
Abstract
The promotion of tumour metastasis by platelets may occur through several mechanisms including the induction of a more metastatic phenotype in tumour cells and assisted extravasation of circulating tumour cells. Whilst the mechanisms underlying platelet-assisted extravasation have been extensively studied, much less attention has been paid to the mechanisms underlying platelet promotion of an aggressive phenotype within a tumour cell population. Herein, we demonstrate in vitro that MDA-MB-231 breast carcinoma cells incubated with washed thrombin-activated platelet membranes adopt a Matrigel-degrading phenotype in a dose- and contact time-dependent manner. The same phenotypic change was observed with three other human tumour cell lines of diverse anatomical origin. Moreover, tumour cell lines that had been cultured with washed thrombin-activated platelet membranes had a greater metastatic capacity when injected into mice. This in vivo effect was reliant upon a co-incubation period of >2 h implying a mechanism involving more than platelet membrane binding that occurred within 5 min. Upon further investigation it was found that simultaneous blocking of the platelet-membrane proteins P-selectin and GPIIb/IIIa prevented interactions between platelet membranes and MDA-MB-231 cells but also significantly reduced the ability of tumour cells to degrade Matrigel. These results confirm that platelets induce a more aggressive phenotype in tumour cells but also identify the platelet proteins involved in this effect. P-selectin and GPIIb/IIIa also play a role in assisting tumour cell extravasation and, thus, are ideal targets for the therapeutic intervention of both stages of platelet-assisted metastasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The pathophysiological role of platelets in cancer metastasis has long been recognised and was first demonstrated by Gasic et al. in 1968 [1]. In these definitive experiments, platelet numbers were reduced in mice prior to the injection of tumour cells with a dramatic reduction in lung metastases observed. Since then, a large body of work has been published confirming these findings and demonstrating the underlying mechanisms of platelet-assisted tumour cell extravasation (for a review see [2] and [3]). Much less attention, however, has been given to the pro-metastatic effect of platelets at the level of the tumour cell. Janowska-Wieczorek et al. showed that platelet microvesicles activate pathways involved in tumour cell proliferation, chemotaxis and metastasis, and in transfer of adhesion proteins and chemokine receptors to the tumour cell surface [4, 5]. More recently Labelle et al. demonstrated that the platelet-derived soluble factor, TGF-β, as well as direct contact between platelets and tumour cells enhanced metastasis through the activation of pathways that induced an epithelial-to-mesenchymal transition [6]. Platelet-derived TGF-β was also demonstrated to enhance the proliferation of ovarian cancer cells in vitro and in vivo [7]. The aim of the study described herein was to investigate the mechanisms underlying the direct contact effect of platelets on the metastatic phenotype of tumour cells as distinct from the pro-metastatic role of soluble factors released from platelets. We demonstrate that contact of several tumour cell lines with washed activated platelet membranes alters tumour cell behaviour, increasing extracellular matrix (ECM) degradation potential in vitro and in vivo. Furthermore, we show that these effects on tumour cell behaviour can be inhibited when platelet P-selectin and GPIIb/IIIa are simultaneously blocked, implying that these two platelet proteins play a cooperative role in stimulating tumour cell invasiveness.
Materials and methods
Mice
Female nude mice on a Swiss background were obtained from the pathogen-free facilities at the John Curtin School of Medical Research, Australian National University, Canberra, Australia. All experimental procedures were approved by the Australian National University Animal Experimentation Ethics Committee.
Antibodies
The platelet adhesion molecule-specific antibodies used in this study were a generous gift from Professor Michael Berndt, College of Medicine and Health, University College Cork, Ireland. They were against human P-selectin (rabbit polyclonal), PECAM-1 (CD31) (rabbit polyclonal), GPIIb/IIIa (clone CRC64), GPIbα (clone AK2) and α2β1 integrin (clone AK7).
Tumour cell lines
Human tumour cell lines studied included MDA-MB-231 breast carcinoma (ATCC HTB-26), MM170 melanoma (CBA-1346), U937 histiocytic lymphoma (ATCC CRL-1593.2), KJD keratinocytes (from P.G. Parsons, Queensland Institute of Medical Research), MOLT-4 T-lymphoma (ATCC CRL-1582), Jurkat T-lymphoma (DSMZ—ACC 282), K562 erythroleukaemia (ATCC CCL-243), COLO397 colonic carcinoma (from R. Whitehead, Ludwig Institute for Cancer Research, Melbourne, Australia) and PE.01 ovarian carcinoma (ECACC—10032308). The MDA-MB-231 breast carcinoma cell lines were cultured in DMEM (Gibco, BRL, Life Technologies, Gaithersburg, MD) while other human tumour cell lines were cultured in RPMI 1640 medium (Gibco, BRL, Life Technologies, Gaithersburg, MD) supplemented with 10 % (v/v) heat-inactivated foetal calf serum (Sigma Chemical Co., St Louis, MO) and 30 μg/ml penicillin (Gibco, BRL, Life Technologies, Gaithersburg, MD) at 37 °C in a 5 % CO2 atmosphere.
