
Abstract
Purpose
To understand the mechanism of nano-crystalline drug formation in Pluronic® (i.e., poly(ethylene oxide-block-propylene oxide) triblock copolymers) based drug-polymer solid dispersions.
Materials and Methods
Four polymers, Pluronic® F127, F108, F68 and PEG 8000, which have different poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO) ratio and chain length, were co-spray dried with BMS-347070, a COX-2 inhibitor, to form 50/50 (w/w) drug-polymer solid dispersions. The solid dispersions were analyzed by powder X-ray diffraction (PXRD), modulated differential scanning calorimetry (mDSC), and hot-stage microscopy. Average size of drug crystallites in different polymers was calculated by the Scherrer equation based on peak-broadening effect in PXRD. Two other drug compounds, BMS-A and BMS-B, were also spray dried with Pluronic® F127, and the solid dispersions were analyzed by PXRD and mDSC.
Results
The average size of BMS-347070 crystallites in PEG 8000, F127, F108 and F68 polymers was 69, 80, 98 and 136 nm, respectively, and the degree of BMS-347070 crystallinity is the lowest in PEG 8000. Hot-stage microscopy showed that 50/50 drug-polymer dispersions crystallized in a two-step process: a portion of the polymer crystallizes first (Step 1), followed by crystallization of drug and remaining polymer (Step 2). The T g value of the BMS-347070/Pluronic® dispersions after Step 1 (i.e., T g1) was measured and/or calculated to be 15–26°C, and that of BMS-347070/PEG 8000 was 60°C. Solid dispersions of BMS-A and BMS-B in Pluronic® F127 have T g1 of 72 and 3°C, respectively; and PXRD showed BMS-A remained amorphous after ∼3 weeks under ambient condition, while BMS-B crystallized in F127 with an average crystallite size of 143 nm.
Conclusions
The size of drug crystallites in the drug-polymer solid dispersions is independent of polymer topology, but is caused kinetically by a combined effect of nucleation rate and crystal growth rate. When drug-Pluronic® solid dispersions crystallize at room temperature, that is close to the T g1 of the systems, a fast nucleation rate and a relatively slow crystal growth rate of the drug synergistically produced small crystallite size. While the much higher T g1 value of drug-PEG 8000 led to a slower nucleation rate and an even slower crystal growth rate at room temperature, therefore, small crystallite size and low drug crystallinity were observed. Results from BMS-A/Pluronic® and BMS-B/Pluronic® systems confirmed this kinetic theory.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Oral formulation development for poorly water-soluble compounds represents one of the most frequent challenges to pharmaceutical scientists. Common strategies to overcome low aqueous solubility and slow dissolution rate include particle size reduction of crystalline drugs, crystal lattice destruction by forming stabilized amorphous drug-polymer solid dispersions, and solublized systems such as lipid-based formulations (1–7). The rationale behind these strategies can be readily explained by the Noyes-Whitney relationship (8), and the selection of one strategy over the other depends on various factors, such as the physiochemical properties of the drug, dose to be delivered, scalability, robustness and manufacture of the formulations, and drug product stability.
One of the more frequently reported strategies in the pharmaceutical literature has been the amorphous drug-polymer solid dispersions (3,5,6). In this type of system, drug and polymers are co-processed, often by spray drying or hot-melt extrusion, to form an amorphous solid solution or dispersion, wherein polymers usually act as crystallization inhibitors to the amorphous drug. However, there are relatively few commercial products comprising an amorphous drug solid dispersion, in large part because the amorphous formulations are thermodynamically metastable which can result in product stability issues during long-term storage and/or excursions in temperature and humidity. Recently, our laboratory reported a nano-crystalline solid dispersion that significantly enhanced the in vitro dissolution rate and the in vivo bioavailability (9). The nano-crystalline solid dispersion was made by co-spray drying BMS-347070, a COX-2 inhibitor, with Pluronic® F127, a tri-block copolymer with a poly(propylene oxide) (PPO) segment sandwiched by two poly(ethylene oxide) (PEO) segments (9). PXRD measurement indicated the average drug crystallite size in the polymer matrix was less than 100 nm. Furthermore, the drug in Pluronic® F127 appeared to have a stable crystal size and degree of crystallinity over a time frame of several years, which could be a crucial advantage over the amorphous formulations.
