
Abstract
Chordoma is a relatively rare, slow-growing, primary bone tumor with an overall incidence of approximately one per million population and accounts for 1–4% of all malignant bone lesions [1, 2]. Although rare, it represents the most frequent primary malignant bone tumor affecting the sacrum [3]. It has a slowly aggressive and locally invasive behavior, and it is considered a low-grade malignant neoplasm. In fact, it is poorly sensitive to conventional radiotherapy and chemotherapy. Surgical resection of sacral chordoma remains the standard for local disease control, even if it is associated with significant morbidity and repercussions for patient’s quality of life due to the close relationship with relevant neurovascular structures [4]. An increasing number of novel (radio)surgical and pharmacological strategies are currently being investigated [5–7] and may have a role in addressing microscopic disease.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Chordoma is a relatively rare, slow-growing, primary bone tumor with an overall incidence of approximately one per million population and accounts for 1–4% of all malignant bone lesions [1, 2]. Although rare, it represents the most frequent primary malignant bone tumor affecting the sacrum [3]. It has a slowly aggressive and locally invasive behavior, and it is considered a low-grade malignant neoplasm. In fact, it is poorly sensitive to conventional radiotherapy and chemotherapy. Surgical resection of sacral chordoma remains the standard for local disease control, even if it is associated with significant morbidity and repercussions for patient’s quality of life due to the close relationship with relevant neurovascular structures [4]. An increasing number of novel (radio)surgical and pharmacological strategies are currently being investigated [5,6,7] and may have a role in addressing microscopic disease.
2 Embryology
The current line of thinking is that chordoma cells originate from remnants of the embryonic notochord [8,9,10,11]. For the purpose of clarifying the pathophysiology, one must first gain an understanding of the ontogenesis of the axial skeleton as it matures from the notochord to its ultimate configuration in the adult. During embryogenesis, in the third week of human development, gastrulation takes place with the formation of the three primary germ layers (ecto-, meso-, and endoderm). The stem cells for all major structural elements of the vertebral column are derived from the mesoderm [12]. One of the key events following this phenomenon is the formation of the notochord in the Carnegie stages 7 through 9 (crown-rump [CR] length of 0.4–2.5 mm), an elongated rod of cells subadjacent to the neural tube with a caudo-cranial extension [13]. The notochord plays a critical role by producing and secreting important signaling factors (e.g., sonic hedgehog [14], bone morphogenetic protein [15, 16]) to the surrounding tissue in order to guide organogenesis and the formation of the axial skeleton [17]. Primary neurulation, the process of the flat neural plate folding into the cylindrical neural tube, occurs in response to soluble growth factors secreted by the notochord (Fig. 15.1). As a result of the cellular shape changes, the neural plate folds creating the U-shape neural groove. This neural groove sets the boundary between the right and left sides of the embryo. Then, the closure of the neural tube disconnects the neural crest from the epidermidis, and segmentation commences in the axial and paraxial mesenchyme with the formation of somites (Carnegie stage 11, CR 2.5–4.5 mm) [18,19,20,21]. In axial section, the mesoderm-derived tissue which bilaterally propagates alongside the notochord evolves by an epithelial-to-mesenchymal transition to form the ventral sclerotomes [22]. The mesenchymal cells are specified based on their location within the somite: they retain the ability to become any kind of somite-derived structure until relatively late in the process of somitogenesis [23] and gradually assume a concentric arrangement around the notochord, forming a perichordal sheath [18, 24, 25]. These mesenchymal cells of the perichordal sheath become cartilaginous (chondroblasts/chondrocytic cells) via a condensation process guided by secreted factors derived from the notochord originating primordial vertebral body [26, 27]. Concurrently with the vertebral body morphogenesis, the confined notochordal cells progressively degenerate and probably undergo apoptosis or differentiate into the chondrocyte-like cells [28]. Occasionally notochord cells remain in the nucleus pulpous of the mature intervertebral disk (Fig. 15.2) or can be witnessed in notochord-like tissue in the intravertebral region, in which case they are described as “benign notochordal cell tumors” (BNCT) [29]. The complete formation of the vertebral segments is expected to be at Carnegie stage 20–22 (CR 18–30 mm) (Fig. 15.3).


Artistic drawings show embryogenesis of the vertebral column: primary neurulation. (a) Shaping. Schematic transverse sections showing neuroectodermal tissues differentiate from the ectoderm and thicken into the neural plate (I); (b) Folding and elevation. The neural plate bends dorsally creating the U-shaped neural groove. Notochord is shown at the ventral part of the neural groove (II). The two ends eventually joining at the neural plate borders, which are now referred to as the neural crest (III); (c) Convergence. Bending of the neural plate with convergence of the neural folds up to the complete closure of the neural tube (IV); (d) Closure. The closure of the neural tube disconnects the neural crest from the epidermidis (V). Neural crest cells differentiate to form most of the peripheral nervous system and notochord degenerates (VI)

Artistic drawing show notochord cells remain in the nucleus pulposus of the mature intervertebral disk. During resegmentation of the sclerotomes to form the vertebrae, each one splits into cranial and caudal segments, and cells remaining in the plane of division coalesce to form the annulus fibrous of the intervertebral disk

Artistic drawing of a human embryo at Carnegie stage 20 show all of the vertebral segments have formed
3 Pathogenesis
The pathophysiology underlying this lethal disease is demonstrated to be complex. Starting from the histological characterization performed by Virchow in 1857 [30] up to the discovery of brachyury’s involvement, numerous progresses have been performed in the pathogenesis of this tumor. Examination of human embryos and fetus showed that notochordal cell nests topographically correspond and distribute to the sites of occurrence of chordoma and histological appearance of tumor cells led to hypothesize the notochordal origin [31, 32]. Molecular research has so far yielded significant findings on the mechanisms underlying the initiation and further progression of chordoma cells. The most compelling evidence of the notochordal hypothesis derived from researches focused on a transcription factor named “brachyury.” It is an important transcription factor in notochord development, but duplicated regions contained only the brachyury gene have been discovered in familial chordoma [33,34,35,36]. The remarkable overexpression revealed this transcription factor to be a crucial aspect of chordoma, although it is still unclear what role brachyury has in the pathogenesis [37]. Some other notochordal factors (Shh, Wnt, galectin-3, NCAM) seem to be relevant in notochord formation and in chordoma, as well as the other overexpression of cell cycle regulatory pathways and an activated receptor tyrosine kinase pathway [11]. The molecular biology process behind the initiation and progression of a chordoma needs to be revealed for a better understanding of the disease and to develop more effective therapies [38].