Preparation of thrombin-activated human platelet membranes
Expired human platelet-rich plasma (PRP) was centrifuged for 15 min at 800×g at room temperature. The washed platelet pellet was resuspended in PBS and incubated with 1 unit/ml of thrombin (Sigma Chemical Co., St Louis, MO) at 37 °C for 5 min. To stop the reaction, 10 ml of cold PBS was added and the platelet membranes were pelleted by centrifugation at 3000×g for 5 min at 4 °C. The thrombin-activated human platelet membranes were washed twice with cold PBS, resuspended in 10 ml of PBS (approximately 2.2 × 1010 platelet equivalents/ml) and stored at 4 °C ready for use or stored frozen in aliquots at −80 °C. To determine the extent of contamination of the thrombin-activate platelet membranes with soluble platelet proteins, platelet factor 4 (PF4) was measured using an ELISA assay (abcam, Cambridge, UK) and compared with the PF4 levels in the platelet releasate and whole platelets solubilised by the addition of 0.1 % Triton-X 100. The PF4 content of platelet membranes was 20 %, and the platelet releasate 105 % of that measured in solubilised whole platelets (Fig. S1).
Morphological studies of tumour cell behaviour on Matrigel
Matrigel BM solution (Becton Dickinson Labware, Bedford, MA) was thawed overnight on ice, diluted 1:1 with ice-cold serum free DMEM, aliquoted into a pre-cooled 12-well tissue culture plate (1 ml/well) then incubated at 37 °C in a 5 % CO2 atmosphere for 30 min. The solid Matrigel was then overlaid with 2 ml of complete DMEM containing 1 × 107 MDA-MB-231 tumour cells in either the presence or absence of thrombin-activated human platelet membranes at 5 × 108 platelet equivalents/ml. The culture plate was incubated at 37 °C in a 5 % CO2 atmosphere for 120 h with tumour cell behaviour being examined at selected time intervals by phase contrast microscopy and selected fields being photographed with a Photometric Sensys Digital Camera (Olympus International Ltd, Sydney, Australia).
Matrigel degradation assay
A Matrigel degradation assay was developed using flat-bottomed 96-well culture plates. Matrigel coated plates were prepared as described above except that a flat-bottomed 96-well culture plate was used and 35 μl of diluted Matrigel added per well. Tumour cells (3 × 104) in DMEM were added (200 μl/well) in either the presence or absence of different concentrations of thrombin-activated human platelet membranes (5 × 101–5 × 108 platelet equivalents/ml) and the plates incubated at 37 °C in a 5 % CO2 atmosphere for up to 120 h. Matrigel-containing wells cultured with tumour cells alone or with platelet membranes alone were included as controls. After 120 h, the culture medium containing the tumour cells and platelet membranes was removed, the plate was washed twice with cold PBS, 100 μl of cold PBS was added to each well and the Matrigel was thawed overnight on ice. The matrix protein content of the thawed Matrigel was measured using a Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA). Briefly, solutions of Matrigel proteins were serially diluted twofold in 100 μl/well of PBS, then 100 μl of Dye Reagent Concentrate was added to each well. The plate was mixed, incubated at room temperature for 5 min, then measured for Matrigel protein content by a Thermo-Max microplate reader (Molecular Devices Corporation, Sunnyvale, CA) with a test wavelength of 570 nm and a reference wavelength of 450 nm. Matrigel protein concentrations were expressed as mg/ml relative to a BSA standard curve. Data are presented as % Matrigel solubilisation based on the formula: 100 − (protein concentration of Matrigel containing wells exposed to tumour cells or platelets/protein concentration of Matrigel containing wells cultured without tumour cells or platelets × 100).
Stimulation of human tumour cells with thrombin-activated human platelet membranes
Sub-confluent (90 % confluent) tumour cells were washed twice prior to serum-free culture medium containing thrombin-activated human platelet membranes at a concentration 5 × 108 platelet equivalents/ml being added. The cells were then cultured at 37 °C in a 5 % CO2 atmosphere for either 2 or 24 h. After stimulation, the culture medium was removed, and the adherent cells washed thoroughly with PBS to remove platelet membranes.
Pulmonary metastasis assays
Following stimulation of various human tumour cell lines with thrombin-activated human platelet membranes for 2 or 24 h, the tumour cell monolayer was washed three times to remove platelet membranes and dead cells. Tumour cells were released from the flasks with 0.125 % trypsin/PBS treatment for 2 min at 37 °C, pelleted by centrifugation (300×g) and a single-cell suspension of the human tumour cells was prepared in serum-free culture medium prior to i.v. injection of 1 × 106 platelet membrane-stimulated tumour cells into the lateral tail vein of immunodeficient nude mice. The same number of untreated tumour cells was injected into nude mice as controls.