While the potential application of this nano-crystalline solid dispersion is encouraging, the mechanism through which the drug crystallizes into nano-sized crystallites in the Pluronic® polymer was not fully established in the previous work. Considering the unique topology of the PEO-PPO-PEO tri-block polymers, and the fact that PEO segments crystallize readily and PPO segment remains amorphous, a “cage” hypothesis was proposed by our laboratory to explain the nano-crystalline drug formation (9). According to the reported “cage” hypothesis, the PEO segments in Pluronic® polymer crystallized and assembled into nano-sized “cages” with PPO segments forming the core, and the subsequent drug crystallization was geometrically constrained in the amorphous PPO regions, thus, nano-sized crystals formed.
Although interesting, the “cage hypothesis” remained to be tested experimentally. Thus, a systematic evaluation is reported in the current work whereby a series of drug/polymer dispersions using different grades of Pluronic® polymers and PEG 8000 were prepared and characterized. These new results lead us to propose an alternative mechanism where the size of drug crystallite is primarily controlled by a combined effect of nucleation rate and crystal growth rate; both processes are dependent on the “effective T g” of the drug-polymer solid dispersions which is influenced by the addition of polymers. The current work lends further insight to the crystallization mechanism that influences the size of the drug crystallized in the polymer matrix, so that some guidance can be established to develop nano-crystalline solid dispersions for other poorly soluble drug candidates.
MATERIALS AND METHODS
Materials
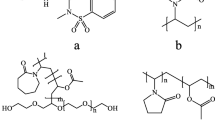
Three drug compounds, BMS-347070 (MW 407), BMS-A (MW 447) and BMS-B (MW 437) were synthesized in-house, each possessing very low aqueous solubility (∼μg/mL) across the physiological pH range. Three grades of Pluronic® polymers, F68 (MW 8400), F108 (MW 14600) and F127 (MW 12600) were received as generous gifts from BASF (Ludwigshafen, Germany). Poly(ethylene glycol) 8000 (PEG 8000, MW 8000) was received from Union Carbride Chemicals and Plastics Company Inc (Danbury, CT). Additional structural information of these polymers is provided in Table I. The chemical structure of BMS-347070, Pluronic® and PEG polymers is shown in Fig. 1. HPLC grade methylene chloride was purchased from EMD Chemicals (Bibbsons Town, NJ).

Chemical structures of BMS-347070, a COX-2 inhibitor; Pluronic®, a tri-block copolymer, with poly(propylene oxide) (PO) segment sandwiched by two poly(ethylene oxide) (EO) segments; and Poly(ethylene glycol) (PEG).
Preparation of Drug-polymer Solid Dispersions by Spray Drying
Drug-polymer solid dispersions (50/50, w/w) were prepared by spray drying a methylene chloride solution of the drug and the polymer using a Buchi B-191 mini spray dryer (Brinkmann Instruments, Westbury, NY). The total solid concentration of drug and polymer in methylene chloride was 5% w/v. During spray drying, the inlet temperature was kept at room temperature, solution pumping rate was ∼4 ml/min, atomizing N2 flow rate was 500 Nl/h, aspirator was set at 100%. After spray drying, the obtained material was dried in the spray dryer with N2 flow for 24 h and the material was collected and stored under ambient condition. It should be noted that all characterization and evaluation was carried out for the reconditioned material after drying for 24 h in N2, not the freshly spray dried material. Immediately after spray drying, drug remains amorphous and only after further drying for 24 h did the drug crystallize and the crystalline drug-polymer dispersion was formed.
Powder X-ray Diffraction (PXRD)
Powder samples were mounted on a sample holder and PXRD patterns were collected using a Bruker D8 Advance diffractometer (Bruker AXS, Inc. Madison, WI). The diffractometer set up was: Cu Kα radiation, with X-ray wavelength of 1.542 nm, voltage of 40 kV, and current of 40 mA. The samples were scanned from 10° to 30° 2θ at a speed of 0.06° 2θ per min, the step size is 0.02° 2θ. Before analyzing samples, the slit configuration of the instrument was optimized to minimize the peak broadening effect due to the instrument setting. After optimization, the following configuration was used for future studies: the divergence slit 0.6 mm, anti-scatter slit 0.6 mm, detector (receiving) slit 0.1 mm.
Average crystallite size was calculated using the Scherrer equation (10):
where τ is the mean crystallite dimension, K is a constant of 0.9, λ is the X-ray wavelength (1.542 nm), \( \beta _{\tau } \) is the line broadening value due to crystal size reduction, i.e., the Full-Width-at-Half-Maximal (FWHM) difference in radian at a certain Bragg angle, between a nano-crystalline sample and the micro-size drug crystals. θ is the Bragg angle. BMS-347070 crystal size was calculated from three BMS-347070 diffraction peaks: (020), (112) and (013), an average crystallite size from the three peaks was reported. Similar procedure was used to calculate the BMS-B crystal size in solid dispersion.
