli ML, Davis RW, Jeffrey SS (2014) Single cell mutational analysis of PIK3CA in circulating tumor cells and metastases in breast cancer reveals heterogeneity, discordance, and mutation persistence in cultured disseminated tumor cells from bone marrow. BMC Cancer 14:456
Deutsch VR, Tomer A (2013) Advances in megakaryocytopoiesis and thrombopoiesis: from bench to bedside. Br J Haematol 161:778–793
DeWitt DL, el-Harith EA, Kraemer SA, Andrews MJ, Yao EF, Armstrong RL, Smith WL (1990) The aspirin and heme-binding sites of ovine and murine prostaglandin endoperoxide synthases. J Biol Chem 265:5192–5198
Dhillon MS, Behera P, Patel S, Shetty V (2014) Orthobiologics and platelet rich plasma. Indian J Orthop 48:1–9
Djaldetti M, Strauss Z (1982) Emperipolesis by megakaryocytes in patients with non-Hodgkin’s lymphoma and megaloblastic anemia. J Submicrosc Cytol 14:407–413
Dona M, Fredman G, Schwab JM, Chiang N, Arita M, Goodarzi A, Cheng G, von Andrian UH, Serhan CN (2008) Resolvin E1, an EPA-derived mediator in whole blood, selectively counterregulates leukocytes and platelets. Blood 112:848–855
Du XY, Zabel BA, Myles T, Allen SJ, Handel TM, Lee PP, Butcher EC, Leung LL (2009) Regulation of chemerin bioactivity by plasma carboxypeptidase N, carboxypeptidase B (activated thrombin-activable fibrinolysis inhibitor), and platelets. J Biol Chem 284:751–758
Dubinski W, Gabril M, Iakovlev VV, Scorilas A, Youssef YM, Faragalla H, Kovacs K, Rotondo F, Metias S, Arsanious A, Plotkin A, Girgis AH, Streutker CJ, Yousef GM (2012) Assessment of the prognostic significance of endoglin (CD105) in clear cell renal cell carcinoma using automated image analysis. Hum Pathol 43:1037–1043
Ellingsen T, Storgaard M, Moller BK, Buus A, Andersen PL, Obel N, Stengaard-Pedersen K (2000) Migration of mononuclear cells in the modified Boyden chamber as evaluated by DNA quantification and flow cytometry. Scand J Immunol 52:257–263
Emond H, Boyer L, Roy DC, Pineault N (2012) Cotransplantation of ex vivo expanded progenitors with nonexpanded cord blood cells improves platelet recovery. Stem Cells Dev 21:3209–3219
Fang J, Nakamura H, Maeda H (2011) The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 63(3):136–151
Fedosov DA, Noguchi H, Gompper G (2014) Multiscale modeling of blood flow: from single cells to blood rheology. Biomech Model Mechanobiol 13:239–258
Fein MR, Egeblad M (2013) Caught in the act: revealing the metastatic process by live imaging. Dis Model Mech 6:580–593
Ferrati S, Shamsudeen S, Summers HD, Rees P, Abbey JV, Schmulen J, Liu X, Wong ST, Bean AJ, Ferrari M, Serda RE (2012) Inter-endothelial transport of microvectors using cellular shuttles and tunneling nanotubes. Small 8:3151–3160
Fidler IJ (1975) Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res 35:218–224
Fidler IJ (1978) Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res 38:2651–2660
Fidler IJ, Nicolson GL (1978) Tumor cell and host properties affecting the implantation and survival of blood-borne metastatic variants of B16 melanoma. Israel J Med Sci 14:38–50
Fitzpatrick FA, Gorman RR (1977) Platelet rich plasma transforms exogenous prostaglandin endoperoxide H2 into thromboxane A2. Prostaglandins 14:881–889
Folkman J, Watson K, Ingber D, Hanahan D (1989) Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 339:58–61
Fontana P, Zufferey A, Daali Y, Reny JL (2014) Antiplatelet therapy: targeting the TxA2 pathway. J Cardiovasc Transl Res 7:29–38
Fox SB, Harris AL (2004) Histological quantitation of tumour angiogenesis. Acta Pathol Microbiol Immunol Scand 112:413–430
Franchi F, Angiolillo DJ (2015) Novel antiplatelet agents in acute coronary syndrome. Nat Rev Cardiol 12:30–47
Fredman G, Van Dyke TE, Serhan CN (2010) Resolvin E1 regulates adenosine diphosphate activation of human platelets. Arterioscler Thromb Vasc Biol 30:2005–2013
Friedl P, Wolf K, Lammerding J (2011) Nuclear mechanics during cell migration. Curr Opin Cell Biol 23:55–64
Friedl P, Sahai E, Weiss S, Yamada KM (2012) New dimensions in cell migration. Nat Rev Mol Cell Biol 13:743–747
Friedl P, Wolf K, Zegers MM (2014) Rho-directed forces in collective migration. Nat Cell Biol 16:208–210
Fu Q, Cheng J, Gao Y, Zhang Y, Chen X, Xie J (2015) Protease-activated receptor 4: a critical participator in inflammatory response. Inflammation 38:886–895
Fukami MH, Salganicoff L (1977) Human platelet storage organelles. A review. Thromb Haemost 38:963–970
Fukumura D, Jain RK (2008) Imaging angiogenesis and the microenvironment. Acta Pathol Microbiol Immunol Scand 116:695–715
Garraud O, Berthet J, Hamzeh-Cognasse H, Cognasse F (2011) Pathogen sensing, subsequent signalling, and signalosome in human platelets. Thromb Res 127:283–286
Gasic GJ, Gasic TB, Stewart CC (1968) Antimetastatic effects associated with platelet reduction. Proc Natl Acad Sci USA 61:46–52
Gecse A, Kis B, Mezei Z, Telegdy G (1999) Effects of inflammatory neuropeptides on the arachidonate cascade of platelets. Int Arch Allergy Immunol 118:166–170
Geddis AE (2010) Megakaryopoiesis. Semin Hematol 47:212–219
Gleim S, Stitham J, Tang WH, Li H, Douville K, Chelikani P, Rade JJ, Martin KA, Hwa J (2013) Human thromboxane A2 receptor genetic variants: in silico, in vitro and “in platelet” analysis. PLoS One 8:e67314