Histological analysis and quantification of tumour metastatic nodules
Mouse pulmonary metastasis was investigated at either 4 or 6 weeks after tumour cell injection. The lungs were removed and surface metastatic nodules counted under an inverted light microscope prior to fixation in 4 % paraformaldehyde, dehydration in 70 % ethanol and staining with haematoxylin and eosin. The pulmonary metastatic nodules were examined using a Leica Q500MC digital image microscopy analysis system (Leica, Cambridge, UK). The extent of pulmonary metastasis was assessed by measuring the relative proportion of each lung section that was occupied by tumour cells, with 25 fields/recipient mouse being examined at 50 x magnification using NIH Image software (Version 1.62). Briefly, digital images were acquired by the Leica Q500MC digital camera from the 4 corners and the centre of each tissue section (50× magnification). The areas of tumour cells, staining a dense pink colour by haematoxylin and eosin, were traced manually using NIH Image software and the relative area of each section occupied by tumour cells ascertained.
Binding of fluorescein-conjugated thrombin-activated human platelet membranes to tumour cells
Fluorescein-conjugated human platelet membranes were prepared by directly incubating the thrombin-activated human platelet membranes with N-hydroxysuccinimide (NHS)-fluorescein (Pierce, Rockford, IL). 1 ml of thrombin-activated human platelet membranes (2.2 × 1010 platelet equivalent/ml) was centrifuged at 3000×g for 5 min at 4 °C. The pelleted platelet membranes were resuspended in 1 ml of PBS, 5 μl of 20 mM NHS-fluorescein was added and then incubated at 37 °C for 30 min. The fluorescein-conjugated platelet membranes were centrifuged and washed twice (3000×g, 5 min for each wash) with a 20 mM Tris–HCl buffer (pH 7.2), then incubated overnight in the same buffer at 4 °C to allow the dissociation of non-covalently bound fluorescein. The fluorescein-labelled human platelet membranes were then centrifuged, resuspended in 1 ml of PBS and stored at 4 °C ready for use.
Antibody inhibition of platelet membrane binding and Matrigel degradation by MDA-MB-231 cells
Fluorescein-conjugated thrombin-activated human platelet membranes (5 × 108 platelet equivalents/ml) were pre-incubated with individual antibodies against platelet adhesion molecules (50 μg/ml) on ice for 30 min. Either antibody treated-platelet membranes or untreated platelet membranes were added to 3 × 104 MDA-MB-231 tumour cells in 100 μl of DMEM in a 96-well plate and incubated at 37 °C for 30 min (final concentration = 2.5 × 108 platelet equivalents/ml). Each treatment was performed in triplicate. The plate was gently mixed by vortexing and then incubated either at 4 °C or in a 37 °C tissue culture incubator for the times indicated. To prevent further platelet membrane binding before flow cytometric analysis, 100 μl/well of ice cold 0.1 % BSA/PBS was added and flow cytometric analysis was performed immediately using a FACScan® (Becton Dickinson, San Jose, CA). For inhibition of Matrigel degradation assays, the different platelet adhesion molecule-specific antibodies (50 μg/ml) were pre-incubated on ice for 30 min with thrombin-activated human platelet membranes (2.5 × 108 platelet equivalents/ml). The antibody-treated or untreated (control) platelet membranes were then added to MDA-MB-231 tumour cells (3 × 104/well) and cultured in Matrigel containing wells. Platelet membranes alone and tumour cells alone were included as negative controls whereas the combination of thrombin-activated human platelet membranes and tumour cells acted as the positive control. The Matrigel protein content in each well was measured after 120 h of culture as described above.
Statistical analysis
The significance of observed differences between experimental groups was analysed using a Students unpaired t test or one-way ANOVA and, when significant, a post hoc Dunnett’s multiple comparison (MC) test was performed. Values were reported as the mean ± standard error of the mean (SEM). Differences in mean values were considered significant if P < 0.05.
Results
Tumour cells exhibit a modified morphology and degrade Matrigel in vitro following contact with washed thrombin-activated platelet membranes
In initial experiments MDA-MB-231 mammary adenocarcinoma cells were added to a thick layer of Matrigel in 12-well tissue culture plates and the behaviour of the tumour cells monitored when cultured in the presence or absence of washed thrombin-activated platelet membranes. Within 12 h of culture initiation, MDA-MB-231 tumour cells had spread and begun to attach to the Matrigel. Control tumour cells (Fig. 1a) and platelet membrane-stimulated tumour cells (Fig. 1b) displayed a similar morphology except that some visible holes were seen in the Matrigel layer of cultures containing the platelet membrane-treated tumour cells (Fig. 1b, arrowheads). After 24 h of culture, the difference in morphology between the control and the stimulated cells became more pronounced. Whilst the control cells kept growing on the Matrigel surface as a monolayer (Fig. 1c), the platelet membrane-stimulated tumour cells generally displayed a more migratory morphology than the unstimulated tumour cells (Fig. 1d). After 72 h of culture, while the control tumour cells continued to grow on the Matrigel surface, becoming sub-confluent (Fig. 1e), the platelet membrane-stimulated tumour cells formed disorganised cell islands with interconnecting migratory channels (Fig. 1f). At this time, the Matrigel displayed a highly granular appearance, suggesting that the platelet membrane-stimulated tumour cells were degrading the Matrigel.