4 Epidemiology
Chordomas are classified on the basis of their location along the spine in sacrococcygeal, clival, cervical, thoracic, and lumbar (listed by the most frequent site) [1,2,3]. Recent studies reported an almost equal distribution in the clivus (32%), mobile spine (32.8%), and sacrum 29.2% [39]. Other epidemiological studies report that the sacrococcygeal area is the most common affect (40–50%) compared to clivus (35–40%) and vertebral bodies (20–40%) [40, 41]. Sacrococcygeal chordomas have very low incidence in patients below 40 years old and are more frequent in males (male to female ratio 2:1) [42,43,44,45].
5 Presentation and Diagnosis
Chordomas of the sacrum often present with non-specific symptoms which can delay diagnosis, such as localized deep pain or radiculopathies related to the spinal level at which they occur [46,47,48,49]. In advanced disease the tumors can present at the time of diagnosis as a slow-growing palpable mass associated with rectal or urinary dysfunction [46, 47, 50]. The average duration of symptoms is about 14 months (range, 4–24 months) [45]. Diagnosis is further complicated by the fact that lytic sacral lesions might be overlooked on plain radiographs of the pelvis, and CT or MRI studies are often not performed without a clinical suspicion of sacral tumor [45, 49]. Differential diagnosis for lumbosacral chordomas includes pilonidal sinus disease, deep abscesses, rectal sarcomas, and several retrorectal tumors (teratomas, extraperitoneal adenomucinosis, cystic lymphangiomas, neurogenic tumors, and cysts, developmental tailgut cysts, sacral myelomeningocele, rectal duplication) [51,52,53]. Most sacrococcygeal chordomas can protrude anteriorly into the pelvis, and rectal examination may be useful for clinical detection of sacral mass even if the tumor is limited by presacral fascia [54].
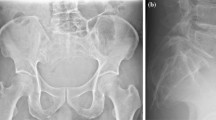
6 Imaging
Chordomas are midline lesions and often appear radiographically as destructive lytic bone lesions. Unlike other primary tumor (osteosarcomas and chondrosarcomas) of the spine, chordomas locally invade the intervertebral disk space as they spread to adjacent vertebral bodies [49]. Computed tomography (CT) and magnetic resonance imaging (MRI) are the gold standard for diagnosis. The tumors are often associated with soft tissue mass (Fig. 15.4). Calcification and bony expansion are present in 30–70% of the cases and appear isointense or hypointense on T1-weighted MRI images and hyperintense on T2-weighted MRI images and enhance with gadolinium [55]. On bone scan, chordomas show reduced or normal uptake of radioisotope when juxtaposed to other bone tumors [56]. Careful preoperative assessment of imaging within the multidisciplinary team is essential in planning surgical approach and discussing strategy of treatment.

Sagittal view of a specimen from a proximal sacral resection show the large involvement of soft tissue
7 Pathology
Chordomas were first described histologically by Virchow in 1857, when he identified the typical “physaliferous” tumor cell chordomas [30]. Physaliferous cells appear as large white cells with round nuclei and abundant vacuolated cytoplasm separated by fibrous septa into lobules [41, 42] and are typical of classic chordomas. Chordomas are classified as classical (or conventional), chondroid, or dedifferentiated [41]. Classic chordomas are pathologically identified by their typical cells and immunoreactivity for S-100, epithelial membrane antigen (MUC1), and cytokeratins [57, 58]. Brachyury staining is used to discriminate chordomas from other chondroid lesions [59]. Chondroid chordoma is a histological variant that account for 5–15% of all chordomas [41]. It shows histological features resembling chondrosarcoma, with hyaline cartilage associated with expression of chordoma markers [60, 61]. Dedifferentiated chordomas account for less than 10% of all chordomas and are characterized by sarcomatous areas with spindle cells such as fibrosarcoma, osteosarcoma, or rhabdomyosarcoma [62,63,64,65,66]. It is characterized by a fulminant clinical course resulting in metastases and/or death within 1 year of diagnosis in most of the cases [64,65,66,67,68].
8 Treatment
The most accredited treatment consists of surgical resection with wide margins, as no chemotherapy has been demonstrated to be effective against chordoma and conventional radiotherapy is only partially effective [5, 11, 69,70,71,72,73,74]. Complex pelvic anatomy coupled with the need of wide margins means that surgery is challenging to preserve essential neural function and avoid injury to visceral and vascular structures during resection. Frequently, margins are positive (marginal or contaminated) [11, 70, 71, 75, 76], and adjuvant treatment strategies must be considered.
8.1 Sacral Resection
In the 1970s, Stener and Gunterberg [77] first introduced the idea of wide en bloc surgical resection for the treatment of sacral tumors. Since then, en bloc resection has remained the mainstay of treatment of sacral chordoma worldwide, as reported by a large multicentric study based on the AOSpine Tumor Knowledge Forum Primary Spinal Tumor database [78]. En bloc sacrectomy is a highly demanding surgical procedure, consisting of a partial or total amputation of the sacrum. It is associated with significant soft tissue and skin defects, which may require reconstruction with myocutaneous flaps to reduce risk of wound infection and breakdown. However, it is attainable in more than 50% of sacral chordomas and offers the best long-term oncological outcomes when wide margins are obtained [69, 79].