Differential Scanning Calorimetry
DSC was performed on a TA DSC Q1000 Differential Scanning Calorimeter (New Castle, DE) with crimped aluminum pans. Approximately 3–5 mg of sample was used for each measurement. Glass transition temperatures of pure drugs, polymers, and the spray dried drug-polymer dispersions were determined by a modulated temperature scanning: the temperature ramp rate was 2°C/min, and the modulation was ±0.50°C every 60 s. The melting enthalpy measurement was performed by a conventional temperature ramp without modulation. The ramp rate was 5°C/min.
Hot-stage Microscopy
Hot-stage microscopy was performed on a Linkam LTS 350 hot-stage (Linkam Scientific Instrument Ltd, Surrey KT20 5HT, UK) coupled with a Nikon Eclipse E600 POL polarized light microscope (Nikon, Melville, NY). Three samples were prepared and examined under the microscope. Sample 1, BMS-347070 drug was melted on a cover glass at 220°C, rapidly quenched to room temperature at 30°C/min, and was sprinkled with PEG 8000 powders. Sample 2, BMS-347070 drug and Pluronic® F127 polymer were melted on a cover glass side by side at 220°C, then the two molten materials were pushed to direct contact by another cover glass. The sample was then cooled down to room temperature at 30°C/min. Sample 3, spray dried 50/50 BMS-347070/Pluronic® F127 was melted between two cover glasses on the hot-stage at 220°C, followed by cooling down to room temperature at 30°C/min.
RESULTS
Average Size of BMS-347070 Crystallites in Pluronic® and PEG Polymers
BMS-347070 was co-spray dried with four different polymers, Pluronic® F68, F108, F127, and PEG 8000, to form solid dispersions. Pluronic® F68, F108, and F127 are PEO-PPO-PEO tri-block copolymers of different segment lengths; and PEG 8000 is a homo-polymer without the PPO segment. The drug/polymer ratio was 50/50 (w/w) in all samples. The average crystal size of BMS-347070 in each drug-polymer solid dispersion was calculated by Scherrer equation based on the peak broadening in PXRD patterns (Fig. 2a) and the results were summarized in Table I.

a PXRD (112) peak of micro-size BMS-347070 drug and spray dried 50/50 BMS-347070/polymers. From inside to outside: micro-size BMS-347070, drug-polymer solid dispersions with F68, F108, F127, and PEG 8000. The peak intensities were normalized for better comparison. The peak broadening of BMS-347070/PEG 8000 is illustrated as an example: B and b are the FWHM values of 50/50 BMS-347070/PEG 8000 and micro-size BMS-347070, respectively, and B-b is the peak broadening value. b Comparison of PXRD (112) peak intensity of spray dried 50/50 BMS-347070/polymers. From top to bottom: drug-polymer solid dispersions with F68, F108, F127, and PEG 8000.
As listed in Table I, the length of PPO segments of Pluronic® F68, F108 and F127 are approximately 13, 22 and 29 nm, respectively. According to the previous “cage” theory (9), it was hypothesized that the nano-sized drug crystals was caused by geometrically constrained PPO “cages,” and the following observation was expected: with its shortest PPO segment, Pluronic® F68 should provide the smallest “cages” and so the smallest drug crystallite size. Pluronic® F127 has the longest PPO segment, therefore it should provide the largest “cages” and the largest drug crystallite size. Since there is no PPO segment and “cages” in PEG 8000, the size of drug crystallites in PEG 8000 should be the largest, should there be drug crystallization.
However, contrary to what the “cage hypothesis” predicts, the peak broadening effect showed an opposite trend (Fig. 2a), and the calculated average size of BMS-347070 crystals was 80, 98 and 136 nm in Pluronic® F127, F108 and F68, respectively. Apparently the crystallite size increased, not decreased, with the decrease of PPO segment length in the Pluronic® polymers. Furthermore, Scherrer equation indicated an average drug crystallite size of 69 nm in PEG 8000, although there is no PPO region in this homopolymer.
Different drug-polymer solid dispersions also showed different intensities of the PXRD reflection peaks (Fig. 2b). This difference is not attributed to a preferred orientation of drug crystals in the solid dispersions because the relative peak intensities in the spectra are comparable to those in the PXRD pattern of the drug. Therefore, the intensity of a certain reflection can serve as a qualitative indication of the degree of crystallinity. Figure 2b shows an overlay of the (112) reflection in PXRD patterns of BMS-347070 in different polymers. Using the peak intensity as a qualitative indication of degree of crystallinity, the amount of BMS-347070 crystals in different polymers can be rank-ordered as: F68>F108>F127>PEG8000, i.e., the drug crystallized the most completely in F68 while the least completely in PEG 8000.