MDA-MB-231 tumor cells exhibit a modified morphology when cultured on Matrigel in the presence of thrombin-activated human platelet membranes. a, c, e Phase contrast images of MDA-MB-231 tumor cells cultured for 12, 24 and 72 h, respectively, on a thin layer of Matrigel. b, d, f Morphology of MDA-MB-231 tumor cells cultured on Matrigel for 12, 24 and 72 h, respectively, in the presence of thrombin-activated human platelet membranes (5 × 108 platelet equivalents/ml). Arrows in b indicate visible holes in the matrix produced by invading tumor cells whereas arrows in d and f highlight long cellular connections between aggregates of tumor cells not seen in control cultures
To investigate this possibility, MDA-MB-231 tumour cells were cultured on a Matrigel layer in 96-well tissue culture plates, the Matrigel protein content in culture wells being determined at various time points over a 120 h culture period. A reduction over time was seen in the Matrigel protein content of wells containing platelet membrane-stimulated MDA-MB-231 tumour cells compared with control wells that contained either unstimulated MDA-MB-231 tumour cells (tumour cells alone) or thrombin-activated platelet membranes alone (P < 0.001) (Fig. 2a).

A number of human tumor cell lines, when exposed to thrombin-activated human platelet membranes, solubilize Matrigel proteins. a, b MDA-MB-231 tumor cells solubilize Matrigel in a time-dependent (a) and platelet membrane concentration-dependent (b) manner. In a tumor cells were cultured with membranes from 5 × 108 platelet equivalents/ml (n = 5). Results in b obtained from 5-day cultures (n = 6). Asterisks indicate a significant reduction in recovered Matrigel proteins when compared with cultures containing 231 cells alone (one-way ANOVA + post hoc Dunnett’s MC test *P < 0.05; ***P < 0.001). c Effect of thrombin-activated platelet membranes on the ability of different human tumor cell lines to solubilize Matrigel. Data presented as percentage solubilization of Matrigel proteins. Cell lines that exhibited significantly enhanced solubilization of Matrigel when exposed to thrombin-activated platelet membranes are indicated by P values in figure. Asterisks refer to cell lines that displayed significant (t-test, P = 0.005) Matrigel solubilization in the absence of platelet membranes. All error bars are standard error of the mean (n = 3)
The concentration of thrombin-activated human platelet membranes required for induction of Matrigel degradation was also evaluated. Figure 2b demonstrates the platelet number-dependent degradation of the Matrigel by the tumour cells. A significant reduction in Matrigel protein content was observed when the MDA-MB-231 tumour cells were co-cultured with thrombin-activated platelet membranes derived from ≥5 × 105 platelet equivalents/ml, with there being a concentration-dependent increase in Matrigel degradation with platelet membrane concentrations up to 5 × 108 platelet equivalents/ml (P < 0.001–0.05).
Following the demonstration that MDA-MB-231 tumour cells could be activated by platelet membranes to degrade Matrigel in vitro, a number of other human tumour cell lines were tested. Significantly enhanced Matrigel degradation was observed with the MM170 malignant melanoma (P = 0.003), KJD keratinocytes (P = 0.008) and MOLT-4 T-lymphoma (P = 0.04) cell lines when cultured with thrombin-activated human platelet membranes (Fig. 2c). It should be noted that the MM170 and KJD tumour cell lines also exhibited an intrinsic ability to degrade Matrigel in the absence of platelet membranes.
Ability of washed thrombin-activated human platelet membranes to facilitate pulmonary metastasis of tumour cells in nude mice
To investigate the stimulatory potential of washed thrombin-activated human platelet membranes on in vivo tumour cell metastasis, the metastatic potential of the MDA-MB-231 tumour cells was examined in immunodeficient nude mice before and after exposure of the tumour cells to platelet membranes. Six weeks following injection of the tumour cells, a highly significant (P < 0.001) increase in lung metastatic nodules was observed in mice injected with tumour cells stimulated for 24 h with thrombin-activated human platelet membranes, compared with mice receiving untreated control tumour cells (Fig. 3a). In these mice, the percentage of each section occupied by tumour cells was 39.7 % compared to 22.6 % in those receiving control tumour cells (Fig. 3b), this almost twofold difference being highly significant (P < 0.0003). An even more pronounced enhancement of tumour metastasis (i.e. fivefold) following 24 h platelet membrane treatment of tumour cells was observed 4 weeks after tumour cell injection (Fig. 3b). Histological examination of these lungs revealed that in mice that were injected with control tumour cells (Fig. 3c, d), peri-alveolar tumour infiltration was limited in comparison to lung sections from mice injected with platelet membrane-treated MDA-MB-231 tumour cells (Fig. 3e, f) where the extent of tumour infiltration caused distortion of the lung anatomy.

