
Abstract
This chapter provides an overview of the biosynthesis and structure of lignin. Moreover, examples of the commercial use of lignin and its promising future implementation are briefly described. Many applications are still hampered by the properties of technical lignins. Thus, the major challenge is the conversion of lignins into suitable building blocks or aromatics in order to open up new avenues for the usage of this renewable raw material. This chapter focuses on details about natural lignin degradation by fungi and bacteria, which harbor potential tools for lignin degradation and modification, which might help to develop eco-efficient processes for lignin utilization.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Biosynthesis and Chemical Structure of Lignin
Detailed knowledge about the structure and biosynthesis of lignin is central for developing new strategies to degrade the complex lignin molecule and precisely modify its single components. Additionally, this information can help to develop lignin with specific characteristics by engineering plants.
The development of tracheids, and thereby water-connecting tissues, is considered to be a keystone process in the evolution of terrestrial plants. This process is associated with the development of the biopolymer lignin. Because of its hydrophobic nature, lignin makes the tracheids impermeable to water, which is essential for water transport. Furthermore, because of its complex structure, it stabilizes the aerial organs and enables erect-growth. Lignin was recently also found in red algae, assuming its original role in ancestors of higher plants and algae was the protection against microbial degradation or UV radiation [1,2,3,4].
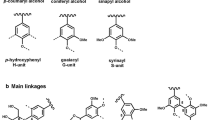
Lignin is mainly composed of the monolignols sinapyl-alcohol, cumaryl-alcohol, and coniferyl-alcohol (see Fig. 1). Generally, lignins from gymnosperms contain mainly coniferyl units (>95%) with a minor amount of cumaryl units (<5%), whereas in angiosperms coniferyl, sinapyl, and marginally cumaryl units occur at 25–50%, 46–75%, and <8%, respectively. Lignins of grasses are also composed of all three components, but differ in higher portions of cumaryl units (up to 33%) [5, 7].

The phenylalanine-derived lignin precursors are synthesized in the cytoplasm and exported to the apoplast. The transport mechanism across the plasma membrane is still unknown. However, reaching the apoplast, the monolignols undergo single-electron oxidation and form reactive radical species. The radicalization process is most likely mediated by laccases and/or peroxidases (see Fig. 1) [8]. In this regard, just recently a triple knock-out of laccases in Arabidopsis thaliana showed an almost completely abolished lignin deposition in roots in vivo, hinting on the participation of laccases in lignin biosynthesis [9].
The lignin polymer is built by the crosslinking of the radical monolignols. Herein the combinatorial random coupling of the radical monomers is the widely accepted mechanism for the formation of lignin. In contrast, a protein-directed synthesis including dirigent proteins has also been hypothesized [10]. The model of directed polymerization of lignin is supported by some experimental findings. For instance, the specificity of a peroxidase for only one monolignol was shown, suggesting a possible regulation of the lignin composition by plants. Accordingly, experiments with knock-out and down-regulation of certain peroxidases resulted in an alteration of sinapyl or coniferyl unit content in lignin [11]. In contrast, a deletion of sinapyl or coniferyl alcohol-delivering enzymes resulted in lignins with high amounts of coniferyl or sinapyl units, contradicting a strict control of monolignol assembly [6]. Additionally, formation of a defined primary lignin structure mediated by protein templates was suggested [12]. This finding is under discussion as lignin shows no optical activity and genetic data are missing [6, 13]. A precise delivery of laccases to the secondary cell wall within the apoplast was shown recently [8]. This indicates a protein-directed synthesis of lignin, especially for the localization of polymerization. Despite that, the dirigent role of proteins within the selectivity for monolignols and building of determined bonds remains in doubt.
According to the random coupling hypothesis, the monomers are coupled either one by one or different oligomers are linked together at the same time, termed “endwise polymerization” and “bulk polymerization.” Herein the availability of the monolignols may influence the prevalence of the two mechanisms. However, the nature of the monolignol probably exerts the most influence. Besides being reactive at the phenolic oxygen (hydroxyl group at the C4-position), the monolignol-derived radicals are reactive at the C1, 2, 3, 5, and at the β–C (see Fig. 2), although steric hindrance may limit or prevent the reactivity in some of these sites [15]. Conferring to the highest computed electron spin density, the p-coumaryl radical in the C1-position has supposedly the highest reactivity, followed by the phenolic oxygen, C3, β–C, and C5. In coniferyl and sinapyl radicals, phenolic oxygen has the highest spin density succeeded by C1, C3, β–C, and C5. Nevertheless, C1-coupling occurs at low frequency in natural lignin and coupling at the methoxylated C-positions happens with an irrelevant frequency, maybe because of steric hindrance. Generally, the phenolic hydroxyl group and the β–C appear as the most likely coupling sites [14].

Cumaryl, coniferyl, and sinapyl alcohols with computed electron spin densities (modified with permission from [14], Copyright 2012, American Chemical Society)
Coupling at the β-position is preferred for coniferyl and even more for sinapyl alcohol monomers. In dimerization reaction experiments, the coniferyl alcohol led to dimers with β–β, β–O–4 and β–5 linkages. The portion of the β–O–4 bond was less than one-third with coniferyl and only about 9% when sinapyl alcohol was used as substrate [16]. In contrast to dimerization, the oligomers are unable to couple only at the β-position during lignification. The cross-coupling of the monolignols coniferyl or sinapyl alcohol with a guaiacyl unit of the lignin polymer (where the β–C site is already coupled) gives two possible bonds: β–O–4– and β–5. Moreover, in coupling reactions of monolignols with a sinapyl unit (where the β–C site is already coupled) only a β–O–4 unit occurs (see Fig. 3b). This shows why β-ethers are formed more frequently in lignification than in monolignol dimerization experiments. Additionally, these findings explain why lignins with higher amounts of sinapyl monomers contain more β-ether bonds. In an approach of peroxidase-mediated in vitro polymerization of coniferyl monomers, addition of sinapyl monomer to the reaction led to a switch from a bulk to an endwise mechanism [11]. The coupling of two lignin oligomers is uncommon in lignins with high sinapyl unit content, but is often found in lignins with high coniferyl unit ratios, where, for example, 5–5 coupling occurs more often [5].

Lignin is a very complex molecule and the analysis of its structure and linkages is a considerable challenge, even with modern NMR methods. The most abundant linkage in lignin is the β–O–4 ether bond. However, the composition of the numerous bond types varies significantly between different plants [19]. The structure of the major bonds in lignin is illustrated in Fig. 3a. In Table 1 the occurrence of these bonds in beech and spruce lignin is shown, which represent hard and soft wood. The proportions of β–1 and β-ether linkages in beech lignin are considerably higher than those in spruce lignin, whereas the content of 5–5 and 4–O–5 linkages is lower. This means that spruce lignin is more condensed, resulting in lower solubility and degradability [17].
A better understanding of the lignin biosynthesis mechanism is important for the development of lignocellulose biomass-utilizing technologies [15]. Detailed knowledge about lignin synthesis pathways is essential for the engineering of plants with a modified lignin polymer structure. A less recalcitrant lignin makes lignocellulose biomass more accessible by treatment methods [21]. Moreover, the lignin structure may be designed in a way that makes lignin itself more suitable for fuel and chemical applications by enhancing its homogeneity and optimizing its chemical properties [22].
2 Technical Lignins
Here, a short overview of existing lignocellulose pretreatment processes and the resulting lignin types is given.
Kraft lignin (KL) and lignosulfonates are the most common commercially available technical lignins [23]. Both these lignins contain sulfur residues as a result of the underlying lignocellulose pretreatment processes [24]. Within the kraft process, biomass is cooked in the presence of sodium hydroxide and sodium sulfide and the lignin is degraded and solubilized in the alkali solution. In contrast, lignosulfonates are gained by cooking with sulfite, at which sulfonating, degrading, and solubilizing of the lignin occurs [25]. Additionally, the soda process and the organosolv process, both working without sulfur, are also described as main industrial lignocellulose pretreatment processes, although the latter is currently not operated on a commercial scale [26]. The soda process was the first lignocellulose pulping method and is similar to the kraft process, but uses solely sodium hydroxide [25, 27]. Within the organosolv process biomass is cooked in a mixture of organic solvents and water and the separation of lignin is performed via solubilization. Common solvents for this method are ethanol, formic acid, and acetic acid.
By means of soda and organosolv pulping processes sulfur-free lignins are obtained [24], which are in many respects superior to sulfur-containing lignins. Sulfur-free lignin has an advantage in environmental applications [23], can be heat-processed without odor release, and is preferred as raw material for several products [28]. For instance, in carbon fiber production, sulfur leads to inferior melt spinning characteristics [28, 29]. Sulfur-free lignin is, moreover, rather suitable as raw material for activated carbon and other aromatic added-value chemicals and does not cause air pollution problems [30].
Besides organosolv and soda pulping, sulfur-free lignin can be obtained by other processes such as steam explosion, ball milling, pyrolysis, and processing with ionic liquids, which have however not yet reached the marketplace [31]. Organosolv processes deliver lignin with the highest purity in the above-named processes [32]. Because of its relatively high homogeneity, purity, and reactivity, organosolv lignin may be the most promising technical lignin for further processing and direct applications [33]. Commercializing of sulfur-free lignins opens new potentials for utilizing lignin in value-added products [34].
Nevertheless, in spite of their lower homogeneity and purity, sulfur-containing lignins can be preferable in certain applications because of sulfur or their higher molecular weight. Thus, for example the addition of lignosulfonates in gypsum paste resulted in a better dispersibility, especially at higher molecular weights and sulfur content [35].
Summing up, the technical lignins vary strongly in their physical and chemical features. Therefore, a specific type of lignin has to be chosen depending on the particular application [36].
3 Lignin Applications
In this section an overview of current industrial lignin usage, details of the challenges in processing technical lignins, and an outlook on potential future applications are given. Thereby, biotechnological and chemical methods for processing lignin components are described.
The utilization of lignin in an economically viable manner is one of the most important tasks of lignocellulose biorefineries. Currently, only about 2% of the 50 million tons of lignin produced by the pulp and paper industry every year are used for industrial purposes, and the residual lignin is burned [18]. In most biorefinery concepts, which focused mainly on hydrolysis and sugar fermentation to ethanol or other fuels, lignin was also underutilized. Remarkably, the refineries produce 60% more lignin than necessary for their own power supply, resulting in a high amount of unused lignin [22]. However, a separation process for KL from pulp and paper mills, called lignoboost, was recently launched on an industrial scale in Finland and the USA. Lignoboost is able to separate lignin efficiently with higher quality, meaning low ash contents. Traditional pulp mills can be transformed into combined biorefineries [37]. Consequently, new high value applications for lignin are needed [22].
A few industrial applications for lignin do already exist, such as the production of synthetic vanillin and dimethyl sulfoxide. However, lignin is mostly used for its chemical properties as electrolytic material, or advantage is taken of its polymeric structure. Thus, lignin is utilized as sequestrant, binder, dispersant, and emulsifier, or, to a lesser extent, used as a filler and in adhesives. For these types of applications, lignin is not modified or modified only slightly, and therefore used in its naturally-occurring structure [38]. A successful example for value-added commercializing of lignin is its processing by injection molding, extrusion, and compression molding, using polymeric lignin, wood meal, and additives such as flax fibers. The thermoplastic material named “arboform” can be used as shells for mobile phones or computers and in components of cars such as steering wheels [39, 40].
Moreover, the use of lignin as carbon fibers, polymeric modifiers, resins, and the expansion of lignin adhesives bears a high potential for a valorization of this raw material [41].
Numerous lignin-converting processes are still on the road toward commercialization. Perez-Cantu et al. were able to prepare an aerogel with lignin as the only phenolic component. Lignin was crosslinked with oligo (ethylene or propylene glycol)-diglycidyl ethers, which results in gels with promising properties for thermal insulation [42]. Engelmann and Ganster also used glycerol-diglycidyl ether for crosslinking low molecular weight fraction lignin. They produced solvent-free resins with lignin contents up to 50%. The lignin resins exhibited a better thermal stability than conventional resins made with pyrogallol [31].
Because of its properties, lignin may not be suitable for applications requiring thermal stability and melting processes, and therefore many studies were carried out on the incorporation of lignin into polymer blends with other synthetic or other bio-based polymers. For example, a combination of lignin and fish protein or wheat gluten was described [43]. Lignin could successfully be introduced in styrene-butadiene rubber as a lignin-layered double hydroxide (L-LDH). Mechanical analyses indicated that L-LDH/styrene-butadiene rubber was superior to LDH/styrene-butadiene rubber concerning elongation at break, modulus, tensile strength and hardness [44]. Recently, Spiridon et al. produced a polylactic acid material with an organosolv lignin content of 7%, which showed improved thermal stability and mechanical properties compared to neat polylactic acid [45]. Chung et al. were able to produce a lignin-g-poly(lactic acid) copolymer, which can additionally be blended with polylactic acid, leading to a material with UV-blocking properties and improved mechanical features. The polymer length could be influenced by an acetylation pretreatment of the lignin [46]. Although much research on transformation of lignin into chemicals, materials, and fuels was carried out, realization into a commercial process is still rare [22].
The most striking difficulties, which restrict the conversion of lignin into high-value products, are the non-uniform structure, chemical reactivity, and impurities of technical lignins. The lignin polymers vary in their size, polymer composition, and degree of crosslinking, as well as in the abundance of functional groups, which results in non-uniform structures. This issue could be overcome by controlled depolymerization of the lignin, which can be performed chemically or by enzymatic pathways. Additionally, technical lignins often have different reactive groups, causing several diverse reactions. When technical lignins are used for polymer applications such as for producing resins, only one of these reactions might be desired and the other side reactions might hamper the polymerization process. Moreover, lignins are limited in chemical reactivity because of their small number of ortho and para reactive sites. Degradation of lignin might help to gain access to the reactive groups but this does not improve the general reactivity of lignin. The introduction of reactive sites into the lignin molecule might be another possibility to increase the reactivity of this inert molecule [25].
One feasible method for increasing the reactive sites of lignin, for instance, is the grafting of functional molecules onto lignin. An example for functionalization of lignin via hydroxyl groups is the esterification with oleic acid. Thereafter, it is possible to epoxidize the double bounds of the fatty acids followed by a ring-opening reaction to produce polyols. These building blocks, together with isocyanate prepolymers, were used to produce polyurethanes, showing advanced properties, and may be utilized as durable materials in the building and automotive industry [47].
Lignin can be functionalized chemically, but eco-efficient biotechnological approaches are also feasible. Some potential examples for a functionalization of lignin by ligninolytic enzymes are given here. Laccases are well-known to generate radicals and can be used to link phenolic compounds, such as vanillic acid diisocyanate or acrylamide, to the lignin polymer. Horseradish peroxidase can also be used in this way and lead to copolymers when incubated with straw pulp lignin and cresol. This process might replace the use of phenolic resins [48,49,50]. Other examples of enzymatic approaches with industrial lignins are the manufacturing of paints or polymer–template complexes and the optimization of chelating properties of lignin. For producing paints, including protective coatings, lignin is mixed with a dye or a pigment. There it reacts with a peroxidase or laccase and the process is stopped when the desired viscosity is reached. Polymer–template complexes are obtained by polymerization of a template (e.g., lignosulfonate) and a monomer through an enzyme (e.g., peroxidases). These complexes can be used for various applications as lightweight energy storage devices (e.g., rechargeable batteries). A polyphenol oxidase was used to improve the chelating capacity of acetosolv lignin. An increase of hydroxyl and carbonyl groups and an improvement of 110% in the chelating capacity were detected [49]. An interesting approach is the coating of materials such as starch-based films with laccase and lignin, which can be used as oxygen-scavenging active packaging for bread, cheese, meat, and various fruits. Alkali lignin, hydrolytic lignin, and organosolv lignin and lignosulfonates were tested. Organosolv lignin and lignosulfonates achieved the best results for oxygen-scavenging [51].
Additionally, lignin represents a promising renewable source of aromatic chemicals [52]. The most current commercial approaches utilize the lignin macromolecule, whereas the main potential can be seen in the depolymerization of lignin into aromatics such as vanillin, phenol, toluene, and benzene. The degradation processes are still in the early stages of development. Approaching research aims at increasing yield and selectivity [53]. Besides the degradation processes, the methods for separating mixtures of lignin derived chemicals such as vanillin and syringaldehyde are challenging and still under investigation [54]. One successful approach of utilizing these mixtures directly was made by Fache et al. They functionalized a mix of vanillin, acetovanillon, vanillic acid, p-hydroxybenzaldehyde, syringaldehyde, and acetosyringone by oxidation and subsequent glycidylation. The mixtures were polymerized and the epoxy resins obtained displayed remarkably good thermomechanical properties [55].
Together with chemical and physical methods (for further information see reviews [56,57,58]), there is an interest in eco-efficient biological methods for lignin degradation. For instance, biological processes could be used to generate aromatic chemicals from lignin. However, a deeper understanding of the natural degradation of lignin is needed to develop lignocellulosic biorefineries [52].
4 Lignin Degradation in Nature
In this section, details of fungal lignin degradation are described. First, the fungal degradation mechanisms are explained, including white-rot, brown-rot, and soft-rot decay. The ligninolytic enzymes participating in these processes, such as laccases, peroxidases, peroxygenases, as well as involved accessory enzymes, are also characterized. Second, the lignin-degrading strategies of bacteria are addressed, which were so far underestimated in their ligninolytic capacity. Bacteria degrade lignin to a far lesser extent compared to fungi. However, as their bioengineering potential is much higher, new methods to find lignolytic bacteria are of interest. Herein, some known lignolytic bacterial strains are described, although the enzymatic background is so far less clear. Potentially, DyP-type peroxidases or laccases could be engaged in the bacterial degradation of lignin.
4.1 Fungal Lignin Degradation
Lignocellulosic biomass is mainly degraded by particular fungi belonging to the basidiomycetes. These specialists can be divided into two main classes termed white-rot and brown-rot fungi [59]. Brown-rot fungi consume primarily carbohydrates, resulting in a brown dry rot of brittle consistency. In contrast, white-rot fungi degrade both lignin and carbohydrates and leave soft spongy debris of light appearance [60]. The wood-rotting fungi often co-exist in a similar ecological niche, which leads to several interactions. Synergistic interactions such as cooperative degradation of substrates are found [61, 62]. Synergetic effects in the production of lignin modifying enzymes were observed when different white-rot fungi species were combined [63, 64]. However, these effects seem to be dependent on the used species, their interaction, and nutritional conditions and environmental factors [64]. The hindrance of further growth of other organisms by occupying a territory and antagonistic effects as nutrition competition can occur. Adverse interactions of fungi may lead to deadlock or replacement [65]. When wood composition and physical properties change during the decaying process, better adapted species replace the prior species [66]. Thus specific patterns of colonialization are found during the decay, partially depending on the wood type [66]. Soft rot fungi often appear as pioneers followed by white-rot and brown-rot fungi [67].
Despite their different decaying mechanisms, further data about nuclear and mitochondrial DNA prove a phylogenetical relation between white-rot and brown-rot fungi [68]. Consequently, the separation of white-rot and brown-rot into two classes is disputed, for the two species Botryobasidium botryosum (found in a genetic tree between Auriculariales and Dacrymycetes) and Jaapia argillacea (probably a sister group of the Gloeophyllales) as these combine features of both categories (see Fig. 4). Analysis of the genomic sequences showed that both lack class II peroxidases and possess reducing polyketide synthase genes, both typical of brown-rot fungi. However, they are closely related to the white-rot fungus Phanerochaete chrysosporium and both strains were able to degrade all polymeric components of wood. Moreover, genes coding for cellobiohydrolases were present, which is characteristic of white-rot fungi [70]. By an investigation of 31 fungal genomes it was suggested that the ancestor of all Agaricomycetes was a white-rot fungus owning class II peroxidases, DyP-type peroxidases, and H2O2-supplying enzymes. Thus, it was assumed that on the one hand, an expansion of class II peroxidase genes leads to the white-rot orders Auriculariales, Hymenochaetales, Corticiales, Russulales, and Polyporales. On the other hand, a parallel decline of class II peroxidases was suggested, finally resulting in the brown-rot fungi Dacryopinax sp., Gloeophyllum trabeum, the Boletales, and the brown-rot species inside the Polyporales (Fig. 4).