Gleissner CA, von Hundelshausen P, Ley K (2008) Platelet chemokines in vascular disease. Arterioscler Thromb Vasc Biol 28:1920–1927
Goldstein LB, Rothwell PM (2012) Advances in prevention and health services delivery 2010-2011. Stroke 43:298–299
Goubran HA, Stakiw J, Radosevic M, Burnouf T (2014) Platelets effects on tumor growth. Semin Oncol 41:359–369
Goubran HA, Burnouf T, Stakiw J, Seghatchian J (2015) Platelet microparticle: a sensitive physiological “fine tuning” balancing factor in health and disease. Transfus Apher Sci 52:12–18
Grandis JR, Argiris A (2009) Targeting angiogenesis from premalignancy to metastases. Cancer Prev Res (Phila Pa) 2:291–294
Greinacher A (2015) Clinical practice. Heparin-induced thrombocytopenia. N Engl J Med 373:252–261
Gritsenko PG, Ilina O, Friedl P (2012) Interstitial guidance of cancer invasion. J Pathol 226:185–199
Grossi IM, Fitzgerald LA, Kendall A, Taylor JD, Sloane BF, Honn KV (1987) Inhibition of human tumor cell induced platelet aggregation by antibodies to platelet glycoproteins Ib and IIb/IIIa. Proc Soc Exp Biol Med 186:378–383
Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, Lam SS, Korman AJ, Allison JP, Freeman GJ, Sharpe AH, Pearce EL, Schumacher TN, Aebersold R, Rammensee HG, Melief CJ, Mardis ER, Gillanders WE, Artyomov MN, Schreiber RD (2014) Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515:577–581
Gunning AP, Chambers S, Pin C, Man AL, Morris VJ, Nicoletti C (2008) Mapping specific adhesive interactions on living human intestinal epithelial cells with atomic force microscopy. FASEB J 22:2331–2339
Haeger A, Krause M, Wolf K, Friedl P (2014) Cell jamming: collective invasion of mesenchymal tumor cells imposed by tissue confinement. Biochim Biophys Acta 1840:2386–2395
Hamet P (1996) Cancer and hypertension. An unresolved issue. Hypertension 28:321–324
Hammarstrom S, Falardeau P (1977) Resolution of prostaglandin endoperoxide synthase and thromboxane synthase of human platelets. Proc Natl Acad Sci USA 74:3691–3695
Hamzeh-Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O (2015) Platelets and infections - complex interactions with bacteria. Front Immunol 6:82
Han H, Cao FL, Wang BZ, Mu XR, Li GY, Wang XW (2014) Expression of angiogenesis regulatory proteins and epithelial-mesenchymal transition factors in platelets of the breast cancer patients. ScientificWorldJournal 2014:878209
Harris NR, Carter PR, Watts MN, Zhang S, Kosloski-Davidson M, Grisham MB (2011) Relationship among circulating leukocytes, platelets, and microvascular responses during induction of chronic colitis. Pathophysiology 18:305–311
Hart IR, Fidler IJ (1980) Role of organ selectivity in the determination of metastatic patterns of B16 melanoma. Cancer Res 40:2281–2287
Hatami J, Andrade PZ, Alves de Matos AP, Djokovic D, Lilaia C, Ferreira FC, Cabral JM, da Silva CL (2015) Developing a co-culture system for effective megakaryo/thrombopoiesis from umbilical cord blood hematopoietic stem/progenitor cells. Cytotherapy 17:428–442
Haynes CM, Fiorese CJ, Lin YF (2013) Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol 23:311–318
Hellberg C, Ostman A, Heldin CH (2010) PDGF and vessel maturation. Recent Results Cancer Res Fortschritte der Krebsforschung Progres dans les recherches sur le cancer 180:103–114
Herszenyi L, Barabas L, Miheller P, Tulassay Z (2015) Colorectal cancer in patients with inflammatory bowel disease: the true impact of the risk. Dig Dis 33:52–57
Hitchcock IS, Kaushansky K (2014) Thrombopoietin from beginning to end. Br J Haematol 165:259–268
Holla VR, Backlund MG, Yang P, Newman RA, DuBois RN (2008) Regulation of prostaglandin transporters in colorectal neoplasia. Cancer Prev Res 1:93–99
Hong SH, Tilan JU, Galli S, Izycka-Swieszewska E, Polk T, Horton M, Mahajan A, Christian D, Jenkins S, Acree R, Connors K, Ledo P, Lu C, Lee YC, Rodriguez O, Toretsky JA, Albanese C, Kitlinska J (2015) High neuropeptide Y release associates with Ewing sarcoma bone dissemination - in vivo model of site-specific metastases. Oncotarget 6:7151–7165
Honn KV, Cicone B, Skoff A (1981) Prostacyclin: a potent antimetastatic agent. Science 212:1270–1272
Honn KV, Menter D, Cavanaugh PG, Neagos G, Moilanen D, Taylor JD, Sloane BF (1983) A review of prostaglandins and the treatment of tumor metastasis. Acta Clin Belg 38:53–67
Honn KV, Tang DG, Crissman JD (1992) Platelets and cancer metastasis: a causal relationship? Cancer Metastasis Rev 11:325–351
Honn KV, Tang DG, Grossi IM, Renaud C, Duniec ZM, Johnson CR, Diglio CA (1994) Enhanced endothelial cell retraction mediated by 12(S)-HETE: a proposed mechanism for the role of platelets in tumor cell metastasis. Exp Cell Res 210:1–9
Horejsova M, Pavlickova V, Koukolik F, Strritesky J (1995) Morphologic verification of neoplastic portal vein obstruction. Casopis lekaru ceskych 134:655–657
Huang S, Yi FM, Zhou R, Chen M, Lei Y, Zhao JZ, Zhang H, Xia B (2013) The utility of platelet, mean platelet volume, and red cell distribution width in the diagnosis of active Crohn’s disease and intestinal tuberculosis. Saudi Med J 34:1161–1166
