
Abstract
A structured development approach is presented to guide the development of stable and commercially viable polymer based amorphous formulations. The proposed approach should not only enable the delivery of poorly soluble drugs but also help to reduce the API needs, reduce in vivo screening, minimize risks for late-stage development, and should ensure consistent quality. During initial assessment, a guided evaluation of the physicochemical properties of the API helps to assess the degree of difficulty for the development. A range of tests including in silico evaluation, high-throughput screening assays, and miniaturized screening tools provide a road map for selecting the appropriate polymer, drug loading, and suitable manufacturing process. A dedicated section provides a review of the characterization tools to assess and quantify the crystallinity, understanding the phase behavior of amorphous solid dispersions, and designing the in vitro dissolution methods. Finally, a reference chart is provided that summarizes the key concepts proposed as part of the structured development approach that can serve as a blueprint for the development of amorphous formulations. The current authors would like to thank and acknowledge the significant contribution of the previous authors of this chapter from the first edition. This current second edition chapter is a revision and update of the original authors’ work.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- In silico evaluation
- High-throughput screening assays
- In vitro dissolution methods
- Amorphous solid dispersions (ASD)
- Miscibility
- Solubility parameter calculations
- Molecular modeling
- Molecular dynamic simulation
- Flory-Huggins theory
- Miniaturized systems
- Supersaturation screening
- X-ray powder diffraction (XRPD)
- Microscopy
8.1 Introduction
Over the last decade amorphous solid dispersions (ASD), in particular those stabilized with polymers have emerged as a method of choice for improving the dissolution behavior and bioavailability of poorly water-soluble active pharmaceutical ingredients (Chiou and Riegelmann 1970; Hancock and Parks 2000; Six et al. 2004; Mishra et al. 2015). However, amorphous compounds are thermodynamically unstable and may crystallize over pharmaceutically relevant timescales, negating any solubility advantage. Amorphous compounds can often be stabilized by combining the active ingredient with a carrier polyme r to form an amorphous, molecular-level solid dispersion, as described in several comprehensive reviews (Leuner and Dressman 2000; Serajuddin 1999; Van den Mooter et al. 2001; Janssens et al. 2010; Mishra et al. 2015; He and Ho 2015). The properties of the resultant solid dispersions are influenced by the physicochemical properties of both the active pharmaceutical ingredient and the carrier polymer. Based on the physical state and the composition of the carrier, ASDs have been classified into different categories: first generation ASDs contain crystalline carriers (e.g. urea, mannitol) while second generation ASDs consist of amorphous carriers (mostly polymers). For some time, surface active agents or self-emulsifiers are used as carriers or additives to improve the dissolution rate and to further reduce the risk of API precipitation and recrystallization (third generation ASDs). In fourth generation ASDs, water insoluble or swellable polymers were introduced to enhance the solubility as well as to ensure the drug release in a controlled manner (controlled release solid dispersions) (Mishra et al. 2015). Recently, new carrier systems such as mesoporous silica (see Chap. 13 “Emerging technologies”) or amino acids (Löbmann et al. 2013; Jensen et al. 2015; Lenz et al. 2015) were used, or functional additives such as plasticizers, solubilizers, wetting agents, superdisintegrants, antioxidants, and pH modifiers were added. Over the past years, the characterization methods as well as the manufacturing technologies further advanced. Despite the expanded theoretical and practical knowledge of amorphous systems with respect to thermodynamic and kinetic stability and the availability of various modern instrumental techniques to qualitatively and quantitatively characterize the amorphous system, only a few amorphous drug products have been introduced into the market place (Table 8.1).
Interestingly, the number of marketed products using an amorphous solid dispersion is recently increasing, which might be a result of a more structured development approach in comparison to the empirical approach used in the past. The formulation scientist is aware of the fact that each drug candidate has its own unique physical and chemical properties, and that therefore no universal formulation can exist (Liu et al. 2015). Therefore, his primary focus in the early stage of development is the selection of a suitable carrier system and drug load, followed by the manufacturing of some prototype formulations for pharmacokinetic testing in animals. Based on the results of the in vivo behavior of the formulation, an intensive analytical characterization and stability testing of the final formulation composition can be defined. For the next step of the development (e.g. process development and up-scaling), the quality by design (QbD) approach was successfully used by a number of researchers so far (Sanghvi et al. 2015). As amorphous solid dispersions are often used for formulating BCS II or IV compounds (BCS = Biopharmaceutical Classification System), the critical quality attributes (CQAs) for the product in development are dissolution, bioavailability and solid-state stability (Siew 2014). Once an acceptable target is identified for each CQA design of experiments can be performed in order to identify the relationship between the critical process parameters and the CQAs, and allows thus setting a design space acceptable for consisted product quality. The focus of this chapter is to describe a structured approach for the development of amorphous formulations that should help bolster confidence and provide a formulation with the best chance for success.
8.2 Ideal Amorphous Formulation: Structured Development
An ideal amorphous formulation should provide the maximum physical stability during processing and storage and maintain supersaturation while the drug is being dissolved and absorbed in the gastrointestinal tract. Among various factors, the selection of polymer and drug loading are two key aspects in the development of an ideal amorphous formulation. The inhibitory effects of polymers against crystallization in the solid state have been attributed to various mechanisms including anti-plasticization by the polymers (Van den Mooter et al. 2001; Oksanen and Zografi 1990), interactions between the API and polymers in solid dispersions (Aso et al. 2002; Taylor and Zografi 1997; Miyazaki et al. 2004), a reduction in local molecular mobility due to coupling between the polymer and API motions (Aso and Yoshioka 2006), and an increase in the activation energy for nucleation (Marsac et al. 2008). On the other hand, maintaining supersaturation during the dissolution process has been attributed to the inhibition of API crystallization from the supersaturated solution by the polymer (Gupta et al. 2004; Tanno et al. 2004) and increased equilibrium solubility of the API due to complexation with the polymer (Usui et al. 1997; Acartürk et al. 1992; Loftsson et al. 1996). Therefore, the ideal formulation should provide solid-state stability during downstream processing and storage as well as maintenance of supersaturation during dissolution (generally about 2–4 h) not only initially but also throughout the product’s shelf life.
The following steps are described in the chapter as part of the structured development approach to support and strengthen amorphous formulations:
-
Initial assessment of physicochemical drug properties to evaluate the suitability of the drug substance for amorphous solid dispersion development,
-
Definition of the formulation composition, e.g. by polymer-excipient-screening,
-
Manufacturing of prototype formulations,
-
Characterization of the amorphous solid dispersion, and
-
Downstream processing of the amorphous formulation.
We believe that using a structured approach to amorphous formulation development consisting of an evaluation of drug substance properties, the selection of a suitable polymer and concentration, and the use of a proper process will enable the development of amorphous formulations with optimum solid-state stability and dissolution performance. An overview of the different stages of the structured approach is shown in Table 8.2.
8.3 Initial Assessment
The aim of the initial assessment is to assess the suitability of the drug substance for amorphous solid dispersion development in terms of the potential solubility advantage of the amorphous over the crystalline form, and the stability of the amorphous form. Furthermore, it should provide the formulator with some criteria to pre-select a manufacturing technology.
8.3.1 Initial Assessment in Terms of Bioavailability
Over the years many researchers tried to predict the solubility advantage of the amorphous form of a drug substance in comparison to the solubility of the crystalline form in order to estimate the potential effect on the bioavailability. In a recent publication Paus et al. compared the temperature-dependent solubility advantage of an amorphous drug substance versus its crystalline form predicted using the perturbed-chain statistical associating fluid theory (PC-SAFT) with the results obtained from the Gibbs-energy-difference (GED) method and experimental data. Overall, the results obtained from this research group indicated that the predictions from PC-SAFT were more accurate than the results obtained from the GED method for the five investigated compounds (Paus et al. 2015). Nevertheless, still quite some deviations were observed between the calculated and measured values, which make the use of such predictions questionable.
8.3.2 Initial Assessment in Terms of Stability
Other key factors for successful ASD development are the glass forming ability (GFA) as well as glass stability (GS) of the compound. These properties can either be predicted based on measured and calculated parameters or they can be experimentally evaluated. The ease of vitrification of a liquid on cooling is described as glass forming ability, whereas the resistance of a material to crystallization is known as glass stability in the literature (Baird et al. 2010). Baird et al. developed a classification system based on a fast DSC screening method to assess the glass forming ability of organic molecules. The sample is first heated at 10 °C/min to approx. 10 °C above the melting temperature of the compound, held at this temperature for 3 min, cooled to −75 °C with a cooling rate of 20 °C/min, and finally reheated at 10 °C/min to temperatures above the melting point. Although it is known that the ability of a compound to form a glass is strongly dependent on the cooling rate applied, and that a minimum cooling rate is required, they fixed the cooling rate to 20 °C/min for the screening method. Compounds, which recrystallized upon cooling are classified as class I compounds (weak glass formers), whereas molecules which showed a crystallization upon reheating are put into class II. Class III is comprised of compounds which didn’t show recrystallization at all (strong glass formers). Although not explicitly discussed, class I compounds may represent a higher risk for the development of stable amorphous formulations, while class III compounds are the ideal candidates for successful ASD formation. This DSC-based classification system provides an early readout from a simple experiment to assess ASD feasibility (with exception of thermally labile compounds). Overall, Baird et al. investigated 51 structurally diverse organic molecules and tried to relate the GFA and GS with physicochemical properties of the compounds. Compounds, which are weak glass formers are usually low molecular weight compounds with rigid structure, whereas strong glass formers tend to have a higher molecular weight and a more complex structure (Baird et al. 2010). A follow-up investigation by the same research group investigated the crystallization tendency of the same set of compounds following rapid solvent evaporation. Overall, the comparison showed identical classifications in 68 % of all cases. Furthermore, they could confirm that high molecular weight compounds with a large number of rotatable bonds are strong glass formers (Van Eerdenburgh et al. 2010).
The relationship between molecular weight and glass forming ability was confirmed by other research groups as well. Mahlin and Bergström (2013) showed that the glass forming ability can be easily predicted from the molecular weight of the compound. Molecules with >300 g/mol are expected to be strong glass formers that can be easily transformed into the amorphous state. The glass stability was originally related to the glass transition temperature, but newer investigations by Alhalaweh et al. (2015) showed that the physical stability of the amorphous drug is related to π–π interactions and aromaticity.
8.3.3 Initial Assessment in Terms of Manufacturability
The physicochemical properties of the compound, usually evaluated during pre-formulation, can be used to pre-select the manufacturing technology. The assessment is usually based on:
-
Melting point and heat of fusion of the crystalline form and the glass transition temperature of the amorphous form (if it can be determined),
-
Thermal stability of the crystalline and amorphous form, and
-
Solubility in organic solvents (low and high boiling point).
For solvent based processes, e.g. spray drying or microprecipitation, the compounds need a reasonable solubility in organic solvents. For spray drying organic solvents with low boiling point are used (volatile solvents, e.g. ethanol, acetone), whereas for the microprecipitation process solvents with high boiling point are used (non-volatile solvents, e.g. dimethylacetamide, dimethylformamide). Hot-melt extrusion is usually difficult for compounds with high melting point (>200 °C) or compounds which are thermally unstable.
In summary, the knowledge of the basic drug substance properties is of utmost importance for assessing the suitability of the compound for an successful ASD development. As all the necessary data for this evaluation are usually determined in the early phases of drug development (preformulation), there is no additional effort expected. In addition, the clinical dose of the drug substance is of course also of critical importance for the feasibility of developing a stable amorphous system.
8.4 Polymer Screening : Part I (Theoretical)
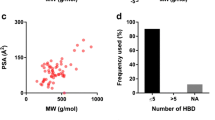
Due to the thermodynamically unstable nature of the amorphous form, many drug substances are stabilized by forming a polymeric amorphous solid dispersion (Baghel et al. 2016). Preformulation data of the drug substance such as type and number of hydrogen-bond donor/acceptor groups, ionic groups, partition coefficient, hygroscopicity, and ratio of T m/T g provide some key characteristics for developing a polymeric amorphous solid dispersion. Friesen et al. (2008) systematically showed the ability to form amorphous systems for a large variety of drug substances based on the T m/T g ratio and the octanol-water partition coefficient (log P). Figure 8.1 provides an overview on a couple of drug substances relating the drug load of the amorphous solid dispersion to the physical stability of the amorphous solid dispersions as well as their dissolution performance.

