
Abstract
Purpose of Review
This review presents an overview of the current knowledge of tumor necrosis factor receptor (TNF-R)-associated factor (TRAF) molecules in inflammation with an emphasis on available human evidence and direct in vivo evidence of mouse models that demonstrate the contribution of TRAF molecules in the pathogenesis of inflammatory diseases.
Recent Findings
The TRAF family of cytoplasmic proteins was initially identified as signaling adaptors that bind directly to the intracellular domains of receptors of the TNF-R superfamily. It is now appreciated that TRAF molecules are widely employed in signaling by a variety of adaptive and innate immune receptors as well as cytokine receptors. TRAF-dependent signaling pathways typically lead to the activation of nuclear factor-κBs (NF-κBs), mitogen-activated protein kinases (MAPKs), or interferon-regulatory factors (IRFs). Most of these signaling pathways have been linked to inflammation, and therefore, TRAF molecules were expected to regulate inflammation and inflammatory responses since their discovery in the 1990s. However, direct in vivo evidence of TRAFs in inflammation and especially in inflammatory diseases had been lacking for many years, partly due to the difficulty imposed by early lethality of TRAF2−/−, TRAF3−/−, and TRAF6−/− mice. With the creation of conditional knockout and lineage-specific transgenic mice of different TRAF molecules, our understanding about TRAFs in inflammation and inflammatory responses has rapidly advanced during the past decade.
Summary
Increasing evidence indicates that TRAF molecules are versatile and indispensable regulators of inflammation and inflammatory responses and that aberrant expression or function of TRAFs contributes to the pathogenesis of inflammatory diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The tumor necrosis factor receptor (TNF-R)-associated factor (TRAF) family of cytoplasmic proteins was initially identified as signaling adaptors that bind directly to the intracellular domains of receptors of the TNF-R superfamily [1, 2, 3••]. It is now recognized that TRAF molecules are widely employed in signaling by a variety of adaptive and innate immune receptors as well as cytokine receptors [3••, 4•, 5]. Adaptive immune receptors that can directly recruit TRAF proteins include T cell receptor, CD28, and co-stimulatory receptors of the TNF-R superfamily (such as CD40, BAFF-R, TACI, BCMA, 4-1BB, OX-40, GITR, CD27, CD30, DR3, HVEM, and TNF-R2) [3••, 4•, 5]. Innate immune receptors that can indirectly employ TRAF proteins in signaling include Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors, and C-type lectin receptors [3••, 5]. Cytokine receptors that can directly or indirectly recruit TRAF proteins include receptors for IL-1β, IL-2, IL-6, IL-17, IL-18, IL-33, type I IFNs, type III IFNs, GM-CSF, M-CSF, and TGF-β [3••, 4•, 5].
Upon ligand engagement, one major role of TRAF molecules is to serve as adaptor proteins in the assembly of receptor-associated signaling complexes, linking upstream receptors to downstream adaptor proteins and effector enzymes [3••, 4•, 5]. This is mediated by the C-terminal TRAF domain, a distinct feature of all TRAF proteins except TRAF7 (a figure of TRAF1-7 structures is provided in a previous review [3••]). The TRAF domain is further divided into N-terminal coiled-coil region (TRAF-N) and a C-terminal β-sandwich domain (TRAF-C or MATH domain) [2, 3••, 6]. The uniqueness and specificity of the binding of each TRAF molecule to various receptors is mainly mediated by minor structural differences in the TRAF-C domain that recognizes major and minor consensus sequences of the cytoplasmic tails of receptors or their associated adaptor proteins [7, 8]. However, a recent study points out that the binding preferences of TRAF proteins may be more complicated than previously appreciated as TRAF molecules also exhibit binding preferences beyond the established core motifs [9], thus warranting further investigation on this aspect. In addition to their role as adaptor proteins, TRAFs (including TRAF2, 3, 5, and 6) also act as E3 ubiquitin ligases [3••, 4•, 5]. TRAF-dependent signaling pathways typically lead to the activation of nuclear factor-κBs (NF-κB1 and NF-κB2), mitogen-activated protein kinases (MAPKs: ERK1/2, JNK1/2, and p38), or interferon-regulatory factors (IRFs: IRF3, IRF5, and IRF7). Therefore, TRAFs function as both adaptor proteins and E3 ubiquitin ligases to regulate receptor signaling in adaptive and innate immune responses as well as other biological processes [3••, 4•, 5].
Because of the prominent importance of the TNF superfamily in inflammation, TRAF molecules were expected to regulate inflammation and inflammatory responses since their discovery as TNF-R-interacting proteins in the 1990s [1, 2]. However, the in vivo functions of TRAFs in inflammation and especially in inflammatory diseases had remained elusive for many years, partly due to the difficulty imposed by early lethality and multiple organ abnormalities of TRAF2−/−, TRAF3−/−, and TRAF6−/− mice [3••]. With the creation of conditional knockout and lineage-specific transgenic mice of different TRAF molecules, our understanding about TRAFs in inflammation and inflammatory responses has rapidly advanced during the past decade. Here, we provide an overview of current knowledge of TRAF molecules in inflammation with an emphasis on available human evidence and direct in vivo evidence of mouse models that demonstrate or implicate the contribution of TRAF molecules in the pathogenesis of inflammatory diseases.
TRAF1
Increasing evidence indicates that as a signaling adaptor, TRAF1 regulates inflammatory responses to the pro-inflammatory cytokine TNFα and microbial ligands of TLRs [3••, 10,11,12,13, 14••]. Expression of TRAF1 is restricted to the spleen, lung and testis under normal conditions [10, 15, 16]. As a direct NF-κB target gene, TRAF1 expression is often upregulated by TNFα and other inflammatory stimuli [17,18,19,20]. When expressed in cells, TRAF1 protein regulates inflammation by directly interacting with TNF-R2, TRAF2, TRIF, IKK2, NIK, and ASK1 [3••, 10, 13, 15, 20,21,22,23,24,25]. Therefore, TRAF1 is able to regulate both canonical and non-canonical NF-κB pathways as well as activation of the MAP kinases (JNK, p38, and ERK) to influence pro-inflammatory cytokine production and inflammatory responses [3••, 10, 13, 20, 22,23,24,25,26]. Recent human and animal studies provide evidence implicating TRAF1 in rheumatoid arthritis, lung inflammation, liver inflammation, and atherosclerosis.
Sepsis and Rheumatoid Arthritis
Genome-wide association studies first identified single-nucleotide polymorphisms (SNPs) at the TRAF1-C5 locus (encoding TRAF1 and complement component 5) on chromosome 9 as risk factors for rheumatoid arthritis (RA) in human patients [27,28,29,30,31,32]. It was subsequently found that SNPs of the TRAF1-C5 locus predict the clinical response to anti-TNF therapy in RA patients [33, 34] and that increased serum levels of TRAF1 correlate with disease activity and autoantibodies in RA patients [35]. In particular, the TRAF1/C5 SNP rs3761847 GG homozygote status is associated with an increased risk of death from sepsis and malignancies in RA patients [36]. Interestingly, it was recently revealed that this SNP (rs3761847 GG) leads to reduced levels of TRAF1 protein in monocytes and T cells [14••]. Monocytes from healthy human subjects with SNP rs3761847 GG of the TRAF1 gene produce increased amounts of pro-inflammatory cytokines in response to lipopolysaccharide (LPS) due to enhanced NF-κB activation [14••]. Mechanistically, TRAF1 inhibits TLR4-induced NF-κB activation by interfering with the linear ubiquitination of NEMO through direct interaction of TRAF1 with three components of the linear ubiquitination (LUBAC) complex, SHARPIN, HOIP, and HOIL-1 [14••]. Consistent with the negative role of TRAF1 in LPS-induced inflammation observed in human monocytes, TRAF1−/− mice are more susceptible to LPS-induced septic shock [14••]. Furthermore, in a genetic mouse model of inflammatory arthritis, KRN/I-A(g7) (KxB/N) mice, the production of anti-GPI autoantibody is markedly impaired by TRAF1 deficiency [37]. These new findings elucidate how this RA-associated TRAF1 SNP could contribute to the increased incidence and severity of sepsis, arthritis, and other inflammatory diseases.
Lung Inflammation
TRAF1 has been shown to be involved in lung inflammation with several different mouse models, including intratracheal TNFα-induced inflammation, LPS inhalation-induced inflammation, and allergic lung inflammation. Intratracheal TNFα-treated TRAF1−/− mice exhibit more severe TNFRI-dependent liver injury due to increased production of TNFα by bronchoalveolar cells, suggesting a negative role of TRAF1 in TNFα-induced lung inflammation [38]. Inhaled LPS induces an inflammatory response that may contribute to the pathogenesis of asthma and other airway diseases. Interestingly, TRAF1−/− mice are deficient in recruiting lymphocytes to the lower respiratory tract after inhalation of LPS, due to decreased expression of ICAM1, VCAM1, CCL17, and CCL20 in the lungs [39]. Experiments of bone marrow chimeras demonstrate that TRAF1 in resident lung cells, but not hematopoietic cells, is responsible for this phenotype [39]. Mice lacking TNFR1 but not TNFR2 show a phenotype similar to the TRAF1−/− mice, suggesting a positive role of the TNFR1-TRAF1 pathway in the induction of chemokines and adhesion molecules in resident lung cells after LPS inhalation [39]. Similarly, TRAF1 expressed in resident lung cells is also required for the development of allergic lung inflammation as revealed by adoptive transfer experiments of ovalbumin (OVA)-immune wild type (WT) CD4+ T cells. In response to OVA exposure, TRAF1−/− recipient mice fail to display eosinophilic inflammation and airway hyperresponsiveness in this model of asthma [40]. In sharp contrast, transfer of OVA-immune TRAF1−/− T cells into naive WT recipients confers significantly more intense pulmonary inflammation and higher airway hyperresponsiveness following inhaled OVA challenge [41]. This is caused by the T cell-intrinsic bias of TRAF1−/− T cells to produce increased amount of the Th2 cytokines (IL-4, IL-5, and IL-13) in response to antigen stimulation [41]. Biochemical analyses revealed that TRAF1 inhibits the induction of Th2 responses by associating with NIP45 in the cytoplasm and thereby preventing the nuclear translocation of NIP45, a Th2 cell-associated transcription factor [41]. Therefore, TRAF1 proteins expressed in resident lung cells and T cells play differential roles in lung inflammation.