The surgical approach is often planned according to tumor extension and level of resection. Usually all resections distal to S3 level could be approached posteriorly only [70, 80, 81], whereas proximal resections need a combined anterior-posterior approach [69,70,71, 78, 79, 82]. Advantages of the posterior approach are a single stage procedure and shorter operating time, whereas the combined approach enables the visceral organs to be dissected away from the tumor and protected during the osteotomy. Some exceptions have been reported in literature, using tools and innovative surgical techniques to perform proximal resections by a posterior approach only in selected cases [83, 84].
Unilateral or bilateral sacrifice of the nerve roots is necessary distal to resection, with corresponding functional damage [77, 82, 85,86,87,88]: motor and sensory deficits in the lower limbs are mainly related to the sacrifice of S1 and L5 nerve roots; sacrectomies that spare the S2 nerve root are associated with abnormal bladder and bowel function, even if better results can be expected if an S3 nerve root is also preserved; sexual dysfunction with relative saddle anesthesia is associated with bilateral S3 lesions, whereas unilateral sacrifice from S2 to S5 reduces but does not abolish urogenital and rectal functions. Numerous complications other than neurological deficits have been reported in literature, such as wound dehiscence, infection, iatrogenic visceral injury, hematoma, massive bleeding, liquoral fistula, flap necrosis, stress fractures, and other less frequently reported [70, 89]. Chen et al. [90] reported that albumin <3.0 g/dL, operating time (>6 h), and previous surgery were statistically significant risk factors for wound infection.
8.2 Radiotherapy
The use of radiotherapy (RT) as primary or adjuvant treatment for chordoma has been debated for several years and remains controversial. The majority of older publications that used conventional photon radiotherapy did not exceed 60 Gy, and investigators report poor local control in sacral chordomas [91, 92]. In fact, the problem of tolerance dose of the organs and tissues surrounding the sacrum results in the limitation of total RT dose that can safely be delivered to the tumor [93, 94]. Advances in radiation technology and treatment have led to more strategic targeting of neoplasms with higher doses of radiation. There is some consensus that the combination therapy of surgical resection and radiation therapy may be associated with higher rates of local control and overall survival [95].
-
Intensity-modulated radiation therapy (IMRT) and stereotactic delivery techniques can custom modulate each photon beam to conform to the tumor volume by minimizing the dose to surrounding tissues [92]. Some authors report the experience in sacral chordomas, finding that local control was significantly higher in patients treated with radiation dose delivered higher than 60 Gy (range 60–78 Gy) [96, 97].
-
Radiosurgery is a technique characterized by a very high dose of RT in a single fraction (or hypofractionated) thanks to the use of image-guided technology coupled with IMRT. The use of radiosurgery for chordoma has shown promising preliminary results for local control [98, 99].
-
Proton beam and heavy-ion particle radiation therapy are characterized by delivering a high-specific radiation dose to the tumor target volume and small dose to uninvolved normal tissue (low risk of radiation toxicity to neural tissue) [94, 100, 101]. Hadrons (high-dose protons or charged particles, including carbon ions, helium, or neon) provide biological and physical advantages in terms of their high relative biological effectiveness and reduced oxygen-enhancement ratio in the tumor region [102]. In fact, studies exploiting the use of hadron therapy in chordomas of the sacrococcygeal region show local control at 5 years of 60–70% [99, 103,104,105]. Compared with protons and photons, carbon ions have a relative higher biological effectiveness with a larger mean energy per unit length of their trajectory [91, 106]. Therefore, carbon-ion radiotherapy has been considered for treatment of unresectable chordoma [107]. Promising local control rate and better preservation of bladder-bowel function with the use of carbon-ion radiotherapy has been reported compared with surgery [107,108,109], whereas good results have also been reported as adjuvant treatment [110]. Unfortunately, the availability of hadron-based therapy is limited because of the associated construction and operational expenses [111, 112], even if the cost is expected to decrease rapidly [113].
Although there is limited literature comparing the effectiveness of newer radiation therapy modalities coupled with surgery, preliminary promising results have been reported with hadron therapy than with photon-based radiation [114,115,116]. However, at this time, single-fraction photon RT and proton-beam and carbon-ion RT with wide en bloc excision both are the accepted treatment standard in the management of chordomas at many quaternary-care cancer centers, showing higher local control rates than conventional IMRT [116]. One of the critiques of RT is that it can cause pathological fracture of residual sacral bone [69, 70, 117].
8.3 Chemotherapy and Medical Treatment
No conventional chemotherapy has proven to be effective in terms of overall survival and local control in patients with sacral chordoma. The advent of molecular targeted therapies and the discovery of molecular profiling of chordomas have offered some encouraging alternatives to conventional chemotherapy for the management of advanced disease. Chordomas overexpress platelet-derived growth factor receptor (PDGFR)B, PDGFRA, and KIT receptors, suggesting a role for imatinib therapy [118,119,120], a tyrosine kinase inhibitor (TKI) with specificity for the kinase domain of PDGFR and KIT receptors. Another TKI, sunitinib, has shown clinical efficacy [121], even if reports are limited by small patient numbers and short follow-up. Additional molecular pathways such as mTOR and MAPK signaling pathways seem to be involved, meaning a possible role of other targeting therapies such as mTOR inhibitors (everolimus, temsirolimus) [122, 123].
In a small series of patients with chordoma, strong expression of epidermal growth factor receptor (EGFR) and c-MET was described [124]. EGFR is a tyrosine kinase receptor implicated in cell proliferation through the binding of several ligands. This led to the report of a patient’s response to cetuximab and gefitinib [125]. Newer EGFR inhibitors, erlotinib and lapatinib, confirmed this efficacy [126, 127]. A recent analysis showed that activation of phosphorylated signal transducer and activator of transcription 3 (STAT3) is associated with poor prognosis [128]. The use of STAT3 inhibitors in chordoma cell lines in vitro showed strong inhibition of cell growth and proliferation [129]. Phase II studies are now ongoing, combining imatinib and everolimus or using lapatinib in HER2-positive advanced chordomas.