Tm/Tg vs. log P diagram adapted from Friesen et al. (2008). Group 1 compounds (circles): high Tg allows high drug loadings in ASD (≥50 % w/w). Group 2 (diamonds): typically 35–50 % w/w drug loading in ASD. Group 3 (squares): drug propensity to crystallize may limit drug loading in ASD (10–35 % w/w). Group 4 (triangles): drug loading can be limited by dissolution rate (10–35 % w/w)
In order to pre-select the most promising polymer(s) for amorphous drug stabilization the miscibility of the drug substance and polymer as well as the potential interactions between the drug and polymer can theoretically be assessed by the chemical structure of the compound and the polymers. The phase behavior of polymer-stabilized amorphous formulations depends on the specific interactions between the polymer and the drug. A key to the selection of polymer and optimal drug loading is to maximize the interactions between the drug and the polymer. As shown in subsequent sections, specific interactions between the drug and polymer are the strongest, and if favorable, have the best probability of achieving the desired stability.
The list of polymers can sometimes be further narrowed by excluding manufacturing technologies which cannot be pursued due to specific properties of the drug substance. The data used during this assessment either are based on the structure or are usually measured during early phases of drug development (preformulation). The key steps considered during initial assessment include:
-
Assessing the miscibility of drug and polymer (e.g. solubility parameters, glass transition temperature, phase diagrams).
-
Assessing the potential of specific interactions between the compound and polymer.
-
Further restriction of polymers based on the pre-selected manufacturing technology.
A list of commonly used pharmaceutical polymers is compiled in Table 8.3, along with some relevant properties. As one can imagine, the list of pharmaceutically acceptable polymers for amorphous formulations is somewhat limited. Although new polymers are being added continuously to the list, a mechanistic understanding of polymer properties needed to stabilize the amorphous system remains somewhat elusive. Other properties, such as: composition of polymer, polydispersity, and monomer levels, are also essential and should be evaluated during development mainly as part of critical material attributes during the Quality by Design phase (QbD) to ensure consistent processing and performance. Some of the key factors related to polymer selection in the design of stable amorphous formulations are discussed in the following sections.
8.4.1 Assessing the Miscibility of Drug and Polymer
The drug solubility determines the upper limit of the drug concentration in the drug-polymer mixture, in which the drug exists in a molecularly dispersed state and no phase separation or crystallization will occur during storage. Any drug amount higher than that solubility will exist in the metastable state and is prone to revert to a low-energy crystalline state under normal stresses of temperature, pressure, and humidity. Furthermore, these systems may not provide consistent dissolution (or in vivo performance) due to the chaotic nature of the reversion process. Increasing the drug loading further, e.g. above the miscibility, will lead to spontaneous phase separation and further down the line to crystallization of the compound.
Although significant efforts have been made to understand and determine drug solubility (crystalline drug) and miscibility (amorphous) in polymers, it still remains a challenge to estimate these values due to low diffusivity and high-molecular weight even at high temperatures, or the relaxation of the amorphous system at lower temperature (Qian et al. 2007; Marsac et al. 2008; Huang et al. 2008). Commonly used methods for estimating the solubility of drugs in polymers include solubility parameter calculations, molecular modeling, molecular dynamic simulation (Gupta et al. 2011), and Flory-Huggins interaction parameter (using thermal analysis and solubility).
8.4.1.1 Solubility Parameters
The evolution of solubility parameters to predict the solubility of organic compounds originated from the Hildebrand solution theory in 1936, and the symbol delta (δ) was adopted in 1950 to represent the term solubility parameter. The solubility parameter, or cohesive energy density (CED) of a material, is the energy which holds that substance together. It is the amount of energy required to separate the constituent atoms or molecules of the material to an infinite distance, and hence it is a direct measure of the attraction that its atoms or molecules have for one another. Cohesive energy is the net effect of all the interatomic/intermolecular interactions including Van der Waals interactions, covalent bonds, ionic bonds, hydrogen bonds, and electrostatic interactions, induced by dipole and permanent dipole interactions (Hancock et al. 1997). Three main components of the solubility parameters include dispersion, polar, and hydrogen-bonding forces and are frequently calculated using group contribution methods. In a more recent publication an improved group contribution parameter set for hot-melt extrusion application was published (Just et al. 2013). These calculated solubility parameters are used to predict the miscibility of drugs with polymers. Greenhalgh et al. observed miscibility between ibuprofen and several excipients when the drug substance and the polymer had a difference in the total solubility parameter of less than 7 MPa1/2, and immiscibility when the difference in the solubility parameter was above 10 MPa1/2 (Greenhalgh et al. 1999). In order to reflect that hydrogen bonding has a stronger effect on solubility compared to dispersion and polar components Albers used the Bagley plot (δh vs. δv (=square root(δp 2 + dd 2))) to predict miscibility. She showed that, from the data set investigated, it could be deduced with few exceptions that two substances are miscible if their distance in the Bagley plot is ≤5.6 MPa1/2, or if the difference in their total solubility parameters is ≤5.4 MPa1/2 (Albers 2008). Overall, it should be mentioned that there is no clear cut-off value for the difference in solubility parameters below which systems are completely miscible in all proportions. However, there exists a significant body of evidence suggesting that the ranges suggested by Greenhalgh et al. (1999) could provide good guidance. Furthermore, the absolute difference between solubility parameters of a drug and polymer should not be considered as an exclusion criterion because other aspects, such as formation of ionic interactions, may help overcome the solubility limitations.
With increasing computational power, molecular dynamic (MD) simulations can be used to calculate the solubility parameter as shown by Gupta et al. (2011). The authors see a clear advantage of these MD simulations as the solubility parameter can be calculated as a function of temperature, and additional functional groups as well as secondary interactions that are not covered in the group contribution methods can also be calculated.
8.4.1.2 Glass Transition Temperature
Amorphous solids are frequently characterized by the glass transition temperature (T g) that corresponds to the temperature at which an amorphous material undergoes a transition from a “glassy state” to a “rubbery state”. Unlike a melting endotherm, this transition is a second order transition and is associated with continuous changes (as opposed to abrupt changes) in thermodynamic properties, such as heat capacity, viscosity, entropy, and volume (see Fig. 8.2). Due to the nature of the transition, its measurement is sensitive to many factors including sample history, rate of cooling, and the presence of impurities.

Pictorial representation of thermodynamic changes depicting the thermal transitions
A molecular dispersion of an amorphous drug with low Tg in a polymer with high Tg will lead to a ASD with a Tg intermediate of the Tg of the two components. As a general guiding principle, amorphous systems with high T gs are preferred to improve stability as they can exist in a glassy state at room temperature, which has substantially high viscosity (>1013 P) limiting the configurational changes and rendering the system immobile. A low-T g system can be considered only if there are significant interactions between the materials (Chokshi et al. 2008).
Based on free volume theory, the T g of a mixture as a function of polymer concentration is generally expressed as a weighted average of the T gs of the pure components and can be calculated using the Gordon–Taylor equation (Gordon and Taylor 1952).
In this equation, T g is the T g of the blend, T g1 and T g2 are the T gs of the pure components, w 1 and w 2 are the weight fractions of each component in the blend, and k is a constant calculated using true density (ρ) and the difference between expansion coefficients of the melt and the glass (Δα) (k = ρ 1Δα 1/ρ 2Δα 2). For early assessments, k is generally considered a constant. A simplified version of this equation for an ideal system, when k = 1, is known as the Fox equation.
As an initial assessment and rough rule of thumb, polymers with high T gs are preferred especially those that can provide amorphous solid dispersions with a single composite T g of 75 °C or higher (i.e., 50 °C above the storage temperature).
8.4.1.3 Prediction of Phase Diagrams
In recent years several attempts are made in order to construct temperature–composition phase diagrams to support the rational selection of drug load and polymer type during the formulation design phase. The temperature-composition phase diagram contains usually the solubility curve, the miscibility curve and the glass transition curve. Tian et al. have predicted the phase diagram for a solid dispersion containing cinnarizine (CIN) and Soluplus® using the method described below and compared their predictions with results obtained from hot-melt extruded dispersions of the same components. The solubility curve was calculated in their work using the solid-liquid equilibrium (SLE) equation considering the polymer behaves like a solvent. In order to solve the equation the activity coefficient of the drug is necessary, which can be estimated on the basis of the extended Hansen model by calculating the Hansen solubility parameter. The miscibility curve, which corresponds to the spinodal curve, was predicted based on the Flory-Huggins theory (Tian et al. 2015). The Flory–Huggins solution theory is used to describe the free energy change of mixing. The Gibbs free energy change accompanying mixing at constant temperature and pressure is written as:
where ΔG m is the free energy of mixing, ΔH m is the enthalpy of mixing, and ΔS m is the entropy of mixing at absolute temperature T. For most solid dispersions, entropy is usually positive; however, it is the enthalpic term that is critical to achieve the negative free energy required for a stable system.
Flory–Huggins applied the solution theory to represent the enthalpic and entropic terms for mixing process as shown below:
where n is the number of moles, φ is the volume fraction, R is the gas constant, and χ is the interaction parameter. The index 1 represents the polymer, whereas 2 represents the drug substance.
The value of χ can be estimated by Hildebrand solubility parameter as follows:
where v′ is the volume of the polymer chain segment and δs are the solubility parameters of the drug and polymer, respectively. The estimation of “χ” can alternatively be done by using the experimentally determined values of solubility parameters or using thermal analysis such as the melting point depression method. The estimated or determined value of “χ” is used to construct the temperature composition phase diagram that provides the miscibility regions. A schematic of free energy of mixing versus composition diagram is shown in Fig. 8.3. Note that the negative “χ” value favors mixing. The minimum in the free energy diagram corresponds to maximum stability for the system. This temperature–composition phase diagram can be further transformed using the first derivative and second derivative to generate the binodal and spinodal decomposition curves that are used to indicate regions of stability, instability, and metastability.

Free energy of mixing as function of composition at different interaction parameters
Tian et al. (2015) used the solubility parameter in order to calculate the Flory-Huggins-Interaction-Parameter χ. The solubility parameter can easily be determined by using the group contribution method, but it is limited to systems were no specific interactions between the drug substance and the polymer are present. Finally, the glass transition curve was estimated using the Fox equation. They showed a good agreement between the theoretically calculated and experimentally determined solubility of CIN in Soluplus® as well as the miscibility level (Tian et al. 2015).
Alternatively, thermal analysis methods can be used for the determination of the Flory-Huggins-Interaction-Parameter χ, including the recrystallization method, the dissolution end point method and the melting point depression method, or the estimation of the solubility of a drug in a polymer from the solubility of the drug in a liquid low molecular weight analogue of the polymer (Knopp et al. 2015). All these methods were compared by Knopp et al. recently, and they came to the conclusion that the magnitude of the predicted solubilities from the solubility in the liquid analogue correlated well with the results from the recrystallization and melting point depression method (Knopp et al. 2015). In a follow up investigation Knopp et al. (2016) critically reviewed the use of the melting point depression method for calculating the interaction parameter χ. The authors performed a statistical analysis of the method proposed by Lin and Huang and showed that the predicted miscibility curve could not be trusted with statistical confidence. They rather propose that the DSC measurements, which are used to make miscibility predictions, should be examined by deriving an objective function. This would result in an unbiased, minimum variance property of the least square estimator. Nevertheless, they also state that additional arguments are needed to prove the underlying physical assumptions, such as the temperature dependence of χ in order to fully believe in the predictions (Knopp et al. 2016). Another alternative in order to determine the Flory-Huggins-Interaction-Parameter χ is the use of molecular dynamic simulations or alternative approaches such as the extended Flory-Huggins theory as implemented in the Material Studio Blends module from Accelrys Inc. (Pajula et al. 2010). The latter was used by Pajula et al. to predict the phase stability of small molecule binary mixtures.
8.4.2 Specific Interactions
It is well recognized that interactions between a drug and polymer have significant effect on the stability of a high-energy amorphous system. The interactions between a drug and a polymer can result from several types of intermolecular interactions , e.g., hydrophobic interactions (due to dispersions forces), hydrogen bonding, or electrostatic (polar or induced-dipole) interactions. As shown in Table 8.4, electrostatic interactions, being strong forces, can provide stability to amorphous solid dispersions.
An understanding of the interactions and their effect on the solubility of a drug in a polymer and the resultant phase diagram determine the space within which high-energy systems can provide maximal benefit. A theoretical basis for the calculation of thermodynamic solubility and kinetic miscibility has been discussed by several researchers (Zhao et al. 2011; Janssens et al. 2010; Paudel et al. 2010).
As shown in Table 8.3, polymers with ionizable groups present opportunities for the formation of hydrogen bonding and/or ionic interactions. It has been generally recognized that proton transfer and exact stoichiometry may not be a requirement for drug–polymer blends due to large differences in the molecular weight. Nevertheless, the presence of ionizable groups can provide secondary structures that are sterically stabilized in addition to reducing mobility. Yoo et al. (2009) investigated the miscibility of polymers and highly crystalline additives. They showed that the likelihood of obtaining a miscible system was the highest in the case where an acid–base ionic interaction is involved in the formation of the amorphous state. In the absence of ionic interactions, systems with similar solubility parameters and partition coefficients showed miscibility. Similar interactions are expected between drugs and polymers. Additionally, (Forster et al. 2001a, b) also showed that ionic interactions and solubility parameters play a role in the formation of the amorphous state.