Huber HJ, Holvoet P (2015) Exosomes: emerging roles in communication between blood cells and vascular tissues during atherosclerosis. Curr Opin Lipidol 26:412–419
Hubertus K, Mischnik M, Timmer J, Herterich S, Mark R, Moulard M, Walter U, Geiger J (2014) Reciprocal regulation of human platelet function by endogenous prostanoids and through multiple prostanoid receptors. Eur J Pharmacol 740:15–27
Hull RD, Raskob GE, Hirsh J, Jay RM, Leclerc JR, Geerts WH, Rosenbloom D, Sackett DL, Anderson C, Harrison L et al (1986) Continuous intravenous heparin compared with intermittent subcutaneous heparin in the initial treatment of proximal-vein thrombosis. N Engl J Med 315:1109–1114
Ilan N, Mohsenin A, Cheung L, Madri JA (2001) PECAM-1 shedding during apoptosis generates a membrane-anchored truncated molecule with unique signaling characteristics. FASEB J 15:362–372
Jackson SP, Mistry N, Yuan Y (2000) Platelets and the injured vessel wall – “rolling into action”: focus on glycoprotein Ib/V/IX and the platelet cytoskeleton. Trends Cardiovasc Med 10:192–197
Jedlitschky G, Greinacher A, Kroemer HK (2012) Transporters in human platelets: physiologic function and impact for pharmacotherapy. Blood 119:3394–3402
Joosse SA, Gorges TM, Pantel K (2015) Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol Med 7:1–11
Kahaleh MB, Osborn I, Leroy EC (1982) Elevated levels of circulating platelet aggregates and beta-thromboglobulin in scleroderma. Ann Intern Med 96:610–613
Kahlert C, Kalluri R (2013) Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med 91:431–437
Kanz L, Lohr GW, Fauser AA (1987) Human megakaryocytic progenitor cells. Klinische Wochenschrift 65:297–307
Kemal Y, Yucel I, Ekiz K, Demirag G, Yilmaz B, Teker F, Ozdemir M (2014) Elevated serum neutrophil to lymphocyte and platelet to lymphocyte ratios could be useful in lung cancer diagnosis. Asian Pac J Cancer Prev 15:2651–2654
Keskin D, Kim J, Cooke VG, Wu CC, Sugimoto H, Gu C, De Palma M, Kalluri R, LeBleu VS (2015) Targeting vascular pericytes in hypoxic tumors increases lung metastasis via angiopoietin-2. Cell Rep 10:1066–1081
Kilincalp S, Coban S, Akinci H, Hamamc M, Karaahmet F, Coskun Y, Ustun Y, Simsek Z, Erarslan E, Yuksel I (2015) Neutrophil/lymphocyte ratio, platelet/lymphocyte ratio, and mean platelet volume as potential biomarkers for early detection and monitoring of colorectal adenocarcinoma. Eur J Cancer Prev 24:328–333
Kim KH, Barazia A, Cho J (2013) Real-time imaging of heterotypic platelet-neutrophil interactions on the activated endothelium during vascular inflammation and thrombus Formation in live mice. J Vis Exp. doi:10.3791/50329
Kisucka J, Butterfield CE, Duda DG, Eichenberger SC, Saffaripour S, Ware J, Ruggeri ZM, Jain RK, Folkman J, Wagner DD (2006) Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc Natl Acad Sci USA 103:855–860
Kofman AV, Letson C, Dupart E, Bao Y, Newcomb WW, Schiff D, Brown J, Abounader R (2013) The p53-microRNA-34a axis regulates cellular entry receptors for tumor-associated human herpes viruses. Med Hypotheses 81:62–67
Kraemer BF, Borst O, Gehring EM, Schoenberger T, Urban B, Ninci E, Seizer P, Schmidt C, Bigalke B, Koch M, Martinovic I, Daub K, Merz T, Schwanitz L, Stellos K, Fiesel F, Schaller M, Lang F, Gawaz M, Lindemann S (2010) PI3 kinase-dependent stimulation of platelet migration by stromal cell-derived factor 1 (SDF-1). J Mol Med 88:1277–1288
Kraemer BF, Schmidt C, Urban B, Bigalke B, Schwanitz L, Koch M, Seizer P, Schaller M, Gawaz M, Lindemann S (2011) High shear flow induces migration of adherent human platelets. Platelets 22:415–421
Krishnamoorthy N, Burkett PR, Dalli J, Abdulnour RE, Colas R, Ramon S, Phipps RP, Petasis NA, Kuchroo VK, Serhan CN, Levy BD (2015) Cutting edge: maresin-1 engages regulatory T cells to limit type 2 innate lymphoid cell activation and promote resolution of lung inflammation. J Immunol 194:863–867
Kumar A, Graham MD (2012) Mechanism of margination in confined flows of blood and other multicomponent suspensions. Phys Rev Lett 109:108102
Labelle M, Hynes RO (2012) The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov 2:1091–1099
Labelle M, Begum S, Hynes RO (2011) Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20:576–590
Langley RE, Rothwell PM (2013) Potential biomarker for aspirin use in colorectal cancer therapy. Nat Rev Clin Oncol 10:8–10
Langley RE, Rothwell PM (2014) Aspirin in gastrointestinal oncology: new data on an old friend. Curr Opin Oncol 26:441–447
Lee SY, Ferrari M, Decuzzi P (2009) Design of bio-mimetic particles with enhanced vascular interaction. J Biomech 42:1885–1890
Levin J, Conley CL (1964) Thrombocytosis associated with malignant disease. Arch Intern Med 114:497–500
Li JY, Li Y, Jiang Z, Wang RT, Wang XS (2014) Elevated mean platelet volume is associated with presence of colon cancer. Asian Pac J Cancer Prev 15:10501–10504
Li H, Liu K, Boardman LA, Zhao Y, Wang L, Sheng Y, Oi N, Limburg PJ, Bode AM, Dong Z (2015) Circulating prostaglandin biosynthesis in colorectal cancer and potential clinical significance. EBioMedicine 2:165–171
Liebig C, Ayala G, Wilks JA, Berger DH, Albo D (2009) Perineural invasion in cancer: a review of the literature. Cancer 115:3379–3391