9 Oncologic Outcome
Several prognostic factors associated with poor survival have been reported: age [78, 130, 131], tumor size [46, 71, 72, 131,132,133], preoperative C-reactive protein >1.0 mg/dL [134], tumor site regarding the proximal extent of the tumor, local invasion into other tissues [131, 135], inadequate surgical margins [46, 69, 70,71,72, 75, 133, 136,137,138], content of extracellular matrix and high Ki-67 index [138], histological category such as dedifferentiated chordomas [41, 139], and local recurrence [46, 138, 140]. Patient survival seems to be less affected by distant metastasis than by local progression of the disease, underlining the role of local control to improve oncological outcomes.
Local recurrence (LR) after surgical treatment is common (43–85%) despite resection with adequate margins [71, 72]. Some authors suggested that infiltration of the musculature adjacent to the sacrum and/or involvement of the sacroiliac joints increases the tendency to local recurrence, even after apparently successful en bloc resection [135, 141, 142]. This hypothesis could also justify also the observation of similar recurrence rate between major resections of proximal part of the sacrum and small resections distal to S3 level [70, 71]. Therefore, in cases of infiltration of the sacroiliac joint, we should consider the tumor at an advanced stage with increased risk of satellite lesions which may promote disease recurrence. The factors associated with higher risk of local recurrence are: old age [82, 140], higher sacral localization [140], inadequate surgical margins [46, 69, 70, 78], and previous intralesional surgery [69,70,71, 78]. Adjuvant radiotherapy may improve oncological outcomes in patients with inadequate surgical margins or dedifferentiated disease, but optimal radiotherapeutic regimens with long-term survival have not been developed. In fact, despite combining en bloc resection with particle radiation therapy, frequent local recurrence remains a reality, and the 5- and 10-year overall survival rates are circa 65% and 35%, respectively [39, 143, 144].
References
Healey JH, Lane JM. Chordoma: a critical review of diagnosis and treatment. Orthop Clin North Am. 1989;20:417–26.
Smoll NR, Gautschi OP, Radovanovic I, Schaller K, Weber DC. Incidence and relative survival of chordomas: the standardized mortality ratio and the impact of chordomas on a population. Cancer. 2013;119:2029–37.
Fabbri N, Ruggieri P. Chordoma. In: Picci P, et al., editors. Atlas of musculoskeletal tumors and tumor like lesions. Switzerland: Springer; 2014. p. 233–8.
Casali PG, Stacchiotti S, Sangalli C, Olmi P, Gronchi A. Chordoma. Curr Opin Oncol. 2007;19:367–70.
Garofalo F, di Summa PG, Christoforidis D, Pracht M, Laudato P, Cherix S, Bouchaab H, Raffoul W, Demartines N, Matter M. Multidisciplinary approach of lumbo-sacral chordoma: from oncological treatment to reconstructive surgery. J Surg Oncol. 2015;112(5):544–54.
Amichetti M, Cianchetti M, Amelio D, Enrici RM, Minniti G. Proton therapy in chordoma of the base of the skull: a systematic review. Neurosurg Rev. 2009;32:403–16.
Staab A, Rutz HP, Ares C, et al. Spot-scanning-based proton therapy for extracranial chordoma. Int J Radiat Oncol Biol Phys. 2011;81:e489–96.
Luschka H. Die Altersveränderungen der Zwischenwirbelknorpel. Virchows Arch A Pathol Anat Histol. 1856;9:311–27.
Kyriakos M, Totty WG, Lenke LG. Giant vertebral notochordal rest: a lesion distinct from chordoma: discussion of an evolving concept. Am J Surg Pathol. 2003;27:396–406.
Walcott BP, Nahed BV, Mohyeldin A, Coumans JV, Kahle KT, Ferreira MJ. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012;13(2):e69–76.
Yakkioui Y, van Overbeeke JJ, Santegoeds R, van Engeland M, Temel Y. Chordoma: the entity. Biochim Biophys Acta. 2014;1846(2):655–69.
O’Rahilly R, Meyer DB. The timing and sequence of events in the development of the human vertebral column during the embryonic period proper. Anat Embryol. 1979;157(2):167–76.
Schoenwolf GC, Larsen WJ. Third week: becomming trilaminar and establishing body axis, Larsen’s human embryology. Philadelphia: Elsevier/Churchill Livingstone; 2009.
Bumcrot DA, McMahon AP. Somite differentiation. Curr Biol. 1995;5:612–4.
Bami M, Mavrogenis AF, Angelini A, Milonaki M, Mitsiokapa E, Stamoulis D, Soucacos PN. Bone morphogenetic protein signaling in musculoskeletal cancer. J Cancer Res Clin Oncol. 2016;142(10):2061–72.
Liem Jr KF, Jessell TM, Briscoe J. Regulation of the neural patterning activity of sonic hedgehog by secreted BMP inhibitors expressed by notochord and somites. Development. 2000;127:4855–66.
Stemple DL. Structure and function of the notochord: an essential organ for chordate development. Development. 2005;132:2503–12.
Dietrich S, Schubert FR, Gruss P. Altered Pax gene expression in murine notochord mutants: the notochord is required to initiate and maintain ventral identity in the somite. Mech Dev. 1993;44:189–207.
Pourquie O, Coltey M, Teillet MA, Ordahl C, Le Douarin NM. Control of dorsoventral patterning of somitic derivatives by notochord and floor plate. Proc Natl Acad Sci U S A. 1993;90:5242–6.
Alvares LE, Lours C, El-Hanfy A, Dietrich S. Microsurgical manipulation of the notochord. Methods Mol Biol. 2008;461:289–303.
Claudio DS. Grafting of somites. Methods Mol Biol. 2008;461:277–87.
Resende TP, Ferreira M, Teillet MA, Tavares AT, Andrade RP, Palmeirim I. Sonic hedgehog in temporal control of somite formation. Proc Natl Acad Sci U S A. 2010;107(29):12907–12.
Gilbert SF. Developmental biology. Sunderland: Sinauer Associates; 2010.
Moore KL, Persaud TVN, Torchia MG. The developing human: clinically oriented embryology. Philadelphia: Saunders/Elsevier; 2008.
Bogduk N. Embryology and development, Clinical anatomy of the lumbar spine and sacrum. Edinburgh: Elsevier/Churchill Livingstone; 2005. p. 149–63.