8.4.3 Hygroscopicity and Water Activity
Hygroscopicity of a polymer plays an important role in determining the physical stability of an amorphous product, especially during storage. Adsorption of water can act in many ways to destabilize amorphous systems, such as by weakening the interactions between the drug and polymer, lowering the solubility or miscibility of the drug in the polymer, and lowering the glass transition temperature of the polymer (plasticization). Although the effect of water on the interactions between the drug and polymer is not easy to assess, several authors have evaluated the effect of water on drug solubility and glass transition temperature. Rumondor et al. (2009b) showed that a small amount of water can significantly lower the solubility of felodipine in PVP. Similarly, in another study, it was shown by differential scanning calorimetry (DSC), atomic force microscopy (AFM), and transmission electron microscopy (TEM) measurements that water can irreversibly disrupt the favorable interactions between a drug and a polymer, thus resulting in phase separation that eventually leads to crystallization (Marsac et al. 2008). A modified Flory–Huggins equation considering a water–drug–polymer ternary system was developed by Rumondor et al. (2009b) to estimate the effect of water on the interaction parameters. It was shown that ingress of water can weaken the interactions between hydrophobic drug and hydrophilic polymer, thus resulting in drug-rich phases that induce crystallization. In a study where different polymers were compared, the authors concluded that use of a hydrophobic polymer, such as hypromellose acetate succinate (HPMC-AS), can be beneficial over more hydrophilic polymers, such as povidone or copovidone, to achieve better stability. A similar finding has been shown by Friesen et al. (2008) where the use of HPMC-AS was shown to provide better supersaturation during dissolution that was related to the formation of aggregated structures by HPMC-AS (Friesen et al. 2008). The aforementioned discussion clearly shows that water can interfere with amorphous systems in many different ways. Therefore, polymers with low water activity are expected to provide the best stability for amorphous systems. On several occasions, we have shown that polymers with ionic groups, such as HPMC-AS or methacrylates (Eudragit L100, Eudragit L100-55, Eudragit E100), can help stabilize amorphous systems by not only providing an opportunity for ionic interactions but also by providing low water activity as they are insoluble in water (Chokshi et al. 2008).
8.4.4 Further Restriction of Polymers Based on the Manufacturing Technology
Once the formulator has pre-selected those polymers that provide miscibility and/or specific interactions with the drug substance he/she can narrow down the list further by considering the pre-selected manufacturing technology (see Sect. 8.3.3). For solvent based processes the polymer needs to have a reasonable solubility in organic solvents (e.g. for spray drying in organic solvents with low boiling point and for the microprecipitation process solvents with high boiling point). For hot-melt extrusion the polymer should have a reasonable low melt viscosity in the temperature range needed for processing the drug substance and it needs to be thermally stable. In summary, the knowledge of the basic polymer properties is very helpful for assessing the suitability of the polymer for a certain manufacturing technology.
8.4.5 Summary of Initial Assessment
As described in this section, it is very important to know the physicochemical properties of the drug substance with respect to its thermal behavior, hydrogen-bond acceptor and donor groups, molecular weight, hydrophobicity, melting point, glass transition temperature, solubility parameter, hygroscopicity, logP, and T m/T g ratio. The basic preformulation data can help assess the degree of difficulty a molecule may present in being converted to and maintained in an amorphous form (Friesen et al. 2008). To best match the properties of a drug substance, it is also important to know the physicochemical properties of polymers such as their thermal behavior, hydrogen-bond acceptor and donor groups, molecular weight, hydrophobicity, melting point/glass transition temperature, solubility parameter, and hygroscopicity. A comprehensive summary of polymer properties is provided in Table 8.3. The initial selection of polymers should be based on solubility parameters, functional groups, and thermal behavior including melting point and T g. Thermal stability and solubility in organic solvents are critical for the selection of the processing method (further discussed in Sect. 8.6). The initial screening will help to narrow down the list of polymers, e.g., considering similar solubility parameters or the possibility of ionic interactions between a weakly basic drug and enteric polymers. The next step in the process of structured development is the conduction of small-scale experiments to further narrow down the choice of polymer and drug loading that has the potential to provide the best stability and supersaturation.
8.5 Miniaturized Methods for ASD Screening
After a careful evaluation of drug and polymer properties and in silico assessment , the next phase in the development of amorphous dispersions is the evaluation of different compositions by miniaturized or small-scale experiments to narrow down the choice of polymer and to get an assessment of drug loading. In the past, these systems were prepared by different small-scale low-throughput methods, including: rotary evaporator (Moneghini et al. 1998), spray-drying (Corrigan et al. 1985), freeze drying (Engers et al. 2010), amorphous film preparation by the ultra-rapid freezing technique (Overhoff et al. 2007; DiNunzio et al. 2008), or small-scale ball milling experiments (Patterson et al. 2007), hot-plate mixing (Chokshi et al. 2005), hot-melt extrusion (Miller et al. 2008), or beaker melt method (Forster et al. 2001a, b). However, a new trend has recently emerged where miniaturized experimental screening systems are being utilized for the evaluation of suitable polymers and additives (or mixtures thereof) for amorphous formulations. These systems use typically less than 10 mg of compound per test sample and work in the 96-well format. The use of these miniaturized systems has the potential to facilitate amorphous formulation development by saving valuable time and resources (manpower and compound). Ideally, the miniaturized assays are partly or fully automated by an assembly of different modules, for instance: robotic systems, high-throughput analytical systems, and specific software and database tools, allowing dozens or hundreds of screening experiments in a short period of time. As discussed before, the development of suitable amorphous systems requires miniaturization of the preparation methods and characterization tools including the analytical methods to evaluate the API supersaturation upon dissolution, as well as to assess the solid state and physical stability of the systems.
The ASD processing technologies can generally be divided into two main classes, solvent based and fusion based methods. Solvent-based technologies include spray drying, fluid bed layering, coprecipitation (e.g. MBP = Microprecipitated Bulk Powder) etc. Fusion-based technologies include melt extrusion, melt granulation, or Kinetisol® technology, etc. The selection of an appropriate processing technology strongly depends on the API properties. Understanding the physicochemical properties of API, therefore, will streamline the amorphous formulation screening design. Most of ASD screening approaches described in the literature are focused either on the assessment of the supersaturation potential of the API-polymer combination in solution or on the evaluation of amorphous drug stabilization in the solid state. So far, only a few papers have considered the use of a combination of different miniaturized assays to identify polymers with appropriate dual function, that is, (1) generation and maintenance of supersaturation and (2) stabilization of the amorphous drug in the formulation matrix. Examples for such combined approaches include the 96-well plate vacuum dry system for ASD screening described by Chiang et al. (2012), the SPADS approach (Wyttenbach et al. 2013) or the MiCoS method (Hu et al. 2013). A comprehensive overview of various miniaturized ASD screening methods is shown in Table 8.5.
At an early development stage, when API supply is limited, a step-by-step ASD screening strategy is recommended: (1) Drug candidates suitable for spray-drying (SDP candidates) with good solubility in volatile solvents (API solubility in volatile solvents >20 mg/mL) are first screened with solvent casting methods for selecting appropriate polymers and drug loads. (2) HME candidates with low melting point (MP < 200 °C) can be evaluated by small scale fusion based methods, e.g. drug-polymer mixtures treated by heat-cool-heat cycling in DSC experiments. If HME candidates have good solubility in volatile solvents, solvent casting methods can also be used. (3) Coprecipitation (MBP) candidates (with poor solubility in volatile organic solvents and high melting point) with good solubility in non-volatile water-miscible solvents (API solubility in non-volatile solvents ideally >50 mg/mL) are screened by miniaturized coprecipitation screening (e.g. MiCoS) for ASD feasibility. In addition, the generally very simple and fast solvent-shift method can be used as a supportive method for all ASD processing technologies (e.g. SDP, HME, MBP) for a first assessment of the supersaturation potential of different compositions. If drug candidates are amendable for different ASD processing technologies and if API supply is not the limiting factor, a parallel screening approach with different screening methods (solvent-casting, melt-fusion, coprecipitation, etc.) can also be adopted. The most important miniaturized ASD screening methods are described in more detail in the following subsections.
8.5.1 Supersaturation Screening
The concept of generating and maintaining supersaturation has been described as the “spring and parachute approach” by Guzmán et al. (2007). In the case of a molecular dispersion (solid solution), release of drug molecules is dictated by dissolution of the hydrophilic carrier (spring function) and leads to a supersaturated state of the drug in solution. Two different miniaturized methods have been applied to assess the supersaturation potential of excipients with poorly water-soluble drugs: co-solvent quenching (solvent-shift method) and amorphous film dissolution (solvent casting method). Examples for both methods described in the literature are shown in Table 8.6.
The co-solvent quench (solvent-shift) method is currently the most common method used for initial polymer screening. In this method, drug is dissolved in a water-miscible solvent with high drug solubility. A small aliquot of the solution is then dispersed in the aqueous phase to create a supersaturated system. In order to determine the extent of drug precipitation, the concentration of the dissolved drug within the aqueous phase or the mass of drug precipitated can be assayed or measured indirectly by turbidity measurement. Alternatively, the film dissolution method involves parallel dissolution screening of solid dispersions with different compositions and drug loads prepared by solvent casting. Amorphous drug films are prepared from mixtures of drug and excipient(s) dissolved in a volatile organic solvent. The organic solvent is then evaporated resulting in a thin film of the formulation. The dissolution medium is added and the drug concentration is determined as a function of time.
8.5.2 Solid State Screening
Miniaturized methods used to experimentally evaluate the homogeneity and stability of an amorphous system include amorphous sample preparation by solvent casting, coprecipitation, melt fusion, freeze drying, or spin coating (Table 8.5) and analysis for recrystallization or phase separation by different analytical techniques, e.g., by polarized light microscopy, DSC, XRPD, FT-IR spectroscopy, Raman spectroscopy, or atomic force microscopy (AFM) . Most commonly, amorphous films are prepared by evaporation of organic drug–excipient solutions on different carrier systems, such as glass slides, cover slips, 96-well microplates, or aluminum pans. The stability of the amorphous systems is assessed by reanalysis of the samples after storage at accelerated conditions (temperature and humidity). Different miniaturized methods to evaluate amorphous drug stability are presented in Table 8.7.
With respect to the amorphous drug stability, drug–polymer miscibility (homogeneity) is essential because immiscibility can result in the formation of drug-rich domains that may be prone to recrystallization. Other important factors strongly affecting the stability of amorphous systems include solid-state solubility and drug–polymer molecular interactions (Konno and Taylor 2006; Konno et al. 2008; Wyttenbach et al. 2013).
Qualitatively, the miscibility of drug and polymer can be assessed by simple DSC measurements, in which mixtures of drug and polymer are mixed and treated by heat–cool–heat cycling in a DSC pan to observe the change of glass transition temperatures (Sarode et al. 2013). Generally, miscibility of drug and polymer are assessed on the second heating scan after the system has initially been heated beyond the melting point of the drug. If the drug is thermally labile, amorphous drug and polymer can be casted in a DSC pan followed by evaporation of the solvent. A general rule of thumb is that a single T g suggests miscibility, observation of the two T gs corresponding to individual components suggests complete immiscibility, and two T gs in between the two individual T gs suggests partial miscibility.
This DSC technique is simple and rapid in obtaining essential information on miscibility but is very crude in that quantitative miscibility information is lacking and the miscibility is given only near the glass transition temperature. In addition, if the drug and polymer have similar T gs, or the change in heat capacity around the T g is small, then this method does not provide much insight into the miscibility of the drug and the polymer.
8.5.3 The SPADS Approach (Screening of Polymers for Amorphous Drug Stabilization): Example for a combined ASD Screening Approach
The SPADS approach has been developed at Roche for solid dispersion screening by Wyttenbach et al. (2013). It combines the assessment of supersaturation potential, the evaluation of drug–polymer miscibility, and the stability of amorphous systems. The aim is to rapidly identify prototype amorphous compositions suitable for preclinical studies and early-stage clinical trials.
The SPADS approach consists of three different miniaturized assays (1) SPADS dissolution, (2) SPADS imaging, and (3) SPADS interaction assay. It is a two-step approach; only the combinations showing promising dissolution behavior are further characterized with respect to their solid-state properties (SPADS imaging and interaction assays). The three SPADS assays will be described in more detail in the following subsections.
