
Abstract
Dogs develop behavioral and cognitive dysfunction with age. Interestingly, as with humans, not all aged dogs become impaired, and there can be significant individual variability. Studies of the brains of cognitively characterized aged dogs suggest several possible underlying neurobiological mechanisms for observed impairments. In this chapter, changes in canine brains associated with atrophy, neuron loss, accumulation of beta-amyloid (Aβ), mitochondrial dysfunction, and resulting accumulation of oxidative damage are described. There are many important features of brain aging in dogs that overlap significantly with human brain aging, suggesting they are a useful model system in which to test interventions that may lead to healthy aging.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Dogs develop behavioral and cognitive dysfunction with age. Interestingly, as with humans, not all aged dogs become impaired, and there can be significant individual variability. Studies of the brains of cognitively characterized aged dogs suggest several possible underlying neurobiological mechanisms for observed impairments. In this chapter, changes in canine brains associated with atrophy, neuron loss, accumulation of beta-amyloid (Aβ), mitochondrial dysfunction, and resulting accumulation of oxidative damage are described. There are many important features of brain aging in dogs that overlap significantly with human brain aging, suggesting they are a useful model system in which to test interventions that may lead to healthy aging.
5.1 Introduction
Several comprehensive reviews of the neuropathology of canine brain aging are available; this chapter will serve as an overview of key features that are relevant for cognitive decline (Bosch et al. 2012; Cotman and Head 2008; Head 2000, 2011, 2013; Schutt et al. 2016). The features include brain atrophy, neuron loss, and accumulation of Alzheimer’s disease (AD) like neuropathology, vascular pathology, oxidative damage, and inflammation. Most of these neuropathologies increase with age in dogs and in several studies of cognitively characterized animals are also observed to be correlated with the extent of cognitive decline.
5.2 Structural Brain Changes and Neuron Losses
A consistent feature of aging in humans is progressive brain atrophy, which is particularly striking in AD. Magnetic resonance imaging (MRI) studies have provided useful outcome measures reflecting changes in brain structure in vivo. Both generalized cortical atrophy (Su et al. 1998) and ventricular widening (Gonzalez-Soriano et al. 2001; Kimotsuki et al. 2005; Su et al. 1998) can be observed in older animals. However, as with various cognitive functions being differentially affected by the aging process, MRI studies suggest not all brain regions atrophy at the same rate.
Losses in tissue volume occur early with age in the prefrontal cortex around 8 – 11 years in beagles (this may differ with various breeds, given that larger breeds tend to have shorter life spans) (Tapp et al. 2004). The hippocampus atrophies in beagles after 11 years of age (Tapp et al. 2004). Paralleling correlations in cognition and frontal cortex volume in people with dementia (Du et al. 2006; Ezekiel et al. 2004), canine prefrontal cortex atrophy is also associated with impaired cognition (Rofina et al. 2006).
Changes in neuronal number or density are observed in normal human brain aging (Šimić et al. 1997; West 1993) and are more extensive in AD (Bobinski et al. 1997; West et al. 2000). In beagles, a selective loss of neurons is observed within the hilus of the hippocampus (up to 30% loss) when comparing young dogs (3 – 5 years) to older dogs (13 – 15 years) (Siwak-Tapp et al. 2008). In addition, hilar neuron number was correlated with cognition; higher numbers of neurons were associated with fewer errors on a visual discrimination task (Siwak-Tapp et al. 2008).
The reason for losses of neurons in the hippocampus may be due to slower replacement by neurogenesis. Interestingly, neurogenesis decreases by up to 90 – 95% in aging beagles (Hwang et al. 2007; Pekcec et al. 2008; Siwak-Tapp et al. 2007). Further, animals with fewer new neurons had higher error scores in measures of learning and memory, suggesting a link between neurogenesis and cognition with age (Siwak-Tapp et al. 2007). Neuron loss and cortical atrophy in vulnerable brain regions of the aged dog may be due to multiple neurodegenerative processes associated with the up- or downregulation of molecular pathways (Swanson et al. 2007). One of these pathways may be a decrease in growth factors such as brain-derived neurotrophic factor (Fahnestock et al. 2010).