Overview of Dicarya fungi and illustration of the relationship between white-rot and brown-rot fungi (modified after [69])
By molecular clock analyses the origin of fungal lignin degradation could be dated to the late Carboniferous period. Remarkably, coal formation, which is mainly caused by lignin burial, has strongly decreased since this period. Accordingly, a correlation of both these events was hypothesized [69].
The woody plant cell walls are structured in different layers. The cells themselves are linked by the middle lamella, which possesses the highest concentration of lignin and pectins and builds the outer layer of the cell wall [71]. Then, from the outside to the inside an S1 layer, a thick S2 layer, a thin inner S3 layer, and a bumpy layer consisting of aromatic precursor molecules are attached to the middle lamella. Therein the lignin content decreases in the same order (see Fig. 5) [73].

Modes of fungal wood decay (modified after [72] with permission from Elsevier, Copyright 2007 The British Mycological Society, published by Elsevier Ltd. All rights reserved)
Rotting fungi colonialize deadwood through hyphal growth. The accession and spreading is enabled by the organization of the tracheids and vessels in the axial direction and in the radial orientation of the xylem ray parenchyma. The joining cells are invaded either by pit apertures or directly through penetration of the cell wall [72].
As already described, there are three modes of lignocellulose degradation: soft rot, brown-rot, and white-rot. White-rot fungi can be further divided on the basis of two types of decay. Some white-rot species consume lignin and structural carbohydrates simultaneously, whereas others selectively degrade lignin and hemicellulose first [74]. These four types of fungal wood decay are illustrated schematically in Fig. 5. The species Fomitopsis pinicol (Polyporales) is an example of brown-rot decay. It starts degradation by secreting low molecular weight substances, which diffuse into the S3 layer of the cell wall. At a later stage, enzymes are involved in the excessive breakdown of hemicellulose and cellulose in the whole secondary wall, whereon the cells contract, resulting in numerous cracks in the cell walls. However, the S3 layer stays intact throughout the whole process and a skeleton of modified lignin is preserved. At an early stage of soft-rot decay by Kretzschmaria deusta (Ascomycota) the hyphae penetrate the S2 layer. There they branch and grow parallel to the cellulose microfibrils. At the end of the decay process the entire secondary cell wall is perforated by holes with conically formed ends and therefore almost completely broken down. Only the guaiacyl-rich middle lamella remains intact. Fomes fomentarius (Polyporales) is a white-rot fungus that causes simultaneous rot. Here, degradation of the cell wall close to the hyphae takes place first. Then hyphae enter the cell wall at right angles to the cell axis. The cell wall is degraded from the lumen toward the outside. Later, the cell wall becomes thinner and holes appear between neighboring cells. At an advanced stage the degradation process is limited by the strong lignified middle lamella. In the other mode of white-rot decay, the so-called selective delignification the middle lamella is also attacked. Herein, low molecular weight substances start the decaying process when diffusing from the hyphae into the secondary cell wall. The degradation of hemicellulose and lignin occurs within the secondary cell wall and even in the middle lamella. In the later stages, the favored degradation of pectin and lignin leads to separation of cells from each other. The compound cellulose is not degraded. This kind of decay is, for example, found in Heterobasidion annosum [72], belonging to the Russulales.
White-rot fungi are of special interest for the biotechnological industry because of their ligninolytic enzymes, which could be used in several industrial processes, such as pulp bleaching and decolorization of dyes in waste water [75]. It was shown that the production of ligninolytic enzymes takes place within the secondary metabolism and depends mainly on the limitation of carbon or nitrogen. However, the expression patterns can differ according to the microorganism and the type of enzyme [76]. Besides that, the gene regulation of these enzymes depends on several factors, such as the presence of xenobiotics, the temperature, day length, or metal ion concentration. A better understanding of these molecular mechanisms is needed to develop an efficient production process for ligninolytic enzymes [75].
To degrade lignin, white-rot fungi secrete class II peroxidases, dye-decolorizing peroxidases (DyP-type peroxidases), laccases, and several accessory enzymes such as aryl alcohol oxidases and glyoxal oxidases [77, 78]. In addition, cellobiohydrolases and lytic polysaccharide monooxygenases for the depolymerization of crystalline cellulose and other carbohydrate active enzymes are found [77]. Lignin peroxidases, manganese peroxidase, versatile peroxidases (class II peroxidases), and laccases have been investigated and stated to be involved in lignin degradation [79]. White-rot fungi secrete one or more of these ligninolytic enzymes [80]. More recently, two additional heme peroxidase families, the dye decolorizing peroxidases (DyPs) and the aromatic peroxygenases, were discovered in the secretome of fungi. The latter catalyze oxyfunctionalization reactions, such as epoxidations or the hydroxylation of aromatic rings and alkyl chains. Furthermore, oxidations of alcohols, aldehydes, and phenols and cleavage of ether bonds were observed. DyPs catalyze the oxidation of aromatics and recalcitrant dyes [79]. The physiological role of these enzymes is not yet fully understood, although the catalytic reactions of DyPs and aromatic peroxygenases seem to be linked to the fungal conversion of lignin [81]. By transcriptome analyses the occurrence of peroxidase expression in different forest soils (oak, beech, spruce, aspen, and sugar maple) was studied. Class II peroxidases were detected in 90% of the tested samples and aromatic peroxygenases were identified in 85%. DyPs were found in 55% of the soil samples. All the peroxidase classes were found in all forest types, with the exception of DyPs which were absent in spruce. Interestingly, within the group of the class II peroxidases, numerous manganese peroxidases but no typical lignin peroxidases or versatile peroxidases were found [79]. Lignin peroxidases were the first ligninolytic enzymes to be discovered, but their essential role in lignin degradation is uncertain as they are not found in all ligninolytic fungi [82]. Additionally, it has been suggested that DyPs may substitute the rarely-found lignin peroxidases in the biodegradation process of non-polyporous white-rot fungi [83].
Non-enzymatic processes are less widespread in white-rot fungi, whereas hydroxyl radicals generated by the Fenton reaction seem to play a major role in the initial stages of polysaccharide degradation in brown-rot fungi. In the Fenton reaction, hydrogen peroxide is reduced by Fe2+, resulting in Fe3+, a hydroxyl ion, and a hydroxyl radical. After the chemical attack, an enzymatic decomposition of pectin and hemicelluloses, and further degradation of cellulose, take place. In contrast to white-rot fungi, the genome of the brown-rot type mostly does not contain genes of cellobiohydrolases, which are important factors in converting crystalline cellulose. In the course of the brown-rot degrading process, the lignin molecule itself is also affected, but is modified rather than degraded [9, 70, 77, 84].
The soft-rot fungi, which are of minor importance, are similar to brown-rot fungi in terms of the chemical processes during the decay [59]. In wet environments soft brownish debris is left by these organisms, which belong primarily to the Ascomycota. In addition, some basidiomycetes and bacteria may cause soft rot as well [60, 85]. Soft rot frequently occurs in environments with extreme conditions, such as high pH and low moisture, where white-rot and brown-rot fungi would be unable to survive [86].
In view of the remarkable differences in the expression pattern of ligninolytic enzymes between diverse fungi [87], all so far well-known enzymes of several species are described below. The diffusion of enzymes into the lignin molecule is limited. Thus a direct attack on lignin is questioned. Accordingly, it was suggested that low molecular weight compounds are required to initiate the oxidative attack on lignin [88]. Thus, besides the features of the ligninolytic enzymes, the mediation of lignin degradation by mediator molecules, as well as other assisting mechanisms, has been considered (see Fig. 6).

Lignin degradation mechanism in fungal species. Lac laccase, MnP manganese peroxidase, VP versatile peroxidase, LiP lignin peroxidase, DyP DyP-type peroxidase, APO aromatic peroxigenase, CDH cellobiose dehydrogenase, GLX glyoxal oxidase, AAO arylalcohol oxidase, AAD aryl aldehyde/aryl alcohol dehydrogenase, Mn3+-chelators: for example, oxalate; mediator molecules: PR phenolic radical, FR fatty acid radical, VA veratryl alcohol; lignin degradation products: (1) β-aryl-ether model compound, (2) p-anisaldehyde, (3) glycolaldehyde, (4) p-methoxybenzy alcohol, (5) glyoxal, (6) glyoxylic acid (modified after [60, 88,89,90,91,89])
Details about the ligninolytic enzyme laccases and class II peroxidases are presented in the following section. Facts about accessory enzymes which provide H2O2 are specified. Additionally, new enzyme classes, the DyP-type peroxidases and peroxygenases, are described, which are associated with lignin degradation.
4.2 Fungal Ligninolytic Enzymes
Herein, fungal enzymes associated with lignin degradation and their mechanisms are described. For visualizing the interaction and function of the enzymes described below, please see Fig. 6.
4.2.1 Laccases
The first laccase was discovered in 1883 in exudates of the Japanese lacquer tree Rhus vernicifera. Since that time, laccases have also been identified in numerous basidiomycetes and ascomycetes [93]. Meanwhile, laccases have been isolated from bacteria and even insects and thus occur almost everywhere. Laccases are the largest group within the so-called multi-copper oxidase enzymes [94]. An exact definition of laccases has not been given. Generally, multi-copper oxidases are considered to be laccases if at least some phenol oxidase activity can be measured and the copper ions are present in the correspondent position [95].
The physiological role of laccases is diverse. For example, laccases catalyze the biosynthesis of a spore pigment in the bacterium B. subtilis and they are involved in the production of the external cuticle of insects. The biological functions of laccases in plants and fungi apparently include the biosynthesis and degradation of lignin [95]. Moreover, fungal laccases take part in stress defense, morphogenesis, plant pathogen/host interaction, and the detoxification of phenol compounds [93, 96]. Fungal laccases are mostly extracellular monomeric proteins of about 60–70 kDa [94].
Characteristically, the laccases contain four copper ions: one of type-1 (Cu1), one of type-2 (Cu2), and two of type-3 (Cu3). The two Cu3 ions and the Cu2 ion are arranged in a triangle and form the trinuclear copper cluster (TNC) (see Fig. 7) [94]. A strong absorption at about 600 nm is caused by the Cu1 copper and results in a characteristic blue color of these enzymes [94]. Laccases catalyze the following overall reaction [96]:

Catalytic cycle of laccases (modified with permission from [97], Copyright 2010 American Chemical Society)
In the first step the substrate donates four electron equivalents (4e−) and in the native intermediate all four copper ions Cu(II) are reduced to Cu(I). The electrons enter thorough the Cu1(T1) site and are transferred to the Cu2(T2) and the two Cu3(T3) (TNC). The single steps of the Cu(II) reduction are not fully understood. The TNC is afterward fully reduced, which is required for the following reaction with dioxygen [97]. The dissociation of two water molecules during this reducing step is suggested [94]. The dioxygen is then reduced successively in two two-electron transfer steps [98]. The TNC reacts with dioxygen and forms a peroxide intermediate with the Cu2 and one of the two Cu3 ions. In this step, two electrons are transferred from Cu2 and the β-Cu3 copper on the dioxygen molecule.
An aspartic acid residue (D94), which is close to the β-Cu3 and Cu2 sites seems to support the reaction of the dioxygens with the Cu2 and the β-Cu3 by providing a negative charge. Furthermore the β-Cu3, in comparison to the α-Cu3, lies near a glutamic acid residue. Next, the bond between the dioxygen is cleaved. The reaction is supported by the glutamic acid residue providing a proton. Finally, the enzyme is in the native intermediate state (NI), which is oxidized completely and is catalytically relevant [97]. In the presence of a substrate the catalytic cycle starts again. If not enough substrate equivalents are available, a resting state can occur as well [94].
The laccases possess a broad substrate range. They can directly oxidize polyphenols, diphenols, aminophenols, polyamines, and aryl diamines. They are also able to catalyze the oxidation of inorganic ions [94]. However, most of the laccases are unable to oxidize non-phenolic compounds directly because of their high redox potentials above 1.3 V vs “normal hydrogen electrode” (NHE), whereas the laccases’ redox potentials are below 0.8 V [99]. Thus, the participation of laccases in lignin degradation of white-rot fungi was questioned, as more than 80% of the total lignin consists of non-phenolic units. Several discoveries, however, affirmed laccases as important participants of the lignin-degrading system of fungi. Laccases were shown to be able to degrade phenolic lignin substructure model compounds. Moreover, the addition of redox mediators enables laccases to oxidize non-phenolic molecules. Numerous fungal strains do not possess lignin and manganese peroxidases but are nevertheless able to degrade lignin. In addition, laccase-deficient mutant strains were inhibited in lignin degradation [100].
Redox mediators are small molecules which act as electron shuttles. Thus, bulky substrates, which cannot be oxidized directly because of steric hindrances, can be converted as well. Furthermore, the different mechanism of the mediator system allows an oxidation of high redox potential molecules [99]. Redox mediators are either artificial molecules such as 2,2′-azino-di-(3-ethylbenzthiazolin-6-sulfonic acid) (ABTS) or 1-hydroxybenzotriazol (HOBt) or can be natural mediators. The latter include phenolic molecules derived from the fungal or plant secondary metabolism or lignin degradation products. The effect of lignin-degrading products as mediator is of course difficult to prove when lignin at the same time is used as substrate [100]. However, the molecules syringaldehyde, acetosyringone, vanillin, acetovanillone, methyl vanillate, and p-coumaric acid enabled the degradation of recalcitrant dyes by laccase [99].
For the oxidation of non-phenolic substrates by laccase-mediator systems, two different mechanisms are proposed. Although ABTS reacts through an electron transfer (ET) route, the mediator HOBt follows a hydrogen atom transfer (HAT) mechanism. Here, a hydrogen atom is subtracted from the –N–OH mediator and an –N–O· radical is formed [94]. For natural phenolic mediators an analogous mechanism is suggested, which was investigated by using phenol red as model substrate for phenolic compounds. Herein, a hydrogen atom is abstracted and a phenoxyl radical (PhO·) is generated.
Comparing natural with artificial mediators, inactivation of the laccase was reduced using natural mediators, but pulp bleaching was more efficient with the HOBt mediator system [99]. However, in consideration of the natural mediators, laccases gain prominence in the fungal lignin degradation mechanism. Moreover, these mediators are of interest for industrial processes as they can be gained easily from lignocellulose material and are also environmentally friendly [99].
The yellow laccases are an interesting exception within the laccases. These laccases do not show the typical blue color, which is caused by another adsorption spectrum of the Cu1 atom. An explanation for the color shift in earlier reports was the incorporation of a lignin-derived mediator in the catalytic center, whereas more recently a variation of the coordinating sphere of Cu1 was postulated [101]. As yellow laccases have a higher redox potential and were shown to oxidize non-phenolic compounds without mediator, they have a high potential to perform several industrial applications [102].
4.2.2 Class II Peroxidases
Peroxidases can generally be described as a group of enzymes that reduce peroxide and oxidize numerous substrates [103]. Genes of heme peroxidases have been identified in nearly all kingdoms of life. They used to be divided into two main superfamilies. The so-called animal peroxidases (recently termed peroxidase-cyclooxygenases) are primarily found in animals, fungi, and bacteria. The plant peroxidases (now peroxidase-catalase superfamily) mainly occur in bacteria, fungi, and plants (see Fig. 8) [104, 105].