Pre-incubation of human tumor cells with thrombin-activated platelet membranes can render some tumor cell lines more metastatic in nude mice. a MDA-MB-231 tumor cells, following 24 h pre-incubated with thrombin-activated platelet membranes (5 × 108 platelet equivalents/ml), produced significantly greater numbers of macroscopic pulmonary metastatic nodules (t-test, P < 0.001) than control tumor cells 6 weeks after tumor cell injection (n = 5 mice/group). b Effect of pre-incubating MDA-MB-231 tumor cells with or without thrombin-activated platelet membranes (5 × 108 platelet equivalents/ml) on the percentage of lung tissue occupied by tumor cells 4 weeks (4wk) and 6 weeks (6wk) after tumor cell injection into nude mice (n = 5 fields/mouse and 5 mice/group). Values in brackets represent the time (2 or 24 h) the tumor cells were pre-incubated with platelet membranes prior to injection into animals. c–f Hematoxylin and eosin staining of lung tissue sections from nude mice 4 weeks after receiving either control MDA-MB-231 tumor cells (c, d) or MDA-MB-231 cells pre-incubated for 24 h with platelet membranes (e, f). Tumor cell infiltration into the alveolar wall is more evident on the left-hand side of dotted line in e in comparison to that seen in c, and between all alveolar spaces in f in comparison to d. Arrows indicate tumour infiltration around blood vessels (c) and within alveolar walls (d, f). g Effect of pre-incubating various human tumor cell lines with or without thrombin-activated platelet membranes (5 × 108 platelet equivalents/ml) on the percentage of lung tissue occupied by tumor cells 4 weeks after tumor cell injection into nude mice (n = 5 fields/mouse and 5 mice/group). All error bars are standard error of the mean
It was important to determine whether platelet membranes simply bound to the tumour cell surface enhanced metastasis or whether long-term activation of the tumour cells by the platelet membranes was required. Thus, the effect of pre-incubating MDA-MB-231 tumour cells with platelet membranes for 2 h, rather than 24 h, on tumour cell metastasis was investigated. Histological assessment of the relative lung tumour area of mice injected with control or 2 h platelet membrane-stimulated tumour cells revealed a relative tumour area of 25.6 and 27.1 %, respectively (Fig. 3b), indicating that prolonged incubation with platelet membranes was required for enhanced metastasis to occur.
The effect of thrombin-activated human platelet membranes on the in vivo pulmonary metastasis of a number of other human tumour types, namely MM170 melanoma, KJD keratinocyte carcinoma, MOLT-4 T-lymphoma, U937 histiocytic lymphoma, K562 erythroleukaemia and COLO397 colonic carcinoma was also investigated. Each of the tumour cell lines was cultured for 24 h with thrombin-activated platelet membranes and lung metastatic nodules were quantified histologically at 4 weeks following injection into recipient immunodeficient nude mice. Similar to the results found with the MDA-MB-231 tumour cells (reproduced in Fig. 3g), enhanced pulmonary metastasis was also observed in mice injected with platelet membrane-stimulated MM170 melanoma cells (Fig. 3g). The lungs from mice receiving control tumour cells showed 8.9 % of the lung section being invaded by tumour cells compared with 22.8 % in the lung tissue from mice that were injected with platelet membrane-treated tumour cells (P < 0.0001). No difference was observed in the lung tumour area in mice injected with the other tumour cell lines following their exposure to platelet membranes although there was a similar trend with the MOLT-4 T lymphoma cell line that did not quite reach statistical significance (P = 0.07) (Fig. 3g). Table 1 summarises the in vitro and in vivo interactions and effects of thrombin-activated platelet membranes on the various human tumour cell lines tested.
The experiments described above demonstrated that thrombin-activated platelet membranes enhanced the capacity of tumour cells to degrade the ECM in vitro and colonise the lungs in vivo. To further probe this phenomenon, the adhesion of fluorescein-conjugated thrombin-activated platelet membranes to tumour cells was investigated using flow cytometry. Following incubation of MDA-MB-231 tumour cells with fluorescein-labelled thrombin-activated platelet membranes at 37 °C for 30 min, there was a significant increase in the fluorescence intensity of the tumour cell population due to the binding of platelet membranes (Fig. S1 A–C). To determine the optimal conditions for the binding of fluorescein-labelled thrombin-activated human platelet membranes to the MDA-MB-231 tumour cells, a time-course of binding was performed. As expected, the binding at 37 °C was rapid, occurring within 5 min. Longer incubation times resulted in greater membrane binding, with substantial binding being observed after 60 min (Fig. S1 D). The optimal platelet membrane concentration required for binding at 37 °C after 30 min incubation was also evaluated using a twofold series of dilutions of thrombin-activated human platelet membranes from 5 × 108 to 3.13 × 107 platelet equivalents/ml in PBS. This clearly demonstrated that platelet membrane binding to MDA-MB-231 tumour cells was concentration dependent with maximal binding being obtained with 2.5 and 5 × 108 platelet equivalents/ml and binding above background still being observed at the lowest concentration used (Fig. S1 E).