5.3 Plaques and Aβ Accumulation
Plaques are extracellular deposits that contain the Aβ peptide, which is a 40–42 amino-acid-long cleavage product of the longer amyloid precursor protein (Selkoe 1994). Aβ accumulation can occur in different types of plaques (e.g., diffuse, neuritic), but also can form structures called oligomers, which are particularly toxic to synapses (Haass and Selkoe 2007; Lesné et al. 2006; Selkoe 2008; Walsh et al. 2002). A useful feature of the aged canine model is that the Aβ sequence is identical to that of humans (Johnstone et al. 1991; Selkoe et al. 1987). Indeed, it was this finding that led to interest in studying aging dogs as an approach to understand human aging and AD (Wisniewski et al. 1990).
In dogs, as shown in Fig. 5.1, Aβ accumulates in the cortex with a relatively well-described pattern that parallels observations in the human brain (Braak and Braak 1991; Braak et al. 1993; Giaccone et al. 1990; Head et al. 2000; Ishihara et al. 1991; Selkoe et al. 1987; Thal et al. 2002; Wisniewski et al. 1970, 1990). Aβ deposition occurs earliest in the prefrontal cortex of the dog and later in the temporal and occipital cortex (Head et al. 2000), similar to previous reports in humans (Thal et al., 2002). Further, several groups have characterized the age-dependent maturation of Aβ deposits within the canine cortex into several phases (Satou et al. 1997; Schutt et al. 2016). Importantly, the extent of Aβ plaque deposition in the dog brain is linked to the severity of cognitive deficits (Colle et al. 2000; Cummings et al. 1996; Head et al. 1998; Rofina et al. 2006). Interestingly, not all studies show a correlation between Aβ and the presence of canine cognitive dysfunction (CCD) (Chambers et al. 2011; Ozawa et al. 2016). However, studies that show a link between the extent of Aβ and cognition also indicate that the location of the deposition is important. For example, dogs with reversal learning deficits indicative of executive dysfunction tend to show more extensive Aβ deposition in the prefrontal cortex (Cummings et al. 1996; Head et al. 1998). In contrast, poor size discrimination learning ability is associated with large amounts of Aβ in the entorhinal cortex (Head et al. 1998). Soluble Aβ can also be measured in the cerebrospinal fluid (CSF) of dogs, making it a useful marker for aging and cognition intervention studies (Head et al. 2010; Sarasa et al. 2013). The ratio of Aβ42/Aβ40 in the CSF is a good predictor of the extent of Aβ measured biochemically in the brain and also declines linearly with age (Head et al. 2010).

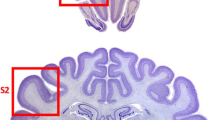
Neuropathology in aging dogs. (a) Aβ immunostaining (6E10 antibody) in the parietal cortex of an aged pet dog (15-year-old Siberian Husky) shows extensive plaque deposition affecting deep cortical layers. (b) CAA (6E10 immunostaining—arrows) in the parietal cortex of an aged dog (14-year-old Sheltie) shows that vascular pathology can be extensive and tends to occur in clusters. Diffuse plaques are also identified by arrowheads. (c) Low-power photograph showing extensive microglial cell labeling (IBA-1 antibody) in gray matter along with intense labeling in the white matter (area below arrows) of a 15-year-old Shih Tzu. (d) Higher magnification photograph from (c) showing that individual microglia contain phagocytic vacuoles and have thickened processes suggesting that some have an activated morphology
In studies characterizing Aβ deposits in plaques, the primary conformation is fibrillar, but Aβ not only exists in this or linear conformations but can also assemble into soluble states that are toxic to synapses and thus neuronal function. Aβ oligomers are small soluble assembly states that interfere with synaptic function and cognition (Kayed et al. 2003; Walsh et al. 2002). Interestingly, Aβ oligomers in the CSF of dogs are inversely related to the amount of total Aβ measured biochemically in the brain, suggesting that oligomers are sequestered into plaques (Head et al. 2010).