Crystallization of BMS-347070 in the Presence of Polymers as “Foreign Seeds”
To investigate if PEG or Pluronic® polymers served as a favorable crystallization template or heterogeneous nucleation site for BMS-347070, crystallization of BMS-347070 in the presence of PEG or Pluronic® polymers as “foreign seeds” was studied by polarized microscopy coupled with hot-stage.
Shown in Fig. 3a, PEG 8000 powders were sprinkled onto the super-cooled BMS-347070 melt at room temperature. The sample was then stored in ambient condition for crystallization. After 9 days, isolated BMS-347070 crystals were observed. Interestingly, the drug crystals all occurred spontaneously within the super-cooled drug liquid, instead of heterogeneously from the PEG 8000 seeds. Figure 3b shows a similar experiment with BMS-347070 and Pluronic® F127. The drug and polymer were first melted next to each other, and then the molten liquids were allowed to merge with each other to form a drug-polymer interface. The sample was then cooled to room temperature, where F127 crystallized in minutes. An interface between the amorphous drug melt and crystalline F127 polymer was formed. The sample was stored in ambient condition, and no crystallization of BMS-347070 was observed at the drug-polymer interface after 2 weeks.

Crystallization of BMS-347070 in presence of PEG 8000 (a) and Pluronic® F127 (b). Scale bar in both pictures represent 30 μm.
Kinetics of Melt Crystallization of BMS-347070/ Pluronic® F127
As shown by a chronological PXRD study reported previously (9), within 15 min after spray drying, a portion of Pluronic® F127 polymer crystallized first from the spray dried 50/50 BMS-347070/Pluronic® F127, followed by the crystallization of BMS-347070, and perhaps more Pluronic® F127 polymer. To understand the crystallization kinetics of the drug-polymer binary system, the material was investigated by hot-stage polarized light microscopy and thermal analysis.
A quench-cooled 50/50 BMS-347070/Pluronic® F127 melt was studied under polarized light microscopy chronologically under ambient conditions. Consistent with the earlier PXRD findings, a two-step crystallization kinetics was observed. In Step 1, crystallization of a portion of F127 was observed within minutes. It is noteworthy that Pluronic® F127 grew into two distinctive morphologies: large spherulites in the center, and small randomly oriented crystallites at the periphery (Fig. 4a). No further crystallization of Pluronic® F127 was observed under the microscope after 1 h. In Step 2, crystals of BMS-347070 appeared and grew along the periphery within 24 h. Identification of drug and polymer crystals was straightforward due to the different melting points of Pluronic® F127 and BMS-347070, which are 56°C and 207°C, respectively. The rich concentration of BMS-347070 along the edge of the sample indicated that BMS-347070 is not miscible with Pluronic® F127 at 50/50 ratio, and the binary dispersion undergoes phase separation after melting.

Melt crystallization of 50/50 BMS-347070/F127. a Hot-stage microscope pictures. Left: after 1 h at room temperature F127 crystallized into two different morphologies; Right: after 24 h drug crystallized. The same scale bar represents 25 and 50 μm, respectively. b Schematic illustration of the two-step crystallization process.
Results of DSC study confirmed the two-step crystallization. Within an hour after quench cooling, samples of 50/50 BMS-347070/F127 showed a relatively small melting enthalpy of F127 and no melting of BMS-347070 upon heating to 220°C. However, several days after quench cooling, the samples showed a larger F127 melting enthalpy and the melting of BMS-347070. Obviously, the initial crystallization of F127 in Step 1 was incomplete.
Figure 4b provides an illustration of the two-step crystallization kinetics. The material started as a homogenous melt of 50/50 BMS-347070/polymer at time zero, and the glass transition temperature (T g) of this material is defined as T g0. Due to the limited miscibility between the two components the liquid separated into two phases. Within several minutes, Step 1 started when a portion of polymer crystallized first. The remaining amorphous phase became more concentrated with the drug, and thus had a different T g from T g0, which is defined as T g1. Step 2 starts after hours or days, when drug and more polymer crystallized.