Inhibition of the binding of thrombin-activated platelet membranes to tumour cells by platelet adhesion molecule-specific antibodies
In order to characterise the possible cell surface molecules involved in the adhesion of thrombin-activated platelet membranes to MDA-MB-231 tumour cells, a number of platelet membrane adhesion molecule-specific antibodies were used to determine their ability to inhibit the platelet membrane-tumour cell interaction. None of the antibodies tested individually demonstrated inhibitory activity, interestingly, however, a combination of the CRC64 monoclonal antibody (mAb) specific for GPIIb/IIIa and the P-selectin polyclonal antibody resulted in substantially reduced fluorescent platelet membrane binding to the MDA-MB-231 tumour cells to a level that was not significantly different to the background fluorescent signal of tumour cells alone (P < 0.0001, Fig. 4a). In contrast, combining the P-selectin specific antibody with antibodies against CD31, GPIbα or the α2β1 integrin had no effect on the highly significant platelet membrane binding (P < 0.0001, Fig. 4a). These results suggest that both P-selectin and the GPIIb/IIIa integrin may participate in the interaction of thrombin-activated human platelet membranes with the MDA-MB-231 tumour cells.

Binding of thrombin-activated platelet membranes to MDA-MD-231 tumor cells and activation of Matrigel solubilization involves P-selectin and the integrin, GPIIb/IIIa. a, b Ability of a range of platelet binding antibodies, either alone or in combination with an anti-P-selectin antibody, to block the binding of fluorescein-labeled thrombin-activated platelet membranes to MDA-MD-231 tumor cells, as measured by flow cytometry (n = 7/group) (a), or to inhibit platelet membrane induced solubilization of Matrigel by MDA-MD-231 tumor cells (n = 7/group) (b). Dotted line in a represents background autofluorescence of tumor cells (negative control) and in b represents Matrigel solubilization by the platelet membrane activated tumor cells in the absence of any anti-platelet antibodies (positive control). One-way ANOVA + post hoc Dunnett’s MC test, *P < 0.05; ***P < 0.001, all error bars are standard error of the mean
Effects of platelet adhesion molecule-specific antibodies on platelet membrane-induced Matrigel degradation by MDA-MB-231 tumour cells
As some of the platelet adhesion molecule-specific antibodies were able to inhibit the binding of platelet membranes to tumour cells, these same antibodies were evaluated for their ability to inhibit platelet membrane-induced Matrigel degradation by MDA-MB-231 tumour cells. Similar to the results seen in the platelet membrane binding assay, none of the antibodies tested individually showed any significant inhibitory effects, however, the combination of the P-selectin specific antibody and the CRC64 mAb specific for GPIIb/IIIa completely inhibited platelet membrane-induced Matrigel degradation by the MDA-MB-231 tumour cells (P < 0.05, Fig. 4b). These results are consistent with engagement of both P-selectin and GPIIb/IIIa for optimal activation of MDA-MB-231 tumour cells by thrombin-activated platelet membranes.
Discussion
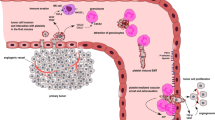
Platelets play an important role in facilitating tumour metastasis by virtue of their ability to interact with tumour cells within the vascular microenvironment. These interactions may occur at the primary tumour level, as tumour-associated blood vessels are poorly formed and ‘leaky’. Furthermore, tumour cells may form a mosaic alongside endothelial cells lining these vessels [8]. Alternatively, platelets may interact with tumour cells that have entered the bloodstream in the process of haematogenous metastasis. Contact between platelets and tumour cells may promote metastasis in two ways: firstly, by promoting the metastatic behaviour of tumour cells, and secondly, by assisting circulating tumour cell extravasation. A significant body of work has been performed elucidating the mechanisms of platelet-assisted tumour cell extravasation [9–15], however, much less attention has been paid to the molecular mechanisms underpinning the promotion by platelets of an aggressive phenotype in a tumour cell population.
Platelets play a seminal role in the wound healing process releasing a multitude of proteins including chemokines, cytokines, degradative enzymes and growth factors that promote cell proliferation and migration, extracellular matrix remodelling and angiogenesis [16]. These effects are not limited to ‘normal’ cells but also to tumorigenic cells. Studies by others have shown that platelet membranes can enhance the secretion of matrix metalloproteinases by MDA-MB-231 tumour cells [17, 18]. Furthermore, it was observed that platelet-derived microvesicles enhanced lung and breast cancer metastasis, invasiveness and angiogenesis [4, 5]. More recently studies by Labelle et al. revealed that direct platelet-tumour cell contact and platelet-derived TGF-β synergistically induced an epithelial to mesenchymal transition and enhanced metastasis of breast and colon carcinoma cells [6]. The role of platelet-derived TGF-β on tumour cell proliferation was subsequently confirmed [7].
The study described herein investigated the direct contact effect of washed thrombin-activated platelet membranes on ECM degradation and metastatic behaviour of a variety of human tumour cell lines. Following thrombin activation, platelets express adhesion molecules (e.g. P-selectin) on their surface and inactive GPIIb/IIIa receptors become activated. It is known that the platelet membrane is a cellular element bearing the highest density of adhesion molecules per unit surface area among the known blood cells [19].