8.5.3.1 SPADS Dissolution Assay
The SPADS dissolution assay is performed on amorphous drug–polymer films prepared in 96-well plates by solvent-based film casting. Specific drug–polymer premixtures are prepared from excipient and drug stock solutions in volatile solvents (e.g., acetone, ethanol, and methanol). The premixtures are distributed into a 96-well plate according to a predetermined filling scheme and the amorphous films are generated by solvent evaporation. To illustrate the method, an example of a possible filling scheme is given in Fig. 8.4. In this example, binary drug–polymer systems with seven polymers at different drug–excipient ratios are tested.

Example of a 96-well plate-filling scheme used in the SPADS dissolution assay
The dissolution screening is performed by adding a stirring bar and a 200 μL dissolution medium (e.g., FaSSIF, pH 6.5 and/or SGF, pH 1.2 for polymers with pH-independent solubility) to each well of the 96-well plate . The microtiter plate is closed and mixed by head-over-head rotation at 37 °C. One 96-well plate is prepared per time point. Generally, two time points, 60 min and 180 min, are considered adequate to describe the dissolution profile. After mixing the plate for the desired time, 100 μL of the solution are collected and transferred to a 96-well filter plate. Filtration is performed by centrifugation. The filtrates are collected in a new 96-well plate. Drug content in the filtrates is determined by UPLC after appropriate dilution. Figure 8.5 shows the results of an SPADS dissolution screening experiment with a poorly soluble Roche development compound using the filling scheme presented in Fig. 8.4. In this example, the most promising polymer from the supersaturation perspective appeared to be Eudragit E PO, since highly supersaturated solutions of the drug were observed at all drug loads for at least 3 h.

Results of a SPADS dissolution experiment in Fassif pH 6.5 with a poorly soluble Roche development compound
8.5.3.2 SPADS Imaging Assay
Optical microscopy and AFM are applied to analyze the molecular homogeneity and stability of promising amorphous API–polymer combinations on micrometer and nanometer scales using the method developed by Lauer et al. (2011). For AFM investigations, glassy film fracture surfaces on glass slides are generated to discriminate between homogeneously and heterogeneously mixed drug–polymer combinations. The homogeneous combinations are further analyzed for physical stability after exposure of the samples to stress conditions (accelerated temperature and humidity, e.g., at 40 °C/75 % RH) for some hours.
8.5.3.3 SPADS Interaction Assay
The SPADS interaction assay is used to study molecular interactions between drug and polymer. Amorphous films are prepared in commercially available 100 μL aluminum pans (used for DSC measurements) by solvent casting. The procedure of film preparation is identical to the procedure used in the SPADS dissolution assay with the only difference that the films are prepared in aluminum pans distributed into a 96-well plate. FTIR spectra are measured in reflection mode using a standard FTIR microscope. FTIR spectroscopy is sensitive to changes in the hydrogen-bonding network and protonation status of the drug, polymer, and water present in the system. FTIR spectra of the solid dispersions are compared with the spectra of both the pure amorphous drug (rather than the crystalline forms) and the pure polymers. Hydrogen-bonding significantly influences peak shapes and intensities, generally resulting in peak broadening and peak shifts.
8.5.4 Summary of the Small-Scale Experiments: Selection of Polymer and Drug Load
All analytical results obtained from initial assessment, miniaturized or small-scale experiments, are collected, assessed, and compared. The collected data are used to preselect the most promising prototype amorphous compositions and drug loads that will be subjected to small-scale solid dispersion preparation for further in vitro and in vivo assessment (dissolution and solid state characterization, animal PK studies). These scaled-down processes include mini-extrusion for hot-melt extrusion, mini-fluid bed systems for coated bead systems, or mini-spray dryers if spray drying will be an option. Based on our experience, this step is very important since various amorphous solid dispersions often have different stability and biopharmaceutical properties based on how they were generated, even when containing the same composition and drug load.
8.6 Selection of Most Suitable Technology
After having identified suitable polymers and drug loads , selecting the right processing technology is essential. The selection of the most appropriate process for the manufacture of a solid dispersion depends on the physicochemical properties of the API and the polymer (solubility, thermal stability, melting point and T g). The manufacturing process directly impacts the complexity of further downstream processing (e.g., particle size, and bulk density), and properties of the finished drug product (e.g., stability and dissolution). Additional criteria that should be taken into consideration are the availability of the equipment, the robustness of the manufacturing process, the impact on the cost of goods (energy consumption and equipment foot print) and the intellectual property considerations. These properties should be assessed to allow the formulator to rank order different technologies. In a second step, the ranking should be confirmed by manufacturing trials on small-scale equipment.
A physically stable solid dispersion of an amorphous API in a polymer can be achieved if the drug and the polymer are intimately mixed at the molecular-level, i.e., a solid solution. Molecular-level mixing is achieved either by dissolution of both components in a solvent (solvent based technologies) followed by solvent removal or by directly mixing both components as liquids as is accomplished by melting (fusion) methods (Chiou and Riegelman 1971). Currently the most relevant technologies for the manufacture of solid dispersions in the pharmaceutical industry are (1) spray-drying, (2) melt extrusion, (3) and co-precipitation. The following section discusses these technologies including a brief overview, schematic of the physical processes, and a summary table with the associated advantages and disadvantages (Table 8.8). Several other technologies are described to produce amorphous solid dispersions such as: spray granulation, fluid bed layering, spray congealing, co-grinding, spin coating, electrostatic spinning, microwave technology, KinetiSol® dispersing, ultra-rapid freezing and supercritical fluid processing (He and Ho 2015), but with the exception of fluid bed layering they have not yet been applied for marketed products.
8.6.1 Overview of Key Manufacturing Technologies
8.6.1.1 Spray-Drying
Spray-drying is well established as an industrial process for transforming solutions, emulsions, or suspensions into a dry powdered form. This is accomplished by pumping a feed solution (e.g., drug and polymer dissolved in a volatile solvent) into an atomizer inside the drying chamber. The atomizer breaks the solution into a plume of small droplets (normally less than 100 μm in diameter). In the drying chamber, the droplets are mixed with a hot drying-gas stream (air or nitrogen). The heat is transferred from the hot drying gas to the droplets to provide the latent heat of vaporization required for rapid evaporation of the solvent from the droplets. By controlling the inlet and outlet temperatures of the spray dryer, together with the spray rate and air volume that are introduced to the spray dryer, the morphology, particle size, and density of the resulting solid dispersion powder can be controlled. The solid powder is typically collected from the gas stream using a cyclone or a filter. The spray-drying process can be performed over a wide range of scales (from milligrams/grams to tons of drugs) (Friesen et al. 2008).
The underlying principle for the manufacture of a solid dispersion by spray drying is to dissolve a physical mixture of the drug and the polymer in a volatile solvent followed by the evaporation of the solvent. In order to produce a molecular dispersion of the drug and the polymer after evaporation of the solvent, both components have to fully dissolve in the solvent or the solvent mixture. Therefore the key parameter is the selection of a suitable organic solvent or solvent mixture. Suitable solvent or solvent mixture must meet the following criteria (Table 8.9):
As described in Table 8.9, the solvent or solvent mixture may have an impact on the characteristics of the obtained spray dried solid dispersion (physical stability and dissolution). Therefore small scale experiments with different solvents and solvent combination are recommended in order to select an appropriate solvent. Besides the solvent selection, also process related criteria have to be considered. For instance the thermal stability of the API at spray drier outlet temperature should be given or in case the outlet temperature is higher than the T g of the spray-dried product, material may stick to the walls of the spray drier, resulting in a low yield that is not acceptable for commercial production (Patterson et al. 2007, 2008). Various approaches have been used in the past in order to scale-up spray drying processes. Further, more in depth information are provided for instance in Gil et al. (2010); Thybo et al. (2008); Dobry et al. (2009); and Kalb et al. (2013).
Spray-dried material is often very fine and contains a high amount of residual solvents. This may have a negative effect on the downstream process (flow and density of particles) and the stability of the spray-dried solid dispersions (recrystallization of the amorphous drug). In addition the residual solvent content can easily exceed ICH limits. Solvent reduction would require a secondary drying step which very often transforms the continuous spray drying process into a batch process. In order to improve the properties of the spray-dried material, more advanced spray-dryer layouts with an additional external fluid bed or an internal fluid bed are available to improve the drying efficiency (avoiding secondary drying) and to agglomerate the fines (Masters 1991). A thorough discussion of spray-drying technology and its application to the formulation of poorly water-soluble drugs is provided in Chap. 10.
8.6.1.2 Hot-Melt Extrusion
Hot-melt extrusion (HME) is frequently used in the pharmaceutical industry for developing solid dispersion formulations. HME is the process of pumping raw materials with a rotating screw(s) under elevated temperatures through a die to form a product of uniform shape. The intense mixing and agitation imposed by the rotating screws results in a uniform dispersion. Generally, the extruder consists of one (single-screw) or two (twin-screw) rotating screws in a stationary barrel. Twin-screw extruders can be operated in co-rotating or counter-rotating modes. Co-rotating twin-screw extruders are widely used since they can be operated at high screw speeds, yielding high output, good mixing, and good conveying characteristics. A fully intermeshing type of screw design is self-wiping, where it minimizes the stationary zone and prevents localized overheating of material in the extruder. The screw is typically modular and is divided into three sections along the lengths of the barrel: feeding, melting or compression, and metering. In the feeding section, the material is transferred from the hopper to the barrel. In the compression zone, the polymer usually starts to melt due to the thermal energy that is generated by shear forces imposed by the rotating screw and from conduction from the barrel via electrical heating bands. The temperature of the melting zone is normally set 10–60 °C above the melting point of semi-crystalline polymers or the glass transition temperature of amorphous polymers to ensure consistent flow. The metering zone is designed to reduce pulsating flow of the molten polymer and provide uniform delivery of the material through the die. The screw configuration can be modified by changing the screw elements to optimize the shear, residence time distribution, and/or product characteristics (Crowley et al. 2007). HME requires pharmaceutical grade polymers that can be processed at relatively low temperatures due to the thermal sensitivity of drugs and polymers. For most polymers, temperatures between 120 and 180 °C have been used. For a complete discussion of HME technology, the reader is referred to Chap. 9.
The viability of melt extrusion depends on the ability to form a one-phase solid solution. API and polymers are subject to elevated temperatures, high pressure, and intensive mixing during the HME process (Patterson et al. 2008). Depending on the process conditions, the crystalline drug either melts or becomes solubilized in the molten polymer. The latter allows the manufacture of a solid dispersion at temperatures below a drug’s melting point. The recrystallization of the drug during the cooling of the extrudate is retarded due to reduced solute migration and the reduction of nucleation kinetics by the viscous polymer medium. Nevertheless the cooling rate has to be carefully evaluated as it may impacts the quality (physical stability) of the obtained extrudates.
The selection of optimal melt extrusion conditions depends on the chemical stability of the drug and the polymer and the physical properties of the polymer. The key considerations to establish the most appropriate process parameters for HME are shown in Table 8.10.
T g analysis by DSC as a function of the polymer concentration provides a baseline for setting the extrusion temperature and helps to assess the miscibility of the system. In addition, rheological properties (zero rate viscosity and activation energy) of the material as a function of shear rate and temperature are key considerations in establishing the HME process. In case of a new product (API/polymer combination) with unknown properties, one approach to identify feasible HME process conditions and investigate the plasticizing effect of the API is to use a torque rheometer for preliminary experiments. It allows running temperature and screw speed ramps in order to investigate the thermic and mechanical impact on the given system. This allows the formulator to identify process temperatures and the required mechanical energy input (screw design).
In 2004 the Food and Drug Administration (FDA) encouraged the pharmaceutical industry to introduce quality by design (QbD) approach and usage of process analytical tools (PAT) in order to improve process understanding and ensure the quality of final drug product. HME as a continuous process is an obvious choice to introduce PAT tools (Patil et al. 2016; Martin 2016). Over the years, in-line NIR and in-line Raman spectroscopy were implemented in order to understand drug-polymer interactions, to continuously quantify the API amount and to monitor the solid state property of the API in solid dispersions (Saerens et al. 2011; Repka et al. 2013). Due to the non-invasive nature of PAT tools, they can be used to investigate the impact of potential critical process parameters on critical quality attributes of the final product. In addition the real-time monitoring of an HME process allows to immediately adjust the process parameters if process deviations are observed (Islam et al. 2015).
8.6.1.3 Co-precipitation
The term co-precipitation has been used in the literature to describe amorphous solid dispersions produced when the drug and the polymer are precipitated together by changing the solubility conditions, either by addition of an anti-solvent or by evaporating the solvent (Simonelli et al. 1969). Further attempts have been made to co-precipitate a solution of drug and polymer in ethanol by addition into aqueous solution; however, the characteristic crystalline and thermal peaks were still present, indicating incomplete conversion to the amorphous form (Kislalioglu et al. 1991).