Atherosclerosis
A recent study by Hessler et al. identified a SNP rs2416804 (GC alleles) of the TRAF1 gene as being associated with carotid intima-media thickness, a marker for subclinical atherosclerosis that predicts subsequent clinical cardiovascular events [42]. TRAF1 expression is significantly upregulated in atherosclerotic plaques of patients with atherosclerosis and also in the blood of patients with acute coronary syndrome [43, 44]. Consistent with the human evidence, TRAF1 deficiency in mice attenuates atherogenesis and impairs monocyte recruitment to the vessel wall as demonstrated by studies with TRAF1−/−LDLR−/− mice on a high-cholesterol diet [44]. Bone marrow transplantations revealed that TRAF1 deficiency in both hematopoietic and vascular resident cells contributes to the observed reduction in atherogenesis in mice [44]. Impaired monocyte recruitment is associated with decreased expression of the adhesion molecules ICAM1 and VCAM1 on endothelial cells [44]. Together, the above evidence warrants further investigation of TRAF1 in the pathogenesis of atherosclerosis.
Liver Inflammation and Hepatic Steatosis
Xiang et al. recently reported that TRAF1 expression is elevated in the livers of human patients with non-alcoholic fatty liver disease (NAFLD) [45]. Interestingly, both high-fat diet (HFD)-treated mice and genetic obese mice (ob/ob) exhibit an increase in TRAF1 expression in the liver compared with lean controls [45]. Palmitate, an inducer of lipid accumulation and insulin resistance in hepatocytes, also increases TRAF1 expression in hepatocytes [45]. Xiang et al. further investigated the role of TRAF1 in insulin resistance, inflammation, and hepatic steatosis using mice with global TRAF1 deficiency or liver-specific TRAF1 overexpression. In response to HFD treatment or in ob/ob mice, TRAF1 deficiency is hepatoprotective, whereas TRAF1 overexpression in hepatocytes exacerbates the pathological development of insulin resistance, inflammatory response and hepatic steatosis. A similar liver protective role of TRAF1 deficiency was also demonstrated in a mouse hepatic ischemia/reperfusion (I/R) injury model by Zhang et al. [25]. Mechanistically, hepatocyte TRAF1 directly interacts with ASK1 to promote hepatic steatosis through enhancing the activation of ASK1-mediated P38 and JNK cascades in response to HFD or palmitate stimulation [45]. Indeed, overexpression of a constitutively active form of ASK1 in the liver reverses TRAF1 deficiency-mediated amelioration of obesity and insulin resistance, while overexpression of a dominant negative form of ASK1 in the liver reverses TRAF1 overexpression-induced exacerbation of inflammation and hepatic steatosis [45]. Thus, the TRAF1-ASK1 axis acts to induce insulin resistance, inflammation, and hepatic steatosis in response to HFD or palmitate stimulation [45].
Brain Inflammation and Ischemic Stroke
In an experimental I/R stroke model, TRAF1 expression is markedly induced in the cortex and stratum of WT mice at 6 h after middle cerebral artery occlusion (MCAO)-induced stroke onset [24]. In cultured primary cortical neurons, oxygen and glucose deprivation also rapidly increases TRAF1 protein levels [24]. Neuron-specific TRAF1 transgenic mice exhibit enlarged inflammatory stroke lesions and detrimental behavioral and neurological dysfunction following MCAO, which is attributable to the enhancement of neuronal apoptosis [24]. Conversely, TRAF1−/− mice have reduced inflammatory stroke lesions and ameliorated behavioral and neurological dysfunction associated with reduced neuronal apoptosis [24]. Mechanistically, TRAF1 directly interacts with ASK1 to induce the MKK4/7-JNK1 pro-apoptotic pathway while inhibiting the Akt-mTOR-CREB pro-survival pathway in neurons [24]. Together, these in vivo studies demonstrate the pathogenic roles of TRAF1 in neurons during ischemic stroke.
TRAF2
TRAF2 is ubiquitously expressed in various cell types and is especially important for inflammation in keratinocytes, macrophages, dendritic cells (DCs), T cells, hepatocytes, epithelial cells, and fibroblasts [3••, 4•, 5, 46, 47••, 48,49,50]. As a signaling adaptor protein and an E3 ubiquitin ligase, TRAF2 regulates inflammatory responses mediated by receptors of the TNF-R superfamily, TLRs, NLRs, RIG-I, cytokine receptors, S100A8 receptors, and S100A9 receptors [3••, 4•, 5, 46, 49, 51, 52]. TRAF2 transduces receptor signals to induce the activation of both canonical and non-canonical NF-κB pathways as well as the activation of the MAPKs (JNK, ERK1/2 and p38) and IRFs (IRF3, IRF5 and IRF7) [3••, 4•, 5,46,49]. Intracellular TRAF2 is also required for ER stress-induced inflammatory responses via the IRE1α-TRAF2-Nur77 signaling axis [53,54,55]. TRAF2 directly interacts with a variety of receptors, adaptor proteins, and enzymes that regulate inflammation. These include receptors of the TNF-R superfamily, IL-17Rs, IL-15Rα, IFNAR1, EMMPRIN, TRAF3, TRAF5, cIAP1/2, Ubc13, TRADD, TRIF, RIP, Act1, MAVS, SOCS3, TAK1, IKKε, A20, CYLD, MCPIP1, HGK, MLKL, IRE1α, and Nur77, among others [3••, 4•, 5, 46, 49,50,51,52, 54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74]. Current human and mouse evidence indicates that TRAF2 plays complex roles in skin inflammation, cardiovascular inflammation, inflammatory bowel diseases, liver inflammation, and autoimmune inflammatory diseases (such as lupus, arthritis, and multiple sclerosis).
Skin Inflammation
In primary human keratinocytes, exposure to ultraviolet (UV) light triggers association of TRAF2 with TNF-R1 to induce NF-κB activation and inflammation [75]. In mice, keratinocyte-specific deletion of TRAF2 (K-TRAF2−/−) causes epidermal hyperplasia and psoriatic skin inflammation with excessive leukocyte infiltration and apoptotic death in the inflamed area by 15 weeks of age [76]. This phenotype partially depends on TNF-induced apoptosis in keratinocytes, as compound deletion of TNF in K-TRAF2−/− mice reduces apoptotic death and delays the development of skin inflammation [76]. Another essential pathway underlying this phenotype is the constitutive NF-κB2 activation and increased expression of inflammatory molecules observed in TRAF2−/− keratinocytes, including M-CSF, IL-23, 4-1BBL, CR1L, and CXCL-16 [76]. Indeed, compound deletion of TNF and NF-κB2 in K-TRAF2−/− mice prevents the skin inflammation caused by TRAF2 deficiency in keratinocytes [76]. Interestingly, S100A8 and S100A9, two inflammatory proteins highly upregulated in skin lesions of human patients with atopic dermatitis, are found to bind to receptors named Neuroplastin-β and EMMPRIN, which recruit GRB2 and TRAF2 to induce NF-κB1 activation, keratinocyte proliferation, and skin inflammation [51, 52]. Consistent with the critical roles of TRAF2 in skin inflammation, mutations of the TRAF2-deubiquitinating enzyme CYLD are identified in patients with familial cylindromatosis (with benign tumors of skin appendages), and CYLD−/− mice are highly susceptible to chemically induced skin tumors [72]. Therefore, TRAF2 expressed in keratinocytes plays protective roles in skin inflammation through multiple pathways.
Cardiovascular Inflammation
Increasing evidence indicates an important role for inflammation in cardiac hypertrophy and failure [77, 78]. TRAF2 expression is upregulated in human atherosclerotic plaques and failing mouse hearts [43, 79]. Two groups independently demonstrated that cardiac-specific TRAF2 overexpression in mice leads to remarkably enhanced cardiac hypertrophy, left ventricular dysfunction, and adverse cardiac remodeling, which are associated with increased activation of NF-κB, JNK and Akt-GSK3 β [79, 80]. It would be interesting to further verify these findings using cardiac-specific TRAF2−/− mice and to investigate the roles of TRAF2 in mouse models of atherosclerosis.
Inflammatory Bowel Diseases
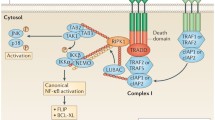
In humans, the expression of TRAF2 is significantly higher in inflamed and non-inflamed tissues of IBD patients than those in healthy control [81, 82]. TRAF2 expression is also higher in inflamed colonic mucosa tissues than in non-inflamed tissues in patients with Crohn’s disease (CD), ulcerative colitis (UC), and nonspecific colitis [81, 82]. Moreover, higher expression of TRAF2 has been identified as a prognosis factor of UC relapse [82]. Liu et al. recently reported that TRAF2 overexpression may result from EZH2 downregulation-mediated epigenetic mechanism in IBD patients [83]. In mice, the roles of TRAF2 in colitis have been demonstrated in both germline TRAF2−/− and myeloid cell-specific TRAF2−/− (M-TRAF2−/−) mice. Germline TRAF2−/− mice spontaneously develop severe colitis and succumb within 3 weeks after birth, which result from TNFα-TNFR1-mediated apoptosis of TRAF2−/− colonic epithelial cells and altered colonic microbiota (Fig. 1) [48]. In the absence of TRAF2, compromised epithelial barrier allows commensal bacteria to induce the accumulation of IL-10-secreting neutrophils in the bone marrow, peripheral blood, and lamina propria (Fig. 1) [84]. Combined treatment with neutralizing antibodies against TNFα and IL-10 substantially ameliorates colitis and prolongs survival in TRAF2−/− mice [84]. Myeloid cell-specific TRAF2 ablation promotes DSS-induced colitis in mice, which is associated with increased production of pro-inflammatory cytokines (TNFα, IL-1 β, IL-6, and IL-12) and decreased levels of IL-10 [47••]. The anti-inflammatory function of TRAF2 is independent of NIK-NK-κB2 and is mediated by elevated protein levels of c-Rel and IRF5 in macrophages [47••]. Mechanistically, c-Rel and IRF5 are constitutively targeted for K48-linked ubiquitination and proteasome-mediated degradation by the TRAF3-TRAF2-cIAP1/2 complex similar to that previously described for NIK [47••]. Thus, TRAF2 plays protective roles against colitis by acting in both epithelial cells and myeloid cells (Fig. 1).