Posttranslationally modified Aβ has also been assessed in aging dog brains. These modified forms of Aβ are thought to represent deposits of Aβ that are chronobiologically older (Azizeh et al. 2000). Pyroglutamyl-modified Aβ increases with age in dogs (Frost et al. 2013; Schutt et al. 2016) as does the amount of racemized Aβ (Azizeh et al. 2000). In addition to modified Aβ, truncated Aβ that is observed in AD brain is also observed in the aged canine brain (Chambers et al. 2011).
5.4 Vascular Neuropathology
A frequent pathology detected in both normal human brain aging and particularly in AD is the presence of cerebral amyloid angiopathy (CAA), which is described as the accumulation of Aβ in the walls of cerebral vessels (Attems 2005; Attems et al. 2005; Herzig et al. 2006). Aged dogs are also vulnerable to vascular pathology with perivascular abnormalities; CAA is observed frequently in aged dogs (Giaccone et al. 1990; Ishihara et al. 1991; Shimada et al. 1992; Uchida et al. 1990, 1991, 1992, 1993, 1997; Yoshino et al. 1996). The consequences of CAA accumulating in the brains of aging dogs may be a compromise to the function of the blood-brain barrier and impaired vascular function (Prior et al. 1996). In turn, vascular dysfunction and BBB disruptions may lead to microhemorrhages (Deane and Zlokovic 2007; Uchida et al. 1990, 1991). The distribution of CAA in dog brain is similar to humans, with particular vulnerability in the occipital cortex (Attems et al. 2005). However, in a systematic study of the extent of CAA in cognitively characterized pet dogs, CAA increased with age but did not correlate with cognition (Ozawa et al. 2016). Thus, aged dogs develop cerebrovascular abnormalities that may not contribute to cognitive decline but are otherwise consistent with those reported in humans.
5.5 Oxidative Damage and Mitochondrial Dysfunction
There are several reviews of the potential role for oxidative damage and mitochondrial dysfunction on brain aging in dogs (Cotman et al. 2002; Dowling and Head 2012). The production of free radicals during the aging process can lead to damaged proteins, lipids, and nucleotides, which may cause neuronal dysfunction and degeneration. The aging dog brain accumulates carbonyl groups, a measure of oxidative damage to proteins (Head et al. 2002; Skoumalova et al. 2003). Typically, the activity of endogenous antioxidants balances the production of free radicals. However, several of these protective mechanisms decline with age. Carbonyl groups are associated with reduced endogenous antioxidant enzyme activity/protein levels, including those of glutamine synthetase and superoxide dismutase (SOD) (Head et al. 2002; Hwang et al. 2008; Kiatipattanasakul et al. 1997; Opii et al. 2008). The end products of lipid peroxidation (oxidative damage to lipids), including 4-hydroxynonenal (4HNE) (Hwang et al. 2008; Papaioannou et al. 2001; Rofina et al. 2004, 2006), lipofuscin (Rofina et al. 2006), lipofuscin-like pigments (Papaioannou et al. 2001; Rofina et al. 2004), or malondialdehyde (Head et al. 2002); all of these increase with age in the canine brain. In addition, there are several reports of increased oxidative damage to DNA or RNA in the aged dog brain (Cotman and Head 2008; Rofina et al. 2006).