Oka Y, Sato Y, Tsuda H, Hanaoka K, Hirai Y, Takahashi Y. Epimorphin acts extracellularly to promote cell sorting and aggregation during the condensation of vertebral cartilage. Dev Biol. 2006;291(1):25–37.
Rodrigo I, Hill RE, Balling R, Münsterberg A, Imai K. Pax1 and Pax9 activate Bapx1 to induce chondrogenic differentiation in the sclerotome. Development. 2003;130(3):473–82.
Hunter CJ, Matyas JR, Duncan NA. The notochordal cell in the nucleus pulposus: a review in the context of tissue engineering. Tissue Eng. 2003;9(4):667–77.
Yamaguchi T, Suzuki S, Ishiiwa H, Ueda Y. Intraosseous benign notochordal cell tumours: overlooked precursors of classic chordomas? Histopathology. 2004;44(6):597–602.
Virchow R. Untersuchungen uber die Entwickelung des Schadelgrundes im gesunden und krankhaften Zustande, und uber den Einfluss derselben auf Schadelform, Gesichtsbildung und Gehirnbau. Berl: G. Rimer; 1857. p. 47.
Ribbert H. Uber die Eccondrosis physaliphora spheno-occipitalis. Zentralbl Allg Pathol Anat. 1894;5:457.
Salisbury JR. The pathology of the human notochord. J Pathol. 1993;171:253–5.
Vujovic S, Henderson S, Presneau N, et al. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol. 2006;209:157–65.
Shen J, Li C-D, Yang H-L, et al. Classic chordoma coexisting with benign notochordal cell rest demonstrating different immunohistological expression patterns of brachyury and galectin-3. J Clin Neurosci. 2011;18:96–9.
Choi K-S, Cohn MJ, Harfe BD. Identification of nucleus pulposus precursor cells and notochordal remnants in the mouse: implications for disk degeneration and chordoma formation. Dev Dyn. 2008;237:3953–8.
Yang XR, Ng D, Alcorta DA, et al. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat Genet. 2009;41:1176–8.
Nibu Y, José-Edwards DS, Di Gregorio A. From notochord formation to hereditary chordoma: the many roles of Brachyury. Biomed Res Int. 2013;2013:826435.
Gulluoglu S, Turksoy O, Kuskucu A, Ture U, Bayrak OF. The molecular aspects of chordoma. Neurosurg Rev. 2016;39(2):185–96.
McMaster ML, Goldstein AM, Bromley CM, Ishibe N, Parry DM. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12:1–11.
Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66.
Chugh R, Tawbi H, Lucas DR, et al. Chordoma: the nonsarcoma primary bone tumor. Oncologist. 2007;12:1344–50.
Papagelopoulos PJ, Mavrogenis AF, Galanis EC, Savvidou OD, Boscainos PJ, Katonis PG, Sim FH. Chordoma of the spine: clinicopathological features, diagnosis, and treatment. Orthopedics. 2004;27(12):1256–63.
Anson KM, Byrne PO, Robertson ID, Gullan RW, Montgomery AC. Radical excision of sacrococcygeal tumours. Br J Surg. 1994;81:460–1.
Smith J, Ludwig RL, Marcove RC. Sacrococcygeal chordoma. A clinic radiological study of 60 patients. Skeletal Radiol. 1987;16:37–44.
Kayani B, Hanna SA, Sewell MD, Saifuddin A, Molloy S, Briggs TW. A review of the surgical management of sacral chordoma. Eur J Surg Oncol. 2014;40(11):1412–20.
Bergh P, Kindblom LG, Gunterberg B, Remotti F, Ryd W, Meis-Kindblom JM. Prognostic factors in chordoma of the sacrum and mobile spine: a study of 39 patients. Cancer. 2000;88:2122–34.
Kaiser TE, Pritchard DJ, Unni KK. Clinicopathologic study of sacrococcygeal chordoma. Cancer. 1984;53:2574–8.
Gray SW, Singhabhandhu B, Smith RA, Skandalakis JE. Sacrococcygeal chordoma: report of a case and review of the literature. Surgery. 1975;78:573–82.
Fourney DR, Gokaslan ZL. Current management of sacral chordoma. Neurosurg Focus. 2003;15:9.
Ozaki T, Hillmann A, Winkelmann W. Surgical treatment of sacrococcygeal chordoma. J Surg Oncol. 1997;64(4):274–9.
Dahan H, Arrive L, Wendum D, et al. Retrorectal developmental cysts in adults: Clinical and radiologic-histopathologic review, differential diagnosis, and treatment. Radiographics. 2001;21:575–84.
Williams LS, Rojiani AM, Quisling RG, et al. Retrorectal cyst-hamartomas and sacral dysplasia: MR appearance. AJNR Am J Neuroradiol. 1998;19:1043–5.
Lim KE, Hsu WC, Wang CR. Tailgut cyst with malignancy: MR imaging findings. AJR Am J Roentgenol. 1998;170:1488–90.
Bjornsson J, Wold LE, Ebersold MJ, Laws ER. Chordoma of the mobile spine. A clinicopathologic analysis of 40 patients. Cancer. 1993;71:735–40.
Llauger J, Palmer J, Amores S, et al. Primary tumors of the sacrum: diagnostic imaging. AJR Am J Roentgenol. 2000;174:417–24.
Rossleigh MA, Smith J, Yeh SD. Scintigraphic features of primary sacral tumors. J Nucl Med. 1986;27:627–30.
Crapanzano JP, Ali SZ, Ginsberg MS, Zakowski MF. Chordoma: a cytologic study with histologic and radiologic correlation. Cancer. 2001;93:40–51.
Mitchell A, Scheithauer BW, Unni KK, Forsyth PJ, Wold LE, McGivney DJ. Chordoma and chondroid neoplasms of the spheno-occiput. An immunohistochemical study of 41 cases with prognostic and nosologic implications. Cancer. 1993;72:2943–9.