Glass Transition Temperatures of BMS-347070/Polymer Solid Dispersions
Glass transition temperatures (T g) of BMS-347070, various polymers, and 50/50 BMS-347070/polymer at different crystallization stages (i.e., T g0 and T g1) were measured by mDSC and/or estimated by the Fox equation (11) and reported in Table II. The T g of pure BMS-347070 and Pluronic® F127 are 77°C and −66°C, respectively. The low T g of Pluronic® F127 significantly reduced the overall T g of the BMS-347070/ Pluronic® F127 solid dispersion. The initial T g0 value of 50/50 BMS-347070/F127 was measured to be −19°C, fairly close to the Fox equation calculation of −13°C. After Step 1, a portion of the polymer crystallized, thus reducing the polymer amount in the remaining drug-polymer amorphous phase. This polymer portion was calculated by the \( {\Delta H^{\prime }_{f} } \mathord{\left/ {\vphantom {{\Delta H^{\prime }_{f} } {\Delta H_{f} }}} \right. \kern-\nulldelimiterspace} {\Delta H_{f} } \) value, where \( \Delta H^{\prime }_{f} \) is the Pluronic® F127 melting enthalpy in the drug-polymer solid dispersion after Step 1, and ΔHf is the melting enthalpy of the pure polymer that underwent the same DSC cycle. The \( {\Delta H^{\prime }_{f} } \mathord{\left/ {\vphantom {{\Delta H^{\prime }_{f} } {\Delta H_{f} }}} \right. \kern-\nulldelimiterspace} {\Delta H_{f} } \) value indicated that approximately 64% of Pluronic® F127 crystallized after Step 1, which led to a drug/polymer ratio of ∼74/26 in the remaining amorphous phase. This ratio allowed the calculation of the T g1 value of the remaining amorphous drug-polymer phase by the Fox equation. The calculated value of 23°C is consistent with the value of 16°C measured by mDSC.
DSC and microscopy studies showed that solid dispersions between BMS-347070 and other polymers also follow the same two-step crystallization kinetics. Similarly, the T g0 and T g1 values of these systems were measured and/or calculated (Table II). It is interesting to note that the T g1 values of the three 50/50 BMS-347070/ Pluronic® systems were all around room temperature, while the T g1 value of the 50/50 BMS-347070/ PEG 8000 is much higher at 60°C, apparently due to two reasons: the much higher T g of PEG 8000 (−27°C) than that of the three Pluronic® polymers (−66 to −62°C), and more PEG 8000 crystallized in Step 1 (∼87%) than the Pluronic® polymers (∼65%).
Solid Dispersions of Two Other Drug Compounds, BMS-A and BMS-B, in Pluronic® F127
Spray dried drug-polymer solid dispersions (50/50 w/w) between two other drug compounds, BMS-A and BMS-B, and Pluronic® F127 were prepared and characterized by mDSC and PXRD, the results are listed in Table III. Compared to the T g of BMS-347070 (77°C), the T g of BMS-A is much higher at 144°C, while that of BMS-B is lower at 56°C. The three drug compounds also have different miscibility with Pluronic® F127: BMS-A is less miscible with the highest portion of Pluronic® F127 (74%) crystallized after Step1, whereas BMS-B is more miscible with the lowest percentage of the polymer (56%) crystallized after Step 1. The T g1 values of the remaining BMS-A/Pluronic® F127 and BMS-B/Pluronic® F127 amorphous phases were 72 and 6°C, respectively. After 3 weeks under ambient condition, PXRD showed that BMS-A remained amorphous in the solid dispersion; while BMS-B in the solid dispersion crystallized overnight, with an average crystallite size of 143 nm.
DISCUSSION
Crystal Size Measurement by PXRD Peak Broadening
In this work, the average size of drug crystallites was calculated based on PXRD peak broadening. Since this methodology has not been established to determine the crystal size of organic compounds in drug/polymer mixture, it is prudent to note the following: First, PXRD peak broadening could be caused by factors other than crystal size reduction. Such factors include the mechanical strain in crystal lattice and crystal defects. However, those events usually happen with preferred orientations causing different extents of peak broadening at different PXRD diffraction peaks. In this work, the calculated size of drug crystals from three PXRD diffraction peaks and the results from different peaks were in close agreement with each other, which indicates that factors like crystal lattice strain and defects are unlikely to be the major cause of the PXRD peak broadening. Second, we examined the effect of polymer addition on the XRD pattern of the drug. Physical blends of 50/50 BMS-347070 and F127 were prepared and scanned with X-ray (data not shown). When compared with the powder pattern of pure BMS-347070, the XRD pattern of the physical mixtures did not show any line broadening. This confirmed that the presence of polymer did not affect the peak width of the drug.