The consequences of increasing oxidative damage with age in dogs may be compromised neuronal function leading to deficits in cognition. In aged pet dogs, higher levels of oxidative end products are correlated with more severe behavioral changes (Rofina et al. 2004, 2006; Skoumalova et al. 2003). Similarly, in studies of laboratory beagles, higher protein oxidative damage (3-nitrotyrosine) and lower endogenous antioxidant capacity (SOD and glutathione-S-transferase activities) in aging animals are associated with poorer prefrontal-dependent learning and spatial learning (Opii et al. 2008). The mitochondria are a critical contributor to oxidative damage, being a source of free radicals that damage proteins, lipids, and DNA/RNA (Shigenaga et al. 1994). Mitochondria isolated from young beagle brain have lower levels of reactive oxygen species production than those isolated from older beagles (Head et al. 2009). Due to the similarities in oxidative damage in dogs and humans, several research groups have suggested that the canine is particularly well suited for studies focused on this mechanism of neurodegeneration (Dowling and Head 2012; Romanucci and Della Salda 2015).
5.6 Inflammation
Neuroinflammation in the aged human and AD brain may lead to the exacerbation of cognitive decline or potentially mediate other neuropathological events causing dementia (Heneka et al. 2015; Wilcock 2013). Although not as well characterized as neuroinflammation in the human brain, there are several small studies in aged pet dog brains. In a recent study, Schutt and colleagues (Schutt et al. 2016) measured canine cytokines in the prefrontal cortex of 15 aged dogs as compared with 2 young dogs. Pro-inflammatory cytokines were generally at low levels and were not associated with the extent of cognitive dysfunction. However, using measures of glial activation (microglial cells and astrocytes), increasing numbers of both types of cells were associated with more extensive CCD in a study of 37 dogs with various breeds included (Ozawa et al. 2016). Similarly, the level of S100β astrocytosis, a putative measure of inflammation, is also correlated with cognitive deficits in pet dogs (Pugliese et al. 2006).
5.7 White Matter Pathology
White matter degeneration can contribute to cognitive decline in humans (Bartzokis 2004; Gold et al. 2012). In a study of myelin protein levels as a function of age in dogs, Chambers et al. report a loss of prefrontal myelin protein (Chambers et al. 2012) that was also associated with some Aβ deposition in CAA. In another study, the extent of ubiquitin labeling in aging dog brains (n = 37) was associated with CCD and is thought to reflect failures in the protein homeostasis in the synapse or in myelin (Ozawa et al. 2016).
5.8 Summary
Identifying neurodegenerative mechanisms underlying cognitive dysfunction in aging dogs will provide novel targets for future intervention studies. These treatments may involve lifestyle changes (e.g., exercise can lead to enhanced neurogenesis and benefit cognition in dogs (Intlekofer and Cotman 2013; Snigdha et al. 2014)), diet changes (e.g., antioxidant diets (Cotman et al. 2002)), removal of Aβ by vaccines (Cotman and Head 2008), or pharmacological manipulations. As more of the gaps in our knowledge are filled, we may learn more of the role of vascular factors and inflammation in canine brain aging, which are also modifiable and may lead to cognitive benefits.
References
Attems J (2005) Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol 110(4):345–359