Cell- and receptor-specific roles of TRAF molecules in inflammatory bowel diseases. The cell types with direct in vivo evidence of TRAFs in IBDs include macrophages (MØ), DCs, neutrophils, TH2 cells, TH17 cells, and epithelial cells (EPC). TRAF molecules (TRAF2, 3, 5, and 6) with anti-inflammatory roles in IBDs are depicted in blue in each specific cell type, and TRAF6 with pro-inflammatory roles in IBDs is depicted in pink in TH17 cells. The dominant TRAF-dependent receptors, TRAF-interacting transcription factors, and TRAF-dependent downstream cytokines and chemokines are shown for each specific cell type as revealed by in vivo evidence obtained from mouse models of IBDs. In addition, TRAF6-mediated regulation of DCs indirectly controls the differentiation of TH1 and Treg cells, which also contribute to the protective effects of DC TRAF6 against enteritis. The known roles of TRAFs in TCR signaling, the development and homeostasis of iNKT cells and T cell subsets, and the activation of CD4 and CD8 T cells are not included in the figure, as their contribution in IBDs has not been directly tested in vivo in whole animal models.
Liver Inflammation
Expression of TRAF2 is downregulated during hepatic differentiation of human pluripotent stem cells, and infection with hepatitis B virus (HBV) induces the expression of TRAF2 in normal primary human hepatocytes [85, 86]. Studies of mouse models revealed that TRAF2 regulates liver inflammation by inhibiting apoptosis and necroptosis in hepatocytes. Tamoxifen-induced TRAF2 deletion in adult mice leads to increased hepatic necroptosis and rapid lethality, which is delayed by co-deletion of RIPK3 or treatment with blocking reagents for TNF-R1 and IFNAR1 [50]. Hepatic TRAF2 depletion by siRNA injection in mice exacerbates Fas-induced hepatic apoptosis and accelerates lethality, as TRAF2 mediates K48-linked ubiquitination and degradation of caspase 8 [87]. Interestingly, hepatocyte-specific deletion of TRAF2 in mice does not alter insulin signaling under normal or HFD conditions, but does attenuate HFD-induced hyperglycemia and obesity due to decreased hepatic gluconeogenesis, resulting in glucagon resistance [88]. In mouse primary hepatocytes, TRAF2 directly promotes glucose production by enhancing glucagon-induced CREB phosphorylation and the expression of PEPCK and G6Pase [88]. Furthermore, specific deletion of TRAF2 from liver parenchymal cells in mice leads to mild and focal spontaneous ductular reaction, while co-deletion of TRAF2 and RIP in these cells results in spontaneous hepatocyte apoptosis, hepatitis, and hepatocellular carcinoma [57]. Taken together, TRAF2 plays complex and indispensable roles in regulating hepatocyte survival and function as well as liver inflammation.
Brain Inflammatory Diseases
Upregulated expression of TRAF2 is detected in the brains of patients with Alzheimer’s disease (AD) and Parkinson’s disease (PD) as well as in the hippocampi of patients with mesial temporal lobe epilepsy [89,90,91]. Interestingly, the 3′ UTR SNP rs7852970 GG of the Traf2 gene is significantly protective against AD as revealed by SNP association studies [89]. TRAF2 proteins are present within plaque-associated neurites and some neurofibrillary tangles in human AD brains [89]. However, the causal role of TRAF2 in brain inflammatory diseases has not been demonstrated with in vivo models and needs further investigation.
Autoimmune Inflammatory Diseases
In human, both down- and upregulated expression of TRAF2 has been detected in peripheral blood mononuclear cells (PBMCs) of patients with systemic lupus erythematosus (SLE) [92, 93]. TRAF2 expression is elevated in PBMCs of patients with RA and relapsing-remitting multiple sclerosis (MS) [94, 95]. Studies of mouse models revealed the critical role of TRAF2 in regulating T cell homeostasis and T cell tolerance, which are essential for controlling autoimmune inflammatory diseases. T cell-specific TRAF2−/− (T-TRAF2−/−) mice display decreased naïve and memory CD8 T cell subsets and NKT cells in the spleen and liver due to reduced sensitivity to IL-15 [49]. TRAF2−/−TNFα−/− mice develop an inflammatory disorder characterized by lymphocyte infiltration in multi-organs and accumulation of anti-dsDNA and anti-histone autoantibodies [96]. The pathogenic TRAF2−/−TNFα−/− T cells show constitutive NF-κB2 activation and produce elevated levels of TH1 and TH17 cytokines, including IFNγ, IL-17, IL-21, and IP-10 [96]. Interestingly however, T cell-specific deficiency of HGK, a MAP4K that directly phosphorylates TRAF2 and targets TRAF2 for lysosomal degradation, also leads to systemic inflammation involving multi-organs and insulin resistance in mice, which are associated with increased levels of IL-6, IL-17, and TH17 cells [74]. Therefore, a delicately balanced level of TRAF2 proteins in T cells is required to sustain T cell tolerance, and both increased or decreased TRAF2 protein levels can lead to autoimmune inflammatory diseases [74, 96].
TRAF3
TRAF3 is closely related to TRAF2 in terms of both structure and function [5, 9]. TRAF3 is ubiquitously expressed in various cell types and is pivotal in regulating inflammation in macrophages, dendritic cells (DCs), neutrophils, B cells, T cells, hepatocytes, cardiocytes, osteoclasts, microglia, and neurons (Fig. 2) [3••, 4•, 5, 47••, 97, 98•, 99,100,101,102,103,104,105,106,107,108,109,110]. Similar to TRAF2, TRAF3 acts as both a signaling adaptor protein and an E3 ubiquitin ligase in inflammatory responses mediated by receptors of the TNF-R superfamily, TLRs, NLRs, RIG-I, and cytokine receptors [3••, 4•, 5, 100, 111]. TRAF3 has overlapping functions with TRAF2 in inducing the activation of both canonical and non-canonical NF-κB pathways as well as the activation of the MAPKs (JNK1/2, ERK1/2 and p38) and IRFs (IRF3, IRF5 and IRF7) [3••, 5, 100, 111]. However, TRAF3 also has distinct roles and unique binding proteins, and therefore cannot be substituted by TRAF2 in inflammation and inflammatory responses. Direct TRAF3-interacting proteins that are important for inflammation include: (1) receptors: members of the TNF-R superfamily that do not contain death domains, NLRP12, and IL-17R [3••, 5, 97, 112]; (2) adaptor proteins: TRAF2, TRAF5, MyD88, TRIF, RIP, RIP2, MAVS, Act1, STING, and ASC [3••, 5, 113,114,115]; (3) enzymes: NIK, IKKε, TAK1, TBK1, Peli1, A20, DUBA, OTUB1, OTUD7B, USP25, CHIP, MYSM1, PTPN22, Syk, CK1ε, DNA-PKc, and NDR1, among others [3••, 5, 116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131]; (4) transcription factors: c-Rel and IRF5 (Fig. 2) [47••]. Increasing human and mouse evidence indicates that alterations in TRAF3 expression or function contribute to the pathogenesis of systemic inflammation, cardiac hypertrophy, inflammatory bowel diseases, liver inflammation, diabetes, bone inflammatory diseases, brain inflammation, and autoimmune inflammatory diseases (such as lupus, arthritis, and multiple sclerosis) (Fig. 2).

Cell-specific roles and signaling networks of TRAF3 in inflammatory diseases. Direct in vivo evidence not only demonstrated the major anti-inflammatory roles of TRAF3 in macrophages (MØ), DCs, neutrophils (NTP), microglia (MG), osteoclast precursors (OP), B cells, and Treg cells but also revealed the pro-inflammatory roles of TRAF3 in hepatocytes, cardiomyocytes, and neurons. The proximal TRAF3-interacting complexes that are critical to inflammatory responses are depicted for each specific cell lineage, with the symbol “-” depicting direct interactions and the symbol “---” depicting indirect connections. The inflammatory disease phenotype of each cell lineage-specific TRAF3 knockout (KO) and transgenic (Tg) mice are also shown, respectively. Spontaneous inflammation or exacerbated inflammatory diseases are depicted in red, and delayed or reduced inflammatory diseases are depicted in green. Evidence shows that the function of TRAF3 in myeloid cells is dynamically modulated according to the metabolic states. In lean mice, TRAF3 plays anti-inflammatory roles in myeloid cells. However, under genetic (ob/ob) or HFD-induced obese state, TRAF3 plays pro-inflammatory roles and promotes metabolic inflammation in myeloid cells. It is also noticed that a delicate balance level of TRAF3 proteins is required to maintain B cell homeostasis and B cell tolerance, as either TRAF3 deficiency or overexpression in B cells leads to systemic autoimmune inflammation in mice. The known roles of TRAF3 in TCR and CD28 signaling, the development and homeostasis of iNKT cells and CD8 memory T cells, and the activation of CD4 and CD8 T cells are not shown in the figure, as their contribution in inflammatory diseases has not been directly demonstrated in vivo in whole animal models.
Systemic Multi-organ Inflammation
We recently investigated the roles of TRAF3 in myeloid cells during inflammatory responses using myeloid cell-specific TRAF3−/− (M-TRAF3−/−) mice [98•]. We found that myeloid cell-specific deletion of TRAF3 leads to increased serum levels of the pro-inflammatory cytokines IL-6 and IL-12 but decreased serum levels of the anti-inflammatory cytokine IL-10 in response to LPS (TLR4 ligand) or polyI:C (TLR3 ligand) challenge [98•]. Interestingly, aging M-TRAF3−/− mice (15-22 months old) spontaneously develop chronic inflammation often affecting multiple organs, including the liver, spleen, intestines, lung, pancreas, and heart [98•]. These findings point to an indispensable anti-inflammatory role for TRAF3 in myeloid cells.
Cardiovascular Inflammation
TRAF3 expression is upregulated in human atherosclerotic plaques and failing human hearts [43,107]. Epigenetic modifications of the Traf3 gene have been observed in cardiovascular inflammation, including DNA methylation that has been associated with vascular recurrence after ischemic stroke in patients treated with clopidogrel and histone acetylation that has been associated with cardiac hypertrophy in mice [132, 133]. Consistent with human evidence, cardiac-specific TRAF3−/− mice exhibit reduced cardiac hypertrophy, fibrosis, and dysfunction [107]. Conversely, transgenic mice overexpressing TRAF3 in the heart develop exaggerated cardiac hypertrophy in response to pressure overload [107]. TRAF3 also promotes an angiotensin II- or phenylephrine-induced hypertrophic response in isolated cardiomyocytes [107]. Mechanistically, TRAF3 directly binds to TBK1, causing increased TBK1 phosphorylation and Akt activation in response to hypertrophic stimuli [107]. The above evidence identified TRAF3 as a key regulator of cardiac hypertrophy.