Superfamilies of heme peroxidases and selected subfamilies, which are associated with lignin degradation (according to [104])
The plant peroxidases superfamily is again subdivided into different classes. Class I includes peroxidases, which are located in eukaryotic organelles, catalase-peroxidases, or bacterial peroxidases. Class III contains the secreted plant heme peroxidases [104, 106]. Prominent examples for class I are the cytochrome c peroxidase and class III includes the horseradish peroxidase [107]. Class II contains the secreted fungal heme peroxidases. These peroxidases are found solely in fungi, more precisely in Agaricomycetes (formerly homobasidiomycetes) [103, 108]. Class II mainly consists of the enzyme type manganese peroxidase (MnP), lignin peroxidase (LiP), and versatile peroxidase (VP) [109].
LiP and MnP were discovered in the 1980s and, because of their high redox potential, described as true ligninases. Later the versatile peroxidases, an additional type of class II peroxidases, were discovered. The versatile peroxidases were shown to combine the catalytic features of MnP and LiP, oxidizing Mn2+ and veratryl alcohol [60]. These enzymes are classified by molecular models, including data of LiP, VP, and MnP from Phanerochaete chryosporium and Pleurotus eryngii. There is an exposed tryptophan residue described, which is typical for LiP. Furthermore, a putative Mn2+-oxidation site was found in MnP. Despite this, there are also class II peroxidases known, which lack both of these sites [110]. These so-called generic peroxidases have a low redox potential and are therefore unlikely to participate in lignin degradation [70, 111]. In a study which included 10 genomes of Polyporales, a reconstruction of the ancestral state showed that a generic peroxidase appears to have evolved an Mn2+-oxidation site, which implies that this gene could be the ancestor of all the class II lignin degrading peroxidases. Furthermore, the development of an exposed tryptophan site supposedly led to the first versatile peroxidase. It was also suggested that the loss of Mn2+-oxidation by an early versatile peroxidase is the origin of all lignin peroxidases (LiP) [112]. The three ligninolytic class II enzymes—manganese, lignin, and versatile peroxidase—are specified below.
4.2.2.1 Manganese Peroxidases
Manganese peroxidases are the most widespread lignin-modifying peroxidases and are secreted by nearly all wood-colonializing fungi [91]. Manganese peroxidases (MnP) are monomeric glycosylated enzymes with a molecular mass of about 30–60 kDa and contain one molecule of heme as iron protoporphyrin XI [88]. The pH optimum of these enzymes lies between 2.5 and 6.8 and the redox potential of MnP is about 1.0–1.2 V (vs normal hydrogen electrode) [88, 113]. MnP catalyzes the following overall reaction:
Interestingly, MnP is the only heme peroxidase, which catalyzes a one-electron Mn2+-oxidation [91]. The Mn2+ ion is oxidized at a binding site close to the heme cofactor. Three ambient acid residues mediate the binding of Mn2+ by their carboxylates, facilitating a direct transfer of electrons to one heme propionate side chain [60].
The catalytic cycle of MnP starts by the formation of an iron–peroxide complex with hydrogen peroxide binding at the native ferric enzyme. For the cleavage of the O–O bond of the hydrogen peroxide, two electrons are essential. The heme transfers these two electrons, which results in the formation of a Fe4+-oxo-porphyrin-radical complex, called compound I. After receiving two electrons, the O–O-bond is cleaved heterolytically and one molecule of water is excluded. Compound I is afterward reduced to compound II by Mn(II). Compound II is likewise reduced by a second Mn(II), resulting in the generation of another molecule of water and the native enzyme. Compound II is dependent on Mn(II) as electron donor, whereas compound I can in turn be reduced by alternative electron donors such as phenolic compounds [93, 114] (see Fig. 9).

Catalytic cycle of manganese peroxidase. Mn Manganese, Fe iron, RH organic molecules (modified after [114] with permission from Elsevier, Copyright 2002 Elsevier Science Inc. All rights reserved)
Mn3+ is a strong diffusible oxidizer, but is not stable under aqueous conditions. However, chelators such as oxalate and malonate can stabilize Mn3+ against dissociation to Mn2+ and insoluble Mn4+. The complexed Mn(III) can diffuse into the lignin molecule, where it acts as reactive redox-mediator [76, 82]. The complexing of the chelates, however, lowers the electron potential of Mn3+, resulting in a mild oxidant. MnP is primarily considered to be an enzyme that oxidizes phenolic compounds because the Mn3+-complex oxidizes monomeric and dimeric phenols as well as phenolic lignin model substances. In contrast, it cannot directly attack non-phenolic compounds of lignin. However, it was supposed that MnP is able to oxidize non-phenolic lignin structures in the presence of additional secondary mediators [76, 114]. MnP-derived Mn3+ has been shown to oxidize thiols and saturated fatty acids to form secondary mediators such as thiyl or lipid radicals [114, 115].
Furthermore, another possible oxidation mechanism of non-phenolic lignin compounds with participation of MnP was suggested. Cellobiose dehydrogenase-generated OH radicals react with non-phenolic lignin structures, thereby introducing hydroxyl groups and enabling Mn3+-chelates to perform a further oxidation step [116].
4.2.2.2 Lignin Peroxidases
Lignin peroxidases (LiP) are similar to MnP monomeric glycosylated enzymes. Their molecular mass ranges between 35 and 55 kDa and they contain one molecule of heme as iron protoporphyrin XI [85].Their pH optimum is quite low and ranges from 1 to 5. The redox potential of LiP is remarkably high at about 1.4–1.5 V [85, 110]. The overall reaction of LiP is represented by the following equation [93]:
Herein, the substrate is a non-phenolic lignin model dimer [117]. The favored substrate for LiP, though, is the non-phenolic monomer veratryl alcohol, whose conversion is used in photometric assays for determination of LiP enzyme activity [96].
The first reaction of the catalytic cycle is the two-electron oxidation of the native ferric enzyme, which results in compound I (see Fig. 10). There, the iron appears as Fe(IV) and a free radical exists on the tetrapyrrole ring. Herein, hydrogen peroxide is reduced and cleaved at the O–O bond and a molecule H2O is released. Instead of a radical in the tetrapyrrole ring, a tryptophan 171 radical state was suggested for compound I (see below) [96]. Next, two successive one-electron transfer steps of compound I from an electron donating substrate (e.g., veratryl alcohol) take place [91]. First, compound I oxidizes a donor substrate by taking one electron and releases a free-radical substrate and compound II, which still contains an Fe(IV) but no tetrapyrrole radical. Then compound II oxidizes a second donor molecule and again a free-radical substrate is formed. LiP is returned to the native ferric oxidation state, which completes the catalytic cycle [82, 91]. In the absence of an electron donor substrate, compound II can react with H2O2, which results in a catalytically inactivate state of LiP (compound III). This ferric-superoxo form can be reset to the native form by oxidation with a veratryl alcohol radical cation or by spontaneous auto oxidation [76]. The oxidation of phenolic compounds by LiP is associated with an assimilation of compound III and thus an inactivation of LiP, because phenolic compounds are unable to reduce compound III to the native state [96].

Catalytic cycle of lignin peroxidase. VA veratryl alcohol, VAD veratryl aldehyde (modified after [96] with permission of Springer, Copyright Humana Press 2008)
Although their catalytic cycle is similar to other peroxidases, LiP demonstrates unique features in oxidizing high-redox potential substrates. Structural aspects that distinguish LiP and other peroxidases were found by crystal structure analysis. One of the nitrogen atoms of the proximal histidine residue (see Fig. 12b) forms a hydrogen bond with the iron of heme peroxidases. In LiP this histidine is quite distant from the heme iron, resulting in a significantly longer hydrogen bond. Thereby the electron deficiency of the heme iron is increased, which leads to the higher redox potential of the oxo-ferryl complex [60, 76].
Another unique structural property of LiP is the exposed tryptophan residue (W171), which seems to play a central role in the oxidation of veratryl alcohol and other non-phenolic substrates [115]. The role of the tryptophan residue in veratryl alcohol oxidation was revealed by its substitution with Phe or Ser. The mutant showed no essential residual activity toward veratryl alcohol, whereas unaltered activity with two artificial dye substrates was detected. In addition, a tryptophan residue, which was introduced into a manganese peroxidase, located equivalent to the 171 position of LiP, resulted in an MnP with an oxidation activity for non-phenolic aromatics [121]. Through so-called long-range electron transfer, which is mediated by the exposed tryptophan at the protein surface, even bulky substrates such as lignin can be oxidized directly [76].
Nevertheless, the oxidation efficiency of LiP decreases significantly as the size of the lignin structure increases. For instance, the catalytic efficiency for a lignin model trimer was only around 4% compared to the activity against a monomeric lignin model compound. Synthetic lignins, which consist of about 20 subunits, were still oxidized, but the presence of veratryl alcohol was required [82]. Veratryl alcohol is secreted by ligninolytic fungi together with LiP and its role in the lignin degradation by LiP has been discussed [96]. Regarding the low efficiency of direct LiP attack on large lignin structures, veratryl alcohol (VA) might act as diffusible mediator and oxidize lignin at distant locations. However, the stability of the VA radical is still disputed. Alternatively, VA could be necessary for reducing LiP during slow cleavage processes of bulky lignin structures. Thereby, VA helps rescue LiP from inactivation by avoiding too long phases in an oxidized state. Furthermore, compound II is not as reactive as compound I and only the latter might be involved in oxidizing methoxylated lignin structures. Veratryl alcohol could be essential to reduce compound II and complete the catalytic cycle. If VA is not a diffusible mediator, the physiological role of LiP may be restricted to the oxidation of smaller lignin breakdown products released by other mechanisms, or their site of action remains solely at the surface of the plant cell walls [82].
4.2.2.3 Versatile Peroxidase
Versatile peroxidases share typical features of the LiP and MnP, showing a hybrid catalytic function. Consequently, they can directly oxidize Mn2+ as well as high redox potential aromatic compounds, including both phenolic and non-phenolic lignin models. Interestingly, different pH optima for Mn2+ (pH 5.0) and aromatic substances (pH 3.0) were observed [76]. VP not only combines the substrate spectrum of LiP and MnP, but actually oxidizes reactive black 5. This reactive dye can neither be oxidized by Mn3+ tartrate, because of its high redox potential, nor by LiP (without VA) because of a rapid inactivation of LiP [122]. Versatile peroxidases (VP) are known to possess an Mn2+-oxidation site and a tryptophan residue for the oxidation of high redox potential compounds via long-range electron transfer. Moreover, an additional oxidation site for low redox potential substrates (0.6–0.8 V) was described for VP. This site is located at the main heme-access channel [123]. The long-range electron pathway of VP is comparable to the pathway found in LiP, but the tryptophan (W164) occurs as a neutral radical. VP show a tenfold lower catalytic efficiency for veratryl alcohol compared to LiP, but act on Mn2+ as efficiently as MnP [91]. The redox potential for VP was determined to be around 1.4–1.5 V, which is equal to LiP [113].
Versatile peroxidases have unique catalytic features, including the oxidation of Mn2+, veratryl alcohol, phenolic and non-phenolic compounds, and high molecular weight compounds, such as dyes (without Mn2+-mediation).Therefore, versatile peroxidases are the most interesting candidates for biotechnological applications amongst basidiomycetes peroxidases [88]. However, the commercial application of VP, and other ligninolytic peroxidases (e.g., in a biocatalytic process for lignin depolymerization), is mainly hampered by their limited availability in large quantities [88, 124].
4.2.3 Aromatic Peroxigenases and DyP-Type Peroxidases Subfamily D
In recent years, two other heme peroxidase families, secreted by saprobic basidiomycetes, have come into focus: The aromatic peroxygenases (APO) and the dye-decolorizing peroxidases (DyP-type peroxidases, DyPs). Lately, a new phylogenetic nomenclature has been suggested. According to Zamocky et al., the DyPs cluster with chlorite dismutases, sharing a common heme binding scaffold, were classified as a peroxidase–chlorite dismutase superfamily [104]. The APO and the chloroperoxidases (CfuCPO) represent the peroxidase–peroxygenase superfamily [125] (see Fig. 8).
CfuCPO was discovered in the 1960s and oxidizes halides such as Cl- to HOCl. These molecules are, in turn, able to halogenate organic molecules. CfuCPO shows a limited peroxygenase activity with indole or p-xylene, but does not act on unactivated carbons, in contrast to APO [126]. The latter catalyze a broad range of reactions such as epoxidations, hydroxylation of aromatic rings and alkyl chains, and ether cleavages, as well as alcohol, aldehyde, and phenol oxidations under consumption of hydrogen peroxide [79]. Following the discovery of these heme thiolate peroxidases, it was obvious that they differ from all so far known peroxidases and are a combination of “classical” heme peroxidases and cytochrome P450 monooxygenases [104].
The first dye-decolorizing peroxidase (DyPs) was discovered in 1995 in the fungus Geotrichum candidum by screening for organisms able to decolorize synthetic dyes [125]. DyPs are abundant in many bacterial phyla, in fungi, and, albeit less widespread, in archaea. They can be divided into subfamilies A, B, C, and D [127]. The ancestor of the peroxidase–chlorite dismutase superfamily was most likely a member of the DyP-subfamily A. DyPs probably evolved in thermophilic facultatively anaerobic Firmicutes, which are believed to be of very old origin. One branch that arose from the DyP A group is the subfamilies DyP C and D, whereas a second branch led to the subfamilies of the shortened DyP B and probably further to subfamilies of the chlorite dismutases [104]. However, the hypothesis of a common ancestor for DyPs and chlorite dismutases is disputed by Sugano and Yoshida [128]. DyP C and D cluster quite closely, although DyP C peroxidases are found in proteo-, actino-, and cyanobacteria, and DyP D peroxidases are only present in fungi. An explanation is the development of the DyP D clade by horizontal gene transfer from cyanobacterial ancestors on dikarya fungi. This theory is supported by the lack of DyP-type peroxidases in genomes of ancestral (early dividing) fungi (e.g., Mucorcircinelloides) [104].
The reactions, which are catalyzed by DyP-type peroxidases and aromatic peroxygenases, seem to be relevant for the conversion of lignin in the nature. However, the physiological function of these enzymes is still an open question [81].
4.2.3.1 Aromatic Peroxygenases
Since the first aromatic peroxygenase was discovered in 2004 in the fungus Agrocybe aegerita, two more similar enzymes of the fungi Coprinellus radians (ink cap) and Marasmius rotula were isolated and characterized. The APO showed molecular weights of 32–46 kDa, were highly glycosylated (up to 40%), and occur extracellularly. The APO catalyzes oxygen transfer reactions at a pH range of 3–10, with a maximum rate at around pH 7 [129]. The sequence homology of APO and chloroperoxidases is only about 30%. However, APO is even less of a homologue to p450s or lignin peroxidases [130]. The prosthetic heme group is linked via iron to an exposed cysteine and shows a soret band at 445–450 nm, which is comparable to the heme signal of cytochrome p450 monooxygenases [126].
Cytochrome P450 monooxygenases can catalyze several industrial relevant oxy-functionalizing reactions. The requirement of redox equivalents such as NADH, which is a main drawback, can be overcome by adding hydrogen peroxide as a co-substrate. Thereby, a side reaction of p450 enzymes is observed, which is termed “shunt pathway.” Nevertheless, the p450 enzymes still suffer from low stability and moderate turnover numbers. APO could overcome these limitations because of their superior stability and high catalytic efficiencies [126, 130].
A catalytic cycle for APO was suggested, which combines elements of the heme peroxidases with the peroxide cycle and the “shunt pathway” of p450 monooxygenases [129]. At the resting state a water molecule is bound at the ferric heme of the enzyme. The catalytic cycle starts with hydrogen peroxide which reacts with the ferric heme, resulting in a negatively charged ferric peroxo-complex (only shown for chloroperoxidase). After a heterolytical cleavage of the peroxide O–O bond, compound I is built [126]. Depending on the substrate, its binding site and the reaction conditions, compound I can react in a monooxygenase or a peroxidase manner [129]. In the monooxygenase pathway, ferryl oxygen is transferred to the substrate and accepts two electrons [129]. In particular, hydrogen is abstracted from the substrate (e.g., an alkane) by compound I, resulting in a protonated compound II (ferryl hydroxide complex) and a substrate radical. The alkyl radical then recombines with an ·OH-equivalent through a rebound mechanism and builds the corresponding alcohol whilst the ferric enzyme is restored. After association of one water molecule to the catalytic center, the cycle starts again (see Fig. 11b) [126, 131]. However, the oxygenation of aromatic rings and other alkenes seems to involve π-bonds instead of a direct insertion of oxygen into a C–H bond. Thus, a hydrogen abstraction does not take place in this case [126, 129].