Attems J, Jellinger KA, Lintner F (2005) Alzheimer’s disease pathology influences severity and topographical distribution of cerebral amyloid angiopathy. Acta Neuropathol 110(3):222–231
Azizeh BY, Head E, Ibrahim MA, Torp R, Tenner AJ, Kim RC, Lott IT, Cotman CW (2000) Molecular dating of senile plaques in the brains of individuals with Down syndrome and in aged dogs. Exp Neurol 163(1):111–122
Bartzokis G (2004) Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging 25(1):5–18. author reply 49–62
Bobinski M, Wegiel J, Tarnawski M, Bobinski M, Reisberg B, de Leon MJ, Miller DC, Wisniewski HM (1997) Relationships between regional neuronal loss and neurofibrillary changes in the hippocampal formation and duration and severity of Alzheimer disease. J Neuropathol Exp Neurol 56(4):414–420
Bosch MN, Pugliese M, Gimeno-Bayon J, Rodriguez MJ, Mahy N (2012) Dogs with cognitive dysfunction syndrome: a natural model of Alzheimer’s disease. Curr Alzheimer Res 9(3):298–314
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259
Braak H, Braak E, Bohl J (1993) Staging of Alzheimer-related cortical destruction. Eur Neurol 33(6):403–408
Chambers JK, Mutsuga M, Uchida K, Nakayama H (2011) Characterization of AβpN3 deposition in the brains of dogs of various ages and other animal species. Amyloid 18(2):63–71
Chambers JK, Uchida K, Nakayama H (2012) White matter myelin loss in the brains of aged dogs. Exp Gerontol 47(3):263–269
Colle M-A, Hauw J-J, Crespeau F, Uchiara T, Akiyama H, Checler F, Pageat P, Duykaerts C (2000) Vascular and parenchymal Aβ deposition in the aging dog: correlation with behavior. Neurobiol Aging 21(5):695–704
Cotman CW, Head E (2008) The canine (dog) model of human aging and disease: dietary, environmental and immunotherapy approaches. J Alzheimer’s Dis 15(4):685–707
Cotman CW, Head E, Muggenburg BA, Zicker S, Milgram NW (2002) Brain aging in the canine: a diet enriched in antioxidants reduces cognitive dysfunction. Neurobiol Aging 23(5):809–818
Cummings BJ, Head E, Afagh AJ, Milgram NW, Cotman CW (1996) Beta-amyloid accumulation correlates with cognitive dysfunction in the aged canine. Neurobiol Learning Memory 66(1):11–23
Deane R, Zlokovic BV (2007) Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 4(2):191–197
Dowling AL, Head E (2012) Antioxidants in the canine model of human aging. Biochim Biophys Acta (BBA)-Mol Basis of Dis 1822(5):685–689
Du AT, Schuff N, Chao LL, Kornak J, Jagust WJ, Kramer JH, Reed BR, Miller BL, Norman D, Chui HC, Weiner MW (2006) Age effects on atrophy rates of entorhinal cortex and hippocampus. Neurobiol Aging 27(5):733–740
Ezekiel F, Chao L, Kornak J, Du AT, Cardenas V, Truran D, Jagust W, Chui H, Miller B, Yaffe K, Schuff N, Weiner M (2004) Comparisons between global and focal brain atrophy rates in normal aging and Alzheimer disease: boundary shift integral versus tracing of the entorhinal cortex and hippocampus. Alzheimer Dis Assoc Disorders 18(4):196
Fahnestock M, Marchese M, Head E, Pop V, Michalski B, Milgram WN, Cotman CW (2010) BDNF increases with behavioral enrichment and an antioxidant diet in the aged dog. Neurobiol Aging 2010
Frost JL, Le KX, Cynis H, Ekpo E, Kleinschmidt M, Palmour RM, Ervin FR, Snigdha S, Cotman CW, Saido TC, Vassar RJ, St George-Hyslop P, Ikezu T, Schilling S, Demuth HU, Lemere CA (2013) Pyroglutamate-3 amyloid-β deposition in the brains of humans, non-human primates, canines, and Alzheimer disease-like transgenic mouse models. Am J Pathol 183(2):369–381
Giaccone G, Verga L, Finazzi M, Pollo B, Tagliavini F, Frangione B, Bugiani O (1990) Cerebral preamyloid deposits and congophilic angiopathy in aged dogs. Neurosci Lett 114(2):178–183