Inflammatory Bowel Diseases
TRAF3 expression is significantly higher in the inflamed colonic mucosa, PBMCs, and plasma in human patients with CD and UC than in healthy controls [82, 134]. In the DSS-induced colitis model, M-TRAF3−/− mice exhibit exacerbated colon inflammation with increased levels of IL-1 β, IL-6, TNF, and IL-12, which is mediated by elevated protein levels of c-Rel and IRF5 in TRAF3−/− macrophages [47••]. Mice genetically deficient in NLRP12, a cytosolic NLR protein that directly interacts with TRAF3 and NIK, are also highly susceptible to DSS-induced colitis and exhibit reduced TRAF3 level and increased activation of NF-κB2, ERK, and Akt [112]. Similarly, mice deficient in PTPN22, a protein tyrosine phosphatase that directly binds to TRAF3 and promotes K63-linked polyubiquitination of TRAF3, show increased severity of DSS-induced colitis due to decreased TRAF3-type I IFN-mediated protection of colonic mucosa [126]. Furthermore, mice deficient in NDR1, a TRAF3-interacting protein that prevents the recruitment of TRAF3 to the IL-17R-Act1-TRAF6 complex, are resistant to TNBS-induced colitis with decreased IL-17 signaling and IL-17-mediated inflammation in epithelial cells [130]. Thus, TRAF3 plays protective roles in colon inflammation by acting on multiple signaling pathways in myeloid cells and colonic epithelial cells (Fig. 1).
Liver Inflammation, Hepatic Steatosis, and Diabetes
In humans, TRAF3 expression is decreased in PBMCs of patients with chronic HBV infection and is upregulated in livers of patients with hepatic steatosis or subjected to liver transplantation [106, 135, 136]. The in vivo roles of TRAF3 in liver inflammation have been elucidated in several different mouse models. Myeloid cell-specific deletion of TRAF3 markedly attenuates metabolic inflammation, insulin resistance, glucose intolerance, and hepatic steatosis in mice with either genetic (ob/ob) or HFD-induced obesity [103]. Hepatocyte-specific TRAF3−/− mice also show ameliorated HFD-induced inflammatory responses, hepatic steatosis, insulin resistance, and type 2 diabetes, while transgenic mice overexpressing TRAF3 in hepatocytes exhibit the opposite phenotype [104, 136]. In response to hepatic I/R injury, hepatocyte-specific, but not myeloid cell-specific, TRAF3 deficiency reduces cell death, inflammatory cell infiltration, and cytokine production, whereas hepatic TRAF3 overexpression results in the opposite effects in mice [106]. Mechanistically, glucose directly increases TRAF3 levels in primary hepatocytes, and TRAF3 binds to TAK1 to modulate the NF-κB, JNK, and insulin-AKT signaling cascades in hepatocytes [104, 106, 136]. Therefore, TRAF3 proteins expressed in both myeloid cells and hepatocytes promote liver inflammatory responses in obesity and I/R settings (Fig. 2).
Bone Inflammatory Diseases
TNF and RANKL mediate bone destruction in common bone diseases, such as osteoarthritis and RA, via TRAF3-dependent mechanisms [108, 109, 137]. TNF increases TRAF3 expression in osteoclast precursors (OPs), and RANKL decreases TRAF3 protein levels by inducing lysosome/autophagy-dependent degradation of TRAF3 [108, 109, 137]. Both TNF and RANKL induce more osteoclasts from TRAF3−/− OPs, whereas overexpression of TRAF3 reduces osteoclast formation from WT OPs induced by TNF, RANKL, and TNF + RANKL [109, 137]. OP-specific TRAF3−/− mice have increased osteoclastogenesis and osteoporosis mediated by increased NF-κB1 and NF-κB2 signaling [109]. In particular, the importance of the noncanonical NF-κB2 signaling is highlighted by the evidence that TNF transgenic (TNF-Tg) mice lacking NF-κB2 p100 exhibit more severe joint erosion and inflammation as well as systemic bone loss than TNF-Tg WT mice [108]. Furthermore, osteoclast-lineage expression of a mutant form of NIK that lacks its TRAF3 binding domain results in constitutive NF-κB2 activation, osteoporosis, and enhanced inflammatory osteolysis in mice following injection of serum from arthritic K/BxN mice (a model of serum transfer arthritis) [138]. Thus, TRAF3 expressed in the osteoclast-lineage plays protective roles against bone inflammatory diseases.
Brain Inflammation and Ischemic Stroke
An autosomal dominant mutation of TRAF3 has been identified as a causative mutation in a patient with a history of herpes simplex virus-1 (HSV-1) encephalitis in childhood, which resulted from impaired TLR3-type I IFN signaling [139]. DNA methylation of the Traf3 gene is downregulated in patients with ischemic stroke, and this Traf3 epigenetic regulation is associated with vascular recurrence and also correlated with an increased platelet aggregation [140]. Consistent with human evidence, the in vivo roles of TRAF3 in brain inflammation and ischemic stroke have been demonstrated with several mouse models. Transgenic mice overexpressing TRAF3 show decreased levels of IL-17-induced inflammatory factors IL-6, KC, and MMP3 in the brain and also exhibit delayed onset and reduced incidence and severity of experimental autoimmune encephalomyelitis (EAE) after MOG immunization [141]. Conversely, M-TRAF3−/− mice, which have TRAF3 deleted in microglia and other myeloid cells, display exacerbated EAE [120]. TRAF3 ablation also restores TLR-induced inflammatory responses and EAE severity in mice deficient in Peli1, an E3 ubiquitin ligase that mediates K48-linked ubiquitination and degradation of TRAF3 in microglia [120]. Interestingly, in a mouse model of ischemic stroke, TRAF3 expression is induced in neurons in response to I/R [110]. Neuron-specific TRAF3−/− mice reduces neuronal death and inflammation following I/R, whereas transgenic mice overexpressing TRAF3 in neurons exhibit more severe ischemic stroke [110]. Neuronal effects of TRAF3 are mediated by TAK1-NF-κB/JNK/Rac-1 signaling [110]. Furthermore, mice deficient in CK1ε, a kinase that interacts with and phosphorylates TRAF3 at Ser349, show aggravated and sustained brain inflammation after infection with West Nile virus [129]. Taken together, TRAF3 plays anti-inflammatory roles in microglia/myeloid cells and pro-inflammatory roles in neurons via distinct signaling pathways during brain inflammation (Fig. 2).
Autoimmune Inflammatory Diseases
TRAF3 expression is upregulated in synovial fluid mononuclear cells and PBMCs of patients with juvenile idiopathic arthritis, and genome-wide association studies (GWASs) identified TRAF3 as a susceptibility gene for human multiple sclerosis [142, 143]. A PTP22 variant (PTPN22_R620W) that is strongly associated with human SLE and RA fails to promote TRAF3 ubiquitination [126]. Mechanistically, TRAF3 is a critical regulator of B cell homeostasis and activation, Treg and iNKT development and function, CD4 and CD8 T cell activation, and medullary thymic epithelial cell (mTEC) development [101, 102, 127, 144,145,146,147], all of which could impact autoimmune responses. Indeed, B cell-specific TRAF3−/− (B-TRAF3−/−) mice exhibit an autoimmune inflammatory phenotype affecting multi-organs, including the liver and kidney [101]. Paradoxically, B cell-specific TRAF3 transgenic mice also develop systemic autoimmune inflammation in multi-organs, including pancreas, kidney, and joints [105]. Treg-specific TRAF3 ablation leads to mild tissue inflammation mostly seen in the lung and liver in mice [144]. Furthermore, TRAF3 plays a central role in inhibiting the NF-κB2 pathway during mTEC development, and mice lacking NF-κB2 components fail to develop mTECs and exhibit systemic autoimmune inflammation [147]. Therefore, TRAF3 dysregulation plays causal roles in the pathogenesis of various autoimmune inflammatory diseases.
TRAF4
Unique among the TRAF family, TRAF4 is mainly involved in developmental, morphogenic, and oncogenic processes [148,149,150,151,152,153]. However, TRAF4 also regulates inflammation and inflammatory responses mediated by GITR, TLRs, NOD2, IL-17R, and IL-25R [3••, 153,154,155,156,157,158,159,160,161]. TRAF4 modulates inflammatory responses by directly interacting with GITR, NOD2, IL-17R, IL-25R, TRIF, Act1, TRAF6, IKKα, MEKK3, MEKK4, SMURF2, and p47phox (a component of cytosolic NADPH oxidase) to control NF-κB, JNK, and reactive oxygen species (ROS) pathways [149, 153,154,155,156,157,158,159,160,161,162]. Current human and mouse evidence implicates TRAF4 in airway inflammation, inflammatory bowel diseases (IBD), and autoimmune encephalomyelitis.
Airway and Pulmonary Inflammation
Initial characterization of TRAF4−/− mice revealed that loss of TRAF4 expression results in developmental defects of the upper respiratory tract, respiratory air flow abnormalities, and increased rates of pulmonary inflammation [150, 151]. However, Zepp et al. recently found that TRAF4−/− mice exhibit blunted airway eosinophilia and Th2 cytokine production in response to IL-25 administration [161]. Mechanistically, IL-25/IL-25R ligation induces the recruitment of TRAF4, which is required for the Act1/IL-25R interaction as well as the recruitment the E3-ligase SMURF2 to degrade the IL-25R inhibitory molecule DAZAP2 [161]. Thus, TRAF4 plays a negative role in pulmonary inflammation during development, but a positive role in IL-25-induced airway inflammation.
Inflammatory Bowel Diseases
TRAF4 is overexpressed in patients with IBDs, and TRAF4 overexpression appears to be an indicator of endoscopic disease activity in UC patients [163]. Interestingly, TRAF4 is identified as a key negative regulator of signaling by the Crohn’s disease susceptibility protein NOD2 [158, 159]. In response to stimulation with bacterial ligands of NOD2, TRAF4 directly binds to NOD2, MEKK4, and IKKα, which leads to phosphorylation of Ser426 on TRAF4 by IKKα and disruption of the NOD2-RIP2 complex [158,159]. Consequently, TRAF4 inhibits NOD2-induced NF-κB activation and bacterial killing in macrophages (Fig. 1) [158, 159]. However, direct in vivo evidence of pathological or protective roles of TRAF4 in IBDs is still lacking and awaits further investigation.