Lima LG, Leal AC, Vargas G, Porto-Carreiro I, Monteiro RQ (2013) Intercellular transfer of tissue factor via the uptake of tumor-derived microvesicles. Thromb Res 132:450–456
Linder AE, Ni W, Diaz JL, Szasz T, Burnett R, Watts SW (2007) Serotonin (5-HT) in veins: not all in vain. J Pharmacol Exp Ther 323:415–421
Liverani E, Kilpatrick LE, Tsygankov AY, Kunapuli SP (2014) The role of P2Y(1)(2) receptor and activated platelets during inflammation. Curr Drug Targets 15:720–728
Liyasova MS, Schopfer LM, Lockridge O (2010) Reaction of human albumin with aspirin in vitro: mass spectrometric identification of acetylated lysines 199, 402, 519, and 545. Biochem Pharmacol 79:784–791
Lonsdorf AS, Kramer BF, Fahrleitner M, Schonberger T, Gnerlich S, Ring S, Gehring S, Schneider SW, Kruhlak MJ, Meuth SG, Nieswandt B, Gawaz M, Enk AH, Langer HF (2012) Engagement of alphaIIbbeta3 (GPIIb/IIIa) with alphanubeta3 integrin mediates interaction of melanoma cells with platelets: a connection to hematogenous metastasis. J Biol Chem 287:2168–2178
Lopez-Novoa JM, Nieto MA (2009) Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med 1:303–314
Macdonald JM, LeBlanc DA, Haas AL, London RE (1999) An NMR analysis of the reaction of ubiquitin with [acetyl-1-13C]aspirin. Biochem Pharmacol 57:1233–1244
Machlus KR, Italiano JE Jr (2013) The incredible journey: from megakaryocyte development to platelet formation. J Cell Biol 201:785–796
Magni P, Motta M (2001) Expression of neuropeptide Y receptors in human prostate cancer cells. Ann Oncol 12(Suppl 2):S27–S29
Marino M, Scuderi F, Ponte E, Maiuri MT, De Cristofaro R, Provenzano C, Rose-John S, Cittadini A, Bartoccioni E (2013) Novel path to IL-6 trans-signaling through thrombin-induced soluble IL-6 receptor release by platelets. J Biol Regul Homeost Agents 27:841–852
Marioni G, Staffieri A, Manzato E, Ralli G, Lionello M, Giacomelli L, Prosenikliev V, Marchese-Ragona R, Busnardo A, Bolzetta F, Blandamura S (2011) A higher CD105-assessed microvessel density and worse prognosis in elderly patients with laryngeal carcinoma. Arch Otolaryngol Head Neck Surg 137:175–180
Mauler M, Bode C, Duerschmied D (2015) Platelet serotonin modulates immune functions. Hamostaseologie 35
McDowell G, Temple I, Li C, Kirwan CC, Bundred NJ, McCollum CN, Burton IE, Kumar S, Byrne GJ (2005) Alteration in platelet function in patients with early breast cancer. Anticancer Res 25:3963–3966
Medeiros PJ, Al-Khazraji BK, Novielli NM, Postovit LM, Chambers AF, Jackson DN (2012) Neuropeptide Y stimulates proliferation and migration in the 4T1 breast cancer cell line. Int J Cancer (Journal international du cancer) 131:276–286
Meinders M, Kulu DI, van de Werken HJ, Hoogenboezem M, Janssen H, Brouwer RW, van Ijcken WF, Rijkers EJ, Demmers JA, Kruger I, van den Berg TK, Suske G, Gutierrez L, Philipsen S (2015) Sp1/Sp3 transcription factors regulate hallmarks of megakaryocyte maturation and platelet formation and function. Blood 125:1957–1967
Melo SA, Sugimoto H, O’Connell JT, Kato N, Villanueva A, Vidal A, Qiu L, Vitkin E, Perelman LT, Melo CA, Lucci A, Ivan C, Calin GA, Kalluri R (2014) Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 26:707–721
Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, LeBleu VS, Mittendorf EA, Weitz J, Rahbari N, Reissfelder C, Pilarsky C, Fraga MF, Piwnica-Worms D, Kalluri R (2015) Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 523(7559):177–182
Menakuru SR, Brown NJ, Staton CA, Reed MW (2008) Angiogenesis in pre-malignant conditions. Br J Cancer 99:1961–1966
Meng R, Wang Y, Yao Y, Zhang Z, Harper DC, Heijnen HF, Sitaram A, Li W, Raposo G, Weiss MJ, Poncz M, Marks MS (2012) SLC35D3 delivery from megakaryocyte early endosomes is required for platelet dense granule biogenesis and is differentially defective in Hermansky-Pudlak syndrome models. Blood 120:404–414
Menter DG, Dubois RN (2012) Prostaglandins in cancer cell adhesion, migration, and invasion. Int J Cell Biol 2012:723419
Menter DG, Harkins C, Onoda J, Riorden W, Sloane BF, Taylor JD, Honn KV (1987a) Inhibition of tumor cell induced platelet aggregation by prostacyclin and carbacyclin: an ultrastructural study. Invas Metastasis 7:109–128
Menter DG, Hatfield JS, Harkins C, Sloane BF, Taylor JD, Crissman JD, Honn KV (1987b) Tumor cell-platelet interactions in vitro and their relationship to in vivo arrest of hematogenously circulating tumor cells. Clin Exp Metastasis 5:65–78
Menter DG, Onoda JM, Moilanen D, Sloane BF, Taylor JD, Honn KV (1987c) Inhibition by prostacyclin of the tumor cell-induced platelet release reaction and platelet aggregation. J Natl Cancer Inst 78:961–969
Menter DG, Steinert BW, Sloane BF, Gundlach N, O’Gara CY, Marnett LJ, Diglio C, Walz D, Taylor JD, Honn KV (1987d) Role of platelet membrane in enhancement of tumor cell adhesion to endothelial cell extracellular matrix. Cancer Res 47:6751–6762
Menter DG, Steinert BW, Sloane BF, Taylor JD, Honn KV (1987e) A new in vitro model for investigation of tumor cell-platelet-endothelial cell interactions and concomitant eicosanoid biosynthesis. Cancer Res 47:2425–2432
Menter DG, Schilsky RL, DuBois RN (2010) Cyclooxygenase-2 and cancer treatment: understanding the risk should be worth the reward. Clin Cancer Res 16:1384–1390