(a) Examples of oxidation reactions of aromatic peroxidases. S substrates, P products; S1 cis-b-methylstyrene, P1 (1R,2S)-cis-b-methylstyrene oxide, S2 benzene, P2/S3 phenol, P3_1 hydroquinone, P3_2 catechol, S4 3,4-dimethoxybenzyl methyl ether, P4 3,4-dimethoxybenzaldehyde and methanol (modified after [126]). (b) Catalytic cycle of APO. Substrate cyclohexane, Cpd compound, RS resting state (modified after [126] with permission from Elsevier, Copyright 2014 Elsevier Ltd. All rights reserved)
In contrast, peroxygenases also catalyze one electron step oxidation of, for example, phenol to a phenoxy radical via the peroxidase pathway. First, an electron is abstracted and one substrate molecule is oxidized, yielding a radical substrate and compound II. In a second step, another electron is transferred, again releasing a molecule of a radical substrate and the ferric oxygen as a water molecule [129].
Examples for reactions catalyzed by APO are illustrated in Fig. 11a.
4.2.3.2 DyP-Type Peroxidases Subfamily D
The dye-decolorizing or DyP-type peroxidases (DyPs) of fungi, represented by the subfamily D, were all found in the supernatant of fungi cultures, hinting at an extracellular occurrence of these enzymes [128]. Fungal DyPs show a molecular weight of 43–69 kDa [119]. Furthermore, the most fungal DyPs are glycosylated monomeric proteins, with a glycosylation degree of 8–38% [118, 132, 133]. They exhibit a typical heme soret band at 406 nm and lack the typical heme binding region of other heme peroxidases, which consists of one proximal histidine, one distal histidine and one additional arginine site. However all DyPs share a common heme binding motive GXXDG (glycine, two variable amino acids, aspartic acid, and glycine) [125]. The distal histidine residue is essential as an acid-base catalyst in common heme peroxidases. Interestingly, DyPs do not possess a catalytic active distal histidine but an aspartic acid (see Fig. 12) [120, 125].

(a) Proposed swing mechanism of compound I formation by DyPs. D171 aspartate 171 (instead of histidine in other peroxidases), OD2 outer carboxylate oxygen atom of aspartate [120]. (b) Heme surrounding in plant heme peroxidases (e.g., MnP) and DyP-type peroxidases; His histidine residue, asp aspartic acid residue (modified after [107, 120], Copyright 2011 FEBS)
In the native conformation, the oxygen atom (OD2) of the aspartic acid residue was shown to be too far away to fulfill its designated role, which is the reception of a proton from the heme-bound H2O2. However, Yoshida et al. postulated that in the presence of H2O2 the aspartic acid is enabled to move toward the heme molecule by a swinging mechanism [120].
The exact details of the catalytic cycle of DyPs remain unknown. However, a similar mechanism as described for the other well-characterized heme peroxidases is assumed [127]. The cycle starts with the oxidation of the resting enzyme by H2O2. Two electron equivalents are transferred and compound I (Fe4+=O and a porphyrin π-cation radical (Por+·)) is built. In two sequential steps the compound I is at first converted to compound II ([Fe4+=O]Por) by reducing a substrate molecule. Then compound II is reduced by another substrate molecule and the resting enzyme state is restored. In contrast to several other investigated heme peroxidases, compounds I and II could not be observed universally in DyPs. For the DyP-type peroxidases DyP B of Rhodococcus jostii and DyP D of Bjerka adjusta, only compound I was detectable. Vice versa, for the peroxidases DyP A of Rhodococcus jostii, DyP A of E. coli, and DyP A of Bacillus subtilis, only compound II was found [134].
DyP-type peroxidases can oxidize bulky substrates that are too large to fit in the active site [127]. Therefore, a long-range electron transfer mechanism, as already described for LiP, was suggested. This mechanism requires an electron pathway from the porphyrin ring to a suitable redox active amino acid (e.g., tyrosine or tryptophan) at the surface of the enzyme [135]. DyP AauDyPI of Auricularia auricula-judae was recently expressed in E. coli. The heterologous enzyme was investigated by multi-frequency EPR spectroscopy with regard to radical-forming amino acid residues (tyrosine and tryptophan) at the surface of the enzyme. The highest signal contribution was found for tryptophan (W377) [136, 137]. Furthermore, mutants of W377 lacked this radical signal. Additionally, the W377-mutant lost the activity for the bulky substrate reactive blue 19, whereas other substrates (e.g., 2,2′-azino-bis(3-ethylthiazoline-6-sulfonate (ABTS)) were still oxidized [137].
The redox potential of several fungal DyPs was determined by measuring the reacting efficiency against a series of phenolic compounds with increasing redox potential. DyPs showed a relatively high redox potential of around 1.1–1.2 V [83]. The characteristic reaction for DyPs is the oxidation of the anthraquinone dye reactive blue 5 (as reactive black 5 for VP) [125]. However, the fungal DyPs have a wide substrate range including 2,6-dimethoxyphenol and ABTS, as well as the high redox potential dyes Reactive Blue 5 and Reactive Black 5. Veratryl alcohol and non-phenolic lignin model compounds were also oxidized by some fungal DyPs, but no activity against Mn2+-ions was detected [118, 119, 138, 139]. Actually, DyP of Irpex lacteus and cellulases acted synergistically on wheat straw, which led to a more efficient hydrolysis and thus an increased glucose yield [118]. Although the optimum of DyPs for the oxidation of phenolic substrates such as 2,6-DMP was found in a range of pH 3.5–4.5, non-phenolic aromatic substrates were converted best under rather acidic conditions (pH 1.4–2.5) [119].
The physiological role of these fungal DyPs is not yet understood, but the catalytic features as well as the secretion under natural conditions (wood cultures) indicate that DyP-type peroxidases might participate in the oxidation of recalcitrant methoxylated aromatics within the lignin polymer. DyPs might take the place of LiPs in species where the latter are absent [83, 125, 127]. The DyP-type peroxidase subfamilies A–C are discussed below.
4.2.4 Accessory Enzymes
Besides the already described ligninases, fungi express “accessory enzymes,” which also play an important role in lignin degradation. These oxidases generate H2O2, which is required by ligninolytic peroxidases and in the Fenton reaction to produce reactive hydroxyl radicals (OH·) [88]. H2O2-producing enzymes include glyoxal oxidases (belonging to the copper-dependent copper radical oxidases family) and several flavoproteins of the glucose-methanol-choline family. The latter includes the aryl-alcohol oxidases, glucose-1-oxidases, alcohol oxidases, and cellobiose dehydrogenases.
The glyoxal oxidases own a broad specificity and catalyze the oxidation of aldehydes to carboxylic acids, thereby reducing dioxygen to H2O2 [140, 141]. The low specificity of this enzyme hampered the determination of the physiological substrate. The enzyme oxidizes, for example, glyoxal, glycolaldehyde, and glyoxalic acid. These molecules could be derived by lignin degradation. It was shown that LiP degraded a β-aryl ether lignin model compound to glycolaldehyde, which is a substrate for the glyoxal oxidase. A further oxidation of glycolaldehyde to oxalate via glyoxal and glyoxylic acid can generate H2O2, which is then recycled by LiP. Moreover, glyoxal could be derived by oxidation of linoleic acid, a fungal metabolite, or by degradation of carbohydrates (e.g., sugars) through hydroxyl radicals (OH·) [142]. Glyoxal oxidase seems to be of importance in the lignin degradation process of P. chrysosporium (see Fig. 6) [143].
Aryl alcohol oxidases and ligninolytic peroxidases are produced simultaneously in Bjerkandera adusta and Pleurotus, which hints at participation of these oxidases in the lignin degradation process. The substrates for the aryl alcohol oxidase could be either lignin-derived compounds or fungal aromatic metabolites [89]. The aryl alcohol oxidases generally catalyze the oxidative dehydrogenation of aromatic and aliphatic polyunsaturated alcohols with a primary hydroxyl group and the oxidation of aldehydes to the corresponding acids [89, 140]. Intracellular aryl alcohol and aryl aldehyde dehydrogenases reduce the aldehydes and acids back to alcohols and aldehydes by consumption of redox equivalents (e.g., NADPH). The reduced molecules are secreted and again serve as substrates for the aryl alcohol oxidase, thus building an H2O2 generating loop. p-Anisaldehyde and the corresponding p-methoxybenzyl alcohol seem to be the physiological substrates for the H2O2-producing redox cycle in Pleurotus species (see Fig. 6) [89].
Brown-rot fungi release methanol during their wood decaying process by demethylation of lignin. The demethylation might be caused by reactive hydroxyl radicals or by an enzymatic process. However, no suitable enzyme has been isolated so far [144, 145]. In Gloeophyllum trabeum a methanol oxidase was identified, which is believed to use the lignin-derived methanol to generate H2O2. Interestingly, although a signal sequence is missing, the methanol oxidase was located extracellularly [89].
Cellobiose dehydrogenases (CDH) consist of two domains: the C-terminal dehydrogenase domain, containing FAD as redox factor and the N-terminal cytochrome domain, which is a heme enzyme. Both modules of CDH evolved parallel as fused genes [140]. The suggested biological roles of CDH are manifold. CDH oxidizes cellobiose to cellobiono-δ-lactone rather inefficiently and this might not be a relevant function. The most common biological role of CDH is the production of Fe2+ and H2O2, which can undergo a Fenton reaction and build a radical hydroxyl (see Fig. 6). The cytochrome subunit perhaps produces reactive oxygen species directly. The generated reactive oxygen species can attack the lignocellulose matrix [90]. Alternatively, other functions of CDH are possible. The reduction of semiquinones to quinones and the reduction of Mn(IV)O2 by CDH was observed. Thereby, dissolved manganese is provided for MnP [146]. The formed quinones can be radicalized by laccase or lignolytic peroxidases. These radicals are reduced to semiquinones, whereby Fe3+ and H2O2 are generated [147].
4.3 Bacterial Lignin Degradation
This section elaborates screening methods for finding new lignolytic bacteria. Moreover, several already known ligninolytic bacteria are described.
Research on lignin degrading organisms has mainly focused on basidiomycetes, especially on white-rot fungi, because of their high ligninolytic activities. In spite of extensive research on lignin degradation, so far no commercial biocatalytic process for lignin depolymerization exists. The challenging protein expression and genetic manipulation in fungi might be one reason. Bacteria might support the progress of industrial lignin utilization. Although the extent of the prokaryotic lignin breakdown is not as complete as in fungi, several bacteria strains have been shown to react on lignin and probably produce small aromatic molecules. It was observed that many soil bacteria, which are able to metabolize aromatic compounds, also show ligninolytic activities. These findings point out the possibility of a reasonable association between lignin degradation and aromatic degradation, considering that lignin is a considerable source for soil-occurring aromatics [124, 148]. The most bacterial lignin degraders known to date belong to the classes actinobacteria, α-proteobacteria, and γ-proteobacteria [52] (see Fig. 13).

Phylogenetic tree of bacteria. Bacteria families with members, which are associated with lignin degradation or potential ligninolytic enzymes are shown in bold (modified after [149], adapted with permission from Macmillan Publishers Ltd Macmillan Publishers Ltd, Copyright 2012)
Two different general approaches are being used for the identification of new ligninolytic organisms: culture dependent and culture independent methods.
Screening of new species by culture-dependent methods is carried out by cultivation steps on lignin model compounds, lignin, or lignin-rich waste (e.g., pulping effluent) as carbon source. Two-step screening methods are increasingly used. Herein, a first enrichment by growth on one or several aromatic compounds is followed by a confirmation of the lignin degradation abilities of selected strains through cultivation on lignin polymers.
A proof of lignin degradation by an organism is possible by showing the growth on lignin model substrates as sole carbon source, detecting modifications of lignin model substrates, or identifying known ligninolytic enzymes. Furthermore, polymeric, more or less natural lignins can be used as screening substrates. The chemical features of the lignin are strongly dependent on the extraction method, with dioxane and klason lignin being close to natural lignin and KL, which is significantly modified. Lignocellulose biomass can also be used as screening substrate. The degradation of lignin can be observed by determining the solid acid-precipitable polymeric lignin (APPL). APPL is built with less methoxyl groups and is complexed with bacterial protein. Moreover, feasible methods are established with synthetic 14C-labeled lignin, which is synthesized from 14C-labeled phenols, or 14C-labeled lignin, which is obtained by growing plants on lignin precursor 14C-phenylalanin (lignin (lignocellulose)). The 14C-labeled lignins or lignin model substances are cultivated with ligninolytic species and the degradation rate is indicated by the amount of generated 14CO2. Less time-consuming screening assays are based on the conversion of monomeric aromatics or synthetic aromatic dyes. The latter methods are apparently less reliable in validating lignin degradation. The use of aromatic lignin model dimers or tetramers is of intermediate effort and reliability [150, 151].
Currently two novel fast spectrophotometric screening methods have been established. The first screening assay with fluorescein isothiocyanate-labeled lignin gives changes in fluorescence over 10 min when lignin breakdown occurs. The second assay is based on chemically nitrated lignin and detects the release of nitrated phenol breakdown products at 430 nm over 20 min [152, 153].
Culture-independent methods include molecular biological methods and bioinformatics tools. These methods allow a direct analysis of the biodiversity of an environmental probe by 16S rRNA or its enzymatic diversity and functionality by screening on specific enzymes, for example lignolytic peroxidases (metagenomics) and their expression (metatranscriptomics). Herein, species that cannot be cultivated are also considered [151]. Interesting sources for screenings on new ligninolytic species are soils, waste water derived from the paper industry, and decaying wood or straw, as well as guts of wood-eating insects such as termites or beetles [124, 151]. For instance, the metagenome of the gut of a wood-boring beetle Anoplophora glabripennis was screened with regard to its microbiome composition and the enzyme functionalities. The microbiome varied between individuals, but Gammaproteobacteria were found to be dominant and Bacilli and Betaproteobacteria were found in equal amounts, although the relative abundances of Actinobacteria, Alphaproteobacteria, Gammaproteobacteria, and Sphingobacteria were inconsistent [154]. No approved ligninolytic enzymes (e.g., MnP) were identified, but bacterial enzymes such as DyP-type peroxidases, copper oxidases, β-etherases, and glutathione-S-transferases, and especially aldo-keto reductases, which are all supposed to be associated with lignin degradation, were described [155].
A considerable amount of ligninolytic bacteria strains were found in the 1970s and 1980s by 14C-labeling methods and the observation of APPL formation during growth on lignocellulose material. For example, soil probes were enriched by cultivation with dioxane lignin as carbon source. Pseudomonas, Xanthomonas, and Acinetobacter strains were isolated and found to change significantly the lignin structure. However, only a small amount (2%) of 14CO2 was found after 150 days of degradation of labeled poplar by a Pseudomonas strain [156]. Moreover, after enrichment of compost and soil on mineral media with straw, KL or ferulic acid selected strains were incubated with labeled straw and investigated on 14CO2 release. A Streptomyces strain was found to evaporate 7.5% of the lignin of labeled wheat straw as CO2 in 15 days, but interestingly was unable to degrade KL or straw. A Thermonospora strain, able to degrade KL and ferulic acid, transformed 8.0% lignin to CO2 [157]. The actinobacteria Nocardia autotrophica, Nocardia corallin, Nocardia opaea, and others were found to release 14CO2 from synthetic dehydropolymer of coniferyl alcohol with about 4–14% in 15 days. Gram-strains as Pseudomonas testosterone, P. putida, and further Pseunomonas species showed a decrease of 0.9–2.2% in 15 days [158]. Strains of the genus Streptomycetes, isolated from termites gut produced 5–15% APPL of the initiate lignin, when they were incubated for 3 weeks with corn stover lignin (lignocellulose). The strain Streptomyces viridosporus T7A, which was the first strain where APPL was observed, solubilized about 7% of the lignocellulose material. For these strains 6–10% of the 14C labeled lignin (in Abies concolor lignocellulose) was found as water-soluble products and only 1–2% as 14CO2 after 3 weeks, hinting at a modification but no complete metabolization of lignin in these strains [159]. However, in other publications values of up to 16% of labeled lignin released as 14CO2 were reported for Nocardia, Rhodococcus, Arthrobacter, Streptomyces, and Thermomonospora strains. The strains Streptomyces cyaneus and Thermonospora mesophila solubilized up to 30% lignin of lignocellulose materials after about 2 days to form APPL. Herein, bacteria showed higher conversion into CO2 when extracted lignin and not lignin incorporated in lignocellulose was used [150, 160, 161]. In short, bacteria can solubilize lignin to high proportions and also the metabolization and the release of 14CO2 was significant, but rather low compared to white-rot fungi such as Phanerochaete chrysosporium, Sporotrichum pulverulentum, and Coriolus versicolor, which were shown to evolve 30–50% of lignin (lignocellulose) as CO2 [162, 163].
More recently, numerous bacterial strains were screened on ligninolytic activity with a nitrogen assay and a fluorescence assay. The strains Streptomyces viridosporus and Pseudonocardia autotrophica were dependent on H2O2 for their activity, and activity could only be detected by the fluorescence assay and not by the nitrogen assay. Via the nitrogen assay, various strains, namely Rhodococcus sp., Acinetobacter sp., Arthrobacter globiformis, Pseudomonas putida, Ochrobactrum pseudogrignonense, and Microbacterium oxydans were found to reveal absorbance signals of around 1–5 milli arbitrary units (mAU). Notably, the three strains Sphingobacterium, Rhodococcus erythropolis, and Streptomyces coelicolor achieved values of 30–70 mAU. For wood-rotting fungi, comparable values of 3–30 mAU were detected, although white-rot fungus Phanerochaete chrysosporium exhibited a very high activity (ca. 700 mAU) [152, 153, 164].
Novel strains Comamonas sp. and Pandoraea sp. isolated from bamboo slips were shown to degrade KL and decolorize KL up to 45% and 40%, respectively. Remarkably, a manganese peroxidase and a laccase activity as well as small molecule degradation products such as cinnamic acid, ferulic acid, 2-hydroxy benzyl alcohol, and vanillyl methyl ketone were detected in culture supernatant of Pandoraea sp. [165, 166].
The participation of inducible extracellular proteins in APPL formation of bacteria was observed. Moreover, it was assumed that these proteins could be extracellular peroxidases [159, 168,167,169]. Thermonospora mesophila, P. autotrophica, Streptomyces sp., and S. viridosporus T7A strains were identified as high peroxidase producers in a screening for extracellular peroxidases in actinobacteria [170]. An extracellular peroxidase of S. viridosporus T7A was purified and characterized. This peroxidase cleaved lignin model compounds, oxidized monomeric aromatics, and was determined to be a heme peroxidase [171]. However, the gene sequence of this so-called lignin peroxidase of S. viridosporus T7A has not been published so far [115]. Members of the new family of the DyP-type peroxidases were associated with lignin degradation in bacteria. Because the extracellular occurrence of these peroxidases has been disputed, a participation in lignin degradation needs to be investigated in detail [148]. Moreover, bacterial laccases have currently come into focus and especially actinobacterial small laccases could be involved in lignin degradation [172]. Both these possibly ligninolytic bacterial enzymes are described in detail in the following section.
4.4 Bacterial Ligninolytic Enzymes
In this section the bacterial laccases and DyP-type peroxidases are addressed. These enzymes might participate in a bacterial lignin decay mechanism.
4.4.1 Bacterial Laccases
Bacterial laccases are not investigated as intensively as fungal and plant laccases. The first bacterial laccase was identified in the plant root associated bacterium Azospirillum lipoferu [173]. Prokaryotic laccases are quite diverse and their molecular weight is 28–180 kDa [174]. Whole genome analysis revealed that laccases are widespread in bacteria and occur, for example, in Bacillus sp., Escherichia coli, Mycobacterium sp., and Pseudomonas sp. Predictions of the three-dimensional structures suggest that all bacterial and fungal laccases consist of three so-called cupredoxin-like domains, which are mainly formed by β-barrels [173]. In addition to these well-described monomeric three-domain laccases, a new kind of laccase was identified, which is only found in prokaryotes. These laccases have two instead of three domains and are active as trimers. The active site is formed at each of the monomers. So far, bacterial laccases were thought to be intracellular. However, a bioinformatics study revealed that 76% of the bacterial laccases have a signal sequence and thus may be extracellular enzymes [175]. Bacterial laccases, in contrast to fungal laccases, are highly active at high temperatures, high pH, and high chloride and copper ion concentrations, making them compatible with a lot of industrial processes [173, 175]. Moreover, crystal structure analysis revealed a large putative substrate binding pocket in prokaryotic laccases compared to fungal and plant laccases [176]. However, bacterial laccases have low reduction potential T1 Cu, with values usually below 0.5 V, limiting their oxidation of high redox potential substrates [173, 174].
All prokaryotic laccases catalyze the reduction of dioxygen to water, but the substrate specificity and thus the assumed physiological roles differ considerably amongst these enzymes [174]. Generally, laccases oxidize aromatic phenols, amines, and inorganic ions such as [Fe(CN)6]. Some of them show activities against metal ions such as Fe2+ and Cu+ [174].
Laccase Cot A of Bacillus subtilis is assumed to be involved in the production of brown spore pigment of the endospore coat [176].
Laccases of Escherichia coli and Bacillus halodurans catalyze the conversion of Cu(I) to Cu(II), which is suggested to be a protection mechanism against the toxic effects of Cu(I) [174, 177].
Pseudomonas putida laccase, was shown to oxidize Mn(II) and Mn(III). In prokaryotes the oxidation of Mn(II) leads mostly to the formation of Mn(IV) oxides [178]. It was speculated that these oxides might protect the bacteria, for example, from UV radiation by binding on the cell surface. Another hypothesis is the oxidation of organic matter by Mn(IV) oxides, which can thus be metabolized by the bacteria [179].
A laccase of the γ-proteobacterium JB was assumed to protect the organism from xenobiotic toxicity, because numerous substances such as p-toluidine and phthalic acid induced the laccase expression [180].
Finally, two-domain laccases of Streptomyces (S. coelicolor, Streptomyces lividans, S. viridosporus T7A) were reported to oxidize a phenolic lignin model substance without mediator, but non-phenolic substances only in the presence of redox mediators. Moreover, wild type strain S. coelicolor and a laccase-deficient mutant were incubated with lignocellulose and both were found to produce APPL. Remarkably, the mutant strain produced 33% less APPL, demonstrating an important role of this laccase in the lignin degradation process of S. coelicolor [172].
Prokaryotic laccases could be of great interest for future industrial biotechnology because they are easily expressed in industrial host organisms. Basically, an increase of their redox potential is required. Further studies on the correlation between T1 copper site ligation, reduction potential, and electronic structure may help to overcome this limitation through directed enzyme engineering [174].
4.4.2 DyP-Type Peroxidases Subfamilies A, B, and C
The subfamilies A, B, and C DyP-type peroxidases mainly contain bacterial enzyme sequences (see Fig. 14) [134].