Based on the principle of solvent-controlled precipitation, a novel technology referred to as microprecipitated bulk powder (MBP) has been developed that provides complete conversion of a drug to the amorphous form, for a large variety of compounds (Albano et al. 2002; Shah et al. 2008). The MBP technology is particularly suitable for highly insoluble compounds for which the utilization of spray drying or hot-melt-extrusion technologies is not feasible. With this technology, a solution of drug and stabilizing polymer is introduced into an anti-solvent that precipitates the drug and the polymer(s) simultaneously to form the MBP. Under appropriate processing conditions, the drug is uniformly embedded in an amorphous form in the polymer. The MBP process is particularly suitable for compounds that may have low-solubility in commonly used solvents, such as acetone or ethanol, but have sufficient solubility in solvents, such as dimethylacetamide (DMA), dimethylsulfoxide (DMSO), dimethylformamide, or N-methylpyrrolidone (NMP) . Based on the fact that usually ionic polymers are used in this process, the solvent-controlled precipitation is carried out under either acidic or basic conditions. The residual solvent is removed by a series of washing steps followed by filtration and drying. Due to the nature of the process, it is used only when other means of making amorphous dispersion are not feasible. The key considerations for the MBP process are listed in Table 8.11. A more detailed discussion of MBP technology and its application to the formulation of poorly water-soluble drugs is provided in Chap. 5.
8.6.2 Summary of Processing Technology
A schematic of key processing technologies is presented in Fig. 8.6. Overall, the selection of the optimal manufacturing process should be based on physicochemical properties of the API and the corresponding polymer partner. HME processing for example is only applicable if thermal stability of API and polymer are given and if the melting temperature of the API is not exceeding the process temperature window too much. If HME is considered, another aspect is the melt viscosity of the selected polymer. Spray drying can only be performed if the API and polymer is soluble in common organic solvents with low boiling point. If this is not the case and solubility is only given in organic solvents with high boiling point like DMSO or DMA MBP technology should be evaluated.

Schematic of key processing technologies for producing amorphous dispersion
Other material characteristics are not as obvious. Many commercial polymers are only available in one particular particle size which makes them more or less favorable for a technology. The throughput in a HME process for example can be very limited if fine and voluminous polymers have to be fed into the extruder. If more than one technology is possible, also the properties of the amorphous intermediate have to be taken into consideration. For example spray dried powder has a much larger surface compared to milled extruded intermediate or MBP powder which could result in decreased physical stability.
8.7 Downstream Processing and Final Product Properties
Once the solid dispersion is manufactured it needs to be further downstream processed to derive the final drug product. The selection of the downstream manufacturing process is based on the properties of the solid dispersion (e.g., particle size and bulk density) as well as the properties of the finished drug product derived from the quality target product profile (QTPP). As an example, further densification of spray dried powders prior to capsule filling or tablet compression is usually necessary due to their low bulk density.
Generally speaking drug product intermediates with small particles and/or low density are normally downstream processed using dry granulation processes such as roller compaction. In addition to the formulation composition which should be optimized with respect to bioavailability and stability, it is of utmost importance that the formulator investigates the impact of potential critical process parameters on the performance of the final drug product, and finally defines the critical process parameters (CPPs) which have an impact on the critical quality attributes (CQAs) of the final drug product. Typical CQAs of solid dispersion based drug products are the amount of residual solvents, impurities, crystallinity (initial and over storage time; physical stability) and the release profile of the drug. For the characterization of the solid dispersion and the finished product, several analytical technologies are available, as described in Sect. 8.8.
8.8 Characterization of the Amorphous Systems
An amorphous material is generally defined as a solid material that lacks long-range symmetry operators (translational, orientational, and conformational operators), which are characteristics of crystalline material. An amorphous system is considered to be a disordered system with random molecular configuration and packing of individual components, in which individual molecules are randomly oriented to one another and exist in a variety of conformational states. Furthermore, amorphous formulations may possess local and short-range order crystallites, residual crystallinity, and different molecular density regions, as exemplified by alpha and beta relaxations. Various levels of micro- and macroheterogeneity may be present in amorphous formulations to add complexity. Consequently, gaining insight into amorphous systems to address the formulation challenge is also an analytical challenge and frequently limits development of a stable amorphous formulation.
For successful amorphous formulation development, it is imperative to have suitable analytical techniques to characterize the materials produced and to assess success at each step. The complexity of analytical testing may vary depending on the developmental stage, from simple qualitative testing at an initial stage to comprehensive and quantitative testing at the clinical manufacturing stage. Regardless, for successful amorphous formulation development, it is critical to understand how the amorphous system is being formed, to determine whether or not the crystalline drug is completely converted to an amorphous state, to understand how the drug and the polymer molecules are arranged in an amorphous solid dispersion, and to determine the uniformity of the formulation produced. Knowledge of these attributes will help scientists to not only assess the risks associated with amorphous formulations but also to mitigate those risks by designing optimal formulations (polymer, drug loading, and process). Moreover, this helps the formulation scientists to predict the in vitro and in vivo performance as well as long-term stability.
In this section, commonly used analytical techniques will be briefly discussed for their merits and limitations in two groups distinguished by the information acquired. First is the set of techniques used to exclude crystallinity in an amorphous system. Obviously, this is the most critical test as the principal impetus of making amorphous systems is to convert a crystalline drug into an amorphous state, and the assurance of complete conversion to amorphous state is of utmost importance. The second set of techniques includes the tests that are used to study the properties of amorphous formulations, i.e., their molecular arrangement and their behaviors. Understanding the molecular arrangement of drug and polymer molecules in amorphous formulations is critical to predict the stability and to understand the in vitro/in vivo performance. Lastly, trends in amorphous formulation stability predictions are briefly discussed, together with experimental techniques that can be employed. Table 8.12 highlights the differences in physiochemical properties between crystalline and amorphous materials that can be further explored.
8.8.1 Detection of Crystallinity in Amorphous System
Many attempts have been made to determine the degree of crystallinity or amorphicity of amorphous formulations. Without doubt, this is the most critical testing in the evaluation of amorphous formulations. Inappropriate selection of polymer, inadequate drug loading (beyond miscibility), and poor process control during pharmaceutical manufacturing will result in incomplete conversion of a crystalline drug to an amorphous state. Any trace level of crystalline drug in amorphous formulations may serve as seeds for recrystallization during in vitro and in vivo dissolution or during storage, which may jeopardize the entire development program. The following techniques are frequently employed to detect the degree of crystallinity.
8.8.1.1 X-Ray Powder Diffraction
X-ray powder diffraction (XRPD) is the most widely used and perhaps the most definitive technique used in detection and quantification of crystallinity. Absence of sharp Bragg’s peaks corresponding to the crystalline drug suggests a formulation is in an amorphous state. It is important, however, to note that XRPD detects the presence of molecular order; therefore, the disorder (amorphous state) is only implied by the absence of the order (long-range symmetry order). The limit of detection of the method ranges from 0.2 to 5 % in an amorphous or excipient mixture and dispersion, and is dependent on the instrument used, the sampling protocol, and its characteristic (Palermo et al. 2012). XRPD is one of the most widely attempted quantification techniques, in which the degree of crystallinity is determined from the intensity of crystalline peaks in the sample. Limit of quantitation is typically in the range of 5 % w/w, depending on intensity of the crystalline peaks.
8.8.1.2 IR and Raman Spectroscopy
Vibrational spectroscopy, such as IR and Raman spectroscopy , can be used to detect the variations in vibrational energy between amorphous and crystalline states. In general, sharp vibrational peaks indicate crystallinity, whereas a broad hump suggests amorphicity, as a result of disorder in molecular arrangements. Having a single crystal structure will aid the interpretation of vibrational spectrum pointing to the region where intermolecular interaction occurs. IR and Raman spectroscopy can be used to determine the amount of crystallinity of an amorphous formulation when a calibration model is used. A major disadvantage of FT-IR spectroscopy is the fact, that the sample preparation is non-trivial for most instrument configurations and most samples are likely non-representative for the entire dosage form (Palermo et al. 2012). IR as well as Raman are more often employed to assess the molecular arrangement of the ASD (see also Sect. 8.8.2.2).
8.8.1.3 Differential Scanning Calorimetry
DSC is probably the most versatile and widely used technique in the characterization of amorphous formulations including quantification of crystallinity. In DSC, samples are heated at constant rate, while heat flow is continuously being monitored, and the temperatures at which thermal events occur are recorded. Thermal events can be glass transition, (re)crystallization, melting, and decomposition. From thermal events, melting and (re)crystallization energy and changes in heat capacity at glass transition can be measured, which can be used to quantitate the crystallinity or the amorphicity. Nevertheless, DSC has several limitations as traditional DSC is not able to detect two Tg’s if they are not at least 10 °C apart from each other and small domains of crystalline material (<30 nm) cannot be detected (Meng et al. 2015). DSC can also be used to assess the molecular arrangement of the ASD (see also Sect. 8.8.2.1).
8.8.1.4 Microscopic Technique
Cross-polarized microscopy is one of the most powerful but largely ignored techniques in amorphous formulation characterization in comparison to more sophisticated instrumental techniques, presumably due to the requirement of experience in the interpretation of data and limited information in this regard. Furthermore, the sample size/volume is rather low and quantification of the amount of crystallinity is nothing which can easily be done. Nevertheless, many researchers have used the cross-polarized microscope to study the kinetics of crystallization and to predict the stability of amorphous systems (Taylor and Zografi 1997; Yu 2001). As stated earlier, the cross-polarized microscope utilizes differences in birefringence between crystalline and amorphous systems, as amorphous material does not exhibit birefringence.
8.8.1.5 Other Techniques
Water vapor sorption can be used to discriminate between amorphous and crystalline materials if hygroscopicity is different, and in the absence of interferences. Isothermal microcalorimetry was one of the earliest techniques used to study amorphous systems with remarkable sensitivity. This is based on the principle that enthalpy changes at constant temperature and relative humidity are associated with (re)crystallization of amorphous material. However, this technique is extremely sensitive to many operational conditions, which makes it difficult for routine use.
8.8.2 Determination of Molecular Arrangement in Amorphous Systems
As discussed earlier in this chapter, an ideal formulation is one that provides the maximum physical stability over the intended period and maintains supersaturation while the drug is being absorbed. Physically, the maximum stability and optimal performance are achieved when drug molecules are molecularly dispersed in a polymer matrix with appropriate intermolecular interactions between the drug and the polymer(s). In recent years, various techniques have been used in order to provide get more insight into the phase behavior of amorphous solid dispersions.
8.8.2.1 Differential Scanning Calorimetry
A first insight into the molecular arrangement of the ASD can be obtained by determining the glass transition temperature of the system using DSC. Despite the difficulty in accurately measuring T g, it is one of the most commonly used tools to characterize amorphous solids and assess the interactions of different materials, particularly miscibility. Binary drug-polymer systems showing a single Tg are generally expected to be miscible, whereas systems with two Tgs or the appearance of a melting endotherm of the drug would generally indicate immiscibility. As described in Sect. 8.4.1.2 the Gordon-Taylor ideal mixing equation can be used to predict the Tg of an amorphous one-phase dispersion, and deviations of the measured Tg from the predicted Tg are indications for molecular interaction between the API and polymer. The popularity of the DSC technique is associated with the fact that it represents changes in the kinetic and thermodynamic properties of amorphous materials and is relatively easy to measure using a small amount of sample.
8.8.2.2 (N)IR and Raman Spectroscopy
Vibrational spectroscopy methods such as IR or Raman spectroscopy can be used to detect crystalline in amorphous material, but more often they are utilized to investigate the molecular arrangement in the ASD. Raman spectroscopy/imaging is one of the most commonly used techniques to investigate the phase behavior on a micrometer scale. In comparison to the traditional Raman techniques, micro-Raman has a much higher detection limit and provides localized compositional information that are distinct from the bulk of the material, however the method has only low spatial resolution (Meng et al. 2015).
IR spectroscopy has been extensively used to study the molecular arrangement of drug and polymer and their interaction. Kaushal et al. studied the differences between intermolecular interactions in amorphous and crystalline phases of celecoxib, valdecoxib, rofecoxib, and etoricoxib using FTIR (Kaushal et al. 2008). Konno et al. studied interactions between amorphous felodipine and PVP, HPMC-AS, and HPMC using FTIR (Konno and Taylor 2006). It was concluded that hydrogen-bonding between drug and polymer were an integral part of amorphous formulation stabilization. In another case study solution cast and quench cooled films of Naproxen-PVP were investigated using FT-IR and subsequent spectral deconvolution, and the results clearly showed that the strength of the drug-polymer H-bonding varied with the composition and process employed (Paudel et al. 2012).