Menter DG, Tucker SC, Kopetz S, Sood AK, Crissman JD, Honn KV (2014) Platelets and cancer: a casual or causal relationship: revisited. Cancer Metastasis Rev 33:231–269
Messerschmidt GL, Makuch R, Appelbaum F, Ungerleider RS, Abrams R, O’Donnell J, Holohan TV, Fontana J, Wright D, Anagnou NP et al (1988) A prospective randomized trial of HLA-matched versus mismatched single-donor platelet transfusions in cancer patients. Cancer 62:795–801
Mezouar S, Mege D, Darbousset R, Farge D, Debourdeau P, Dignat-George F, Panicot-Dubois L, Dubois C (2014) Involvement of platelet-derived microparticles in tumor progression and thrombosis. Semin Oncol 41:346–358
Mezouar S, Darbousset R, Dignat-George F, Panicot-Dubois L, Dubois C (2015) Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int J Cancer (Journal international du cancer) 136:462–475
Milane L, Singh A, Mattheolabakis G, Suresh M, Amiji MM (2015) Exosome mediated communication within the tumor microenvironment. J Control Release 219:278–294
Miller JF, Sadelain M (2015) The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell 27:439–449
Mitrugno A, Williams D, Kerrigan SW, Moran N (2014) A novel and essential role for FcgammaRIIa in cancer cell-induced platelet activation. Blood 123:249–260
Moins-Teisserenc H, Daubord M, Clave E, Douay C, Felix J, Marie-Cardine A, Ram-Wolff C, Maki G, Beldjord K, Homyrda L, Michel L, Bensussan A, Toubert A, Bagot M (2015) CD158k is a reliable marker for diagnosis of Sezary syndrome and reveals an unprecedented heterogeneity of circulating malignant cells. J Invest Dermatol 135:247–257
Nagy JA, Dvorak AM, Dvorak HF (2012) Vascular hyperpermeability, angiogenesis, and stroma generation. Cold Spring Harb Perspect Med 2:a006544
Nishikii H, Kanazawa Y, Umemoto T, Goltsev Y, Matsuzaki Y, Matsushita K, Yamato M, Nolan GP, Negrin R, Chiba S (2015) Unipotent megakaryopoietic pathway bridging hematopoietic stem cells and mature megakaryocytes. Stem Cells 33:2196–2207
Nistico P, Bissell MJ, Radisky DC (2012) Epithelial-mesenchymal transition: general principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb Perspect Med 4
O’Byrne KJ, Steward WP (2001) Tumour angiogenesis: a novel therapeutic target in patients with malignant disease. Expert Opin Emerg Drugs 6:155–174
Ogawa T, Kitamura K, Kangawa K, Matsuo H, Eto T (1992) Platelet neuropeptide Y in spontaneously hypertensive rats. J Hypertens 10:765–771
Oleksowicz L, Dutcher JP, DeLeon-Fernandez M, Etkind P (1997) A GPIb alpha-related protein is expressed by fresh human breast carcinoma tissue and is regulated by a PKC-sensitive mechanism. Exp Cell Res 237:110–117
Orellana R, Kato S, Erices R, Bravo ML, Gonzalez P, Oliva B, Cubillos S, Valdivia A, Ibanez C, Branes J, Barriga MI, Bravo E, Alonso C, Bustamente E, Castellon E, Hidalgo P, Trigo C, Panes O, Pereira J, Mezzano D, Cuello MA, Owen GI (2015) Platelets enhance tissue factor protein and metastasis initiating cell markers, and act as chemoattractants increasing the migration of ovarian cancer cells. BMC Cancer 15:290
Pai VP, Marshall AM, Hernandez LL, Buckley AR, Horseman ND (2009) Altered serotonin physiology in human breast cancers favors paradoxical growth and cell survival. Breast Cancer Res 11:R81
Pallotta I, Lovett M, Rice W, Kaplan DL, Balduini A (2009) Bone marrow osteoblastic niche: a new model to study physiological regulation of megakaryopoiesis. PLoS One 4:e8359
Pang JH, Coupland LA, Freeman C, Chong BH, Parish CR (2015a) Activation of tumour cell ECM degradation by thrombin-activated platelet membranes: potentially a P-selectin and GPIIb/IIIa-dependent process. Clin Exp Metastasis 32:495–505
Pang Q, Liu C, Qu K, Liu S (2015b) Conflicting relationship between platelets and prognosis of hepatocellular carcinoma: is platelet-derived serotonin involved in? Liver Int 35:2484
Panuganti S, Schlinker AC, Lindholm PF, Papoutsakis ET, Miller WM (2013) Three-stage ex vivo expansion of high-ploidy megakaryocytic cells: toward large-scale platelet production. Tissue Eng A 19:998–1014
Pasula S, Cai X, Dong Y, Messa M, McManus J, Chang B, Liu X, Zhu H, Mansat RS, Yoon SJ, Hahn S, Keeling J, Saunders D, Ko G, Knight J, Newton G, Luscinskas F, Sun X, Towner R, Lupu F, Xia L, Cremona O, De Camilli P, Min W, Chen H (2012) Endothelial epsin deficiency decreases tumor growth by enhancing VEGF signaling. J Clin Invest 122:4424–4438
Pawlak D, Malczyk E, Darewicz J, Azzadin A, Buczko W (2000) Platelet serotonergic mechanisms in patients with cancer of the urinary bladder. Thromb Res 98:367–374
Penington DG, Streatfield K, Roxburgh AE (1976) Megakaryocytes and the heterogeneity of circulating platelets. Br J Haematol 34:639–653
Petrucci G, De Cristofaro R, Rutella S, Ranelletti FO, Pocaterra D, Lancellotti S, Habib A, Patrono C, Rocca B (2011) Prostaglandin E2 differentially modulates human platelet function through the prostanoid EP2 and EP3 receptors. J Pharmacol Exp Ther 336:391–402
Pinckard RN, Hawkins D, Farr RS (1968) In vitro acetylation of plasma proteins, enzymes and DNA by aspirin. Nature 219:68–69
Pirman DA, Efuet E, Ding XP, Pan Y, Tan L, Fischer SM, DuBois RN, Yang P (2013) Changes in cancer cell metabolism revealed by direct sample analysis with MALDI mass spectrometry. PLoS One 8:e61379