Oakley GJ, Fuhrer K, Seethala RR. Brachyury, SOX-9, and podoplanin, new markers in the skull base chordoma vs chondrosarcoma differential: a tissue microarray-based comparative analysis. Mod Pathol. 2008;21:1461–9.
Paidakakos NA, Rovlias A, Rokas E, Theodoropoulos S, Katafygiotis P. Primary clear cell chondrosarcoma of the spine: a case report of a rare entity and a review of the literature. Case Rep Oncol Med. 2012;2012:693137.
Jeffrey PB, Biava CG, Davis RL. Chondroid chordoma. A hyalinized chordoma without cartilaginous differentiation. Am J Clin Pathol. 1995;103(3):271–9.
Saito A, Hasegawa T, Shimoda T, Toda G, Hirohashi S, Tajima G, et al. Dedifferentiated chordoma: a case report. Jpn J Clin Oncol. 1998;28:766–71.
Kim SC, Cho W, Chang UK, Youn SM. Two cases of dedifferentiated chordoma in the sacrum. Korean J Spine. 2015;12(3):230–4.
Meis JM, Raymond AK, Evans HL, Charles RE, Giraldo AA. “De-differentiated” chordoma: a clinicopathologic and immunohistochemical study of three cases. Am J Surg Pathol. 1987;11:516–25.
Bisceglia M, D’Angelo VA, Guglielmi G, Dor DB, Pasguinelli. De-differentiated chordoma of the thoracic spine with rhabdomyosarcomatous differentiations: report of a case and review of the literature. Ann Diagn Pathol 2007;11:262–273.
Kishikawa H, Tanaka K. Chordoma: report of an autopsy case with fibrosarcoma. Acta Pathol Jpn. 1974;24:299–308.
Morimitsu Y, Aoki T, Yokoyama K, Hashimoto H. Sarcomatoid chordoma: chordoma with a massive spindle cell component. Skeletal Radiol. 2000;29:721–5.
Fukuda T, Aihara T, Ban S, Nakajima T, Machinami R. Sacrococcygeal chordoma with malignant spindle cell component: a report of two autopsy cases with a review of the literature. Acta Pathol Jpn. 1992;42:448–53.
Fuchs B, Dickey ID, Yaszemski MJ, Inwards CY, Sim FH. Operative management of sacral chordoma. J Bone Joint Surg Am. 2005;87:2211–6.
Ruggieri P, Angelini A, Ussia G, Montalti M, Mercuri M. Surgical margins and local control in resection of sacral chordomas. Clin Orthop Relat Res. 2010;468:2939–47.
Angelini A, Pala E, Calabrò T, Maraldi M, Ruggieri P. Prognostic factors in surgical resection of sacral chordoma. J Surg Oncol. 2015;112(4):344–51.
Jawad MU, Scully SP. Surgery significantly improves survival in patients with chordoma. Spine. 2010;35:117–23.
Romeo S, Hogendoorn PC. Brachyury and chordoma: the chondroid-chordoid dilemma resolved? J Pathol. 2006;209:143–6.
Park JB, Lee CK, Koh JS, Lee JK, Park EY, Riew KD. Overexpressions of nerve growth factor and its tropomyosin-related kinase A receptor on chordoma cells. Spine. 2007;32(18):1969–73.
Hulen CA, Temple T, Fox WP, Sama AA, Green BA, Eismont FJ. Oncologic and functional outcome following sacrectomy for sacral chordoma. J Bone Joint Surg Am. 2006;88-A:1532–9.
Randall RL, Bruckner J, Lloyd C, Pohlman TH, Conrad EU. Sacral resection and reconstruction for tumors and tumor-like conditions. Orthopaedics. 2005;28:307–13.
Stener B, Gunterberg B. High amputation of the sacrum for extirpation of tumors. Principles and technique. Spine. 1978;3:351–66.
Varga PP, Szövérfi Z, Fisher CG, Boriani S, Gokaslan ZL, Dekutoski MB, Chou D, Quraishi NA, Reynolds JJ, Luzzati A, Williams R, Fehlings MG, Germscheid NM, Lazary A, Rhines LD. Surgical treatment of sacral chordoma: prognostic variables for local recurrence and overall survival. Eur Spine J. 2015;24(5):1092–101.
Hsieh PC, Xu R, Sciubba DM, et al. Long-term clinical outcomes following en bloc resections for sacral chordomas and chondrosarcomas: a series of twenty consecutive patients. Spine. 2009;34:2233–9.
Atalar H, Selek H, Yildiz Y, et al. Management of sacrococcygeal chordomas. Int Orthop. 2006;30:514–8.
Devin C, Chong PY, Holt GE, et al. Level-adjusted perioperative risk of sacral amputations. J Surg Oncol. 2006;94:203–11.
Samson IR, Springfield DS, Suit HD, Mankin HJ. Operative treatment of sacrococcygeal chordoma. A review of twenty-one cases. J Bone Joint Surg Am. 1993;75:1476–84.
Angelini A, Ruggieri P. A new surgical technique (modified Osaka technique) of sacral resection by posterior-only approach: description and preliminary results. Spine. 2013;38(3):E185–92.
Osaka S, Yoshida Y, Ryu J. Longitudinal osteotomy of lateral sacrum for malignant iliac tumor using modified threadwire saw. J Surg Oncol. 2007;95(3):258–60.
Gunterberg B, Kewenter J, Petersen I, Stener B. Anorectal function after major resections of the sacrum with bilateral or unilateral sacrifice of sacral nerves. Br J Surg. 1976;63(7):546–54.
Gunterberg B, Norlén L, Stener B, Sundin T. Neurourologic evaluation after resection of the sacrum. Invest Urol. 1975;13(3):183–8.
Gunterberg B, Petersen I. Sexual function after major resection of the sacrum with bilateral or unilateral sacrifice of sacral nerves. Fertil Steril. 1976;27:1146–53.
Zoccali C, Skoch J, Patel AS, Walter CM, Maykowski P, Baaj AA. Residual neurological function after sacral root resection during en-bloc sacrectomy: a systematic review. Eur Spine J. 2016;25. [Epub ahead of print].