Phylogenetic tree of the DyP-type peroxidase family members. YcdB (E. coli), DyPP2 (Amycolatopsis sp.), Ajpl (Auricularia auricula-judae), DyP (Bjerkandera adusta), TyrA (Shewanella oneidensis), DyP B (Rhodococcus jostii) (adapted after [128] with permission from Elsevier, Copyright 2015 Elsevier Inc. All rights reserved)
DyP A, the ancestral clade of the whole superfamily, is probably located extracellularly, and thus twin-arginine translocation (TAT) signal sequences are found in these enzyme sequences. In contrast, DyP C and DyP B peroxidases appear to be involved in the intracellular metabolism [104, 127]. DyP B subfamily is rather divergent. Besides its additional branch, which possibly leads to the chlorite dismutases, DyP B peroxidases were also found in fungi and lower eukaryotes (protists or slime molds). DyP B peroxidases probably first evolved in Proteobacteria and afterward spread via horizontal gene transfer amongst the other bacterial phyla and the eukaryotes [104].
DyP C peroxidases cluster close to the fungal DyP D peroxidases and seem to share their high activity. The molecular weights of DyPs were observed to be around 54 kDa, 44–48 kDa, and about 32 kDa for DyP C, A, and B, respectively [181,182,183,184,185,186]. The quaternary structures of DyPs have been reported to vary from monomers to hexamers [127].
The catalytic cycle for DyP-type peroxidases is already concerned in the above Sect. 4.2.3. For the functionality of fungal DyP-type peroxidases, the essential amino acid residue aspartate of the GXXDG motif was proposed to be essential for the interaction with H2O2 and the formation of compound I [127]. This might be true for DyP-type peroxidases of subfamily D, and aspartate seems to be important for the peroxidase activity of particular subfamily A members [187, 188]. However, for DyP B of actinobacterium Rhodococcus jostii and protobacterium Pseudomonas putida and DyP A peroxidase EfeB of E. coli this aspartate residue does not appear be essential for the peroxidase activity [189, 190]. Moreover, an arginine residue was shown to be essential in DyP B of R. jostii, as a mutant with arginine substituted by leucine had no detectable peroxidase activity [134].
DyPs generally have a wide substrate specificity and a considerable lower pH optimum compared to other plant peroxidases [115]. All DyP-type peroxidases have a peroxidase activity, although for some DyPs, which have inefficient catalytic properties, this activity might not be relevant for their physiological role [134]. For instance, ABTS, which is a general peroxidase substrate, is converted by A- and B-type DyPs three orders of magnitude less efficiently than by C- and D-type DyPs [134]. The exact physiological functions of DyP-type peroxidases are unknown, but some have been proposed [128].
A DyP-type peroxidase of archaebacterium Halobacterium salinarum was suggested to be involved in the protection against oxidative stress. The function of DyP B peroxidase of proteobacterium Francisella tularensis was also suggested to be involved in the defense of oxidative stress [128, 191].
Studies on YcdB (DyP A) and Yfex (DyP B) of E. coli indicate that both these DyP-type peroxidases are able to extract iron from heme and thereby leave the protoporphyrin ring intact [184]. These findings are supported by the genomic context of these enzymes in E. coli. The genes efeU and efeO, which code for an iron transporter and an iron uptake component, occur downstream of Yfex and YcdB [134]. A similar function was described for a DyP A and DyP B peroxidase of Staphylococcus aureus. The cluster was expressed in a YcdB and Yfex deficient E. coli mutant and restored the ability of E. coli to use heme as an iron source. DyP A of S. aureus showed even less peroxide activity as E. coli YcdB [192]. The latter enzymes could be an example of the physiological function for DyPs being independent of the peroxidase activity [128]. In contrast, several members of the DyP-type peroxidases subfamilies B, C, and A were associated with lignin degradation in bacteria.
DyP B from R. jostii was shown to cleave a phenolic lignin model compound. In the presence of Mn2+, which is also a substrate for this peroxidase, this activity was increased. Furthermore, an activity against straw lignocellulose could be measured only by adding MnCl2. Nitrated lignin was incubated with R. jostii wild type and mutant strains. In the wild type strain, R. jostii photometrically measured changes at 430 nm hinted at the evaporation of nitrated phenol compounds. In contrast, in a DyP B-deficient R. jostii mutant, no changes were detected. The heterologous DyP B enzyme also showed activity in the nitrate assay [193]. A binding pocket with three glutamate residues and one threonine was proposed for DyP B of R. jostii. However, in Pseudomonas putida DyP B the same residues are present, but no Mn2+-oxidizing activity was found. Moreover, another Mn2+-oxidizing enzyme (DyP C, Amycolatopsis sp.) did not have the same Mn2+-binding motif [194,193,195]. Engineering of DyP B (N246A) resulted in a 15-fold increase in the catalytic efficiency (kcat/km) against Mn2+. The engineered DyP B was able to catalyze a manganese-dependent conversion of KL and solvent-extracted fractions of KL. Nevertheless, DyP C of actinobacterium Amycolatopsis sp. still oxidizes Mn2+ more efficiently within two orders of magnitude [196]. Paradoxically, the ligninolytic peroxidase of R. jostii lacks, as do most DyP B peroxidases, a signal sequence for export and consequently is supposed to be an intracellular enzyme. This is in contrast to its suggested function, which is the measured ligninolytic activity in the supernatant of R. jostii. It was hypothesized that DyP B might be exported by an unknown mechanism. Downstream of DyP B a gene coding for encapsulin was identified. Encapsulins can form nanocompartments with incorporated DyP B. A higher ligninolytic activity for the complexed DyP B in comparison to free DyP B was measured, but still the export of DyP B remains in question [193, 197].
DyP C of Amycolatopsis sp. (formerly Streptomyces griseus) was also reported in the context of lignin degradation. As already mentioned, a very high Mn2+-oxidizing activity almost comparable to fungal VP was observed. DyP C was able to degrade high redox synthetic dyes with high catalytic efficiency and was able to cleave a phenolic lignin model substance. Interestingly, an oxidase activity with 4-methoxymandelic acid in the presence of Mn2+ was found. Some homolog DyP C peroxidases of other related actinomycetes cluster on operons with other biomass-processing enzymes, though DyP C of Amycolatopsis sp. does not [186]. To date, only DyP C of Amycolatopsis sp. and DyP C of cyanobacterium Anabaena sp. have been characterized. DyP C of Anabaena sp. oxidizes guaiacol, pyrogallol, and anthraquinone dyes such as Reactive Blue 5 with high catalytic efficiencies, the latter with 107, which is equal to activities of fungal DyPs (Auricularia auricular). However, this DyP C peroxidase can neither oxidize veratryl alcohol nor manganese [185, 186]. In general, DyP C peroxidases are found in the neighborhood of various enzymes, such as a doxorubicin resistance gene, halo-acid dehalogenase, or a methyl-accepting chemoreceptor. Overall, the biochemical characterizations and the bioinformatic data indicate that the peroxidase activity of DyP C peroxidases is physiologically relevant and that DyP C peroxidases have diverse biological roles [134].
Just recently two DyP A peroxidases have been characterized, which seem to participate in lignin degradation. DyP A of Bacillus subtilis (KCTC2023) was able to cleave a non-phenolic lignin model dimer without mediator and oxidized the high-redox non-phenolic monomer veratryl alcohol with a high efficiency in comparison to other bacterial DyP-type peroxidases. The anthraquinone dyes reactive black 5 and reactive blue 19 were also oxidized. Interestingly, these substrates differed in their temperature optima. Whereas the high redox substrates veratryl alcohol and the lignin model dimer were converted best at 50 °C, the optimum for dye oxidation was 30 °C [198]. The DyP A peroxidase of Saccharomonospora viridis was shown to bleach eucalyptus kraft pulp (21.8% reduced kappa number, 2.98% increase in brightness). The enzyme performed well over a broad pH range and had a high temperature stability. The pH optimum of this enzyme was determined with triarylmethane dye brilliant green as a substrate. Astonishingly, in contrast to other DyP-type peroxidases, which work mostly in acid pH, the optimum of DyP A of S. viridis was pH 7. Another interesting observation was the eightfold boost of enzyme activity through the reducing agent β-mercaptoethanol, perhaps by preventing the enzyme from forming S–S linkages [183].
Further bioinformatical and biochemical studies on DyP-type peroxidases could help to clarify their physiological roles. Further research should also help to develop their biotechnological potential, which is expected mainly in processes for modification and degradation of lignin and industrial dyes [134].
4.4.3 Accessory Enzymes
Assuming an analogous mechanism of fungal and bacterial lignin degradation, bacteria also require accessory enzymes, providing H2O2 for their extracellular peroxidases. A few H2O2-generating enzymes [199, 200] and analogs to fungal accessory enzymes [201, 202] have been described in bacteria, but to the best of our knowledge no publication which deals with their participation in lignin degradation is available.
4.5 Bacterial Catabolism of Lignin Degradation Intermediates
Although bacteria are able to break down lignin, they generally play a more important role in mineralization of oligomeric and monomeric lignin derivatives, which are derived from fungal attack on the lignin macromolecule [203, 204]. Herein, we encounter the degradation pathways of aromatics by bacteria.
Many bacteria strains are known to metabolize lignin-derived aromatic compounds. For example, biphenyl-degrading species are found in several bacterial genera such as Sphingomonas, Burkholderia, Rhodococcus, Pseudomonas, Achromobacter, Comamonas, Ralstonia, Acinetobacter, and Bacillus [124]. The most intensively studied bacterium regarding the aromatics metabolism is the proteobacterium Sphingobium sp. SYK-6 [52].
A degradation of biphenyl 5,5-dehydrovanillate is described to proceed via a demethylation step, catalyzed by O-demethylase (LigX). The O-demethylase is probably a three-component monooxygenase [204]. One of the aromatic rings is cleaved by a dioxygenase (LigZ) and the meta-cleavage compound is converted into 5-carboxyvanillate and 4-carboxy-2-hydroxypentadienoic acid by hydrolase LigY. 5-Carboxyvanillate is decarboxylated by decarboxylase LigW to result in the central metabolite vanillic acid (Fig. 15B). 4-Carboxy-2-hydroxypentadienoic acid is probably hydrated and cleaved by an aldolase to form two molecules of pyruvate [52, 124].