The most striking observation described in this work was that washed, thrombin-activated platelet membranes dramatically increase the ECM degrading capacity of some tumour cell lines in vitro and lung colonizing ability in vivo. In the case of the MDA-MB-231 mammary adenocarcinoma cells, this effect was shown to be both platelet number- and time-dependent with a co-incubation time of >2 h being required to influence metastasis in vivo. These results strongly indicate that thrombin-activated platelet membranes can induce a more aggressive phenotype in mammary adenocarcinoma cells rather than just simply aiding tumour cell adherence. Eight other human tumour cell lines were also examined for a platelet membrane effect and three lines of diverse anatomical origin (i.e. melanoma, lymphoma and keratinocyte carcinoma) displayed an enhanced ability to degrade Matrigel. The MM170 melanoma cells and MOLT-4 T lymphoma were also more highly metastatic in vivo following incubation with thrombin-activated platelet membranes, although the in vivo results for MOLT-4 did not quite reach statistical significance (P = 0.07). The metastatic behaviour of five tumour cell lines was not observed to increase despite binding to platelet membranes. It is possible that intrinsic differences in these tumour cells, such as reduced surface receptor expression, may result in weaker platelet membrane binding and hence insufficient signalling to drive the behaviour of tumour cells to that of a metastatic phenotype. Other cell lines may bind platelets efficiently but not be pre-programmed to respond to platelet binding. Furthermore, some of the cell lines studied may be at a maximum metastatic capacity and do not require the additional stimulation of platelet binding.
In order to stimulate tumour cells to become metastatic, it is reasonable to assume that platelet membranes must be able to bind to ligands on the surface of the tumour cells and then exert their stimulatory effect via trans-membrane signalling. Using fluorescein conjugated thrombin-activated platelet membranes it was found that thrombin-activated platelet membranes bound to MDA-MB-231 tumour cells within a few minutes at 37 °C, with binding shown to be platelet membrane concentration-dependent. With regard to the latter, the concentration of thrombin-activated platelet membranes required for maximal binding to the MDA-MB-231 cells (i.e. 5 × 108 platelet equivalents/ml) is equivalent to the normal plasma platelet concentration.
Further studies were performed to characterise the possible cell surface molecules that are involved in mediating the platelet membrane-tumour cell interaction and, in particular, that trigger enhanced extracellular matrix invasion by the tumour cells. A number of platelet adhesion molecule-specific antibodies were tested for their ability to inhibit the platelet membrane-tumour cell interaction. Several studies have emphasised the critical role of P-selectin in mediating the interaction of platelets with tumour cells during tumour metastasis [14, 20]. Furthermore, CD24, the major carrier of sialyl-Lewis antigens on tumour cells that interact with P-selectin, has been demonstrated to promote tumour cell proliferation, adhesion and invasion through suppression of tissue factor pathway inhibitor-2 [21]. A P-selectin specific antibody alone, however, did not significantly inhibit either platelet membrane binding to tumour cells or the ability of the platelet membranes to enhance Matrigel degradation by MDA-MB-231 tumour cells. The same results were observed with the CRC64 mAb specific for the platelet GPIIb/IIIa integrin. In contrast, a combination of these two antibodies significantly inhibited both platelet membrane binding to the MDA-MB-231 tumour cells and platelet membrane induced-Matrigel degradation by these cells. Therefore, whilst these results support the current literature which suggests that the GPIIb/IIIa integrin is necessary for tumour-cell-induced platelet aggregation [22] and endothelial cell binding and migration [23], and that P-selectin plays a critical role in initiating platelet mediated-tumour metastasis [24], our data suggest that both P-selectin and GPIIb/IIIa are required for the effective interaction of tumour cells with thrombin-activated platelet membranes. In fact, it seems highly likely that P-selectin and GPIIb/IIIa on platelet membranes act synergistically in increasing the ability of MDA-MB-231 tumour cells to degrade the ECM in vitro and be more metastatic in vivo. This function of platelets may be a normal physiological process involved in wound healing and angiogenesis that has been subverted by some metastatic tumour cells. Based on our findings a combination of P-selectin and GPIIb/IIIa antagonists may represent a novel approach to inhibit tumour metastasis. Finally it should be emphasised that the studies described herein along with evidence provided by Janowska-Wieczorek et al. [4, 5] and Labelle et al. [6] suggest that platelets play a more active role in tumour metastasis than was previously realised.