Gold BT, Johnson NF, Powell DK, Smith CD (2012) White matter integrity and vulnerability to Alzheimer’s disease: preliminary findings and future directions. Biochim Biophys Acta (BBA)-Mol Basis Dis 1822(3):416–422
Gonzalez-Soriano J, Garcia PM, Contreras-Rodriguez J, Martinez-Sainz P, Rodriguez-Veiga E (2001) Age-related changes in the ventricular system of the dog brain. Ann Anatomy-Anatomischer Anzeiger 183(3):283–291
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol 8(2):101–112
Head E (2000) Brain aging in dogs: parallels with human brain aging and Alzheimer’s disease. Veterinary Therapeut: Research Applied Veterinary Med 2(3):247–260
Head E (2011) Neurobiology of the aging dog. Age 33(3):485–496
Head E (2013) A canine model of human aging and Alzheimer’s disease. Biochim Biophys Acta (BBA)-Mol Basis Dis 1832(9):1384–1389
Head E, Callahan H, Muggenburg BA, Cotman CW, Milgram NW (1998) Visual-discrimination learning ability and β-amyloid accumulation in the dog. Neurobiol Aging 19(5):415–425
Head E, McCleary R, Hahn FF, Milgram NW, Cotman CW (2000) Region-specific age at onset of β-amyloid in dogs. Neurobiol Aging 21(1):89–96
Head E, Liu J, Hagen TM, Muggenburg BA, Milgram NW, Ames BN, Cotman CW (2002) Oxidative damage increases with age in a canine model of human brain aging. J Neurochem 82(2):375–381
Head E, Nukala VN, Fenoglio KA, Muggenburg BA, Cotman CW, Sullivan PG (2009) Effects of age, dietary, and behavioral enrichment on brain mitochondria in a canine model of human aging. Exp Neurol 220(1):171–176
Head E, Pop V, Sarsoza F, Kayed R, Beckett TL, Studzinski CM, Tomic JL, Glabe CG, Murphy MP (2010) Amyloid-β peptide and oligomers in the brain and cerebrospinal fluid of aged canines. J Alzheimer’s Dis 20(2):637–646
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14(4):388–405
Herzig MC, Nostrand WE, Jucker M (2006) Mechanism of cerebral β-amyloid angiopathy: murine and cellular models. Brain Pathol 16(1):40–54
Hwang IK, Yoo KY, Li H, Choi JH, Kwon YG, Ahn Y, Lee IS, Won MH (2007) Differences in doublecortin immunoreactivity and protein levels in the hippocampal dentate gyrus between adult and aged dogs. Neurochem Res 32(9):1604–1609
Hwang IK, Yoon YS, Yoo KY, Li H, Choi JH, Kim DW, Yi SS, Seong JK, Lee IS, Won MH (2008) Differences in lipid peroxidation and Cu, Zn-superoxide dismutase in the hippocampal CA1 region between adult and aged dogs. J Veterinary Med Sci 70(3):273–277
Intlekofer KA, Cotman CW (2013) Exercise counteracts declining hippocampal function in aging and Alzheimer’s disease. Neurobiol Dis 57:47–55
Ishihara T, Gondo T, Takahashi M, Uchino F, Ikeda SI, Allsop D, Imai K (1991) Immunohistochemical and immunoelectron microscopical characterization of cerebrovascular and senile plaque amyloid in aged dogs’ brains. Brain Res 548(1):196–205
Johnstone EM, Chaney MO, Norris FH, Pascual R, Little SP (1991) Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Mol Brain Res 10(4):299–305
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300(5618):486–489
Kiatipattanasakul W, Nakamura SI, Kuroki K, Nakayama H (1997) Immunohistochemical detection of anti-oxidative stress enzymes in the dog brain. Neuropathology 17(4):307–312
Kimotsuki T, Nagaoka T, Yasuda M, Tamahara S, Matsuki N, Ono K (2005) Changes of magnetic resonance imaging on the brain in beagle dogs with aging. J Veterinary Med Sci 67(10):961–967
Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Ashe KH (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440(7082):352–357
Opii WO, Joshi G, Head E, Milgram NW, Muggenburg BA, Klein JB, Pierce WM, Cotman CW, Butterfield DA (2008) Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer’s disease. Neurobiol Aging 29(1):51–70