Brain Inflammation and Autoimmune Encephalomyelitis
TRAF4 expression is downregulated in the brain of patients with schizophrenia [164], which is associated with high levels of pro-inflammatory cytokines in the blood and cerebrospinal fluid [165]. Characterization of TRAF4−/− mice revealed that TRAF4 is required for myelin homeostasis in the central nervous system (CNS) [166]. TRAF4−/− mice exhibit myelin perturbation and degeneration of a high number of Purkinje cells, which is linked to the activation of the Nogo receptor-p75NTR-RhoA pathway in the CNS [166]. In a TH17-mediated EAE model, adoptive transfer of MOG35-55-specific WT TH17 cells into irradiated TRAF4−/− recipient mice induces an accelerated onset of disease with increased numbers of immune cell infiltration in the brain [160]. Biochemical analyses revealed that IL-17/IL-17R engagement recruits Act1, TRAF6, and TRAF4 [153]. Interestingly, TRAF4 competes with TRAF6 for binding to Act1 on the same site, thereby inhibiting IL-17-induced production of pro-inflammatory cytokines (such as GM-CSF and IL-6) and chemokines (such as CXCL1 and CCL2) in brain resident cells (such as astrocytes). Taken together, TRAF4 is important in restricting the effects of IL-17 signaling and disease pathogenesis in TH17-mediated autoimmune encephalomyelitis [160].
TRAF5
As a close structural homolog of TRAF2 and TRAF3, TRAF5 regulates inflammatory responses mediated by receptors of the TNF-R superfamily, TLRs, RIG-I, IL-17R, and gp130 (a component of IL-6 receptor) [3••, 116, 167,168,169,170,171,172,173]. TRAF5 is important in the inflammatory responses mounted by CD4+ TH2 and TH17 cells, neutrophils, macrophages, hepatocytes, B cells, endothelial cells, and fibroblasts [67, 168, 169, 171,172,173,174]. Upon receptor engagement, TRAF5 has been shown to induce the activation of both NF-κB1 and NF-κB2 pathways as well as the activation of the MAPKs (JNK and ERK1/2) by directly interacting with the upstream receptors and downstream adaptor proteins or enzymes[3••, 116, 167,168,169]. TRAF5-interacting proteins that participates in inflammation include OX40, IL-17R, gp130, TRAF2, TRAF3, MyD88, MAVS, TAB2, Act1, USP25, and RORγt, among others [3••, 116, 167,168,169,170, 172••, 173,174,175,176,177]. Available human and mouse evidence implicates TRAF5 in lung inflammation, cardiovascular inflammation, inflammatory bowel diseases, liver inflammation, and autoimmune inflammatory diseases (such as encephalomyelitis, arthritis, lupus, and uveitis).
Airway and Pulmonary Inflammation
So et al. reported that in the presence of OX40 stimulation by an agonistic anti-OX40 antibody, TRAF5−/− mice display enhanced TH2 response to immunization with the T-dependent antigen KLH in adjuvant CFA or alum [174]. Similarly, in a model of OVA-induced allergic airway inflammation, in which endogenous OX40/OX40L interactions are essential for the priming of TH2 cells in asthmatic-like responses, TRAF5−/− mice show exaggerated TH2-driven airway and lung inflammation [174]. TRAF5 deficiency leads to increased numbers of inflammatory cells and elevated levels of TH2 cytokines IL-5 and IL-13 in bronchoalveolar lavage (BAL) fluid as well as OVA-specific IgE in the plasma following OVA challenge [174]. Interestingly, Bulek et al. found that in a mouse model of IL-17-induced pulmonary inflammation, the IL-17-Act1-TRAF2-TRAF5-IKKε signaling axis is required for neutrophilia and lung inflammation [67]. This is mediated by IL-17-induced expression of chemokines (CXCL1 and CXCL2) and cytokines (TNF, IL-6 and G-CSF) in the lung tissue and especially in airway epithelial cells [67, 175]. Therefore, TRAF5 plays a negative role in TH2-induced airway and lung inflammation but a positive role in IL-17-induced airway and lung inflammation via distinct cell types and signaling pathways.
Atherosclerosis and Cardiac Fibrosis
TRAF5 is overexpressed in human atherosclerotic plaques and is downregulated in total blood RNA of patients with stable or acute coronary heart disease [43,178]. TRAF5−/−LDLR−/− mice on a high-cholesterol diet exhibit accelerated atherosclerosis with increased rolling and adhesion of inflammatory leukocytes [178]. This is mediated by (1) increased uptake of LDL by TRAF5−/− macrophages likely via their elevated expression of CD36; (2) increased production of chemokines KC and MCP-1 by TRAF5−/− macrophages and endothelial cells; (3) enhanced JNK activation and expression of adhesion molecule ICAM-1 on TRAF5−/− endothelial cells; (4) increased expression of VCAM-1 on TRAF5−/− monocytes; and (5) reduced number of TRAF5−/− Treg cells in the spleen [178]. Interestingly, in a mouse model of cardiac hypertrophy, TRAF5 expression is robustly induced in the heart in response to transthoracic aorta constriction [179]. TRAF5 deficiency promotes cardiac hypertrophy and fibrosis, which is associated with elevated levels of pro-inflammatory cytokines IL-6, TNFα, and MCP-1 in heart tissues and cardiomyocytes [179]. Thus, TRAF5 has a protective role in cardiovascular inflammation by acting in cardiomyocytes, blood vessel endothelial cells, and leukocytes.
Inflammatory Bowel Diseases
Like TRAF2 and TRAF3, TRAF5 expression is significantly upregulated in inflamed colonic mucosa, PBMCs, and plasma of patients with CD and UC [134]. TRAF5−/− mice are more susceptible to DSS-induced colitis with increased frequencies of CD4+ TH2 and TH17 cells as well as enhanced cytokine levels of IFN-γ, IL-4, and IL-17A in the colons after DSS treatment [171]. Thus, TRAF5 deficiency aggravates DSS-induced colitis, most likely by regulating TH2 and TH17-mediated inflammation in mice [171]. These findings are consistent with previous evidence that TRAF5 controls TH2 and TH17 development by regulating the OX40, gp130, and IL-17R signaling as well as RORγt stability [67,172••, 173, 174,176]. Therefore, TRAF5 plays anti-inflammatory roles in colitis mainly by acting in CD4+ TH2 and TH17 cells (Fig. 1).
Liver Inflammation and Hepatic Steatosis
It has recently been reported that TRAF5 expression is decreased in the fatty livers of both NAFLD patients and obese mice and also in palmitate-treated hepatocytes in vitro [180]. TRAF5−/− mice exhibit deterioration of HFD-induced metabolic disorders and have increased levels of pro-inflammatory cytokines (IL-1 β, IL-6, TNFα, and MCP-1) in the liver and serum [180]. Conversely, transgenic TRAF5 overexpression in the liver significantly suppresses nonalcoholic steatohepatitis (NASH)-like phenotypes in mice after HFD treatment and also inhibits the progression of NAFLD in ob/ob mice [180]. Mechanistically, TRAF5 regulates hepatic steatosis by targeting JNK1 signaling, as evidenced by the fact that JNK1 ablation markedly ameliorates the detrimental effects of TRAF5 deficiency on obesity, inflammation, insulin resistance, hepatic steatosis, and fibrosis [180]. These findings identify TRAF5 as a protective factor in liver inflammation and hepatic steatosis.
Brain Inflammation and Ischemic Stroke
Wang et al. reported that TRAF5 protein levels are upregulated by cerebral I/R in the neurons of ischemic mouse brains [181]. TRAF5−/− mice exhibit reduced infarct size and improved neurological function following MCAO-induced ischemic stroke [181]. This is associated with decreased neuronal apoptosis and attenuated blood-brain barrier (BBB) disruption, which is mediated by decreased expression and activity of MMP2 and MMP9 as well as decreased expression of iNOS, COX-2, TNFα, IL-1 β, MCP-1, and ICAM-1 [181]. In contrast, neuron-specific TRAF5 transgenic mice show exacerbated brain injury and edema following cerebral I/R [181]. Mechanistically, TRAF5 induces NF-κB activation and inhibits the Akt/FoxO1 pathway in neurons to promote inflammatory responses and brain injury following ischemic stroke [181].
Autoimmune Encephalomyelitis
TRAF5 has recently been recognized as a critical regulator of TH17 development through multiple mechanisms. TRAF5 inhibits IL-6-induced early TH17 development by directly interacting with gp130 and antagonizing the recruitment of STAT3 to gp130 in CD4+ T cells [172••, 173]. Paradoxically, however, TRAF5 also directly interacts with and ubiquitinates the TH17 lineage-specific transcription factor RORγt via K63-linked polyubiquitination, thereby stabilizing the RORγt protein to promote the expression of TH17-related genes such as IL-17A [176]. Despite the complex negative and positive roles of TRAF5 in TH17 development and function, TRAF5−/− mice exhibit greatly exaggerated TH17-mediated inflammation and autoimmune encephalomyelitis in the EAE model [172••]. On the other hand, in IL-17-responding cells such as epithelial cells and fibroblasts, IL-17R signaling induces Act1-mediated K63-linked ubiquitination of TRAF5 and the subsequent interaction of TRAF5 with the splicing factor SF2 (ASF) to stabilize the mRNA transcripts of chemokines and cytokines [67, 175, 182]. Deficiency in USP25, an enzyme that removes K63-linked ubiquitination of TRAF5 and TRAF6, also leads to exacerbated EAE severity [177]. Together, the above evidence implicates TRAF5 dysregulation in autoimmune encephalomyelitis mediated by TH17 cells and IL-17-responding epithelial cells and fibroblasts.