Abbreviations
- BSA:
-
Bovine serum albumin
- DMEM:
-
Dulbecco’s minimal essential media
- ECM:
-
Extracellular matrix
- GP:
-
Glycoprotein
- mAb:
-
Monoclonal antibody
- PBS:
-
Phosphate buffered saline
- PL-Memb:
-
Thrombin-activated platelet membranes
- PRP:
-
Platelet-rich plasma
- TGF-β:
-
Transforming growth factor beta
References
Gasic GJ, Gasic TB, Stewart CC (1968) Antimetastatic effects associated with platelet reduction. Proc Natl Acad Sci USA 61(1):46–52
Bendas G, Borsig L (2012) Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol 2012:676731. doi:10.1155/2012/676731
Gay LJ, Felding-Habermann B (2011) Contribution of platelets to tumour metastasis. Nat Rev Cancer 11(2):123–134
Janowska-Wieczorek A, Wysoczynski M, Kijowski J, Marquez-Curtis L, Machalinski B, Ratajczak J, Ratajczak MZ (2005) Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer 113(5):752–760
Janowska-Wieczorek A, Marquez-Curtis LA, Wysoczynski M, Ratajczak MZ (2006) Enhancing effect of platelet-derived microvesicles on the invasive potential of breast cancer cells. Transfusion 46(7):1199–1209
Labelle M, Begum S, Hynes RO (2011) Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20(5):576–590
Cho MS, Bottsford-Miller J, Vasquez HG, Stone R, Zand B, Kroll MH, Sood AK, Afshar-Kharghan V (2012) Platelets increase the proliferation of ovarian cancer cells. Blood 120(24):4869–4872. doi:10.1182/blood-2012-06-438598
Chang YS, di Tomaso E, McDonald DM, Jones R, Jain RK, Munn LL (2000) Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc Natl Acad Sci USA 97(26):14608–14613
Gasic GJ (1984) Role of plasma, platelets, and endothelial cells in tumor metastasis. Cancer Metastasis Rev 3(2):99–114
Tanaka NG, Tohgo A, Ogawa H (1986) Platelet-aggregating activities of metastasizing tumor cells. V. In situ roles of platelets in hematogenous metastases. Invasion Metastasis 6(4):209–224
Honn KV, Tang DG, Crissman JD (1992) Platelets and cancer metastasis: a causal relationship? Cancer Metastasis Rev 11(3–4):325–351
Menter DG, Hatfield JS, Harkins C, Sloane BF, Taylor JD, Crissman JD, Honn KV (1987) Tumor cell-platelet interactions in vitro and their relationship to in vivo arrest of hematogenously circulating tumor cells. Clin Exp Metastasis 5(1):65–78
Nierodzik ML, Klepfish A, Karpatkin S (1995) Role of platelets, thrombin, integrin IIb-IIIa, fibronectin and von Willebrand factor on tumor adhesion in vitro and metastasis in vivo. Thromb Haemost 74(1):282–290
Coupland LA, Chong BH, Parish CR (2012) Platelets and P-selectin control tumor cell metastasis in an organ-specific manner and independently of NK cells. Cancer Res 72(18):4662–4671
Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S (2013) Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 24(1):130–137. doi:10.1016/j.ccr.2013.05.008
Blair P, Flaumenhaft R (2009) Platelet alpha-granules: basic biology and clinical correlates. Blood Rev 23(4):177–189
Belloc C, Lu H, Soria C, Fridman R, Legrand Y, Menashi S (1995) The effect of platelets on invasiveness and protease production of human mammary tumor cells. Int J Cancer 60(3):413–417
Lindenmeyer F, Legrand Y, Menashi S (1997) Upregulation of MMP-9 expression in MDA-MB231 tumor cells by platelet granular membrane. FEBS Lett 418(1–2):19–22
Hawiger J (1989) Platelet secretory pathways: an overview. Methods Enzymol 169:191–195
Kim YJ, Borsig L, Varki NM, Varki A (1998) P-selectin deficiency attenuates tumor growth and metastasis. Proc Natl Acad Sci USA 95(16):9325–9330
Bretz N, Noske A, Keller S, Erbe-Hofmann N, Schlange T, Salnikov AV, Moldenhauer G, Kristiansen G, Altevogt P (2012) CD24 promotes tumor cell invasion by suppressing tissue factor pathway inhibitor-2 (TFPI-2) in a c-Src-dependent fashion. Clin Exp Metastasis 29(1):27–38. doi:10.1007/s10585-011-9426-4
Bastida E, Almirall L, Ordinas A (1987) Tumor-cell-induced platelet aggregation is a glycoprotein-dependent and lipoxygenase-associated process. Int J Cancer 39(6):760–763
Zhao F, Li L, Guan L, Yang H, Wu C, Liu Y (2014) Roles for GP IIb/IIIa and alphavbeta3 integrins in MDA-MB-231 cell invasion and shear flow-induced cancer cell mechanotransduction. Cancer Lett 344(1):62–73. doi:10.1016/j.canlet.2013.10.019
Borsig L, Wong R, Feramisco J, Nadeau DR, Varki NM, Varki A (2001) Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci USA 98(6):3352–3357
Acknowledgments
This work was supported by a National Health and Medical Research Council of Australia Program Grant. Thanks to members of the Cancer & Vascular Biology Group and the Microscopy and Cytometry Facility of The John Curtin School of Medical Research for their technical assistance.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pang, J.H., Coupland, L.A., Freeman, C. et al. Activation of tumour cell ECM degradation by thrombin-activated platelet membranes: potentially a P-selectin and GPIIb/IIIa-dependent process. Clin Exp Metastasis 32, 495–505 (2015). https://doi.org/10.1007/s10585-015-9722-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-015-9722-5