Ozawa M, Chambers JK, Uchida K, Nakayama H (2016) The relation between canine cognitive dysfunction and age-related brain lesions. J Veterinary Med Sci 78(6):997–1006
Papaioannou N, Tooten PC, van Ederen AM, Bohl JR, Rofina J, Tsangaris T, Gruys E (2001) Immunohistochemical investigation of the brain of aged dogs. I. Detection of neurofibrillary tangles and of 4-hydroxynonenal protein, an oxidative damage product, in senile plaques. Amyloid 8(1):11–21
Pekcec A, Baumgärtner W, Bankstahl JP, Stein VM, Potschka H (2008) Effect of aging on neurogenesis in the canine brain. Aging Cell 7(3):368–374
Prior R, D’Urso D, Frank R, Prikulis I, Pavlakovic G (1996) Loss of vessel wall viability in cerebral amyloid angiopathy. Neuroreport 7(2):562–564
Pugliese M, Geloso MC, Carrasco JL, Mascort J, Michetti F, Mahy N (2006) Canine cognitive deficit correlates with diffuse plaque maturation and S100β (−) astrocytosis but not with insulin cerebrospinal fluid level. Acta Neuropathol 111(6):519–528
Rofina JE, Singh K, Skoumalova-Vesela A, van Ederen AM, van Asten AJ, Wilhelm J, Gruys E (2004) Histochemical accumulation of oxidative damage products is associated with Alzheimer-like pathology in the canine. Amyloid 11(2):90–100
Rofina JE, Van Ederen AM, Toussaint MJM, Secreve M, Van Der Spek A, Van Der Meer I, Eerdenburg FJ, Gruys E (2006) Cognitive disturbances in old dogs suffering from the canine counterpart of Alzheimer’s disease. Brain Res 1069(1):216–226
Romanucci M, Della Salda L (2015) Oxidative stress and protein quality control systems in the aged canine brain as a model for human neurodegenerative disorders. Oxidative Med Cell Longevity 2015:e940131
Sarasa L, Allué JA, Pesini P, González-Martínez Á, Sarasa M (2013) Identification of β-amyloid species in canine cerebrospinal fluid by mass spectrometry. Neurobiol Aging 34(9):2125–2132
Satou T, Cummings BJ, Head E, Nielson KA, Hahn FF, Milgram NW, Velazquez P, Cribbs DH, Tenner AJ, Cotman CW (1997) The progression of β-amyloid deposition in the frontal cortex of the aged canine. Brain Res 774(1):35–43
Schutt T, Helboe L, Pedersen LO, Waldemar G, Berendt M, Pedersen JT (2016) Dogs with cognitive dysfunction as a spontaneous model for early Alzheimer’s disease: a translational study of neuropathological and inflammatory markers. J Alzheimer’s Dis 52(2):433–449
Selkoe DJ (1994) Normal and abnormal biology of the beta-amyloid precursor protein. Ann Rev Neurosci 17(1):489–517
Selkoe DJ (2008) Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav Brain Res 192(1):106–113
Selkoe DJ, Bell DS, Podlisny MB, Price DL, Cork LC (1987) Conservation of brain amyloid proteins in aged mammals and humans with Alzheimer’s disease. Science 235(4791):873–877
Shigenaga MK, Hagen TM, Ames BN (1994) Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci 91(23):10771–10778
Shimada A, Kuwamura M, Akawkura T, Umemura T, Takada K, Ohama E, Itakura C (1992) Topographic relationship between senile plaques and cerebrovascular amyloidosis in the brain of aged dogs. J Veterinary Med Sci 54(1):137–144
Šimić G, Kostović I, Winblad B, Bogdanović N (1997) Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer’s disease. J Comparative Neurol 379(4):482–494
Siwak-Tapp CT, Head E, Muggenburg BA, Milgram NW, Cotman CW (2007) Neurogenesis decreases with age in the canine hippocampus and correlates with cognitive function. Neurobiol Learning Memory 88(2):249–259
Siwak-Tapp CT, Head E, Muggenburg BA, Milgram NW, Cotman CW (2008) Region specific neuron loss in the aged canine hippocampus is reduced by enrichment. Neurobiol Aging 29(1):39–50