Other Autoimmune Inflammatory Diseases
As a key signal transducer of the IL-17R-Act1 axis, TRAF5 is also involved in other IL-17-associated autoimmune inflammatory diseases, including arthritis, lupus, and uveitis (eye inflammation) [168, 176, 183, 184]. For example, an increase in the TRAF5 mRNA level is detected in CD4+ T cells of patients with SLE [176]. Interestingly, three SNPs (rs6540679 AG, rs12569232 GG, and rs10863888 AG) of TRAF5 are associated with two autoimmune uveitis entities, Behçet’s disease and Vogt-Koyanagi-Harada syndrome in humans [183]. Two of these SNPs (rs6540679 AA and rs12569232 GG) are also associated with pediatric uveitis [184]. However, the causal roles of TRAF5 dysregulation in these autoimmune inflammatory diseases remain to be determined with animal models.
TRAF6
Among all TRAF molecules, TRAF6 has been the most extensively investigated in inflammatory responses. Historically, TRAF6 was initially considered as the only TRAF molecule that can regulate signaling of both the TNF-R and the TLR/IL-1R superfamilies [185, 186]. TRAF6 is ubiquitously expressed and important for inflammation in various cell types, especially in T cells, macrophages, dendritic cells (DCs), osteoclasts, mast cells, epithelial cells, fibroblasts, microglia, astrocytes, and neurons [3••, 90, 185,186,187,188,189,190,191,192,193]. Serving as both a signaling adaptor and an E3 ubiquitin ligase, TRAF6 transduces signals that emanate from receptors of the TNF-R superfamily, TLRs, NLRs, RIG-I, cytokine receptors, and C-type lectin receptors in inflammatory responses [3••, 186]. TRAF6 is required for receptor-induced activation of both canonical and non-canonical NF-κB pathways as well as activation of the MAPKs (JNK1/2, ERK1/2, and p38) and IRFs (IRF3, IRF4, IRF5, and IRF7) [3••, 186, 194,195,196]. TRAF6 directly interacts with a variety of critical regulators of inflammation and inflammatory responses. These include (1) receptors: members of the TNF-R superfamily, NLRX1, NLRC3, IL-17Rs, and TGF β receptors [3••, 186, 197,198,199,200,201]; (2) adaptor proteins: MyD88, TRIF, MAVS, TAB2, NEMO, Ubc13, DAB2, LAT, TRIP6, ECSIT, SOCS1, SOCS2, SOCS3, TAX1BP1, GIT2, and Keratin 8 [3••, 186, 202,203,204,205,206,207,208,209,210,211]; (3) enzymes: TAK1, TBK1, IRAK1, ASK1, Act1, WWP1, IRE1α, A20, CYLD, USP20, USP25, Pellino3, Itch, SHP, MST4, IPMK, and PINK1 [3••, 177, 186, 188, 212,213,214,215,216,217,218,219,220,221,222,223]; (4) transcription factors: IRF4, IRF5, IRF7, STAT1, STAT3, Smad6, HIF1α, Nur77, and DCP1a, among others [3••, 186, 196, 219, 224,225,226,227,228,229,230,231]. Both human and mouse evidence indicates the pivotal importance of TRAF6 in sepsis, lung inflammation, cardiovascular inflammation, inflammatory bowel diseases, liver inflammation, diabetes, pancreatitis, neurodegenerative diseases, and autoimmune inflammatory diseases (such as autoimmune hepatitis, lupus, arthritis, and multiple sclerosis).
Sepsis
TRAF6 expression is downregulated in PBMCs in critically ill patients with sepsis, and an intronic SNP of TRAF6 (rs4755453) is associated with susceptibility to sepsis-induced acute lung injury (a protective role identified for the C allele of rs4755453) [232, 233]. TRAF6 deficiency leads to defective LPS-, IL1β- and CD40- signaling in mice [234, 235]. Multiple TRAF6-interacting proteins, including IRF5, ASK1, Akt2, and CRTC2, are activated by TRAF6 and play important roles in LPS-induced septic shock responses as demonstrated by in vivo evidence in mouse models [236,237,238,239]. Notably, a variety of TRAF6-interacting proteins regulate LPS-induced septic shock responses in mouse models by directly targeting TRAF6 ubiquitination, degradation or complex formation, including IRAK1, IRAK2, IRAK-M, A20, NLRX1, NLRC3, CD204, β-arrestin, GIT2, MST4, Nur77, IPMK, SHP, and Keratin 8, among others [198, 199, 210, 211, 215, 220,221,222, 230, 240,241,242,243,244,245]. Therefore, TRAF6 is a central regulator of sepsis.
Airway Inflammation
In cultured human macrophages, cigarette smoke induces TRAF6 degradation, NF-κB activation and IL-8 production via TLR4-IRAK-dependent signaling [246]. During lung I/R injury, DAMPs induce autophagy to amplify the inflammatory response by enhancing K63-linked ubiquitination of TRAF6 and activation of the downstream MAPK and NF-κB signaling in alveolar macrophages [247]. In mice, high doses of CpG oligodeoxynucleotides protect against allergic airway inflammation by stimulating the TLR9-TRIF-TRAF6-NF-κB2 pathway [195]. In the presence of TGF β, ligation of OX40 induces conventional CD4+ T cell polarization to TH9 cells, and OX40L-transgenic mice develop an extensive autoimmune disease with severe TH9-driven airway inflammation. However, OX40 fails to induce TH9 polarization in CD4 T cells of T cell-specific TRAF6−/− mice due to defective OX40-TRAF6-NIK-NF-κB2 signaling in TRAF6−/− T cells [194]. In contrast, TRAF6 in DCs is required for the maintenance of tolerance in the lung to prevent allergic asthma. Specific deletion of TRAF6 from DCs leads to spontaneous generation of TH2-associated immune responses in the lung and increased susceptibility to the model antigen OVA-induced asthmatic airway inflammation with exacerbated eosinophil infiltration [248]. As a component of the IL-17R-Act1-TRAF6 signaling axis in macrophages, fibroblasts, and epithelial cells, TRAF6 is also required for IL-17 and IL-25-induced airway and pulmonary inflammation [177, 200, 249]. Furthermore, TRAF6−/− mast cells show impaired production of cytokine IL-6, IL-13, TNF, and CCL-9 following FcεRI aggregation, suggesting a positive role for mast cell TRAF6 in allergic inflammatory responses [189]. Thus, TRAF6 is an indispensable regulator of airway inflammation by acting in both immune cells and lung resident cells.
Cardiovascular Inflammation
TRAF6 expression is upregulated in human atherosclerotic plaques and human hearts with hypertrophic cardiomyopathy, dilated cardiomyopathy, and atrial fibrosis [43, 250,251,252]. Using LDLR−/− mice reconstituted with TRAF6−/− fetal liver cells and on a high cholesterol diet, Stachon et al. reported that TRAF6 is not required for atherogenesis. Interestingly, however, Polykratis et al. revealed the opposite protective versus pathogenic roles of TRAF6 in atherosclerosis using two different conditional TRAF6 knockout mouse models [253]. Endothelial TRAF6 deficiency reduces atherosclerosis in female ApoE−/− mice by inhibiting NF-κB-dependent proinflammatory gene expression and monocyte adhesion to endothelial cells [253]. In contrast, myeloid cell-specific TRAF6 deficiency causes exacerbated atherosclerosis in both male and female ApoE−/− mice by increasing ER stress and apoptosis and by reducing IL-10 production and the efferocytosis capacity of TRAF6−/− macrophages [253]. In mouse hypertrophic hearts, elevation of TRAF6 protein levels is induced by ROS generated during hypertrophic progression [250]. Cardiac-specific TRAF6 transgenic mice show exacerbated cardiac hypertrophy in response to pressure overload or angiotensin II challenge, whereas cardiac-specific TRAF6−/− mice exhibit an alleviated cardiac hypertrophy phenotype [250]. Mechanistically, ROS triggers TRAF6 auto-ubiquitination and subsequent TRAF6-TAB2-TAK1 signaling, which regulates cardiac remodeling via the p38 and JNK1/2 pathways [250]. Thus, TRAF6 plays complex roles in endothelial cells, macrophages, and cardiomyocytes via distinct signaling pathways during cardiovascular inflammation.
Inflammatory Bowel Diseases
In humans, epigenetic silencing of TRAF6 by hypermethylation and reduced TRAF6 expression are detected in PBMCs of IBD patients [254]. Paradoxically, Shen et al. reported that TRAF6 expression is elevated in PBMCs and intestinal mucosa of IBD patients and that soluble TRAF6 levels in plasma are also significantly higher in patients with CD and UC than in healthy controls [163]. In mice, dendritic cell-specific deletion of TRAF6 (DC-TRAF6−/−) results in spontaneous eosinophilic enteritis and fibrosis in the small intestine associated with a TH2 inflammatory response in the lamina propria [255•]. The aberrant TH2 response is linked to decreased Treg cell numbers in the small intestine and diminished induction of iTreg cells due to defective expression of IL-2 in TRAF6−/− DCs [255•]. The TH2-associated small intestine inflammation is exacerbated in germ-free DC-TRAF6−/− mice compared to specific pathogen-free DC-TRAF6−/− mice, suggesting that the enteritis phenotype is independent of gut microbiota [256]. In contrast, young mice with T cell-specific TRAF6 deletion show attenuated DSS-induced colitis, which is associated with increased TGF β-Smad2/3 signaling in TRAF6−/− T cells and increased TH17 differentiation [257]. Interestingly, intestinal epithelial cell (IEC)-specific deletion of TRAF6 leads to exacerbated DSS-induced colitis in mice, which is dependent on gut microbiota but independent of TLR-MyD88/TRIF signaling in IECs [258]. Furthermore, activation of NOD2, a major susceptibility gene of CD, protects mice from TNBS-induced colitis by increasing the expression of IRF4, which binds to TRAF6 and RICK to inhibit their K63-linked polyubiquitination [196]. Together, the above evidence indicates that TRAF6 plays diverse and critical roles in IBDs by differentially acting in various cell types in the intestines (Fig. 1).