NIR chemical imaging combines NIR spectroscopy with digital imaging. Due to the synergistic capabilities this method can be used to assess the physical distribution of components in solids. Ma et al. (2013) used the method in order to determine the drug distribution in an ASD by comparing it to that in a homogeneous physical mixture with the same composition, and to facilitate the selection of the formulation composition and manufacturing technology for an ASD.
8.8.2.3 Atomic Force Microscopy
Atomic force microscopy (AFM) allows to get a more in depth view by assessing the phase behavior on a nanometer scale. It is a scanning probe technique which has a high spatial resolution. Phase separation of the ASD leads to differences in the local material properties that can be assessed by using AFM in tapping mode. Lauer et al. (2013) used AFM in order to compare the homogeneity of hot-melt extrudates obtained from a small scale extruder with data obtained from screening experiments. Lamm et al. (2016) investigated the phase behavior of ASD containing copovidone and vitamin TPGS in dependence of various process parameters used to prepare the extrudates. Furthermore, they analyzed the nature of the domains present in some of the samples by generating force-displacement curves. This technology is also known as force spectroscopy (Lamm et al. 2016). Other researchers coupled AFM with local or nanothermal analysis (Qi et al. 2011; Purohit and Taylor 2015), photothermal Fourier-transform infrared microspectroscopy (PT-FTIR: Qi et al. 2011), or nanoscale infrared spectroscopy (Purohit and Taylor 2015) to get further insight into the different phases formed.
8.8.2.4 Solid-State NMR
Solid-state NMR (ssNMR) has been increasingly used to study miscibility and molecular interaction between amorphous drug and polymer. A single relaxation time for the drug and polymer indicates complete miscibility (Meng et al. 2015). Mistry et al. (2015) used FT-IR in combination with ssNMR to probe the interactions between ketoconazole (KTZ) and three different polymers. In case of poly (acrylic acid) (PAA) a proton was transferred from the carboxylic acid group of PAA to the imidazole nitrogen N3 of KTZ indicating that both interact via ionic interaction. For the system with poly(2-hydroxyethyl methacrylate) (PHEMA) they could prove H-bonding between each N3 and C═O of KTZ and the ─OH of PHEMA, whereas the KTZ-PVP system only exhibit weaker interactions (Mistry et al. 2015). In another study by Song et al. (2016) ssNMR was used to proof the protonation of the quinazoline nitrogen in lapatinib (LB) and gefitinib (GB), the protonation of the secondary amine nitrogen in LB and the protonation of the tertiary amine nitrogen atom in GB in ASDs with polystyrene sulfonic acid (PSSA), and thus confirming the strong intermolecular acid-base interactions present between LB and PSSA as well as GB and PSSA.
8.8.2.5 Other Techniques
In order to assess the molecular arrangement/miscibility of the ASD XRPD is often combined with computational methods such as Pair Distribution Functions (PDFs) , Pure Curve Resolution Method (PCRM) and Alternate Least Square (ALS) (Meng et al. 2015). Bates et al. (2006) have used XRPD in combination with the pairwise distribution function to investigate the local structure of amorphous formulations. Mistry et al. (2015) used variable temperature XRPD in order to determine the crystallization onset temperature of various Ketoconazole solid dispersions. They could link an increase in the strengths of interactions between the drug and polymer with a higher crystallization onset temperatures.
Furthermore, Song et al. (2016) used X-ray photoelectron spectroscopy (XPS) to determine the binding energy between lapatinib (LB) and polystyrene sulfonic acid (PSSA) as well as gefitinib (GB) and PSSA. They detected increasing binding energy of the nitrogen atoms in LB and GB, which strongly indicates protonation of these atoms (Song et al. 2016).
8.8.3 Dissolution Method for Amorphous Formulations
The dissolution profile is considered to be a good predictor for the in vivo performance, however it is difficult to establish an in vitro-in vivo correlation due to the complex nature of supersaturation generation and maintenance in combination with the driving force triggering absorption of ASDs (Baghel et al. 2016). In general it is assumed that the drug release mechanism of an ASD depends on the type of polymer used. The dissolution profile of an ASD containing hydrophilic, medium soluble polymers, which dissolve in the medium, are characterized by an initial surge of supersaturation followed by a decrease in drug concentration due to precipitation (Park 2015). The degree of precipitation depends on the type of polymer used. ASDs containing medium insoluble polymers lack the initial fast increase in concentration and supersaturation generation, and their overall release profile is sustained for an extended period of time (Park 2015). For these types of ASDs the rate of supersaturation generation has a huge impact on the kinetic solubility profile as discovered by Sun and Lee (2013).
Based on the above mentioned considerations the development of meaningful dissolution methods for kinetically unstable systems of poorly soluble drugs is tremendous task. Nevertheless, the requirements in terms of dissolution might change during the whole development ranging from predicting the in vivo performance to control batch to batch consistency. In the first phase the physiologically relevant conditions, such as composition of the medium, pH of the medium, dose to GI fluid volume ratio, and exposure time, should be considered (He and Ho 2015). For these studies the use of non-sink conditions, the measurement of the pH at the end of the dissolution test and the characterization of the precipitate should be considered (He and Ho 2015). The use of a diffusion study might also be useful, in order to relate the drug release with the absorption process. Later in the development the focus of the method has changed towards quality control (QC) , and it is vital to demonstrate adequate discriminating power for the QC method.
A perfect sink condition will not help to ascertain the ability of a system to maintain supersaturation. However, an overly nonsink condition may result in overdiscrimination and potential elimination of a viable formulation. There are several publications related to dissolution of amorphous formulations (Law et al. 2004; Chan and Kazarian 2004; Doherty and York 1987; Janssens and Guy 2010). The effect of polymer type on the dissolution rate of amorphous systems has been described by Konno et al. (2008).
Unlike as for crystalline APIs, equilibrium solubility cannot be determined for amorphous materials because, at equilibrium, drug solubility using the crystalline or the amorphous form is similar, by definition. Therefore, the method has to determine the kinetic solubility in physiologically relevant conditions.
The kinetic solubility of amorphous material can be determined in bio-relevant or aqueous medium with or without surfactant. If we accept the fact that precipitation follows supersaturation, then solubility of amorphous material at a 30–60 min time window, or at the peak of the upward slope could be used for determination of kinetic solubility. This is illustrated in Fig. 8.7, where the maximum amount of drug dissolved (C max) at the time to achieve the maximum concentration (T max) could be used for kinetic solubility determination. Furthermore, the dissolution profile is characterized by the duration in which supersaturation is maintained, and by the area under the curve (AUC).

Schematic representation for kinetic solubility determination for amorphous materials
Based on the kinetic solubility, dissolution conditions can be varied with regard to dose, volume of dissolution medium, and surfactant concentration of dissolution medium for screening amorphous formulations. A dissolution medium representing 100 % saturation is the most efficient way of screening formulations as it avoid unnecessary stress during dissolution testing. An example for such an dissolution test is shown in Fig. 8.8 for an research compound. Additionally, a formulation maintaining supersaturation for at least 2–4 h (physiologically relevant) would represent a viable formulation. On the contrary, a formulation maintaining supersaturation for less than 60 min would need careful evaluation.

Effect of surfactant concentration on the dissolution behavior of amorphous solid dispersion and supersaturation
More recent research was done by Lynne S. Taylors group in order to better understand the mechanism of precipitation by investigating the solution phase behavior during dissolution. They investigated for instance the dissolution behavior of danazol from ASDs containing different polymers, and found out that upon dissolution prior to crystallization of the drug liquid-liquid phase separation occurred. The phase behavior of the solutions present during dissolution was monitored using UV and fluorescence spectroscopy (Jackson et al. 2016). In another research article the release profile of Celecoxib ASDs with high drug loading was optimized in terms of rapid release and crystallization inhibition by using a combination of two different polymers. Besides the drug release, which was determined “at sink” conditions the authors also determined the nucleation induction time in a precipitation assay using light scattering in order to optimize the crystallization inhibition (Xie and Taylor 2016).
8.8.4 Stability Prediction
The goal for amorphous formulation stability is to maintain long-term physical stability with respect to solid state properties and maintain supersaturation during the time course of the dissolution process mimicking the in vivo dissolution window, which is generally about 2–4 h. The key to understanding stability is to understand that molecules in an amorphous system can have significant molecular motion both above and below the glass transition temperature. Molecular motion in the form of translational and rotational diffusion can generally be described in terms of molecular relaxation time or annealing time. In addition, the effect of water on molecular mobility and stability must be accounted for. Water can exist in an amorphous state with a T g of about −138 °C (135 K). As such, water can reduce glass transition temperatures of amorphous systems substantially. The effect on T g can be more pronounced for partially amorphous systems. In such cases, most of the water will be located in the disordered region and the T g in that region will be considerably lower (Marsac et al. 2010; Rumondor et al. 2009a, b). Temperature naturally has a direct effect on molecular mobility. Molecular mobility is generally considered one of the key factors in determining amorphous system stability. The molecular mobility of a system can be quantified using dielectric spectroscopy. This technology was employed by Mistry et al. in order to compare ASD of ketoconazole with three different polymers. They could show that the molecular mobility was only moderately reduced in case of PVP and poly(2-hydroxyethyl methacrylate), whereas a strong reduction was observed for poly(acrylic acid) (Mistry et al. 2015).
In spite of intensive research to predict the stability of amorphous systems through molecular mobility concepts, the prediction of physical stability is rather unpredictable. Often, physical instability occurs in a nonlinear fashion after varying induction periods. Although the stability of amorphous systems is not predictable, one can use accelerated stability condition of severe humidity and temperature to rank order the formulations. A commonly used accelerated stability condition for formulation screening is 40 °C/100 % RH or 40 °C/75% RH open. In this case, amorphous solid dispersions are subjected to stress conditions and are evaluated by XRPD, FTIR, and DSC analysis. In addition, the suspension stability that is performed to enable the use of amorphous dispersion for toxicology studies is also helpful to assess physical stability. For toxicology purposes, the preferred vehicle is the one having the lowest solubility for amorphous solid dispersion and should maintain amorphous form stability for at least 4 h at room temperature.
8.9 Overall Summary
In this chapter, we have proposed a structured development approach for amorphous formulations based on sound physicochemical principles of the drug and the polymer with the goal of maximizing success rates and reducing risks. The proposed approach consists of a comprehensive evaluation of the drug substance and polymer properties and understanding the basic principles to help design amorphous dispersions that provide consistent in vitro and in vivo performance. The assessment of basic physicochemical properties of drug substance including its thermal behavior (T g, T m, stability, and recrystallization), hydrogen-bond acceptor and donor groups, molecular weight, hydrophobicity, solubility parameters, solubility (aqueous and organic solvents), and hygroscopicity forms the basis of determining the degree of difficulty a molecule may present in converting to and maintaining an amorphous form. Likewise, a thorough understanding of the physicochemical and mechanical properties of polymers will help in the determination of the suitability of polymer(s) for a given drug for an amorphous solid dispersion. Besides theoretical considerations in terms of solubility and miscibility, miniaturization techniques, such as the SPADS approach help in the selection of specific polymers and drug loads for maximum solid-state stability and supersaturation.
In a second phase, the thorough evaluation of physicochemical properties of drugs and polymers with respect to thermal behavior, solvent and aqueous solubility and stability guide the selection of appropriate processing techniques for amorphous solid dispersions. During the development of the amorphous formulation the impact of various process parameters need to be investigated in order to ensure consisted quality in terms of dissolution performance and stability. Different instrumental techniques are presented to characterize and quantify the crystallinity and for understanding the phase behavior of amorphous solid dispersions. The use of more than one technique is preferred to establish confidence. Amorphous formulation stability is difficult to predict; however, accelerated storage conditions such as 40 °C/100 % RH and suspension stability test provide a useful insight into the stability of these amorphous systems. Similarly, approaches are presented to help develop appropriate dissolution methods based on kinetic solubility and consideration of the supersaturation level to avoid false negatives. Finally, a flowchart shown in Fig. 8.9 is proposed as a blueprint for structured development of amorphous formulations.