Powell AA, Talasaz AH, Zhang H, Coram MA, Reddy A, Deng G, Telli ML, Advani RH, Carlson RW, Mollick JA, Sheth S, Kurian AW, Ford JM, Stockdale FE, Quake SR, Pease RF, Mindrinos MN, Bhanot G, Dairkee SH, Davis RW, Jeffrey SS (2012) Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS One 7:e33788
Qi C, Li B, Guo S, Wei B, Shao C, Li J, Yang Y, Zhang Q, Li J, He X, Wang L, Zhang Y (2015) P-selectin-mediated adhesion between platelets and tumor cells promotes intestinal tumorigenesis in Apc(Min/+) mice. Int J Biol Sci 11:679–687
Raeisossadati R, Farshchian M, Ganji A, Tavassoli A, Velayati A, Dadkhah E, Chavoshi S, Mehrabi Bahar M, Memar B, Rajabi Mashhadi MT, Naseh H, Forghanifard MM, Moghbeli M, Moaven O, Abbaszadegan MR (2011) Quantitative analysis of TEM-8 and CEA tumor markers indicating free tumor cells in the peripheral blood of colorectal cancer patients. Int J Colorectal Dis 26:1265–1270
Rafii S, Cao Z, Lis R, Siempos II, Chavez D, Shido K, Rabbany SY, Ding BS (2015) Platelet-derived SDF-1 primes the pulmonary capillary vascular niche to drive lung alveolar regeneration. Nat Cell Biol 17:123–136
Rafique I, Kirkwood JM, Tarhini AA (2015) Immune checkpoint blockade and interferon-alpha in melanoma. Semin Oncol 42:436–447
Raju NC, Eikelboom JW, Hirsh J (2008) Platelet ADP-receptor antagonists for cardiovascular disease: past, present and future. Nat Clin Pract Cardiovasc Med 5:766–780
Rastogi V, Sharma R, Misra SR, Yadav L, Sharma V (2014) Emperipolesis - a review. J Clin Diagn Res 8:ZM01–ZM02
Ratajczak P, Leboeuf C, Wang L, Briere J, Loisel-Ferreira I, Thieblemont C, Zhao WL, Janin A (2012) BCL2 expression in CD105 positive neoangiogenic cells and tumor progression in angioimmunoblastic T-cell lymphoma. Mod Pathol 25:805–814
Rath D, Chatterjee M, Borst O, Muller K, Stellos K, Mack AF, Bongartz A, Bigalke B, Langer H, Schwab M, Gawaz M, Geisler T (2014) Expression of stromal cell-derived factor-1 receptors CXCR4 and CXCR7 on circulating platelets of patients with acute coronary syndrome and association with left ventricular functional recovery. Eur Heart J 35:386–394
Rath D, Chatterjee M, Borst O, Muller K, Langer H, Mack AF, Schwab M, Winter S, Gawaz M, Geisler T (2015) Platelet surface expression of stromal cell-derived factor-1 receptors CXCR4 and CXCR7 is associated with clinical outcomes in patients with coronary artery disease. J Thromb Haemost 13:719–728
Reynolds AB, Herbert L, Cleveland JL, Berg ST, Gaut JR (1992) p120, a novel substrate of protein tyrosine kinase receptors and of p60v-src, is related to cadherin-binding factors beta-catenin, plakoglobin and armadillo. Oncogene 7:2439–2445
Reynolds AB, Daniel J, McCrea PD, Wheelock MJ, Wu J, Zhang Z (1994) Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol Cell Biol 14:8333–8342
Rivadeneyra L, Pozner RG, Meiss R, Fondevila C, Gomez RM, Schattner M (2015) Poly (I:C) downregulates platelet production and function through type I interferon. Thromb Haemost 114
Rival Y, Del Maschio A, Rabiet MJ, Dejana E, Duperray A (1996) Inhibition of platelet endothelial cell adhesion molecule-1 synthesis and leukocyte transmigration in endothelial cells by the combined action of TNF-alpha and IFN-gamma. J Immunol 157:1233–1241
Robbins PD, Morelli AE (2014) Regulation of immune responses by extracellular vesicles. Nat Rev Immunol 14:195–208
Roop RP, Naughton MJ, Van Poznak C, Schneider JG, Lammers PE, Pluard TJ, Johnson F, Eby CS, Weilbaecher KN (2013) A randomized phase II trial investigating the effect of platelet function inhibition on circulating tumor cells in patients with metastatic breast cancer. Clin Breast Cancer 13:409–415
Ross R, Glomset J, Kariya B, Raines E (1978) Role of platelet factors in the growth of cells in culture. National Cancer Institute Monograph 103–108
Rossaint J, Zarbock A (2015) Platelets in leucocyte recruitment and function. Cardiovasc Res 107(3):386–395
Rothwell PM (2013a) Alternate-day, low-dose aspirin and cancer risk. Ann Intern Med 159:148–150
Rothwell PM (2013b) Aspirin in prevention of sporadic colorectal cancer: current clinical evidence and overall balance of risks and benefits. Recent Results Cancer Res (Fortschritte der Krebsforschung Progres dans les recherches sur le cancer) 191:121–142
Rothwell PM, Price JF, Fowkes FG, Zanchetti A, Roncaglioni MC, Tognoni G, Lee R, Belch JF, Wilson M, Mehta Z, Meade TW (2012) Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 379:1602–1612
Rowlinson SW, Crews BC, Goodwin DC, Schneider C, Gierse JK, Marnett LJ (2000) Spatial requirements for 15-(R)-hydroxy-5Z,8Z,11Z, 13E-eicosatetraenoic acid synthesis within the cyclooxygenase active site of murine COX-2. Why acetylated COX-1 does not synthesize 15-(R)-hete. J Biol Chem 275:6586–6591
Salzman EW, Rosenberg RD, Smith MH, Lindon JN, Favreau L (1980) Effect of heparin and heparin fractions on platelet aggregation. J Clin Invest 65:64–73
Sanduleanu S, Rutter MD (2014) Interval colorectal cancers in inflammatory bowel disease: the grim statistics and true stories. Gastrointest Endosc Clin N Am 24:337–348
Sapino A, Bongiovanni M, Cassoni P, Righi L, Arisio R, Deaglio S, Malavasi F (2001) Expression of CD31 by cells of extensive ductal in situ and invasive carcinomas of the breast. J Pathol 194:254–261