Liver inflammation, Diabetes, and Hepatic Steatosis
TRAF6 expression is decreased in PBMCs of chronic hepatitis B virus-infected patients [259], but is elevated in PBMCs of patients with type 2 diabetes [260, 261]. Significant association is detected between the TRAF6 SNP rs16928973 TT and diabetic nephropathy (DN) in patients with type 2 diabetes [262]. In mice, specific deletion of TRAF6 from medullary thymic epithelial (mTEC) cells leads to autoimmune hepatitis associated with autoantibodies and abnormal hepatic T cell populations and functions, caused by impaired mTEC development and T cell tolerance to liver autoantigens [190]. In a model of concanavalin A-induced murine hepatitis, the OX40-TRAF6-MALT1-caspase 1-gasdermin D signaling is required to induce pyroptosis in hepatic iNKT cells and the subsequent liver inflammation and injury [263]. In mouse primary hepatocytes, insulin induces TRAF6-mediated K63-linked ubiquitination of APPL1 and Akt activation, and TRAF6 silencing leads to insulin resistance in hepatocytes [264]. Interestingly, mice deficient in Pellino3, a negative regulator of TRAF6 activity, show exacerbated HFD-induced inflammation, hepatic steatosis, and insulin resistance with increased expression of IL-1 β, TNF, IL-6, and CCL2 in the liver [219]. These effects of Pellino3 deficiency result from augmented TRAF6-mediated K63-linked ubiquitination and stabilization of HIF1α in the liver [219]. However, direct in vivo evidence of TRAF6 in hepatocytes, insulin resistance, and hepatic steatosis is still lacking and awaits further investigation with hepatocyte-specific TRAF6−/− mice.
Pancreatitis
Nishida et al. found that IL-36α, a cytokine detected in fibrotic tissue of chronic pancreatitis, induces inflammatory mediators from human pancreatic myofibroblasts via the IL-36R-MyD88-TRAF6-IRAK1-TAK1 signaling pathway [265]. In mice, TRAF6 expression is significantly increased in caerulein-induced acute pancreatitis, and TRAF6 plays a protective role in acinar cells against caerulein-induced apoptosis [207]. LPS-induced SOCS1 and SOCS3 exacerbate caerulein-induced pancreatitis by directly interacting with TRAF6 and degrading TRAF6 protein via ubiquitination [207]. As a convergence point of the TLR4-dependent and the TLR4-independent signaling pathways, TRAF6 may play an important role in pancreatitis [207, 266], which remains to be directly elucidated using conditional TRAF6 knockout mice in pancreatitis models.
Brain Inflammatory Diseases
TRAF6 has been implicated in the pathogenesis of a variety of human brain inflammatory diseases, including ischemic stroke, Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington disease (HD). TRAF6 SNP haplotype rs5030416 (allele C)-rs5030411 (allele C) has been associated with susceptibility to ischemic stroke [267], and TRAF6 expression is upregulated in peripheral blood of stroke patients [268]. TRAF6 protein levels are elevated in the brains of PD and HD patients [90, 193]. Interestingly, abnormal protein aggregates of TRAF6 are detected in the brains of patients with AD, PD, and HD [90, 191,192,193]. In human AD brains, TRAF6 associates with the ubiquitin-associating protein sequestosome1/p62 and tau in neurofibrillary tangles, and TRAF6 catalyzes K63-linked polyubiquitination of tau to promote tau aggregation into insoluble tangles [191]. Indeed, tau was recovered as a polyubiquitinated protein in brain lysates from WT mice, but was not polyubiquitinated in brain lysates recovered from the TRAF6−/− mice [191]. In human PD brains, TRAF6 interacts with and ubiquitinates misfolded mutant PARK7/DJ-1 and PARK1/aSYN to promote the accumulation of insoluble aggregates and Lewy bodies [192]. Similarly in human HD brains, TRAF6 binds to mutant N-HTT proteins and also ubiquitinates mutant N-HTT to increase the formation of insoluble aggregates [193]. Interestingly, however, instead of the conventional K63-linked polyubiquitination, TRAF6 promotes atypical ubiquitination of mutant DJ-1, aSYN and N-HTT with K6, K27, and K29 linkage formation [192, 193]. Therefore, TRAF6 appears to act as a key pathogenic E3 ligase in neurodegenerative diseases.
Autoimmune Inflammatory Diseases
TRAF6 expression is elevated in human patients with SLE and RA [4•, 186, 269,270,271]. Downregulated expression of TRAF6 has also been detected in PBMCs of SLE patients [93]. Three TRAF6 SNPs (rs5030437 A allele, rs4755453 C allele, and rs540386 A allele) are associated with lupus, and the SNP rs5030437 A allele is also associated with RA [271, 272]. The causal roles of TRAF6 dysregulation in autoimmune inflammatory diseases have been demonstrated by several mouse models with TRAF6 deficiency. In addition to its essential roles in tolerance induction in DCs and mTECs described above, TRAF6 is also required for normal function of CD4+ T cells and Treg cells. An early study by Chiffoleau et al. showed that chimeric mice reconstituted with TRAF6−/− fetal liver cells develop a progressive lethal inflammatory disease associated with massive organ infiltration and activation of CD4+ T cells in a TH2-polarized phenotype [273]. King et al. verified the findings with T cell-specific TRAF6−/− (T-TRAF6−/−) mice, which also exhibit a systemic autoimmune inflammatory disease affecting multiple organs [274]. This autoimmune inflammatory phenotype is mediated by increased TCR-PI3K-Akt activation and reduced expression of Cbl-b and consequently independence from CD28 co-stimulation and impaired anergy induction in TRAF6−/− CD4+ T cells [274, 275]. Consistent with the notion that TRAF6 is required for Treg development and maintenance [257, 276, 277], Treg-specific TRAF6−/− mice spontaneously develop allergic skin diseases, arthritis, lymphadenopathy, and hyper IgE phenotypes [277]. Mechanistically, TRAF6 deficiency results in reduced stability and protein level of FoxP3 in Treg cells, leading to rapid conversion of FoxP3-expressing Treg cells into TH2-like inflammatory cells in mice [277]. In summary, TRAF6 protects hosts from various autoimmune inflammatory diseases by acting in CD4+ TH cell subsets, Treg cells, DCs, and mTECs.
TRAF7
TRAF7 protein does not have the TRAF homology domain that defines the TRAF family [3••]. Currently, there is no human evidence or direct in vivo evidence about TRAF7 in inflammation and inflammatory diseases.
Here, we summarize genetic variations and alterations in expression of TRAF molecules in human inflammatory diseases in Table 1 and also provide a list of direct in vivo evidence of TRAF molecules in inflammatory diseases in mouse models in Table 2.
Conclusions
During the past decade, our knowledge of TRAF molecules in inflammation and inflammatory diseases has rapidly grown. Compelling evidence has demonstrated the central importance of TRAFs in regulating and controlling inflammation and inflammatory responses in both humans and mice. This is highlighted by mounting evidence of TRAF dysregulation or dysfunction in human patients with various inflammatory diseases. The causal roles of TRAF dysregulation or dysfunction in inflammatory diseases have been verified in a variety of mouse models as reviewed here. It is recognized that although TRAFs have overlapping roles, each TRAF molecule (TRAF1 to 6) also plays distinct and indispensable roles in inflammation and inflammatory responses as demonstrated by studies of mouse models with genetic engineering of the corresponding Traf gene. It is also increasingly clear that for each specific TRAF molecule, its role in inflammation may vary substantially depending on the specific receptor pathways engaged, the cellular and organ context, the functional or metabolic state of the cell, and the stoichiometry of other TRAFs and TRAF-interacting proteins in the cell as revealed by comprehensive analyses of different inflammatory responses and disease models. In conclusion, TRAF molecules are versatile and indispensable regulators of inflammation and inflammatory responses, and aberrant expression or function of TRAFs contributes to the pathogenesis of inflammatory diseases.
Perspective
Given the central importance of TRAFs in inflammation and inflammatory diseases, it would be envisioned that TRAFs and TRAF-dependent signaling pathways represent rational therapeutic targets for human inflammatory diseases. Development of therapeutic strategies and agents that specifically targets TRAFs and TRAF-dependent signaling pathways will thus be the next major challenge in the field. This could be particularly difficult considering the broad and highly diverse roles of TRAFs in inflammation and other immune responses depending on the stimuli, cell type, tissue environment, and functional state of the hosts. In this regard, insights gained into the proximal TRAF signaling complexes and detailed structures of the interactions between each TRAF and its specific signaling partners will guide the design of context-specific therapeutics. In addition, local or cell-specific drug delivery would be beneficial to improve the therapeutic efficacy and avoid systemic side effects of TRAF-targeting therapeutics. To date, only TRAF6-specific therapeutic agents have been tested in inflammatory disease settings. Inhibition of TRAF6 expression using specific small interfering RNA reduces the severity of arthritis and joint inflammation in a mouse RA model [282]. Notably, structure-based in silico approaches have been employed to screen and identify compounds that specifically block TRAF6-CD40 or TRAF6-RANK interactions, which serve as lead compounds for further drug development and optimization [283, 284]. The small molecule inhibitor (SMI) of CD40-TRAF6 interaction, 6877002, effectively reduces in vivo inflammation in mouse models of peritonitis, polymicrobial sepsis, and EAE [283, 285]. Another SMI of CD40-TRAF6 interaction, 6860766, potently ameliorates in vivo inflammation in mouse models of polymicrobial sepsis and HFD-induced obesity [283, 286]. These findings provide proof-of-concept evidence that manipulation of TRAF expression and function holds therapeutic potential for inflammatory diseases. Further development of therapeutic strategies and agents that specifically target other TRAF molecules or signaling complexes will add to our armament to improve the treatment of human inflammatory diseases.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Inoue J, Ishida T, Tsukamoto N, Kobayashi N, Naito A, Azuma S, et al. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp Cell Res. 2000;254:14–24.
Wajant H, Henkler F, Scheurich P. The TNF-receptor-associated factor family. Scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal. 2001;13:389–400.
•• Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal. 2013;8:7. This article provides a comprehensive review of TRAF molecules in receptor signaling and in human diseases
• So T, Nagashima H, Ishii N. TNF receptor-associated factor (TRAF) signaling network in CD4(+) T-lymphocytes. Tohoku J Exp Med. 2015;236:139–54. This article provides an excellent review of the complex signaling network of TRAF molecules in CD4+ T cells
Yang XD, Sun SC. Targeting signaling factors for degradation, an emerging mechanism for TRAF functions. Immunol Rev. 2015;266:56–71.
Pullen SS, Miller HG, Everdeen DS, Dang TT, Crute JJ, Kehry MR. CD40-tumor necrosis factor receptor-associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry. 1998;37:11836–45.
Chung JY, Lu M, Yin Q, Lin SC, Wu H. Molecular basis for the unique specificity of TRAF6. Adv Exp Med Biol. 2007;597:122–30.