Angelini A, Drago G, Trovarelli G, Calabrò T, Ruggieri P. Infection after surgical resection for pelvic bone tumors: an analysis of 270 patients from one institution. Clin Orthop Relat Res. 2014;472(1):349–59.
Chen KW, Yang HL, Lu J, et al. Risk factors for postoperative wound infections of sacral chordoma after surgical excision. J Spinal Disord Tech. 2011;24:230–4.
Pennicooke B, Laufer I, Sahgal A, Varga PP, Gokaslan ZL, Bilsky MH, Yamada YJ. Safety and local control of radiation therapy for chordoma of the spine and sacrum: a systematic review. Spine. 2016;41(Suppl 20):S186–92.
Rich TA, Schiller A, Suit HD, Mankin HJ. Clinical and pathologic review of 48 cases of chordoma. Cancer. 1985;56:182–7.
Catton C, O’Sullivan B, Bell R, et al. Chordoma: long-term follow-up after radical photon irradiation. Radiother Oncol. 1996;41:67–72.
Cummings BJ, Hodson DI, Bush RS. Chordoma: the results of megavoltage radiation therapy. Int J Radiat Oncol Biol Phys. 1983;9:633–42.
Rotondo RL, Folkert W, Liebsch NJ, et al. High-dose proton-based radiation therapy in the management of spine chordomas: outcomes and clinicopathological prognostic factors. J Neurosurg Spine. 2015;23:788–97.
Zabel-du Bois A, Nikoghosyan A, Schwahofer A, et al. Intensity modulated radiotherapy in the management of sacral chordoma in primary versus recurrent disease. Radiother Oncol. 2010;97:408–12.
Hug EB, Fitzek MM, Liebsch NJ, et al. Locally challenging osteo- and chondrogenic tumors of the axial skeleton: results of combined proton and photon radiation therapy using three-dimensional treatment planning. Int J Radiat Oncol Biol Phys. 1995;31:467–76.
Yamada Y, Laufer I, Cox BW, et al. Preliminary results of high-dose single-fraction radiotherapy for the management of chordomas of the spine and sacrum. Neurosurgery. 2013;73:673–80.
Brown JM, Koong AC. High-dose single-fraction radiotherapy: exploiting a new biology? Int J Radiat Oncol Biol Phys. 2008;71:324–5.
Suit HD, Goitein M, Munzenrider J, et al. Definitive radiation therapy for chordoma and chondrosarcoma of base of skull and cervical spine. J Neurosurg. 1982;56:377–85.
Austin-Seymour M, Munzenrider JE, Goitein M, et al. Progress in low-LET heavy particle therapy: intracranial and paracranial tumors and uveal melanomas. Radiat Res Suppl. 1985;8:219–26.
Tobias CA, Blakely EA, Alpen EL, et al. Molecular and cellular radiobiology of heavy ions. Int J Radiat Oncol Biol Phys. 1982;8:2109–20.
DeLaney TF, Liebsch NJ, Pedlow FX, et al. Long-term results of phase II study of high dose photon/proton radiotherapy in the management of spine chordomas, chondrosarcomas, and other sarcomas. J Surg Oncol. 2014;110:115–22.
McDonald MW, Linton OR, Shah MV. Proton therapy for reirradiation of progressive or recurrent chordoma. Int J Radiat Oncol Biol Phys. 2013;87:1107–14.
Indelicato DJ, Rotondo RL, Begosh-Mayne D, et al. A prospective outcomes study of proton therapy for chordomas and chondrosarcomas of the spine. Int J Radiat Oncol Biol Phys. 2016;95:297–303.
Scholz M, Kraft G. Track structure and the calculation of biological effects of heavy charged particles. Adv Space Res. 1996;18:5–14.
Imai R, Kamada T, Tsuji H, et al. Carbon ion radiotherapy for unresectable sacral chordomas. Clin Cancer Res. 2004;10:5741–6.
Nishida Y, Kamada T, Imai R, et al. Clinical outcome of sacral chordoma with carbon ion radiotherapy compared with surgery. Int J Radiat Oncol Biol Phys. 2011;79:110–6.
Imai R, Kamada T, Sugahara S, et al. Carbon ion radiotherapy for sacral chordoma. Br J Radiol. 2011;84(Spec. No. 1):S48–54.
Uhl M, Welzel T, Jensen A, Ellerbrock M, Haberer T, Jäkel O, Herfarth K, Debus J. Carbon ion beam treatment in patients with primary and recurrent sacrococcygeal chordoma. Strahlenther Onkol. 2015;191(7):597–603.
Lundkvist J, Ekman M, Ericsson SR, Jönsson B, Glimelius B. Proton therapy of cancer: potential clinical advantages and cost-effectiveness. Acta Oncol. 2005;44:850–61.
Durante M, Loeffler JS. Charged particles in radiation oncology. Nat Rev Clin Oncol. 2010;7:37–43.
Mohan R, Bortfeld T. Proton therapy: clinical gains through current and future treatment programs. Front Radiat Ther Oncol. 2011;43:440–64.
Goitein M, Cox JD. Should randomized clinical trials be required for proton radiotherapy? J Clin Oncol. 2008;26:175–6.
Park L, Delaney TF, Liebsch NJ, et al. Sacral chordomas: impact of high-dose proton/photon-beam radiation therapy combined with or without surgery for primary versus recurrent tumor. Int J Radiat Oncol Biol Phys. 2006;65:1514–21.
Suit H, Kooy H, Trofimov A, et al. Should positive phase III clinical trial data be required before proton beam therapy is more widely adopted? No. Radiother Oncol. 2008;86:148–53.
Patt JC. CORR Insights(®): sacral insufficiency fractures are common after high-dose radiation for sacral chordomas treated with or without surgery. Clin Orthop Relat Res. 2016;474(3):773–5.
Negri T, Casieri P, Miselli F, et al. Evidence for PDGFRA, PDGFRB and KIT deregulation in an NSCLC patient. Br J Cancer. 2007;96:180–1.
Casali PG, Messina A, Stacchiotti S, et al. Imatinib mesylate in chordoma. Cancer. 2004;101:2086–97.