While PXRD might not be the most accurate tool to estimate the size of the drug crystallites quantitatively, the rank order of the crystallite sizes calculated by Scherrer equation would be expected to be the same. Ideally, size measurement of nano-crystals would come from imaging tools with sufficient spatial resolution, such as Atomic Force Microscopy (AFM) or Transmission Electron Microscopy (TEM). This characterization work is currently under investigation.
Invalidation of the “Cage Hypothesis”
Based on the calculated nano-crystallite size data, the average size of BMS-347070 crystals in different Pluronic® polymers and PEG 8000 is not proportionally related with the length of the PPO segments of the polymers (Fig. 2 and Table I), as predicated by the previous “cage hypothesis.” The length of the PPO segment in the three Pluronic polymers ranges from 13 to 29 nm. Crystallization of the PEO portions in Pluronic® polymers cannot produce geometrically large enough PPO “cages” to accommodate drug crystals with a size range of ∼100 nm.
One could argue that PPO “cages” of ∼100 nm is possible if Pluronic® polymers formed micelles and drug crystallized within them. In fact, it is widely known that as surfactants, Pluronic® polymers do self-assemble into micelles in aqueous medium, with their hydrophobic PPO portion as the inner core and the hydrophilic PEO portions as the shell (12–14). It was also reported that Pluronic® was used as a template in aqueous phase to control the synthesis of nanoparticles (15). However, the self-assembly of Pluronic®polymers occurs in aqueous solution, not in organic solvents such as methylene chloride. The drug-polymer solid dispersions were prepared by spray drying a methylene chloride solution, where no micelle exists (confirmed by laser scattering, data not shown). The subsequent crystallization of drug-Pluronic® dispersion happens in the solid state, there is no theoretical basis or prior observations to support the possibility of Pluronic® self-assembling into micelles in the solid state.
On the other hand, the formation of nano-sized BMS-347070 crystals in PEG 8000, where no PPO segment is present, proved again that the tri-block PEO-PPO-PEO polymer topology in Pluronic® is not a prerequisite for nano-sized drug crystals formation in these solid dispersions.
From a theoretical aspect, “PPO cages” containing nano-sized drug crystallites might be possible if the drug has higher affinity with the PPO segments compared with the PEO segments; and if the drug crystallites were small enough to be encapsulated inside the “PPO cages” (i.e., less than 20–30 nm). The above conditions were not met in the current system: the solubility of BMS-347070 in PEG 400 and poly(propylene glycol) (PPG 3500) is approximately 1%w/w and ≪0.3%w/w, respectively, suggesting higher affinity between the drug and the PEO segments; also, the drug crystallite size exceeded the length of PPO segment.
Polymer Enhanced Heterogeneous Nucleation of Drug
As discussed above, solubility of BMS-347070 in PEG 400 and PPG 3500 are negligible, suggesting very low projected solubility in either the PEO or PPO segments. On the other hand, specific chemical interaction (such as hydrogen bonding) between the drug and polymers is unlikely, since there is no hydrogen bonding donor or acceptor in PEO or PPO segments. We, therefore, conclude the drug solubility in polymers, or the specific chemical interaction between drug and polymers did not meaningfully contribute to the drug’s crystallization behavior.
One possible mechanism that might explain the nano-sized drug crystal formation in polymers is that the polymers may have enhanced heterogeneous nucleation of the drug. The semi-crystalline Pluronic® and PEG polymers could serve as external templates or seeds, to induce a fast heterogeneous nucleation of the drug, thereby producing small size crystals. This led us to study the nucleation of BMS-347070 in the presence of PEG 8000 and Pluronic® F127 as external seeds (Fig. 3). However, no drug crystals formed in contact with either polymers, indicating that these polymers do not serve as “seeds” to enhance the nucleation and crystallization of BMS-347070.
Relationship Between Crystallization Kinetics and Crystal Size
According to the classical nucleation theory (CNT), crystallization begins with nucleation which is followed by crystal growth (16–19). In the nucleation process, a cluster of molecules must reach a critical size by overcoming a certain kinetic barrier induced by surface tension. Lower nucleation temperature, or larger extent of supercooling (ΔT = T m−T, where T m is the melting point and T is the nucleation temperature), leads to a smaller kinetic barrier, and therefore, a higher nucleation rate. However, when the nucleation temperature drops below the T g, the diffusivity of the molecules decreases sharply (an order of ∼106) that the nucleation is inhibited. As a result, the nucleation rate usually peaks around T g. Once nuclei are formed, the subsequent crystal growth results in an increase in size of these nuclei, which is controlled by the combined effect of thermodynamic driving force and molecular mobility. Compared to nucleation rate, crystal growth rate usually peaks at a higher temperature between T g and T m, while it becomes significantly low at T g.