Degradation pathways for lignin derivatives in Sphingobium sp. SYK-6: (A1) degradation of (S)-enantiomer β-aryl ether, (A2) degradation of (R)-enantiomer β-aryl ether, and (B) degradation of biphenyls. Abbreviations: LigD-O dehydrogenases, LigP and LigE/F glutathione-S-transferases, LigG β-thioetherase, VD vanillin dehydrogenase, LigX O-demethylase (monooxygenase), LigZ dioxygenase, LigY hydrolyase, LigW/W2 decarboxylase (modified after [203, 205] with permission from Taylor & Francis Ltd, Copyright Japan Society for Bioscience and Agrochemistry, Copyright 2014 by The American Society for Biochemistry and Molecular Biology, Inc.)
Sphingobium sp. SYK-6 moreover degrades β-aryl ether compounds such as guaiacylglycerol-β-guaiacyl ether (GGE) or guaiacyl-α-veratrylglycerol (VG). The dehydrogenases LigD, LigL, LigN, and LigO dehydrogenate VG at the α-C a-hydroxyl group to form a ketone. Then a β-S-glutathionyl-α-ketothioether is built at the β-ether. Thereby, the cosubstrate glutathione is linked to the β–C and guaiacol is released. Interestingly, this step is stereoselective and catalyzed by different glutathione-S-tranferases. LigF only catalyzes β(S)-ether, resulting in a β(R)-thioether, whereas β(R)-ether is converted by LigP and LigE to form a β(S)-thioether. The β-thioetherase LigG seems to be stereoselective, too, and cleaves preferentially the β-(R)-thioether. In this step a second molecule GSH is consumed, producing glutathione disulfide (GS-SG) and β-deoxy-α-veratrylglycerone [205] (Fig. 15A).
In Rhodococcus jostii an alternative conversion of the β-aryl ether into a ketone cleavage product was suggested, working with a radical β-elimination mechanism via hydrogen abstraction [152]. The ketone intermediate is then metabolized to vanillic acid. It was assumed that this step might be an oxidation of the hydroxyl group into carboxylic acid, followed by a C–C cleavage similar to a β-oxidation [52].
Further pathways of other aromatic compounds such as the monomers eugenol and coniferyl alcohol, as well as dimers pinoresinol and di-aryl ether, were described in Sphingobium sp. SYK-6 and other species, leading to vanillic acid. Vanillic acid is demethylized by a monooxygenase to form protocatechuic acid [124, 203, 206].
Aerobic degradation pathways include, besides catechol and some other aromatics, protocatechuate as their central intermediate [207]. Protocatechuate may originate from lignin breakdown products, as described above, but also from chlorinated aromatics and other materials [208].
In Sphingobium sp. SYK-6, protocatechuic acid is cleaved aerobically by an extradiol dioxygense (protocatechuic acid-4,5-dioxygenase named LigAB), meaning this enzyme cuts the aromatic ring not between the two ring hydroxyl groups, but outside, initiating the meta-cleavage pathway. Other bacteria, for example, Pseudomonas, Acinetobacter, and Rhodococcus, are known to use an alternative designated ortho-cleavage pathway, including an aerobic ring cleavage by an intradiol protocatechuate 3,4-dioxygenase as a first step. Moreover, some bacteria, for example, Rhodococcus equii, own a protocatechuic acid 3,4- and a 4,5-dioxygenase.
The meta-pathway continues with oxidation of the dioxygenase cleavage product 4-carboxy-2-hydroxymuconate semialdehyde by a dehydrogenase (LigC), producing 2-pyrone-4,6-dicarboxylate. This molecule is hereupon hydrolyzed by LigI, resulting in 4-oxalomesaconate, which is transformed by a hydratase LigJ and an aldolase LigK into pyruvate and oxaloacetate, which enter the citric acid cycle and are respired (see Fig. 15) [52, 203].
In Pseudomonas putida, as an example of the ortho-pathway, the cleavage product β-carboxymuconate is transformed by a cycloisomerase into γ-carboxymuconolactone, which is decarboxylated by γ-carboxymuconolactone decarboxylase into β-ketoadipate enol-lactone and then reduced to β-ketoadipate by a hydrolase. The subsequent β-ketoadipate pathway degrades β-ketoadipate to succinyl- and acetyl-CoA via a CoA transferase and a CoA thiolase. Succinyl- and acetyl-CoA can be utilized by the cell in the citric acid cycle or the fatty acid biosynthesis (see Fig. 16) [208].