Skoumalova A, Rofina J, Schwippelova Z, Gruys E, Wilhelm J (2003) The role of free radicals in canine counterpart of senile dementia of the Alzheimer type. Exp Gerontol 38(6):711–719
Snigdha S, de Rivera C, Milgram NW, Cotman C (2014) Exercise enhances memory consolidation in the aging brain. Front Aging Neurosci 6:3
Su MY, Head E, Brooks WM, Wang Z, Muggenberg BA, Adam GE, Sutherland RJ, Cotman CW, Nalcioglu O (1998) Magnetic resonance imaging of anatomic and vascular characteristics in a canine model of human aging. Neurobiol Aging 19(5):479–485
Swanson KS, Vester BM, Apanavicius CJ, Kirby NA, Schook LB (2007) Implications of age and diet on canine cerebral cortex transcription. Neurobiol Aging 30(8):1314–1326
Tapp PD, Siwak CT, Gao FQ, Chiou JY, Black SE, Head E, Muggenburg BA, Cotman CW, Milgram NW, Su MY (2004) Frontal lobe volume, function, and β-amyloid pathology in a canine model of aging. J Neurosci 24(38):8205–8213
Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58(12):1791–1800
Uchida K, Miyauchi Y, Nakayama H, Goto N (1990) Amyloid angiopathy with cerebral hemorrhage and senile plaque in aged dogs. Nippon Juigaku Zasshi 52(3):605–611
Uchida K, Nakayama H, Goto N (1991) Pathological studies on cerebral amyloid angiopathy, senile plaques and amyloid deposition in visceral organs in aged dogs. J Veterinary Med Sci 53(6):1037–1042
Uchida K, Tani Y, Uetsuka K, Nakayama H, Goto N (1992) Immunohistochemical studies on canine cerebral amyloid angiopathy and senile plaques. J Veterinary Med Sci 54(4):659–667
Uchida K, Okuda R, Yamaguchi R, Tateyama S, Nakayama H, Goto N (1993) Double-labeling immunohistochemical studies on canine senile plaques and cerebral amyloid angiopathy. J Veterinary Med Sci 55(4):637–642
Uchida K, Kuroki K, Yoshino T, Yamaguchi R, Tateyama S (1997) Immunohistochemical study of constituents other than β-protein in canine senile plaques and cerebral amyloid angiopathy. Acta Neuropathol 93(3):277–284
Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ (2002) Amyloid-β oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans 30(4):552–557
West MJ (1993) Regionally specific loss of neurons in the aging human hippocampus. Neurobiol Aging 14(4):287–293
West MJ, Kawas CH, Martin LJ, Troncoso JC (2000) The CA1 region of the human hippocampus is a hot spot in Alzheimer’s disease. Ann NY Acad Sci 908(1):255–259
Wilcock DM (2013) Neuroinflammatory phenotypes and their roles in Alzheimer’s disease. Neurodegenerative Dis 13(2-3):183–185
Wisniewski H, Johnson AB, Raine CS, Kay WJ, Terry RD (1970) Senile plaques and cerebral amyloidosis in aged dogs. A histochemical and ultrastructural study. Lab Investig 23:287–296
Wisniewski HM, Wegiel J, Morys J, Bancher C, Soltysiak Z, Kim KS (1990) Aged dogs: an animal model to study beta-protein amyloidogenesis. In: Alzheimer’s disease. Epidemiology, neuropathology, neurochemistry, and clinics. Springer, New York, pp 151–167
Yoshino T, Uchida K, Tateyama S, Yamaguchi R, Nakayama H, Goto N (1996) A retrospective study of canine senile plaques and cerebral amyloid angiopathy. Veterinary Pathol Online 33(2):230–234
Acknowledgment
Funding provided by the NIH/NIA R01AG0031764.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Nichol, J., Head, E. (2017). Brain Aging in the Dog. In: Landsberg, G., Maďari, A., Žilka, N. (eds) Canine and Feline Dementia. Springer, Cham. https://doi.org/10.1007/978-3-319-53219-6_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-53219-6_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-53218-9
Online ISBN: 978-3-319-53219-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)