The temperature dependence of nucleation rate and crystal growth rate is illustrated in Fig. 5 (16–19). When an amorphous liquid crystallizes, nucleation and crystal growth are two competing processes that have different temperature dependence. When the temperature is high, the nucleation rate is low and the crystal growth rate is high. Fewer crystals form and these crystals grow rapidly into larger sizes. As the temperature drops, the nucleation rate increases while crystal growth rate decreases, more crystal nuclei form, but they grow slowly into smaller crystal size. Based on CNT, when no heterogeneous nucleation or secondary nucleation occurs, the formation of nano-sized crystals should come from significantly impeded crystal growth with reasonably high nucleation rate. Such crystallization conditions can be achieved near the T g of the system. We hypothesize that this was exactly what happened in the BMS-347070/Pluronic® solid dispersions (Fig. 4, Table II): after Step 1 when the first portion of the polymer crystallized, the T g1 values of the remaining BMS-347070/Pluronic® amorphous phase were close to room temperature, and crystallization at room temperature enable the synergetic effect of fast nucleation and slow crystal growth, thus, nano-sized drug crystallites formed. The T g1 value of BMS-347070/PEG 8000 is 60°C, much higher than the room temperature where the solid dispersion crystallized, thus, both nucleation rate and crystal growth rate were low, which leads to small crystal size, as well as lower degree of crystallinity (Fig. 2b).

Figure 2b qualitatively compared the degree of BMS-347070 crystallinity in different solid dispersions weeks after preparation. The lowest crystallinity of BMS-347070 in PEG 8000 is attributed to the much higher T g1 value of the system, as discussed earlier. In Pluronic® F68, F108 and F127 solid dispersions, the crystallinity of BMS-347070 decreases, and the drug crystal size decreases with the same order as well. The three Pluronic® polymers have similar T g, similar miscibility with BMS-347070, and their solid dispersions with BMS-347070 have similar T g1 values (Table II). The different degree of crystallinity is unlikely to be T g related. One possible explanation would be different drug diffusivity in these Pluronic® polymers, which needs to be investigated further in future studies.
Some early unpublished data indicated that aged samples of 50/50 BMS-347070/ Pluronic®F127 solid dispersion (∼5 years under ambient conditions) had overlapping PXRD patterns with what was obtained days after the sample preparation, suggesting that the crystallization of these Pluronic® based solid dispersions happens rather fast, presumably due to their low T g. Once the majority of drug crystallized (∼70% in the case of BMS-347070/ Pluronic® F127), the remaining amorphous drug appeared to be rather stable in the system, presumably due to the dilution and stabilization of the drug by the polymers, as well as a higher T g of the remaining drug-polymer phase after more polymer crystallized. A more quantitative, and long-term chronological (months to years) study would be interesting to investigate the kinetics of these drug-polymer systems, especially the drug-PEG 8000 system due to its higher amorphous drug content. However, significant changes in crystal size and degree of crystallinity are not expected in the drug—Pluronic® systems, since most of the drug has already crystallized.
Validation of Hypothesis with Other Drug-polymer Solid Dispersions
Studies of 50/50 BMS-A/Pluronic® F127 and BMS-B/ Pluronic® F127 solid dispersions confirmed the theory that drug crystallite size is kinetically dependent. BMS-A has a high T g of 144°C and a poor miscibility with Pluronic® F127 (Table III), leading to a higher T g1 value of 72°C. According to the CNT, both nucleation rate and crystal growth rate are very low at room temperature, which explains the fact that this system remained amorphous after 3 weeks under ambient conditions. On the other hand, the low T g and better miscibility of BMS-B with F127 led to a low T g1 of 3°C in the BMS-B/ Pluronic® F127 solid dispersion. According to the CNT, the nucleation rate should be relatively slower while crystal growth rate would be higher, therefore, larger crystallite size were expected. This is consistent with the measured average crystallite size of 143 nm in this system.
CONCLUSION
The size of drug crystallites in the drug/Pluronic® solid dispersions is determined by a combined effect of nucleation rate and crystal growth rate. Once mixed with drug molecules, the polymers act as “T g manipulators” to adjust the “effective” T g of the drug-polymer solid dispersions at crystallization, i.e., T g1 as defined in this study. The difference between the T g1 value of a solid dispersion and the temperature where the crystallization of solid dispersion occurs, is a key factor to determine the drug crystallite size in the system.