Satelli A, Mitra A, Brownlee Z, Xia X, Bellister S, Overman MJ, Kopetz S, Ellis LM, Meng QH, Li S (2015) Epithelial-mesenchymal transitioned circulating tumor cells capture for detecting tumor progression. Clin Cancer Res 21:899–906
Schmidt EM, Kraemer BF, Borst O, Munzer P, Schonberger T, Schmidt C, Leibrock C, Towhid ST, Seizer P, Kuhl D, Stournaras C, Lindemann S, Gawaz M, Lang F (2012) SGK1 sensitivity of platelet migration. Cell Physiol Biochem 30:259–268
Schroeder CP, Yang P, Newman RA, Lotan R (2007) Simultaneous inhibition of COX-2 and 5-LOX activities augments growth arrest and death of premalignant and malignant human lung cell lines. J Exp Ther Oncol 6:183–192
Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S (2013) Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 24:130–137
Serhan CN (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510:92–101
Serhan CN, Gotlinger K, Hong S, Arita M (2004) Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: an overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat 73:155–172
Serhan CN, Gotlinger K, Hong S, Lu Y, Siegelman J, Baer T, Yang R, Colgan SP, Petasis NA (2006) Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J Immunol 176:1848–1859
Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, Oh SF, Spite M (2009) Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med 206:15–23
Shenkman B, Brill A, Brill G, Lider O, Savion N, Varon D (2004) Differential response of platelets to chemokines: RANTES non-competitively inhibits stimulatory effect of SDF-1 alpha. J Thromb Haemost 2:154–160
Smith SJ, Tilly H, Ward JH, Macarthur DC, Lowe J, Coyle B, Grundy RG (2012) CD105 (Endoglin) exerts prognostic effects via its role in the microvascular niche of paediatric high grade glioma. Acta Neuropathol 124:99–110
Spectre G, Zhu L, Ersoy M, Hjemdahl P, Savion N, Varon D, Li N (2012) Platelets selectively enhance lymphocyte adhesion on subendothelial matrix under arterial flow conditions. Thromb Haemost 108:328–337
Spite M, Serhan CN (2010) Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res 107:1170–1184
Spite M, Claria J, Serhan CN (2014) Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab 19:21–36
Starke J, Wehrle-Haller B, Friedl P (2014) Plasticity of the actin cytoskeleton in response to extracellular matrix nanostructure and dimensionality. Biochem Soc Trans 42:1356–1366
Staton CA, Chetwood AS, Cameron IC, Cross SS, Brown NJ, Reed MW (2007) The angiogenic switch occurs at the adenoma stage of the adenoma carcinoma sequence in colorectal cancer. Gut 56:1426–1432
Stocks T, Van Hemelrijck M, Manjer J, Bjorge T, Ulmer H, Hallmans G, Lindkvist B, Selmer R, Nagel G, Tretli S, Concin H, Engeland A, Jonsson H, Stattin P (2012) Blood pressure and risk of cancer incidence and mortality in the Metabolic Syndrome and Cancer Project. Hypertension 59:802–810
Stone RL, Nick AM, McNeish IA, Balkwill F, Han HD, Bottsford-Miller J, Rupairmoole R, Armaiz-Pena GN, Pecot CV, Coward J, Deavers MT, Vasquez HG, Urbauer D, Landen CN, Hu W, Gershenson H, Matsuo K, Shahzad MM, King ER, Tekedereli I, Ozpolat B, Ahn EH, Bond VK, Wang R, Drew AF, Gushiken F, Lamkin D, Collins K, DeGeest K, Lutgendorf SK, Chiu W, Lopez-Berestein G, Afshar-Kharghan V, Sood AK (2012) Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med 366:610–618
Stopczynski RE, Normolle DP, Hartman DJ, Ying H, DeBerry JJ, Bielefeldt K, Rhim AD, DePinho RA, Albers KM, Davis BM (2014) Neuroplastic changes occur early in the development of pancreatic ductal adenocarcinoma. Cancer Res 74:1718–1727
Sun YP, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, Colgan SP, Petasis NA, Serhan CN (2007) Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem 282:9323–9334
Tanaka K, Morimoto Y, Toiyama Y, Okugawa Y, Inoue Y, Uchida K, Kimura K, Mizoguchi A, Kusunoki M (2012) Intravital dual-colored visualization of colorectal liver metastasis in living mice using two photon laser scanning microscopy. Microsc Res Tech 75:307–315
Tanaka K, Toiyama Y, Okugawa Y, Okigami M, Inoue Y, Uchida K, Araki T, Mohri Y, Mizoguchi A, Kusunoki M (2014) In vivo optical imaging of cancer metastasis using multiphoton microscopy: a short review. Am J Transl Res 6:179–187
Tang J, Gao X, Zhi M, Zhou HM, Zhang M, Chen HW, Yang QF, Liang ZZ (2015) Plateletcrit: a sensitive biomarker for evaluating disease activity in Crohn’s disease with low hs-CRP. J Dig Dis 16:118–124
Thiele W, Krishnan J, Rothley M, Weih D, Plaumann D, Kuch V, Quagliata L, Weich HA, Sleeman JP (2012) VEGFR-3 is expressed on megakaryocyte precursors in the murine bone marrow and plays a regulatory role in megakaryopoiesis. Blood 120:1899–1907
Thomas CP, Morgan LT, Maskrey BH, Murphy RC, Kuhn H, Hazen SL, Goodall AH, Hamali HA, Collins PW, O’Donnell VB (2010) Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. J Biol Chem 285:6891–6903
Thon JN, Italiano JE (2010) Platelet formation. Semin Hematol 47:220–226
Tijssen MR, Ghevaert C (2013) Transcription factors in late megakaryopoiesis and related platelet disorders. J Thromb Haemost 11:593–604