Structured development approach for amorphous formulation development
References
Acartürk F, Kislal Ö et al (1992) The effect of some natural polymers on the solubility and dissolution characteristics of nifedipine. Int J Pharm 85(1–3):1–6
Adrjanowicz K, Kaminski K et al (2010) Dielectric relaxation studies and dissolution behavior of amorphous verapamil hydrochloride. J Pharm Sci 99(2):828–839
Agrawal AM, Dudhedia MS et al (2016) Hot melt extrusion: development of an amorphous solid dispersion for an insoluble drug from mini-scale to clinical scale. AAPS PharmSciTech 17(1):133–147
Albano AA, Phuapradit W et al (2002) Stable complexes of poorly soluble compounds in ionic polymers. US Patent Office, United States of America, F. Hoffmann-La Roche Ltd, 7
Albers J (2008) Hot-melt extrusion with poorly soluble drugs. Heinrich-Heine-University, Düsseldorf
Alhalaweh A, Alzghoul A et al (2015) Physical stability of drugs after storage above and below the glass transition temperature: Relationship to glass-forming ability. Int J Pharm 495(1):312–317
Al-Obaidi H, Brocchini S et al (2009) Anomalous properties of spray dried solid dispersions. J Pharm Sci 98(12):4724–4737
Aso Y, Yoshioka S (2006) Molecular mobility of nifedipine–PVP and phenobarbital–PVP solid dispersions as measured by 13 C-NMR spin-lattice relaxation time. J Pharm Sci 95(2):318–325
Aso Y, Yoshioka S et al (2002) Effect of water on the molecular mobility of sucrose and poly(vinylpyrrolidone) in a colyophilized formulation as measured by 13 C-NMR relaxation time. Chem Pharm Bull 50(6):822–826
Baghel S, Cathcart H et al (2016) Polymeric amorphous solid dispersions: a review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J Pharm Sci. doi:10.1016/j.xphs.2015.10.008
Baird JA, Van Eerdenburgh B et al (2010) A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J Pharm Sci 99(9):3787–3806
Barillaro V, Pescarmona PP et al (2008) High-throughput study of phenytoin solid dispersions: formulation using an automated solvent casting method, dissolution testing, and scaling-up. J Comb Chem 10(5):637–643
Bates S, Zografi G et al (2006) Analysis of amorphous and nanocrystalline solids from their X-ray diffraction patterns. Pharm Res 23(10):2333–2349, Epub 2006 Sep 22
Breitenbach J (2002) Melt extrusion: from process to drug delivery technology. Eur J Pharm Biopharm 54(2):107–117
Chan KLA, Kazarian SG (2004) FTIR spectroscopic imaging of dissolution of a solid dispersion of nifedipine in poly(ethylene glycol). Mol Pharm 1(4):331–335
Chiang P-C, Ran Y et al (2012) Evaluation of drug load and polymer by using a 96-well plate vacuum dry system for amorphous solid dispersion drug delivery. AAPS PharmSciTech 13(2):713–722
Chiou WL, Riegelman S (1971) Pharmaceutical applications of solid dispersion systems. J Pharm Sci 60(9):1281–1302
Chiou WL, Riegelmann S (1970) Oral absorption of griseofulvin in dogs: increased absorption via solid dispersion in polyethylene glycol 6000. J Pharm Sci 59:937–942
Chokshi RJ, Sandhu HK et al (2005) Characterization of physico-mechanical properties of indomethacin and polymers to assess their suitability for hot-melt extrusion processs as a means to manufacture solid dispersion/solution. J Pharm Sci 94(11):2463–2474
Chokshi RJ, Shah NH et al (2008) Stabilization of low glass transition temperature indomethacin formulations: impact of polymer-type and its concentration. J Pharm Sci 97(6):2286–2298
Corrigan OI, Holohan EM et al (1985) Physicochemical properties of indomethacin and related compounds co-spray dried with polyvinylpyrrolidone. Drug Dev Ind Pharm 11(2–3):677–695
Crowley MM, Zhang F et al (2007) Pharmaceutical applications of hot-melt extrusion: part I. Drug Dev Ind Pharm 33(9):909–926
Curatolo W, Nightingale J et al (2009) Utility of hydroxypropylmethylcellulose acetate succinate (HPMC-AS) for initiation and maintenance of drug supersaturation in the GI milieu. Pharm Res 26(6):1419–1431
De Maesschalk R, Stokbroekx S et al (2010) Development of a 96-well plate dissolution method for screening solid dispersions: Comparison to classical USP methods and its use in predicting oral bioavailability in animals. In: AAPS annual meeting and exposition. Ernest N. Morial Convention Center, New Orleans
Deng W, Majumdar S et al (2013) Stabilization of fenofibrate in low molecular weight hydroxypropylcellulose matrices produced by hot-melt extrusion. Drug Dev Ind Pharm 39(2):290–298
DiNunzio JC, Miller DA et al (2008) Amorphous compositions using concentration enhancing polymers for improved bioavailability of itraconazole. Mol Pharm 5(6):968–980
Dobry DE, Settell DM et al (2009) A model-based methodology for spray-drying process development. J Pharm Innov 4(3):133–142
Doherty C, York P (1987) Mechanisms of dissolution of frusemide PVP solid dispersions. Int J Pharm 34(3):197–205
Engers D, Teng J et al (2010) A solid-state approach to enable early development compounds: selection and animal bioavailability studies of an itraconazole amorphous solid dispersion. J Pharm Sci 99(9):3901–3922
Feng J, Xu L et al (2012) Evaluation of polymer carriers with regard to the bioavailability enhancement of bifendate solid dispersions prepared by hot-melt extrusion. Drug Dev Ind Pharm 38(6):735–743
Forster A, Hempenstall J et al (2001a) The potential of small-scale fusion experiments and the Gordon-Taylor equation to predict the suitability of drug/polymer blends for melt extrusion. Drug Dev Ind Pharm 27(6):549–560
Forster A, Hempenstall J et al (2001b) Selection of excipients for melt extrusion with two poorly water-soluble drugs by solubility parameter calculation and thermal analysis. Int J Pharm 226(1–2):147–161
Friesen DT, Shanker R et al (2008) Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: an overview. Mol Pharm 5(6):1003–1019
Gil M, Vicente J, Gaspar F (2010) Scale-up methodology for pharmaceutical spray drying. Chem Today 28(4):18–22
Gordon M, Taylor JS (1952) Ideal copolymers and the second-order transitions of synthetic rubbers I. Noncrystalline copolymers. J Appl Chem 2:493–500
Greenhalgh DJ, Williams AC et al (1999) Solubility parameters as predictors of miscibility in solid dispersions. J Pharm Sci 88(11):1182–1190
Gupta P, Kakumanu VK et al (2004) Stability and solubility of celecoxib-PVP amorphous dispersions: a molecular perspective. Pharm Res 21:1762–1769
Gupta J, Nunes C et al (2011) Prediction of solubility parameters and miscibility of pharmaceutical compounds by molecular dynamics simulations. J Phys Chem B 115(9):2014–2023
Guzmán HR, Tawa M et al (2007) Combined use of crystalline salt forms and precipitation inhibitors to improve oral absorption of celecoxib from solid oral formulations. J Pharm Sci 96(10):2686–2702
Hadida S, Van Goor F et al (2014) Case history: Kalydeco® (VX-770, Ivacaftor), a CFTR potentiator for the treatment of patients with cystic fibrosis and the G551D-CFTR mutation. Annu Rep Med Chem 49:383–398
Hancock BC, Parks M (2000) What is the true solubility advantage for amorphous pharmaceuticals? Pharm Res 17(4):397–404
Hancock BC, York P et al (1997) The use of solubility parameters in pharmaceutical dosage form design. Int J Pharm 148:1–21
He Y, Ho C (2015) Amorphous solid dispersions: utilization and challenges in drug discovery and development. J Pharm Sci 104(10):3237–3258
Hu Q, Choi DS et al (2013) Highly efficient miniaturized coprecipitation screening (MiCoS) for amorphous solid dispersion formulation development. Int J Pharm 450:53–62
Huang Y, Dai W-G (2014) Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm Sin B 4(1):18–25
Huang J, Wigent RJ et al (2008) Drug-polymer interaction and its significance on the physical stability of nifedipine amorphous dispersion in microparticles of an ammonio methacrylate copolymer and ethylcellulose binary blend. J Pharm Sci 97(1):251–262
Hugo M, Kunath K et al (2013) Selection of excipient, solvent and packaging to optimize the performance of spray-dried formulations: case example fenofibrate. Drug Dev Ind Pharm 39(2):402–412
Islam M, Scoutaris N et al (2015) Implementation of transmission NIR as a PAT tool for monitoring drug transformation during HME processing. Eur J Pharm Biopharm 96:106–116
Jackson MJ, Kestur US et al (2016) Dissolution of danazol amorphous solid dispersions: supersaturation and phase behavior as a function of drug loading and polymer type. Mol Pharm 13(1):223–231
Janssens S, Guy VM (2010) Review: physical chemistry of solid dispersions. J Pharm Pharmacol 12:15
Janssens S, Nagels S et al (2008) Formulation and characterization of ternary solid dispersions made up of Itraconazole and two excipients, TPGS 1000 and PVP VA 64, that were selected based on supersaturation screening study. Eur J Pharm Biopharm 69:158–166
Janssens S, De Zeure A et al (2010) Influence of preparation methods on solid state supersaturation of amorphous solid dispersions: a case study with itraconazole and eudragit E100. Pharm Res 27(5):775–785
Jensen KT, Blaabjerg LI et al (2015) Preparation and characterization of spray-dried co-amorphous drug–amino acid salts. J Pharm Pharmacol. doi:10.1111/jphp.12458
Jijun F, Lili Z et al (2010) Stable nimodipine tablets with high bioavailability containing NM-SD prepared by hot-melt extrusion. Powder Technol 204:214–221
Just S, Sievert F et al (2013) Improved group contribution parameter set for the application of solubility parameters to melt extrusion. Eur J Pharm Biopharm 85(3):1191–1199
Kalb O, Page S et al (2013) Scale-up of solid dispersions. Encyclopedia of pharmaceutical science and technology, fourth edition: doi: 10.1081/E-EPT4-120050349
Kaushal AM, Chakraborti AK et al (2008) FTIR studies on differential intermolecular association in crystalline and amorphous states of structurally related non-steroidal anti-inflammatory drugs. Mol Pharm 5(6):937–945
Kislalioglu MS, Khan MA et al (1991) Physical characterization and dissolution properties of ibuprofen: eudragit coprecipitates. J Pharm Sci 80(8):799–804
Knopp MM, Tajber L et al (2015) Comparative study of different methods for the prediction of drug–polymer solubility. Mol Pharm 12:3408–3419
Knopp MM, Olesen NE et al (2016) Statistical analysis of a method to predict drug–polymer miscibility. J Pharm Sci 105:362–367
Konno H, Taylor LS (2006) Influence of different polymers on the crystallization tendency of molecularly dispersed amorphous felodipine. J Pharm Sci 95(12):2692–2705
Konno H, Handa T et al (2008) Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur J Pharm Biopharm 70(2):493–499
Kwong AD, Kauffman RS et al (2011) Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat Biotechnol 29(11):993–1003
Lamm MS, DiNunzio J et al (2016) Assessing mixing quality of a copovidone-TPGS hot melt extrusion process with atomic force microscopy and differential scanning calorimetry. AAPS PharmSciTech 17(1):89–98
Lauer M, Grassmann O et al (2011) Atomic force microscopy-based screening of drug-excipient miscibility and stability of solid dispersions. Pharm Res 28(3):572–584
Lauer M, Siam M et al (2013) Rapid assessment of homogeneity and stability of amorphous solid dispersions by atomic force microscopy—from bench to batch. Pharm Res 30(8):2010–2022
Law D, Schmitt EA et al (2004) Ritonavir-PEG 8000 amorphous solid dispersions: in vitro and in vivo evaluations. J Pharm Sci 93(3):563–570
Lee T, Lee J (2003) Drug-carrier screening on a chip. Pharm Technol North Am 27(1):40–48
Lemmer HJR, Liebenberg W (2013) Preparation and evaluation of metastable solid-state forms of lopinavir. Pharmazie 68:327–332
Lenz E, Jensen KT et al (2015) Solid-state properties and dissolution behaviour of tablets containing co-amorphous indomethacin–arginine. Eur J Pharm Biopharm 96:44–52
Leuner C, Dressman J (2000) Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm 50(1):47–60
Liu H, Taylor LS et al (2015) The role of polymers in oral bioavailability enhancement; a review. Polymer 77:399–415
Löbmann K, Grohganz H et al (2013) Amino acids as co-amorphous stabilizers for poorly water soluble drugs—part 1: preparation, stability and dissolution enhancement. Eur J Pharm Biopharm 85:873–881
Loftsson T, Fririksdóttir H et al (1996) The effect of water-soluble polymers on aqueous solubility of drugs. Int J Pharm 127(2):293–296
Ma HM, Choi DS et al (2013) Evaluation on the drug–polymer mixing status in amorphous solid dispersions at the early stage formulation and process development. J Pharm Innov 8:163–174
Mahlin D, Bergström CAS (2013) Early drug development predictions of glass-forming ability and physical stability of drugs. Eur J Pharm Sci 49(2):323–332