Meta- and ortho-cleavage pathways in bacteria Sphingobium sp. SYK-6 and Pseudomonas putida. Abbreviations: P3,4O, protocatechuic acid-3,4-dioxygenase, CM β-carboxy-muconate, CMLE β-carboxy-cis,cis-muconate lactonizing enzyme (cycloisomerase), CML γ-carboxy-muconolactone, CMD γ-carboxy-muconolactone decarboxylase, KAEL β-ketoadipate enol-lactone, ELH β-ketoadipate enol-lactone hydrolase, KA β-ketoadipate, TR β-ketoadipate succinyl-CoA transferase, CoA coenzyme A, TH β-ketoadipyl-CoA thiolase, AC acetyl, SC succinyl, LigAB protocatechuic acid 4,5-dioxygenase, CHMS 4-carboxy-2-hydroxymuconate-6-semialdehyde, LigC CHMS dehydrogenase, PDC 2-pyrone-4,6-dicarboxylate, LigI PDC hydrolase, OMA 4-oxalomesaconate, LigJ OMA hydratase, CHA 4-carboxy-2-hydroxy-3-O-methylgallate, LigK CHA aldolase, TCA tricarboxylic acid (modified after [203, 208] with permission from Taylor & Francis Ltd, Copyright Japan Society for Bioscience and Agrochemistry, with permission from Elsevier, Copyright 2012 Elsevier Ltd. All rights reserved)
Catechol may be derived from pollutants (e.g., phenol, toluene, or benzene) and similar sources as protocatechuate. Catechol is also found as central intermediate in bacteria [207, 208]. Its degradation pathway includes mechanisms similar to the protecatechuate pathway and is described here only briefly. Catechol is either cleaved extradiolically to form 2-hydroxymuconic semialdehyde (meta-pathway), which is converted to acetaldehyde and pyruvate, or cleaved intradiolically (ortho-pathway) to form cis,cis-muconate. This intermediate is, analogous to protocatechuate, cyclo-isomerized to muconolactone, followed by a double bond shift, resulting in the enol-lactone, which is hydrolyzed and enters the corresponding pathway as β-ketoadipate [207, 208].
Further research on bacterial lignin degrading pathways should help not only to clarify the carbon cycle on Earth, but also to identify novel useful tools for the conversion of lignin into building blocks and fine chemicals of industrial interest [203]. Recently, the gene of the vanillin dehydrogenase has been deleted in the ligninolytic bacterium Rhodococcus jostii. The mutant strain was grown on wheat straw lignocellulose as a feedstock, which resulted in an accumulation of vanillin in the culture broth [209]. This gives an exemplary approach for biotechnological utilization of lignocellulose and, accordingly, lignin, stimulating further research on bacterial lignin degradation.
References
Martone PT, Estevez JM, Lu F, et al (2009) Discovery of lignin in seaweed reveals convergent evolution of cell-wall architecture. Curr Biol 19:169–175. doi:10.1016/j.cub.2008.12.031
Espiñeira JM, Novo Uzal E, Gómez Ros LV, et al (2011) Distribution of lignin monomers and the evolution of lignification among lower plants. Plant Biol (Stuttg) 13:59–68. doi:10.1111/j.1438-8677.2010.00345.x
Oinonen P, Zhang L, Lawoko M, Henriksson G (2014) On the formation of lignin polysaccharide networks in Norway spruce. Phytochemistry. doi:10.1016/j.phytochem.2014.10.027
Delaux P-M, Nanda AK, Mathé C, et al (2012) Molecular and biochemical aspects of plant terrestrialization. Perspect Plant Ecol Evol Syst 14:49–59. doi:10.1016/j.ppees.2011.09.001
Vanholme R, Demedts B, Morreel K, et al (2010) Lignin biosynthesis and structure. Plant Physiol 153:895–905. doi:10.1104/pp.110.155119
Bonawitz ND, Chapple C (2010) The genetics of lignin biosynthesis: connecting genotype to phenotype. Annu Rev Genet 44:337–363. doi:10.1146/annurev-genet-102209-163508
Ek M, Gellerstedt G, Henriksson G (2009) Wood chemistry and biotechnology. Walter de Gruyter, Berlin
Schuetz M, Benske A, Smith RA, et al (2014) Laccases direct lignification in the discrete secondary cell wall domains of protoxylem. Plant Physiol 166:798–807. doi:10.1104/pp.114.245597
Zhao Q, Nakashima J, Chen F, et al (2013) LACCASE is necessary and nonredundant with PEROXIDASE for lignin polymerization during vascular development in arabidopsis. Plant Cell 25:3976–3987. doi:10.1105/tpc.113.117770
Sangha AK, Petridis L, Smith JC, et al (2012) Molecular simulation as a tool for studying lignin. Environ Prog Sustain Energy 31:47–54. doi:10.1002/ep.10628
Wang Y, Chantreau M, Sibout R, Hawkins S (2013) Plant cell wall lignification and monolignol metabolism. Front Plant Sci. doi:10.3389/fpls.2013.00220
Davin LB, Lewis NG (2005) Lignin primary structures and dirigent sites. Curr Opin Biotechnol 16:407–415. doi:10.1016/j.copbio.2005.06.011
Hatfield R, Vermerris W (2001) Lignin formation in plants. The dilemma of linkage specificity. Plant Physiol 126:1351–1357. doi:10.1104/pp.126.4.1351
Sangha AK, Parks JM, Standaert RF, et al (2012) Radical coupling reactions in lignin synthesis: a density functional theory study. J Phys Chem B 116:4760–4768. doi:10.1021/jp2122449
Sangha AK, Davison BH, Standaert RF, et al (2014) Chemical factors that control lignin polymerization. J Phys Chem B 118:164–170. doi:10.1021/jp411998t
Ralph J, Lundquist K, Brunow G, et al (2004) Lignins: natural polymers from oxidative coupling of 4-hydroxyphenyl- propanoids. Phytochem Rev 3:29–60. doi:10.1023/B:PHYT.0000047809.65444.a4
Saake B, Lehnen R (2000) Lignin. In: Ullmanns encyclopedia of industrial chemistry. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Zakzeski J, Bruijnincx PCA, Jongerius AL, Weckhuysen BM (2010) The catalytic valorization of lignin for the production of renewable chemicals. Chem Rev 110:3552–3599. doi:10.1021/cr900354u
Joffres B, Laurenti D, Charon N, et al (2013) Thermochemical conversion of lignin for fuels and chemicals: a review. Oil Gas Sci Technol – Rev D’IFP Energies Nouv 68:753–763. doi:10.2516/ogst/2013132
Sette M, Wechselberger R, Crestini C (2011) Elucidation of lignin structure by quantitative 2D NMR. Chem Eur J 17:9529–9535. doi:10.1002/chem.201003045
Vanholme R, Morreel K, Darrah C, et al (2012) Metabolic engineering of novel lignin in biomass crops. New Phytol 196:978–1000. doi:10.1111/j.1469-8137.2012.04337.x
Ragauskas AJ, Beckham GT, Biddy MJ, et al (2014) Lignin valorization: improving lignin processing in the biorefinery. Science 344:1246843. doi:10.1126/science.1246843
Matsushita Y (2015) Conversion of technical lignins to functional materials with retained polymeric properties. J Wood Sci 61:230–250. doi:10.1007/s10086-015-1470-2
Klamrassamee T, Laosiripojana N, Cronin D, et al (2015) Effects of mesostructured silica catalysts on the depolymerization of organosolv lignin fractionated from woody eucalyptus. Bioresour Technol 180:222–229. doi:10.1016/j.biortech.2014.12.098
Vishtal A (2011) Challenges in industrial applications of technical lignins. Bioresources 6(3):3547–3468
Strassberger Z, Tanase S, Rothenberg G (2014) The pros and cons of lignin valorisation in an integrated biorefinery. RSC Adv 4:25310–25318. doi:10.1039/C4RA04747H
Doherty WOS, Mousavioun P, Fellows CM (2011) Value-adding to cellulosic ethanol: lignin polymers. Ind Crop Prod 33:259–276. doi:10.1016/j.indcrop.2010.10.022
Lora JH, Glasser WG (2002) Recent industrial applications of lignin: a sustainable alternative to nonrenewable materials. J Polym Environ 10:39–48. doi:10.1023/A:1021070006895
Paul R, Burwell D, Dai X et al (2015) Recent progress in producing #11; lignin-based carbon fibers for functional applications. GrafTech International Holdings Inc., Brooklyn Heights
Gomes FJB, Santos FA, Colodette JL, et al (2014) Literature review on biorefinery processes integrated to the pulp industry. Nat Resour 5:419–432. doi:10.4236/nr.2014.59039
Upton BM, Kasko AM (2015) Strategies for the conversion of lignin to high-value polymeric materials: review and perspective. Chem Rev 116:2275–2306. doi:10.1021/acs.chemrev.5b00345
Berlin A, Balakshin M (2014) Industrial lignins: analysis, properties, and applications. Bioenergy Res Adv Appl 2014:315–336. doi:10.1016/B978-0-444-59561-4.00018-8
De Wild PJ, Huijgen WJJ, Gosselink RJA (2014) Lignin pyrolysis for profitable lignocellulosic biorefineries. Biofuels Bioprod Biorefin 8:645–657. doi:10.1002/bbb.1474
Smaranda A, Tucu D (2011) Different industrial applications of lignin as a sustainable material. Buletinul Agir - Numere Publicate. http://www.buletinulagir.agir.ro/articol.php?id=1293. Accessed 22 Apr 2017
Norgren M, Edlund H (2014) Lignin: recent advances and emerging applications. Curr Opin Colloid Interface Sci 19:409–416. doi:10.1016/j.cocis.2014.08.004
Calvo-Flores FG, Dobado JA, Isac-García J et al (2015) Lignin and lignans as renewable raw materials: chemistry, technology and applications. Wiley, Chichester. http://eu.wiley.com/WileyCDA/WileyTitle/productCd-1118597869.html. Accessed 11 Jan 2016
Zhu W (2015) Precipitation of kraft lignin: yield and equilibrium. Doctoral thesis, Chalmers University of Technology
Rosas JM, Berenguer R, Valero-Romero MJ, et al (2014) Preparation of different carbon materials by thermochemical conversion of lignin. Front Mater. doi:10.3389/fmats.2014.00029
Burkhardt-Karrenbrock A, Seegmüller S, Burk R (2001) Flüssigholz – Ein Überblick. Eur J Wood Wood Prod 59:13–18. doi:10.1007/s001070050465
Nägele H, Pfitzer J, Ziegler L et al (2013) Lignin Matrix Composites from natural resources–ARBOFORM. In: Kabasci S (ed) Bio-based plastics: materials and applications. Wiley, Chichester pp 89–115
White JF (2007) Top value-added chemicals from biomass. In: Results of screening for potential candidates from biorefinery lignin, vol II. U.S. Department of Energy, Oak Ridge
Perez-Cantu L, Liebner F, Smirnova I (2014) Preparation of aerogels from wheat straw lignin by cross-linking with oligo(alkylene glycol)-α,ω-diglycidyl ethers. Microporous Mesoporous Mater 195:303–310. doi:10.1016/j.micromeso.2014.04.018
Duval A, Lawoko M (2014) A review on lignin-based polymeric, micro- and nano-structured materials. React Funct Polym 85:78–96. doi:10.1016/j.reactfunctpolym.2014.09.017
Xiao S, Feng J, Zhu J, et al (2013) Preparation and characterization of lignin-layered double hydroxide/styrene-butadiene rubber composites. J Appl Polym Sci 130:1308–1312. doi:10.1002/app.39311
Ten E, Vermerris W (2015) Recent developments in polymers derived from industrial lignin. J Appl Polym Sci 132:42069. doi:10.1002/app.42069
Chung Y-L, Olsson JV, Li RJ, et al (2013) A renewable lignin–lactide copolymer and application in biobased composites. ACS Sustain Chem Eng 1:1231–1238. doi:10.1021/sc4000835
Laurichesse S, Huillet C, Avérous L (2014) Original polyols based on organosolv lignin and fatty acids: new bio-based building blocks for segmented polyurethane synthesis. Green Chem 16:3958–3970. doi:10.1039/C4GC00596A
Kudanga T, Nyanhongo GS, Guebitz GM, Burton S (2011) Potential applications of laccase-mediated coupling and grafting reactions: a review. Enzyme Microb Technol 48:195–208. doi:10.1016/j.enzmictec.2010.11.007
Sena-Martins G, Almeida-Vara E, Duarte JC (2008) Eco-friendly new products from enzymatically modified industrial lignins. Ind Crops Prod 27:189–195. doi: 10.1016/j.indcrop.2007.07.016
Hüttermann A, Mai C, Kharazipour A (2001) Modification of lignin for the production of new compounded materials. Appl Microbiol Biotechnol 55:387–394
Johansson K, Gillgren T, Winestrand S, et al (2014) Comparison of lignin derivatives as substrates for laccase-catalyzed scavenging of oxygen in coatings and films. J Biol Eng 8:1. doi: 10.1186/1754-1611-8-1
Bugg TDH, Ahmad M, Hardiman EM, Rahmanpour R (2011) Pathways for degradation of lignin in bacteria and fungi. Nat Prod Rep 28:1883–1896. doi:10.1039/c1np00042j
Smolarski N (2012) High-value opportunities for lignin: unlocking its potential - bio-based news–the portal for bio-based economy and industrial biotechnology. In: Bio-based news. http://news.bio-based.eu/high-value-opportunities-for-lignin-unlocking-its-potential/. Accessed 18 May 2015
Mota MIF, Pinto PCR, Loureiro JM, Rodrigues AE (2016) Recovery of vanillin and syringaldehyde from lignin oxidation: a review of separation and purification processes. Sep Purif Rev 45:227–259. doi:10.1080/15422119.2015.1070178
Fache M, Boutevin B, Caillol S (2016) Epoxy thermosets from model mixtures of the lignin-to-vanillin process. Green Chem 18:712–725. doi:10.1039/C5GC01070E
Xu C, Arancon RAD, Labidi J, Luque R (2014) Lignin depolymerisation strategies: towards valuable chemicals and fuels. Chem Soc Rev 43:7485–7500. doi:10.1039/c4cs00235k
Pandey MP, Kim CS (2011) Lignin depolymerization and conversion: a review of thermochemical methods. Chem Eng Technol 34:29–41. doi:10.1002/ceat.201000270
Li C, Zhao X, Wang A, et al (2015) Catalytic transformation of lignin for the production of chemicals and fuels. Chem Rev 115:11559–11624. doi:10.1021/acs.chemrev.5b00155
Papadopoulos AN (2011) Sorption of acetylated pine wood decayed by brown rot, white rot and soft rot: different fungi—different behaviours. Wood Sci Technol 46:919–926. doi:10.1007/s00226-011-0450-y
Martínez AT, Speranza M, Ruiz-Dueñas FJ, et al (2005) Biodegradation of lignocellulosics: microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int Microbiol Off J Span Soc Microbiol 8:195–204
Wang W, Yuan T, Cui B (2014) Biological pretreatment with white rot fungi and their co-culture to overcome lignocellulosic recalcitrance for improved enzymatic digestion. Bioresources 9:3968–3976. doi:10.15376/biores.9.3.3968-3976
Sundman V, Näse L (1972) The synergistic ability of some wood-degrading fungi to transform lignins and lignosulfonates on various media. Arch Mikrobiol 86:339–348. doi:10.1007/BF00424990
Qi-he C, Krügener S, Hirth T, et al (2011) Co-cultured production of lignin-modifying enzymes with white-rot fungi. Appl Biochem Biotechnol 165:700–718. doi:10.1007/s12010-011-9289-9
Chi Y, Hatakka A, Maijala P (2007) Can co-culturing of two white-rot fungi increase lignin degradation and the production of lignin-degrading enzymes? Int Biodeterior Biodegrad 59:32–39. doi:10.1016/j.ibiod.2006.06.025
Boddy L (2000) Interspecific combative interactions between wood-decaying basidiomycetes. FEMS Microbiol Ecol 31:185–194. doi:10.1111/j.1574-6941.2000.tb00683.x
Prewitt L, Kang Y, Kakumanu ML, Williams M (2014) Fungal and bacterial community succession differs for three wood types during decay in a forest soil. Microb Ecol 68:212–221. doi:10.1007/s00248-014-0396-3
Rajala T, Peltoniemi M, Pennanen T, Mäkipää R (2012) Fungal community dynamics in relation to substrate quality of decaying Norway spruce (Picea abies [L.] Karst.) logs in boreal forests. FEMS Microbiol Ecol 81:494–505. doi:10.1111/j.1574-6941.2012.01376.x
Zhou L-W, Wei Y-L, Dai Y-C (2014) Phylogenetic analysis of ligninolytic peroxidases: preliminary insights into the alternation of white-rot and brown-rot fungi in their lineage. Mycology 5:29–42. doi:10.1080/21501203.2014.895784
Floudas D, Binder M, Riley R, et al (2012) The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336:1715–1719. doi:10.1126/science.1221748
Riley R, Salamov AA, Brown DW, et al (2014) Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc Natl Acad Sci U S A 111:9923–9928. doi:10.1073/pnas.1400592111
Raghavan R, Adusumalli R-B, Buerki G, et al (2012) Deformation of the compound middle lamella in spruce latewood by micro-pillar compression of double cell walls. J Mater Sci 47:6125–6130. doi:10.1007/s10853-012-6531-y
Schwarze FWMR (2007) Wood decay under the microscope. Fungal Biol Rev 21:133–170. doi:10.1016/j.fbr.2007.09.001
Zeng Y, Zhao S, Yang S, Ding S-Y (2014) Lignin plays a negative role in the biochemical process for producing lignocellulosic biofuels. Curr Opin Biotechnol 27:38–45. doi:10.1016/j.copbio.2013.09.008
Pandey KK, Pitman AJ (2003) FTIR studies of the changes in wood chemistry following decay by brown-rot and white-rot fungi. Int Biodeterior Biodegrad 52:151–160. doi:10.1016/S0964-8305(03)00052-0
Janusz G, Kucharzyk KH, Pawlik A, et al (2013) Fungal laccase, manganese peroxidase and lignin peroxidase: gene expression and regulation. Enzym Microb Technol 52:1–12. doi:10.1016/j.enzmictec.2012.10.003
Furukawa T, Bello FO, Horsfall L (2014) Microbial enzyme systems for lignin degradation and their transcriptional regulation. Front Biol 9:448–471. doi:10.1007/s11515-014-1336-9
Floudas D, Held BW, Riley R, et al (2015) Evolution of novel wood decay mechanisms in Agaricales revealed by the genome sequences of Fistulina hepatica and Cylindrobasidium torrendii. Fungal Genet Biol 76:78–92. doi:10.1016/j.fgb.2015.02.002
Alfaro M, Oguiza JA, Ramírez L, Pisabarro AG (2014) Comparative analysis of secretomes in basidiomycete fungi. J Proteome 102:28–43. doi:10.1016/j.jprot.2014.03.001
Kellner H, Luis P, Pecyna MJ, et al (2014) Widespread occurrence of expressed fungal secretory peroxidases in forest soils. PLoS ONE 9:e95557. doi:10.1371/journal.pone.0095557
Pointing S (2001) Feasibility of bioremediation by white-rot fungi. Appl Microbiol Biotechnol 57:20–33. doi:10.1007/s002530100745
Christiane Liers TA (2011) Patterns of lignin degradation and oxidative enzyme secretion by different wood- and litter-colonizing basidiomycetes and ascomycetes grown on beech-wood. FEMS Microbiol Ecol 78:91–102. doi:10.1111/j.1574-6941.2011.01144.x
Hammel KE, Cullen D (2008) Role of fungal peroxidases in biological ligninolysis. Curr Opin Plant Biol 11:349–355. doi:10.1016/j.pbi.2008.02.003
Liers C, Aranda E, Strittmatter E, et al (2014) Phenol oxidation by DyP-type peroxidases in comparison to fungal and plant peroxidases. J Mol Catal B Enzym 103:41–46. doi:10.1016/j.molcatb.2013.09.025
Hastrup ACS, Howell C, Larsen FH, et al (2012) Differences in crystalline cellulose modification due to degradation by brown and white rot fungi. Fungal Biol 116:1052–1063. doi:10.1016/j.funbio.2012.07.009
de Boer W, Folman LB, Summerbell RC, Boddy L (2005) Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol Rev 29:795–811. doi:10.1016/j.femsre.2004.11.005
Blanchette RA (2000) A review of microbial deterioration found in archaeological wood from different environments. Int Biodeterior Biodegrad 46:189–204. doi:10.1016/S0964-8305(00)00077-9
Kües U (2007) Wood production, wood technology, and biotechnological impacts. Universitätsverlag Göttingen, Göttingen
Dashtban M, Schraft H, Syed TA, Qin W (2010) Fungal biodegradation and enzymatic modification of lignin. Int J Biochem Mol Biol 1:36–50
Hernández-Ortega A, Ferreira P, Martínez AT (2012) Fungal aryl-alcohol oxidase: a peroxide-producing flavoenzyme involved in lignin degradation. Appl Microbiol Biotechnol 93:1395–1410. doi:10.1007/s00253-011-3836-8
Ludwig R, Harreither W, Tasca F, Gorton L (2010) Cellobiose dehydrogenase: a versatile catalyst for electrochemical applications. ChemPhysChem 11:2674–2697. doi:10.1002/cphc.201000216
Pollegioni L, Tonin F, Rosini E (2015) Lignin-degrading enzymes. FEBS J 282:1190–1213. doi:10.1111/febs.13224
Hammel KE, Mozuch MD, Jensen KA, Kersten PJ (1994) H2O2 recycling during oxidation of the arylglycerol beta-aryl ether lignin structure by lignin peroxidase and glyoxal oxidase. Biochemistry (Mosc) 33:13349–13354
Giardina P, Sannia G (2015) Laccases: old enzymes with a promising future. Cell Mol Life Sci 72:855–856. doi:10.1007/s00018-014-1821-y
Giardina P, Faraco V, Pezzella C, et al (2010) Laccases: a never-ending story. Cell Mol Life Sci 67:369–385. doi:10.1007/s00018-009-0169-1
Reiss R, Ihssen J, Richter M, et al (2013) Laccase versus laccase-like multi-copper oxidase: a comparative study of similar enzymes with diverse substrate spectra. PLoS ONE 8:e65633. doi:10.1371/journal.pone.0065633
Wong DWS (2009) Structure and action mechanism of ligninolytic enzymes. Appl Biochem Biotechnol 157:174–209. doi:10.1007/s12010-008-8279-z
Jones SM, Solomon EI (2015) Electron transfer and reaction mechanism of laccases. Cell Mol Life Sci 72:869–883. doi:10.1007/s00018-014-1826-6
Yoon J, Solomon EI (2007) Electronic structure of the peroxy intermediate and its correlation to the native intermediate in the multicopper oxidases: insights into the reductive cleavage of the O−O bond. J Am Chem Soc 129:13127–13136. doi:10.1021/ja073947a
Cañas AI, Camarero S (2010) Laccases and their natural mediators: biotechnological tools for sustainable eco-friendly processes. Biotechnol Adv 28:694–705. doi:10.1016/j.biotechadv.2010.05.002
Munk L, Sitarz AK, Kalyani DC, et al (2015) Can laccases catalyze bond cleavage in lignin? Biotechnol Adv 33:13–24. doi:10.1016/j.biotechadv.2014.12.008
Daroch M, Houghton CA, Moore JK, et al (2014) Glycosylated yellow laccases of the basidiomycete Stropharia aeruginosa. Enzym Microb Technol 58–59:1–7. doi:10.1016/j.enzmictec.2014.02.003
Chaurasia PK, Bharati SL, Singh SK (2013) Comparative studies on the blue and yellow laccases. Res Plant Sci 1:32–37. doi:10.1007/s12010-011-9289-9
Passardi F, Bakalovic N, Teixeira FK, et al (2007) Prokaryotic origins of the non-animal peroxidase superfamily and organelle-mediated transmission to eukaryotes. Genomics 89:567–579. doi:10.1016/j.ygeno.2007.01.006
Zámocký M, Hofbauer S, Schaffner I, et al (2015) Independent evolution of four heme peroxidase superfamilies. Arch Biochem Biophys 574:108–119. doi:10.1016/j.abb.2014.12.025