Stacchiotti S, Marrari A, Tamborini E, et al. Response to imatinib plus sirolimus in advanced chordoma. Ann Oncol. 2009;20:1886–94.
George S, Merriam P, Maki RG, et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J Clin Oncol. 2009;27:3154–60.
Tamborini E, Virdis E, Negri T, et al. Analysis of receptor tyrosine kinases (RTKs) and downstream pathways in chordomas. Neuro-Oncology. 2010;12:776–89.
Stacchiotti S, Longhi A, Ferraresi V, et al. Phase II study of imatinib in advanced chordoma. J Clin Oncol. 2012;30:914–20.
Weinberger PM, Yu Z, Kowalski D, et al. Differential expression of epidermal growth factor receptor, c-Met, and HER2/neu in chordoma compared with 17 other malignancies. Arch Otolaryngol Head Neck Surg. 2005;131:707–11.
Hof H, Welzel T, Debus J. Effectiveness of cetuximab/gefitinib in the therapy of a sacral chordoma. Onkologie. 2006;29:572–4.
Singhal N, Kotasek D, Parnis FX. Response to erlotinib in a patient with treatment refractory chordoma. Anticancer Drugs. 2009;20:953–5.
Stacchiotti S, Tamborini E, Lo Vullo S, et al. Phase II study on lapatinib in advanced EGFR-positive chordoma. Ann Oncol. 2013;24:1931–6.
Yang C, Schwab JH, Schoenfeld AJ, et al. A novel target for treatment of chordoma: signal transducers and activators of transcription 3. Mol Cancer Ther. 2009;8:2597–605.
Yang C, Hornicek FJ, Wood KB, et al. Blockage of Stat3 with CDDO-Me inhibits tumor cell growth in chordoma. Spine. 2010;35:1668–75.
Thieblemont C, Biron P, Rocher F, Bouhour D, Bobin JY, Gérard JP, et al. Prognostic factors in chordoma: role of postoperative radiotherapy. Eur J Cancer. 1995;31A:2255–9.
McGirt MJ, Gokaslan ZL, Chaichana KL. Preoperative grading scale to predict survival in patients undergoing resection of malignant primary osseous spinal neoplasms. Spine J. 2011;11(3):190–6. doi:10.1016/j.spinee.2011.01.013.
Ozger A, Eralp L, Sungur M, Atalar AC. Surgical management of sacral chordoma. Acta Orthop Belg. 2010;76:243–53.
Lee J, Bhatia N, Hoang B, Ziogas A, Zell J. Analysis of prognostic factors for patients with chordoma with use of the California Cancer Registry. J Bone Joint Surg Am. 2012;94:356–63.
Hobusch GM, Bodner F, Walzer S, Marculescu R, Funovics PT, Sulzbacher I, Windhager R, Panotopoulos J. C-reactive protein as a prognostic factor in patients with chordoma of lumbar spine and sacrum—a single center pilot study. World J Surg Oncol. 2016;14:111.
Hanna SA, Aston WJ, Briggs TW, Cannon SR, Saifuddin A. Sacral chordoma: can local recurrence after sacrectomy be predicted? Clin Orthop Relat Res. 2008;466(9):2217–23.
Boriani S, Bandiera S, Biagini R, Bacchini P, Boriani L, Cappuccio M, Chevalley F, Gasbarrini A, Picci P, Weinstein JN. Chordoma of the mobile spine: fifty years of experience. Spine. 2006;31(4):493–503.
Stacchiotti S, Casali PG, Lo Vullo S, Mariani L, Palassini E, Mercuri M, et al. Chordoma of the mobile spine and sacrum: a retrospective analysis of a series of patients surgically treated at two referral centers. Ann Surg Oncol. 2010;17:211–9.
von Witzleben A, Goerttler LT, Lennerz J, Weissinger S, Kornmann M, Mayer-Steinacker R, von Baer A, Schultheiss M, Möller P, Barth TF. In chordoma, metastasis, recurrences, Ki-67 index, and a matrix-poor phenotype are associated with patients’ shorter overall survival. Eur Spine J. 2016;25:4016–24.
Ridenour 3rd RV, Ahrens WA, Folpe AL, Miller DV. Clinical and histopathologic features of chordomas in children and young adults. Pediatr Dev Pathol. 2010;13(1):9–17.
Cheng EY, Ozerdemoglu RA, Transfeldt EE, Thompson Jr RC. Lumbosacral chordoma. Prognostic factors and treatment. Spine. 1999;24:1639–45.
Chen KW, Yang HL, Lu J, Liu JY, Chen XQ. Prognostic factors of sacral chordoma after surgical therapy: a study of 36 patients. Spinal Cord. 2009;48:166–71.
Ishii K, Chiba K, Watanabe M, Yabe H, Fujimura Y, Toyama Y. Local recurrence after S2-3 sacrectomy in sacral chordoma. Report of four cases. J Neurosurg. 2002;97(1 Suppl):98–101.
Dorfman HD, Czerniak B. Bone cancers. Cancer. 1995;75:203–10.
Mukherjee D, Chaichana KL, Parker SL, Gokaslan ZL, McGirt MJ. Association of surgical resection and survival in patients with malignant primary osseous spinal neoplasms from the surveillance, epidemiology, and end results (SEER) database. Eur Spine J. 2012;22:1375.
Acknowledgements
We would like to thank Pierpaolo Furlano for the production of research drawings which formed the basis of the final drawings in this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
No benefits have been or will be received from a commercial party related directly or indirectly to the subject matter of this article.
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Angelini, A., Ruggieri, P. (2017). Chordoma of the Sacrum. In: Ruggieri, P., Angelini, A., Vanel, D., Picci, P. (eds) Tumors of the Sacrum. Springer, Cham. https://doi.org/10.1007/978-3-319-51202-0_15
Download citation
DOI: https://doi.org/10.1007/978-3-319-51202-0_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-51200-6
Online ISBN: 978-3-319-51202-0
eBook Packages: MedicineMedicine (R0)