Marsac PJ, Konno H et al (2006) A comparison of the physical stability of amorphous felodipine and nifedipine systems. Pharm Res 23:2306–2316
Marsac PJ, Konno H et al (2008) Recrystallization of nifedipine and felodipine from amorphous molecular-level solid dispersions containing poly(vinylpyrrolidone) and sorbed water. Pharm Res 25(3):647–656
Marsac PJ, Rumondor ACF et al (2010) Effect of temperature and moisture on the miscibility of amorphous dispersions of felodipine and poly(vinyl pyrrolidone). J Pharm Sci 99(1):169–185
Martin C (2016) Twin screw extruders as continuous mixers for thermal processing: a technical and historical perspective. AAPS PharmSciTech 17(1):3–19
Masters K (1991) Spray drying handbook, Longman scientific & technical. Wiley, Burnt Mill
Meng F, Dave V et al (2015) Qualitative and quantitative methods to determine miscibility in amorphous drug-polymer systems. Eur J Pharm Sci 77:106–111
Miller D, DiNunzio J et al (2008) Targeted intestinal delivery of supersaturated itraconazole for improved oral absorption. Pharm Res 25(6):1450–1459
Mishra DK, Dhote V et al (2015) Amorphous solid dispersion technique for improved drug delivery: basics to clinical applications. Drug Deliv Transl Res 5:552–565
Mistry P, Mohapatra S et al (2015) Role of the strength of drug–polymer interactions on the molecular mobility and crystallization inhibition in ketoconazole solid dispersions. Mol Pharm 12(9):3339–3350
Miyazaki T, Yoshioka S et al (2004) Ability of polyvinylpyrrolidone and polyacrylic acid to inhibit the crystallization of amorphous acetaminophen. J Pharm Sci 93:2710–2717
Miyazaki T, Yoshioka S et al (2007) Crystallization rate of amorphous nifedipine analogues unrelated to the glass transition temperature. Int J Pharm 336:191–195
Moes J, Koolen S et al (2011) Pharmaceutical development and preliminary clinical testing of an oral solid dispersion formulation of docetaxel (ModraDoc001). J Pharm Sci 420:244–250
Moneghini M, Carcano A et al (1998) Studies in dissolution enhancement of atenolol. Int J Pharm 175:177–183
Oksanen CA, Zografi G (1990) The relationship between the glass transition temperature and water vapor absorption by poly(vinylpyrrolidone). Pharm Res 7(9):654–657
Overhoff KA, Engstrom JD et al (2007) Novel ultra-rapid freezing particle engineering process for enhancement of dissolution rates of poorly water-soluble drugs. Eur J Pharm Biopharm 65(1):57–67
Pajula K, Taskinen M et al (2010) Predicting the formation and stability of amorphous small molecule binary mixtures from computationally determined Flory–Huggins interaction parameter and phase diagram. Mol Pharm 7(3):795–804
Palermo RN, Anderson CA et al (2012) Review: use of thermal, diffraction, and vibrational analytical methods to determine mechanism of solid dispersion stability. J Pharm Innov 7:2–12
Park K (2015) Drug release mechanism from amorphous solid dispersions. J Control Release 211:171
Patel KP, Pathak CJ et al (2015) Nanostructured lipid carrier—a novel dosage form to improve the oral bioavailability of lopinavir. Eur J Biomed Pharm Sci 2(2):295–311
Patil H, Tiwari R et al (2016) Hot-melt extrusion: from theory to application in pharmaceutical formulation. AAPS PharmSciTech 17(1):20–42
Patterson JE, James MB et al (2007) Preparation of glass solutions of three poorly water soluble drugs by spray drying, melt extrusion and ball milling. Int J Pharm 336(1):22–34
Patterson JE, James MB et al (2008) Melt extrusion and spray drying of carbamazepine and dipyridamole with polyvinylpyrrolidone/vinyl acetate copolymers. Drug Dev Ind Pharm 34:95–106
Paudel A, Van den Mooter G (2012) Influence of solvent composition on the miscibility and physical stability of naproxen/PVP K 25 solid dispersions prepared by cosolvent spray-drying. Pharm Res 29:251–270
Paudel A, Van Humbeeck J et al (2010) Theoretical and experimental investigation on the solid solubility and miscibility of naproxen in poly(vinylpyrrolidone). Mol Pharm 7(4):1133–1148
Paudel A, Nies E et al (2012) Relating hydrogen-bonding interactions with the phase behavior of naproxen/PVP K 25 solid dispersions: evaluation of solution-cast and quench-cooled films. Mol Pharm 9(11):3301–3317
Paus R, Ji Y et al (2015) Predicting the solubility advantage of amorphous pharmaceuticals: a novel thermodynamic approach. Mol Pharm 12(8):2823–2833
Purohit HS, Taylor LS (2015) Miscibility of itraconazole-hydroxypropyl methylcellulose blends: insights with high resolution analytical methodologies. Mol Pharm 12(12):4542–4553
Qi S, Belton P et al (2011) Compositional analysis of low quantities of phase separation in hot-melt-extruded solid dispersions: a combined atomic force microscopy, photothermal fourier-transform infrared microspectroscopy, and localized thermal analysis approach. Pharm Res 28(9):2311–2326
Qian F, Tao J et al (2007) Mechanistic investigation of pluronic® based nano-crystalline drug-polymer solid dispersions. Pharm Res 24(8):1551–1560
Read MD, Coppens KA et al (2010) Hot melt extrusion technology for the manufacture of poorly soluble drugs with controlled release dissolution profiles. ANTEC:1203–1206
Repka M, Langley N et al (2013) Melt extrusion; materials, technology and drug product design. Springer, New York
Rowe R, Shesky P et al (2010) Handbook of pharmaceutical excipients, 4th edn. APhA, Washington
Rumondor A, Stanford L et al (2009a) Effects of polymer type and storage relative humidity on the kinetics of felodipine crystallization from amorphous solid dispersions. Pharm Res 26(12):2599–2606
Rumondor ACF, Marsac PJ et al (2009b) Phase behavior of poly(vinylpyrrolidone) containing amorphous solid dispersions in the presence of moisture. Mol Pharm 6(5):1492–1505
Saerens L, Dierickx L et al (2011) Raman spectroscopy for the in-line polymer-drug quantification and solid state characterization during pharmaceutical hot-melt extrusion process. Eur J Pharm Biopharm 77:158–163
Sanghvi T, Katstra J et al (2015) Pharmaceutical amorphous solid dispersions. In: Formulation development of amorphous dispersions, 1st edn. Wiley, Hoboken
Sarode AL, Sandhu H et al (2013) Hot melt extrusion (HME) for amorphous solid dispersions: Predictive tools for processing and impact of drug–polymer interactions on supersaturation. Eur J Pharm Sci 48:371–384
Serajuddin ATM (1999) Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci 88:1058–1066
Shah N, Sandhu H et al (2008) Solid complexes with ionic polymers: pharmaceutical technology is pleased to recognize the winners of its innovations in pharma science awards. Pharm Technol 32(12):2
Shah N, Iyer RM et al (2013) Improved human bioavailability of vemurafenib, a practically insoluble drug, using an amorphous polymer-stabilized solid dispersion prepared by a solvent-controlled coprecipitation process. J Pharm Sci 102(3):967–981
Shanbhag A, Rabel S et al (2008) Method for screening of solid dispersion formulations of low-solubility compounds-miniaturization and automation of solvent casting and dissolution testing. Int J Pharm 351(1–2):209–218
Siew A (2014) Solving poor solubility with amorphous solid dispersions: weighing the pros and cons of hot-melt extrusion and spray drying. Pharm Technol 38(1):30–35
Simonelli AP, Mehta SC et al (1969) Dissolution rates of high energy polyvinylpyrrolidone (PVP)-sulfathiazole coprecipitates. J Pharm Sci 58(5):538–549
Singh H, Atef E et al (2007) High throughput screening of solid dispersion using solvent evaporation technique aaps annual meeting and exposition. San Diego Convention Center, San Diego
Six K, Verreck G et al (2004) Increased physical stability and improved dissolution properties of itraconazole, a class II drug, by solid dispersions that combine fast- and slow-dissolving polymers. J Pharm Sci 93(1):124–131
Song Y, Zemlyanov D et al (2016) Acid–base interactions of polystyrene sulfonic acid in amorphous solid dispersions using a combined UV/FTIR/XPS/ssNMR study. Mol Pharm 13(2):483–492
Sun DD, Lee PI (2013) Evolution of supersaturation of amorphous pharmaceuticals: the effect of rate of supersaturation generation. Mol Pharm 10(11):4330–4346
Swinney K, Herman J et al (2009) Configuration of an automated screening tool to facilitate solid dispersion development AAPS annual meeting and exposition. Los Angeles Convention Center, Los Angeles
Tanno F, Nishiyama Y et al (2004) Evaluation of hypromellose acetate succinate (HPMC-AS) as a carrier in solid dispersions. Drug Dev Ind Pharm 30(1):9–17
Taylor LS, Zografi G (1997) Spectroscopic characterization of interactions between PVP and indomethacin in amorphous molecular dispersions. Pharm Res 14:1691–1698
Thybo P, Hovgaard L et al (2008) Scaling up the spray drying process from pilot to production scale using an atomized droplet size criterion. Pharm Res 25(7):1610–1620
Tian B, Wang X et al (2015) Theoretical prediction of a phase diagram for solid dispersions. Pharm Res 32:840–851
Usui F, Maeda K et al (1997) Inhibitory effects of water-soluble polymers on precipitation of RS-8359. Int J Pharm 154(1):59–66
Van den Mooter G, Wuyts M et al (2001) Physical stabilisation of amorphous ketoconazole in solid dispersions with polyvinylpyrrolidone K25. Eur J Pharm Sci 12(3):261–269
Van Eerdenbrugh B, Taylor LS (2010) Small scale screening to determine the ability of different polymers to inhibit drug crystallization upon rapid solvent evaporation. Mol Pharm 7(4):1328–1337
Van Eerdenburgh B, Baird JA et al (2010) Crystallization tendency of active pharmaceutical ingredients following rapid solvent evaporation—classification and comparison with crystallization tendency from undercooled melts. J Pharm Sci 99(9):3826–3838
Vandecruys R, Peeters J et al (2007) Use of screening method to determine excipients which optimize the extend and stability of supersaturated drug solutions and application of this system to solid formulation design. Int J Pharm 342:168–175
Warren DB, Benameur H et al (2010) Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: a mechanistic basis for utility. J Drug Target 18:704–731
Weuts I, Van Dycke F et al (2011) Physicochemical properties of the amorphous drug, cast films, and spray dried powders to predict formulation probability of success for solid dispersions: etravirine. J Pharm Sci 100(1):260–274
Wyttenbach N, Janas C et al (2013) Miniaturized screening of polymers for amorphous drug stabilization (SPADS): rapid assessment of solid dispersion systems. Eur J Pharm Biopharm 84:583–598
Wyttenbach N, Kirchmeyer W et al (2016) Theoretical considerations of the Prigogine–Defay ratio with regard to the glass-forming ability of drugs from undercooled melts. Mol Pharm 13:241–250
Xie T, Taylor LS (2016) Improved release of celecoxib from high drug loading amorphous solid dispersions formulated with polyacrylic acid and cellulose derivatives. Mol Pharm 13(3):873–884
Yamashita T, Kokubo T et al (2010) Antiprecipitant screening system for basic model compounds using bio-relevant media. J Assoc Lab Automat 15(4):306–312
Yang Z, Han CD (2008) Rheology of miscible polymer blends with hydrogen bonding. Macromolecules 41(6):2104–2118
Yoo S-u, Krill SL et al (2009) Miscibility/stability considerations in binary solid dispersion systems composed of functional excipients towards the design of multi-component amorphous systems. J Pharm Sci 98(12):4711–4723
Yu L (2001) Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv Drug Deliv Rev 48:27–42
Zhao Y, Inbar P et al (2011) Prediction of the thermal phase diagram of amorphous solid dispersions by flory-huggins theory. J Pharm Sci 100(8):3196–3207
Acknowledgments
The authors of the second edition of the book chapter would like to thank their former co-workers Navnit Shah, Harpreet Sandhu, Duk Soon Choi and Oskar Kalb for the great work done together for the first edition.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 American Association of Pharmaceutical Scientists
About this chapter
Cite this chapter
Page, S., Maurer, R., Wyttenbach, N. (2016). Structured Development Approach for Amorphous Systems. In: Williams III, R., Watts, A., Miller, D. (eds) Formulating Poorly Water Soluble Drugs. AAPS Advances in the Pharmaceutical Sciences Series, vol 22. Springer, Cham. https://doi.org/10.1007/978-3-319-42609-9_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-42609-9_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-42607-5
Online ISBN: 978-3-319-42609-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)