Passardi F, Theiler G, Zamocky M, et al (2007) PeroxiBase: the peroxidase database. Phytochemistry 68:1605–1611. doi:10.1016/j.phytochem.2007.04.005
Lundell TK, Mäkelä MR, Hildén K (2010) Lignin-modifying enzymes in filamentous basidiomycetes–ecological, functional and phylogenetic review. J Basic Microbiol 50:5–20. doi:10.1002/jobm.200900338
Sugano Y (2008) DyP-type peroxidases comprise a novel heme peroxidase family. Cell Mol Life Sci 66:1387–1403. doi:10.1007/s00018-008-8651-8
Nakasone KK, Hibbett DS, Goranova G (2009) Neocampanella, a new corticioid fungal genus, and a note on Dendrothele bispora. Botany 87:875–882. doi:10.1139/B09-046
Zámocký M, Gasselhuber B, Furtmüller PG, Obinger C (2014) Turning points in the evolution of peroxidase–catalase superfamily: molecular phylogeny of hybrid heme peroxidases. Cell Mol Life Sci 71:4681–4696. doi:10.1007/s00018-014-1643-y
Fernández-Fueyo E, Ruiz-Dueñas FJ, Miki Y, et al (2012) Lignin-degrading peroxidases from genome of selective ligninolytic fungus Ceriporiopsis subvermispora. J Biol Chem 287:16903–16916. doi:10.1074/jbc.M112.356378
Morales M, Mate MJ, Romero A, et al (2012) Two oxidation sites for low redox potential substrates. J Biol Chem 287:41053–41067. doi:10.1074/jbc.M112.405548
Ruiz-Dueñas FJ, Lundell T, Floudas D, et al (2013) Lignin-degrading peroxidases in Polyporales: an evolutionary survey based on 10 sequenced genomes. Mycologia 105:1428–1444. doi:10.3852/13-059
Harms H, Schlosser D, Wick LY (2011) Untapped potential: exploiting fungi in bioremediation of hazardous chemicals. Nat Rev Microbiol 9:177–192. doi:10.1038/nrmicro2519
Hofrichter M (2002) Review: lignin conversion by manganese peroxidase (MnP). Enzym Microb Technol 30:454–466. doi:10.1016/S0141-0229(01)00528-2
Abdel-Hamid AM, Solbiati JO, Cann IKO (2013) Insights into lignin degradation and its potential industrial applications. Adv Appl Microbiol 82:1–28. doi:10.1016/B978-0-12-407679-2.00001-6
Hildén L, Johansson G, Pettersson G, et al (2000) Do the extracellular enzymes cellobiose dehydrogenase and manganese peroxidase form a pathway in lignin biodegradation? FEBS Lett 477:79–83. doi:10.1016/S0014-5793(00)01757-9
Lundell T, Wever R, Floris R, et al (1993) Lignin peroxidase L3 from Phlebia radiata. Pre-steady-state and steady-state studies with veratryl alcohol and a non-phenolic lignin model compound 1-(3,4-dimethoxyphenyl)-2-(2-methoxyphenoxy)propane-1,3-diol. Eur J Biochem FEBS 211:391–402
Salvachúa D, Prieto A, Martínez ÁT, Martínez MJ (2013) Characterization of a novel dye-decolorizing peroxidase (DyP)-type enzyme from Irpex lacteus and its application in enzymatic hydrolysis of wheat straw. Appl Environ Microbiol 79:4316–4324. doi:10.1128/AEM.00699-13
Liers C, Pecyna MJ, Kellner H, et al (2013) Substrate oxidation by dye-decolorizing peroxidases (DyPs) from wood- and litter-degrading agaricomycetes compared to other fungal and plant heme-peroxidases. Appl Microbiol Biotechnol 97:5839–5849. doi:10.1007/s00253-012-4521-2
Yoshida T, Tsuge H, Konno H, et al (2011) The catalytic mechanism of dye-decolorizing peroxidase DyP may require the swinging movement of an aspartic acid residue. FEBS J 278:2387–2394. doi:10.1111/j.1742-4658.2011.08161.x
Piontek K, Smith AT, Blodig W (2001) Lignin peroxidase structure and function. Biochem Soc Trans 29:111–116. doi:10.1042/0300-5127:0290111
Heinfling A, Ruiz-Dueñas FJ, Martınez MJ, et al (1998) A study on reducing substrates of manganese-oxidizing peroxidases from Pleurotus eryngii and Bjerkandera adusta. FEBS Lett 428:141–146. doi:10.1016/S0014-5793(98)00512-2
Garcia-Ruiz E, Gonzalez-Perez D, Ruiz-Dueñas FJ, et al (2012) Directed evolution of a temperature-, peroxide- and alkaline pH-tolerant versatile peroxidase. Biochem J 441:487–498. doi:10.1042/BJ20111199
Bugg TDH, Ahmad M, Hardiman EM, Singh R (2011) The emerging role for bacteria in lignin degradation and bio-product formation. Curr Opin Biotechnol 22:394–400. doi:10.1016/j.copbio.2010.10.009
Hofrichter M, Ullrich R, Pecyna MJ, et al (2010) New and classic families of secreted fungal heme peroxidases. Appl Microbiol Biotechnol 87:871–897. doi:10.1007/s00253-010-2633-0
Hofrichter M, Ullrich R (2014) Oxidations catalyzed by fungal peroxygenases. Curr Opin Chem Biol 19:116–125. doi:10.1016/j.cbpa.2014.01.015
Dana I, Colpa MWF (2013) DyP-type peroxidases: a promising and versatile class of enzymes. J Ind Microbiol Biotechnol 41:1–7. doi:10.1007/s10295-013-1371-6
Yoshida T, Sugano Y (2015) A structural and functional perspective of DyP-type peroxidase family. Arch Biochem Biophys 574:49–55. doi:10.1016/j.abb.2015.01.022
Martin Hofrichter RU (2009) New trends in fungal biooxidation. Ind Appl 10:425–449. doi:10.1007/978-3-642-11458-8_21
Piontek K, Strittmatter E, Ullrich R, et al (2013) Structural basis of substrate conversion in a new aromatic peroxygenase cytochrome P450 functionality with benefits. J Biol Chem 288:34767–34776. doi:10.1074/jbc.M113.514521
Peter S, Kinne M, Wang X, et al (2011) Selective hydroxylation of alkanes by an extracellular fungal peroxygenase. FEBS J 278:3667–3675. doi:10.1111/j.1742-4658.2011.08285.x
Scheibner M, Hülsdau B, Zelena K, et al (2007) Novel peroxidases of Marasmius scorodonius degrade β-carotene. Appl Microbiol Biotechnol 77:1241–1250. doi:10.1007/s00253-007-1261-9
Vincenza Faraco AP (2007) Identification of a new member of the dye-decolorizing peroxidase family from Pleurotus ostreatus. World J Microbiol Biotechnol 23:889–893. doi:10.1007/s11274-006-9303-5
Singh R, Eltis LD (2015) The multihued palette of dye-decolorizing peroxidases. Arch Biochem Biophys 574:56–65. doi:10.1016/j.abb.2015.01.014
Strittmatter E, Liers C, Ullrich R, et al (2013) First crystal structure of a fungal high-redox potential dye-decolorizing peroxidase: substrate interaction sites and long-range electron transfer. J Biol Chem 288:4095–4102. doi:10.1074/jbc.M112.400176
Strittmatter E, Serrer K, Liers C, et al (2015) The toolbox of Auricularia auricula-judae dye-decolorizing peroxidase–identification of three new potential substrate-interaction sites. Arch Biochem Biophys 574:75–85. doi:10.1016/j.abb.2014.12.016
Linde D, Pogni R, Cañellas M, et al (2015) Catalytic surface radical in dye-decolorizing peroxidase: a computational, spectroscopic and site-directed mutagenesis study. Biochem J 466:253–262. doi:10.1042/BJ20141211
Liers C, Bobeth C, Pecyna M, et al (2010) DyP-like peroxidases of the jelly fungus Auricularia auricula-judae oxidize nonphenolic lignin model compounds and high-redox potential dyes. Appl Microbiol Biotechnol 85:1869–1879. doi:10.1007/s00253-009-2173-7
Fernández-Fueyo E, Linde D, Almendral D, et al (2015) Description of the first fungal dye-decolorizing peroxidase oxidizing manganese(II). Appl Microbiol Biotechnol 99:8927–8942. doi:10.1007/s00253-015-6665-3
Levasseur A, Drula E, Lombard V, et al (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels 6:41. doi:10.1186/1754-6834-6-41
Yamada Y, Wang J, Kawagishi H, Hirai H (2014) Improvement of ligninolytic properties by recombinant expression of glyoxal oxidase gene in hyper lignin-degrading fungus Phanerochaete sordida YK-624. Biosci Biotechnol Biochem 78:2128–2133. doi:10.1080/09168451.2014.946398
Kersten P, Cullen D (2014) Copper radical oxidases and related extracellular oxidoreductases of wood-decay agaricomycetes. Fungal Genet Biol 72:124–130. doi:10.1016/j.fgb.2014.05.011
Ruiz-Dueñas FJ, Martínez AT (2009) Microbial degradation of lignin: how a bulky recalcitrant polymer is efficiently recycled in nature and how we can take advantage of this. Microb Biotechnol 2:164–177. doi:10.1111/j.1751-7915.2008.00078.x
Gibson A, Malek L, Dekker RFH, Ross B (2015) Detecting volatile compounds from Kraft lignin degradation in the headspace of microbial cultures by selected ion flow tube mass spectrometry (SIFT-MS). J Microbiol Methods 112:40–45. doi:10.1016/j.mimet.2015.03.008
Hammel KE, Kapich AN, Jensen KAJ, Ryan ZC (2002) Reactive oxygen species as agents of wood decay by fungi. Enzym Microb Technol 30:445–453. doi:10.1016/S0141-0229(02)00011-X
Henriksson G, Johansson G, Pettersson G (2000) A critical review of cellobiose dehydrogenases. J Biotechnol 78:93–113
Gómez-Toribio V, García-Martín AB, Martínez MJ, et al (2009) Induction of extracellular hydroxyl radical production by white-rot fungi through quinone redox cycling. Appl Environ Microbiol 75:3944–3953. doi:10.1128/AEM.02137-08
Brown ME, Chang MC (2014) Exploring bacterial lignin degradation. Curr Opin Chem Biol 19:1–7. doi:10.1016/j.cbpa.2013.11.015
Wu D, Hugenholtz P, Mavromatis K, et al (2009) A phylogeny-driven genomic encyclopaedia of bacteria and archaea. Nature 462:1056–1060. doi:10.1038/nature08656
Zimmermann W (1990) Degradation of lignin by bacteria. J Biotechnol 13:119–130. doi:10.1016/0168-1656(90)90098-V
Tian J-H, Pourcher A-M, Bouchez T, et al (2014) Occurrence of lignin degradation genotypes and phenotypes among prokaryotes. Appl Microbiol Biotechnol 98:9527–9544. doi:10.1007/s00253-014-6142-4
Ahmad M, Taylor CR, Pink D, et al (2010) Development of novel assays for lignin degradation: comparative analysis of bacterial and fungal lignin degraders. Mol BioSyst 6:815–821. doi:10.1039/b908966g
Taylor CR, Hardiman EM, Ahmad M, et al (2012) Isolation of bacterial strains able to metabolize lignin from screening of environmental samples. J Appl Microbiol 113:521–530. doi:10.1111/j.1365-2672.2012.05352.x
Scully ED, Geib SM, Carlson JE, et al (2014) Functional genomics and microbiome profiling of the Asian longhorned beetle (Anoplophora glabripennis) reveal insights into the digestive physiology and nutritional ecology of wood feeding beetles. BMC Genomics 15:1096. doi:10.1186/1471-2164-15-1096
Scully ED, Geib SM, Hoover K, et al (2013) Metagenomic profiling reveals lignocellulose degrading system in a microbial community associated with a wood-feeding beetle. PLoS ONE 8:e73827. doi:10.1371/journal.pone.0073827
Odier E, Janin G, Monties B (1981) Poplar lignin decomposition by gram-negative aerobic bacteria. Appl Environ Microbiol 41:337–341
McCarthy AJ, Broda P (1984) Screening for lignin-degrading actinomycetes and characterization of their activity against (14C)lignin-labelled wheat lignocellulose. Microbiology 130:2905–2913
Haider K, Trojanowski J, Sundman V (1978) Screening for lignin degrading bacteria by means of 14C-labelled lignins. Arch Microbiol 119:103–106. doi:10.1007/BF00407936
Pasti MB, Pometto AL, Nuti MP, Crawford DL (1990) Lignin-solubilizing ability of actinomycetes isolated from termite (Termitidae) gut. Appl Environ Microbiol 56:2213–2218
Pometto AL, Crawford DL (1986) Effects of pH on lignin and cellulose degradation by Streptomyces viridosporus. Appl Environ Microbiol 52:246–250
Crawford DL (1978) Lignocellulose decomposition by selected Streptomyces strains. Appl Environ Microbiol 35:1041–1045
McCarthy AJ, MacDonald MJ, Paterson A, Broda P (1984) Degradation of [14C]lignin-labelled wheat lignocellulose by white-rot fungi. J Gen Microbiol 130:1023–1030. doi:10.1099/00221287-130-5-1023
Vicuña R (1988) Bacterial degradation of lignin. Enzym Microb Technol 10:646–655. doi:10.1016/0141-0229(88)90055-5
Taylor CR (2013) Isolation of environmental lignin-degrading bacteria and identification of extracellular enzymes. University of Warwick, Coventry
Shi Y, Chai L, Tang C, et al (2013) Biochemical investigation of kraft lignin degradation by Pandoraea sp. B-6 isolated from bamboo slips. Bioprocess Biosyst Eng 36:1957–1965. doi:10.1007/s00449-013-0972-9
Chai L, Chen Y, Tang C, et al (2014) Depolymerization and decolorization of kraft lignin by bacterium Comamonas sp. B-9. Appl Microbiol Biotechnol 98:1907–1912. doi:10.1007/s00253-013-5166-5
Ramachandra M, Crawford DL, Hertel G (1988) Characterization of an extracellular lignin peroxidase of the lignocellulolytic actinomycete Streptomyces viridosporus. Appl Environ Microbiol 54:3057–3063
Mason JC, Richards M, Zimmermann W, Broda P (1988) Identification of extracellular proteins from actinomycetes responsible for the solubilisation of lignocellulose. Appl Microbiol Biotechnol 28:276–280. doi:10.1007/BF00250455
Magnuson TS, Crawford DL (1992) Comparison of extracellular peroxidase- and esterase-deficient mutants of Streptomyces viridosporus T7A. Appl Environ Microbiol 58:1070–1072
Mercer DK, Iqbal M, Miller P, McCarthy AJ (1996) Screening Actinomycetes for extracellular peroxidase activity. Appl Environ Microbiol 62:2186–2190
le Roes-Hill M, Khan N, Burton SG (2011) Actinobacterial peroxidases: an unexplored resource for biocatalysis. Appl Biochem Biotechnol 164:681–713. doi:10.1007/s12010-011-9167-5
Majumdar S, Lukk T, Solbiati JO, et al (2014) Roles of small laccases from Streptomyces in lignin degradation. Biochemistry (Mosc) 53:4047–4058. doi:10.1021/bi500285t
Chandra R, Chowdhary P (2015) Properties of bacterial laccases and their application in bioremediation of industrial wastes. Evnviron Sci Process Impacts 17:326–342. doi:10.1039/C4EM00627E
Santhanam N, Vivanco JM, Decker SR, Reardon KF (2011) Expression of industrially relevant laccases: prokaryotic style. Trends Biotechnol 29:480–489. doi:10.1016/j.tibtech.2011.04.005
Ausec L, Zakrzewski M, Goesmann A, et al (2011) Bioinformatic analysis reveals high diversity of bacterial genes for laccase-like enzymes. PLoS ONE 6:e25724. doi:10.1371/journal.pone.0025724
Dwivedi UN, Singh P, Pandey VP, Kumar A (2011) Structure–function relationship among bacterial, fungal and plant laccases. J Mol Catal B Enzym 68:117–128. doi:10.1016/j.molcatb.2010.11.002
Ladomersky E, Petris MJ (2015) Copper tolerance and virulence in bacteria. Metallomics 7:957–964. doi:10.1039/c4mt00327f
Geszvain K, McCarthy JK, Tebo BM (2013) Elimination of manganese(II,III) oxidation in pseudomonas putida GB-1 by a double knockout of two putative multicopper oxidase genes. Appl Environ Microbiol 79:357–366. doi:10.1128/AEM.01850-12
Tebo BM, Johnson HA, McCarthy JK, Templeton AS (2005) Geomicrobiology of manganese(II) oxidation. Trends Microbiol 13:421–428. doi:10.1016/j.tim.2005.07.009
Singh G, Batish M, Sharma P, Capalash N (2009) Xenobiotics enhance laccase activity in alkali-tolerant γ-proteobacterium JB. Braz J Microbiol 40:26–30. doi:10.1590/S1517-83822009000100004
Santos A, Mendes S, Brissos V, Martins LO (2013) New dye-decolorizing peroxidases from Bacillus subtilis and Pseudomonas putida MET94: towards biotechnological applications. Appl Microbiol Biotechnol 98:2053–2065. doi:10.1007/s00253-013-5041-4
Sturm A, Schierhorn A, Lindenstrauss U, et al (2006) YcdB from Escherichia coli reveals a novel class of tat-dependently translocated hemoproteins. J Biol Chem 281:13972–13978. doi:10.1074/jbc.M511891200
Yu W, Liu W, Huang H, et al (2014) Application of a novel alkali-tolerant thermostable DyP-type peroxidase from Saccharomonospora viridis DSM 43017 in biobleaching of eucalyptus kraft pulp. PLoS One 9:e110319. doi:10.1371/journal.pone.0110319
Létoffé S, Heuck G, Delepelaire P, et al (2009) Bacteria capture iron from heme by keeping tetrapyrrol skeleton intact. Proc Natl Acad Sci U S A 106:11719–11724. doi:10.1073/pnas.0903842106
Ogola HJO, Kamiike T, Hashimoto N, et al (2009) Molecular characterization of a novel peroxidase from the cyanobacterium Anabaena sp. strain PCC 7120. Appl Environ Microbiol 75:7509–7518. doi:10.1128/AEM.01121-09
Brown ME, Barros T, Chang MCY (2012) Identification and characterization of a multifunctional dye peroxidase from a lignin-reactive bacterium. ACS Chem Biol 7:2074–2081. doi:10.1021/cb300383y
van Bloois E, Pazmiño DET, Winter RT, Fraaije MW (2010) A robust and extracellular heme-containing peroxidase from Thermobifida fusca as prototype of a bacterial peroxidase superfamily. Appl Microbiol Biotechnol 86:1419–1430. doi:10.1007/s00253-009-2369-x
Sugano Y, Muramatsu R, Ichiyanagi A, et al (2007) DyP, a unique dye-decolorizing peroxidase, represents a novel heme peroxidase family: ASP171 replaces the distal histidine of classical peroxidases. J Biol Chem 282:36652–36658. doi:10.1074/jbc.M706996200
Singh R, Grigg JC, Armstrong Z, et al (2012) Distal heme pocket residues of B-type dye-decolorizing peroxidase: arginine but not aspartate is essential for peroxidase activity. J Biol Chem 287:10623–10630. doi:10.1074/jbc.M111.332171
Mendes S, Brissos V, Gabriel A, et al (2015) An integrated view of redox and catalytic properties of B-type PpDyP from Pseudomonas putida MET94 and its distal variants. Arch Biochem Biophys 574:99–107. doi:10.1016/j.abb.2015.03.009
Binesse J, Lindgren H, Lindgren L, et al (2015) Roles of reactive oxygen species-degrading enzymes of Francisella tularensis SCHU S4. Infect Immun 83:2255–2263. doi:10.1128/IAI.02488-14
Turlin E, Débarbouillé M, Augustyniak K, et al (2013) Staphylococcus aureus FepA and FepB proteins drive heme iron utilization in Escherichia coli. PLoS ONE 8:e56529. doi:10.1371/journal.pone.0056529
Ahmad M, Roberts JN, Hardiman EM, et al (2011) Identification of DypB from Rhodococcus jostii RHA1 as a lignin peroxidase. Biochemistry (Mosc) 50:5096–5107. doi:10.1021/bi101892z
Strachan C, VanInsberghe D, Williams D (2012) Ligninase activity is not consistently predicted by the presence of manganese coordinating residues in Dyp-like proteins. J Exp Microbiol Immunol 16:66–72
Roberts JN, Singh R, Grigg JC, et al (2011) Characterization of dye-decolorizing peroxidases from Rhodococcus jostii RHA1. Biochemistry (Mosc) 50:5108–5119. doi:10.1021/bi200427h
Singh R, Grigg JC, Qin W, et al (2013) Improved manganese-oxidizing activity of DypB, a peroxidase from a lignolytic bacterium. ACS Chem Biol 8:700–706. doi:10.1021/cb300608x
Rahmanpour R, Bugg TDH (2013) Assembly in vitro of Rhodococcus jostii RHA1 encapsulin and peroxidase DypB to form a nanocompartment. FEBS J 280:2097–2104. doi:10.1111/febs.12234
Min K, Gong G, Woo HM, et al (2015) A dye-decolorizing peroxidase from Bacillus subtilis exhibiting substrate-dependent optimum temperature for dyes and β-ether lignin dimer. Sci Rep 5:8245. doi:10.1038/srep08245
Mai-Prochnow A, Lucas-Elio P, Egan S, et al (2008) Hydrogen peroxide linked to lysine oxidase activity facilitates biofilm differentiation and dispersal in several Gram-negative bacteria. J Bacteriol 190:5493–5501. doi:10.1128/JB.00549-08
Jin J, Mazon H, van den Heuvel RHH, et al (2007) Discovery of a eugenol oxidase from Rhodococcus sp. strain RHA1. FEBS J 274:2311–2321. doi:10.1111/j.1742-4658.2007.05767.x
Phugare SS, Waghmare SR, Jadhav JP (2011) Purification and characterization of dye degrading of veratryl alcohol oxidase from Pseudomonas aeruginosa strain BCH. World J Microbiol Biotechnol 27:2415–2423. doi:10.1007/s11274-011-0714-6
Tamboli DP, Telke AA, Dawkar VV, et al (2011) Purification and characterization of bacterial aryl alcohol oxidase from Sphingobacterium sp. ATM and its uses in textile dye decolorization. Biotechnol Bioprocess Eng 16:661–668. doi:10.1007/s12257-011-0031-9
Masai E, Katayama Y, Fukuda M (2007) Genetic and biochemical investigations on bacterial catabolic pathways for lignin-derived aromatic compounds. Biosci Biotechnol Biochem 71:1–15
Yoshikata T, Suzuki K, Kamimura N, et al (2014) A three-component O-demethylase system essential for catabolism of a lignin-derived biphenyl compound in Sphingobium sp. strain SYK-6. Appl Environ Microbiol 80:7142–7153. doi:10.1128/AEM.02236-14
Gall DL, Kim H, Lu F, et al (2014) Stereochemical features of glutathione-dependent enzymes in the Sphingobium sp. strain SYK-6 β-aryl etherase pathway. J Biol Chem 289:8656–8667. doi:10.1074/jbc.M113.536250
Mishra S, Sachan A, Sachan SG (2013) Production of natural value-added compounds: an insight into the eugenol biotransformation pathway. J Ind Microbiol Biotechnol 40:545–550. doi:10.1007/s10295-013-1255-9
Fuchs G, Boll M, Heider J (2011) Microbial degradation of aromatic compounds–from one strategy to four. Nat Rev Microbiol 9:803–816. doi:10.1038/nrmicro2652
Wells Jr T, Ragauskas AJ (2012) Biotechnological opportunities with the β-ketoadipate pathway. Trends Biotechnol 30:627–637. doi:10.1016/j.tibtech.2012.09.008
Sainsbury PD, Hardiman EM, Ahmad M, et al (2013) Breaking down lignin to high-value chemicals: the conversion of lignocellulose to vanillin in a gene deletion mutant of Rhodococcus jostii RHA1. ACS Chem Biol 8:2151–2156. doi:10.1021/cb400505a
Acknowledgements
We would like to thank Prof. Dr. Thomas Hirth for making this work possible at Fraunhofer IGB and IGVP, University Stuttgart and Dr. Steffen Rupp for the support at the department.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Rais, D., Zibek, S. (2017). Biotechnological and Biochemical Utilization of Lignin. In: Wagemann, K., Tippkötter, N. (eds) Biorefineries. Advances in Biochemical Engineering/Biotechnology, vol 166. Springer, Cham. https://doi.org/10.1007/10_2017_6
Download citation
DOI: https://doi.org/10.1007/10_2017_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-97117-9
Online ISBN: 978-3-319-97119-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)