It should be noted that the crystallization of drugs from a drug-polymer binary system is a complex process, and the crystallization kinetics is dependent on many factors, especially crystallization tendency of the drug compound and drug-polymer interaction. A T g1 value around room temperature only suggests that the nucleation rate of the solid dispersion should be relatively faster at room temperature than the other temperatures; it does not lead to a certainty of drug crystallization within a certain time, nor to the extent of drug crystallization. Actually, in order to produce a crystalline drug-polymer solid dispersion with a stable crystallite size and a constant degree of crystallinity, a fast and complete drug crystallization is preferred. Once the majority of amorphous drug is depleted from the system, less uncertainty remains regarding the long term physical stability. In this aspect, drugs with higher crystallization tendency appear to be better candidates for this system.
The current research reported here has contributed additional understanding of the mechanism of nano-crystallization of drug-polymer binary systems. Further investigations continue to identify the characteristics of the drug, polymer, and process conditions that facilitate the formation of nano-crystalline solid dispersions for potential application in formulating poorly water-soluble drug compounds.
References
O. Chambin and V. Jannin. Interest of multifunctional lipid excipients: case of Gelucire 44/14. Drug Dev. Ind. Pharm. 31:527–534 (2005).
J. Hu, K. P. Johnston, and R. O. Williams, III. Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug Dev. Ind. Pharm. 30:233–245 (2004).
C. Leuner and J. Dressman. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 50:47–60 (2000).
E. Merisko-Liversidge, G. G. Liversidge, and E. R. Cooper. Nanosizing: a formulation approach for poorly-water-soluble compounds. Eur. J. Pharm. Sci. 18:113–120 (2003).
A. T. Serajuddin. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 88:1058–1066 (1999).
S. Sethia and E. Squillante. Solid dispersions: revival with greater possibilities and applications in oral drug delivery. Crit Rev. Ther. Drug Carrier Syst. 20:215–247 (2003).
N. Subramanian and S. K. Ghosal. Enhancement of gastrointestinal absorption of poorly water soluble drugs via lipid based systems. Indian J. Exp. Biol. 42:1056–1065 (2004).
A. Martin. Physical Pharmacy, Lippincott Williams & Wilkins, Baltimore, MD, 1993.
S. X. Yin, M. Franchini, J. Chen, A. Hsieh, S. Jen, T. Lee, M. Hussain, and R. Smith. Bioavailability enhancement of a COX-2 inhibitor, BMS-347070, from a nanocrystalline dispersion prepared by spray-drying. J. Pharm. Sci. 94:1598–1607 (2005).
L. E. Alexander and Klug HP. X-ray Diffraction Procedures-for Polycrystalline and Amorphousmaterials, Wiley, New York, 1974.
T. G. Fox. Influence of diluent and of copolymer composition on the glass transition temperature of a polymer system. Bull. Am. Phys. Soc. 1:123 (1956).
A. Kabanov, J. Zhu, and V. Alakhov. Pluronic block copolymers for gene delivery. Adv. Genet. 53:231–261 (2005).
A. V. Kabanov and V. Y. Alakhov. Pluronic block copolymers in drug delivery: from micellar nanocontainers to biological response modifiers. Crit Rev. Ther. Drug Carrier Syst. 19:1–72 (2002).
G. S. Kwon. Polymeric micelles for delivery of poorly water-soluble compounds. Crit Rev. Ther. Drug Carrier Syst. 20:357–403 (2003).
C.S. Yang, D.D. Awschalom, and G.D. Stucky. Growth of CdS nanorods in nonionic amphiphilic triblock copolymer systems. Chem Mater. 14:1277–1284 (2002).
J. W. Mullin. Crystallization, Butterworth-Heinemann, Oxford, 2001.
D. W. Oxtoby. Nucleation of first-order phase transitions. Acc. Chem. Res. 31:91–97 (1998).
M. Volmer. Kinetik der Phasenbildung, Leipsig, Steinkopff, 1939.
M. C. Weinberg. A few topics concerning nucleation and crystallization in glasses. J. Non-Crystalline Solids 255:1–14 (1999).
Acknowledgement
The authors thank Dr. Shawn Yin and Ms. Anisha Patel for assistance with PXRD and helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors Feng Qian and Jing Tao contributed equally to this study
Rights and permissions
About this article
Cite this article
Qian, F., Tao, J., Desikan, S. et al. Mechanistic Investigation of Pluronic® Based Nano-crystalline Drug-polymer Solid Dispersions. Pharm Res 24, 1551–1560 (2007). https://doi.org/10.1007/s11095-007-9275-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-007-9275-7