Tilan JU, Everhart LM, Abe K, Kuo-Bonde L, Chalothorn D, Kitlinska J, Burnett MS, Epstein SE, Faber JE, Zukowska Z (2013) Platelet neuropeptide Y is critical for ischemic revascularization in mice. FASEB J 27:2244–2255
Tokarev AA, Butylin AA, Ataullakhanov FI (2011a) Platelet adhesion from shear blood flow is controlled by near-wall rebounding collisions with erythrocytes. Biophys J 100:799–808
Tokarev AA, Butylin AA, Ermakova EA, Shnol EE, Panasenko GP, Ataullakhanov FI (2011b) Finite platelet size could be responsible for platelet margination effect. Biophys J 101:1835–1843
Toltl LJ, Nazi I, Jafari R, Arnold DM (2011) Piecing together the humoral and cellular mechanisms of immune thrombocytopenia. Semin Thromb Hemost 37:631–639
Topalian SL, Drake CG, Pardoll DM (2015) Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27:450–461
Tucker SC, Honn KV (2013) Emerging targets in lipid-based therapy. Biochem Pharmacol 85:673–688
Umar A, Steele V, Menter D, Hawk E (2016) Mechanisms of non-steroidal anti-inflammatory drugs (NSAIDs) in cancer prevention. Semin Oncol 43(1):65–77
Unwith S, Zhao H, Hennah L, Ma D (2015) The potential role of HIF on tumour progression and dissemination. Int J Cancer (Journal international du cancer) 136:2491–2503
Uppal A, Wightman SC, Ganai S, Weichselbaum RR, An G (2014) Investigation of the essential role of platelet-tumor cell interactions in metastasis progression using an agent-based model. Theor Biol Med Model 11:17
van Dongen CJ, van den Belt AG, Prins MH, Lensing AW (2004) Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev CD001100
van Es N, Sturk A, Middeldorp S, Nieuwland R (2014) Effects of cancer on platelets. Semin Oncol 41:311–318
von Hundelshausen P, Weber C (2007) Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res 100:27–40
Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39:359–407
Wallace EL, Smyth SS (2013) Targeting platelet thrombin receptor signaling to prevent thrombosis. Pharmaceuticals 6:915–928
Walsh TG, Metharom P, Berndt MC (2015) The functional role of platelets in the regulation of angiogenesis. Platelets 26:199–211
Wang R, Stone R, Kaelber J, Rochat R, Nick A, Vijayan K, Afshar-Kharghan V, Schmid M, Dong J, Sood A, Chiu W (2015) Electron cryotomography reveals ultrastructure alterations in platelets from patients with ovarian cancer. Proc Natl Acad Sci USA 112(46):14266–14271
Weber JS, Yang JC, Atkins MB, Disis ML (2015) Toxicities of immunotherapy for the practitioner. J Clin Oncol 33:2092–2099
Wharin C, Tagalakis V (2014) Management of venous thromboembolism in cancer patients and the role of the new oral anticoagulants. Blood Rev 28:1–8
Woods JR (1964) Experimental studies of the intravascular dissemination of ascitic V2 carcinoma cells in the rabbit, with special reference to fibrinogen and fibrinolytic agents. Bulletin der Schweizerischen Akademie der Medizinischen Wissenschaften 20:92–121
Xia P, Wang S, Guo Z, Yao X (2008) Emperipolesis, entosis and beyond: dance with fate. Cell Res 18:705–707
Yang P, Chan D, Felix E, Madden T, Klein RD, Shureiqi I, Chen X, Dannenberg AJ, Newman RA (2006) Determination of endogenous tissue inflammation profiles by LC/MS/MS: COX- and LOX-derived bioactive lipids. Prostaglandins Leukot Essent Fatty Acids 75:385–395
Yang P, Cartwright C, Chan D, Ding J, Felix E, Pan Y, Pang J, Rhea P, Block K, Fischer SM, Newman RA (2014a) Anticancer activity of fish oils against human lung cancer is associated with changes in formation of PGE2 and PGE3 and alteration of Akt phosphorylation. Mol Carcinog 53:566–577
Yang P, Jiang Y, Fischer SM (2014b) Prostaglandin E3 metabolism and cancer. Cancer Lett 348:1–11
Zacharski LR, Prandoni P, Monreal M (2005) Warfarin versus low-molecular-weight heparin therapy in cancer patients. Oncologist 10:72–79
Zaslavsky A, Baek KH, Lynch RC, Short S, Grillo J, Folkman J, Italiano JE Jr, Ryeom S (2010) Platelet-derived thrombospondin-1 is a critical negative regulator and potential biomarker of angiogenesis. Blood 115:4605–4613
Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, Sahai E, Reis ESC (2015) Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162:1257–1270
Zhang X, Yuan X, Shi H, Wu L, Qian H, Xu W (2015) Exosomes in cancer: small particle, big player. J Hematol Oncol 8:83
Zharikov S, Shiva S (2013) Platelet mitochondrial function: from regulation of thrombosis to biomarker of disease. Biochem Soc Trans 41:118–123
Acknowledgments
Grant and other support: Boone Pickens Distinguished Chair for Early Prevention of Cancer to EH, the Duncan Family Institute, Colorectal Cancer Moon Shot, 1R01CA187238-01, 5R01CA172670-03 and 1R01CA184843-01A1, and CA177909. Many thanks to Kenneth Dunner Jr. for his electron microscopy expertise and effort.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Menter, D.G. et al. (2017). Platelets: “First Responders” in Cancer Progression and Metastasis. In: Gresele, P., Kleiman, N., Lopez, J., Page, C. (eds) Platelets in Thrombotic and Non-Thrombotic Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-47462-5_74
Download citation
DOI: https://doi.org/10.1007/978-3-319-47462-5_74
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-47460-1
Online ISBN: 978-3-319-47462-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)