Ely KR, Kodandapani R, Wu S. Protein-protein interactions in TRAF3. Adv Exp Med Biol. 2007;597:114–21.
Foight GW, Keating AE. Comparison of the peptide binding preferences of three closely related TRAF paralogs: TRAF2, TRAF3, and TRAF5. Protein Sci. 2016;25:1273–89.
ee SY, Choi Y. TRAF1 and its biological functions. Adv Exp Med Biol. 2007;597:25–31.
Speiser DE, Lee SY, Wong B, Arron J, Santana A, Kong YY, et al. A regulatory role for TRAF1 in antigen-induced apoptosis of T cells. J Exp Med. 1997;185:1777–83.
Tsitsikov EN, Laouini D, Dunn IF, Sannikova TY, Davidson L, Alt FW, et al. TRAF1 is a negative regulator of TNF signaling. enhanced TNF signaling in TRAF1-deficient mice. Immunity. 2001;15:647–57.
Su X, Li S, Meng M, Qian W, Xie W, Chen D, et al. TNF receptor-associated factor-1 (TRAF1) negatively regulates Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF)-mediated signaling. Eur J Immunol. 2006;36:199–206.
•• Abdul-Sater AA, Edilova MI, Clouthier DL, Mbanwi A, Kremmer E, Watts TH. The signaling adaptor TRAF1 negatively regulates Toll-like receptor signaling and this underlies its role in rheumatic disease. Nat Immunol. 2017;18:26–35. This article reports the direct in vivo evidence of TRAF1 in LPS-induced septic shock and the human evidence of the signaling mechanisms of RA-associated TRAF1 SNP in monocytes
Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78:681–92.
Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell. 1995;80:389–99.
Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–3.
Schwenzer R, Siemienski K, Liptay S, Schubert G, Peters N, Scheurich P, et al. The human tumor necrosis factor (TNF) receptor-associated factor 1 gene (TRAF1) is up-regulated by cytokines of the TNF ligand family and modulates TNF-induced activation of NF-kappaB and c-Jun N-terminal kinase. J Biol Chem. 1999;274:19368–74.
Pryhuber GS, Huyck HL, Staversky RJ, Finkelstein JN, O’Reilly MA. Tumor necrosis factor-alpha-induced lung cell expression of antiapoptotic genes TRAF1 and cIAP2. Am J Respir Cell Mol Biol. 2000;22:150–6.
Sughra K, Birbach A, de Martin R, Schmid JA. Interaction of the TNFR-receptor associated factor TRAF1 with I-kappa B kinase-2 and TRAF2 indicates a regulatory function for NF-kappa B signaling. PLoS One. 2010;5:e12683.
Arron JR, Pewzner-Jung Y, Walsh MC, Kobayashi T, Choi Y. Regulation of the subcellular localization of tumor necrosis factor receptor-associated factor (TRAF)2 by TRAF1 reveals mechanisms of TRAF2 signaling. J Exp Med. 2002;196:923–34.
O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–64.
Choudhary S, Kalita M, Fang L, Patel KV, Tian B, Zhao Y, et al. Inducible tumor necrosis factor (TNF) receptor-associated factor-1 expression couples the canonical to the non-canonical NF-kappaB pathway in TNF stimulation. J Biol Chem. 2013;288:14612–23.
Lu YY, Li ZZ, Jiang DS, Wang L, Zhang Y, Chen K, et al. TRAF1 is a critical regulator of cerebral ischaemia-reperfusion injury and neuronal death. Nat Commun. 2013;4:2852.
Zhang XF, Zhang R, Huang L, Wang PX, Zhang Y, Jiang DS, et al. TRAF1 is a key mediator for hepatic ischemia/reperfusion injury. Cell Death Dis. 2014;5:e1467.
McPherson AJ, Snell LM, Mak TW, Watts TH. Opposing roles for TRAF1 in the alternative versus classical NF-kappaB pathway in T cells. J Biol Chem. 2012;287:23010–9.
Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med. 2007;357:1199–209.
Kurreeman FA, Padyukov L, Marques RB, Schrodi SJ, Seddighzadeh M, Stoeken-Rijsbergen G, et al. A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis. PLoS Med. 2007;4:e278.
Chang M, Rowland CM, Garcia VE, Schrodi SJ, Catanese JJ, van der Helm-van Mil AH, et al. A large-scale rheumatoid arthritis genetic study identifies association at chromosome 9q33.2. PLoS Genet. 2008;4:e1000107.
Han TU, Bang SY, Kang C, Bae SC. TRAF1 polymorphisms associated with rheumatoid arthritis susceptibility in Asians and in Caucasians. Arthritis Rheum. 2009;60:2577–84.
Zhu J, Zhang D, Wu F, He F, Liu X, Wu L, et al. Single nucleotide polymorphisms at the TRAF1/C5 locus are associated with rheumatoid arthritis in a Han Chinese population. BMC Med Genet. 2011;12:53.
Xu K, Peng H, Zhou M, Wang W, Li R, Zhu KK, et al. Association study of TRAF1/C5 polymorphism (rs10818488) with susceptibility to rheumatoid arthritis and systemic lupus erythematosus: a meta-analysis. Gene. 2013;517:46–54.
Nishimoto T, Seta N, Anan R, Yamamoto T, Kaneko Y, Takeuchi T, et al. A single nucleotide polymorphism of TRAF1 predicts the clinical response to anti-TNF treatment in Japanese patients with rheumatoid arthritis. Clin Exp Rheumatol. 2014;32:211–7.
Canhao H, Rodrigues AM, Santos MJ, Carmona-Fernandes D, Bettencourt BF, Cui J, et al. TRAF1/C5 but not PTPRC variants are potential predictors of rheumatoid arthritis response to anti-tumor necrosis factor therapy. Biomed Res Int. 2015;2015:490295.
Cheng T, Sun X, Wu J, Wang M, Eisenberg RA, Chen Z. Increased serum levels of tumor necrosis factor receptor-associated factor 1 (TRAF1) correlate with disease activity and autoantibodies in rheumatoid arthritis. Clin Chim Acta. 2016;462:103–6.
Panoulas VF, Smith JP, Nightingale P, Kitas GD. Association of the TRAF1/C5 locus with increased mortality, particularly from malignancy or sepsis, in patients with rheumatoid arthritis. Arthritis Rheum. 2009;60:39–46.
Cheng T, Choi Y, Finkel TH, Tsao PY, Ji MQ, Eisenberg RA. Tumor necrosis factor receptor-associated factor 1 influences KRN/I-Ag7 mouse arthritis autoantibody production. J Clin Immunol. 2013;33:759–66.
Pryhuber GS, Huyck HL, Roper JM, Cornejo J, O’Reilly MA, Pierce RH, et al. Acute tumor necrosis factor-alpha-induced liver injury in the absence of tumor necrosis factor receptor-associated factor 1 gene expression. Am J Pathol. 2005;166:1637–45.
Oyoshi MK, Barthel R. Tsitsikov EN. TRAF1 regulates recruitment of lymphocytes and, to a lesser extent, neutrophils, myeloid dendritic cells and monocytes to the lung airways following lipopolysaccharide inhalation. Immunology. 2007;120:303–14.
Oyoshi MK, Bryce P, Goya S, Pichavant M, Umetsu DT, Oettgen HC, et al. TNF receptor-associated factor 1 expressed in resident lung cells is required for the development of allergic lung inflammation. J Immunol. 2008;180:1878–85.
Bryce PJ, Oyoshi MK, Kawamoto S, Oettgen HC, Tsitsikov EN. TRAF1 regulates Th2 differentiation, allergic inflammation and nuclear localization of the Th2 transcription factor, NIP45. Int Immunol. 2006;18:101–11.
Hessler N, Geisel MH, Coassin S, Erbel R, Heilmann S, Hennig F, et al. Linkage and association analysis identifies TRAF1 influencing common carotid intima-media thickness. Stroke. 2016;47:2904–9.
Zirlik A, Bavendiek U, Libby P, MacFarlane L, Gerdes N, Jagielska J, et al. TRAF-1, -2, -3, -5, and -6 are induced in atherosclerotic plaques and differentially mediate proinflammatory functions of CD40L in endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:1101–7.
Missiou A, Kostlin N, Varo N, Rudolf P, Aichele P, Ernst S, et al. Tumor necrosis factor receptor-associated factor 1 (TRAF1) deficiency attenuates atherosclerosis in mice by impairing monocyte recruitment to the vessel wall. Circulation. 2010;121:2033–44.
Xiang M, Wang PX, Wang AB, Zhang XJ, Zhang Y, Zhang P, et al. Targeting hepatic TRAF1-ASK1 signaling to improve inflammation, insulin resistance, and hepatic steatosis. J Hepatol. 2016;64:1365–77.
Borghi A, Verstrepen L, Beyaert R. TRAF2 multitasking in TNF receptor-induced signaling to NF-kappaB, MAP kinases and cell death. Biochem Pharmacol. 2016;116:1–10.
•• Jin J, Xiao Y, Hu H, Zou Q, Li Y, Gao Y, et al. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat Commun. 2015;6:5930. This article provides the direct in vivo evidence of the importance of myeloid cell TRAF2 and TRAF3 in DSS-induced colitis
Piao JH, Hasegawa M, Heissig B, Hattori K, Takeda K, Iwakura Y, et al. Tumor necrosis factor receptor-associated factor (TRAF) 2 controls homeostasis of the colon to prevent spontaneous development of murine inflammatory bowel disease. J Biol Chem. 2011;286:17879–88.
Villanueva JE, Malle EK, Gardam S, Silveira PA, Zammit NW, Walters SN, et al. TRAF2 regulates peripheral CD8(+) T-cell and NKT-cell homeostasis by modulating sensitivity to IL-15. Eur J Immunol. 2015;45:1820–31.
Petersen SL, Chen TT, Lawrence DA, Marsters SA, Gonzalvez F, Ashkenazi A. TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ. 2015;22:1846–57.
Sakaguchi M, Yamamoto M, Miyai M, Maeda T, Hiruma J, Murata H, et al. Identification of an S100A8 receptor neuroplastin-beta and its heterodimer formation with EMMPRIN. J Invest Dermatol. 2016;136:2240–50.
Hibino T, Sakaguchi M, Miyamoto S, Yamamoto M, Motoyama A, Hosoi J, et al. S100A9 is a novel ligand of EMMPRIN that promotes melanoma metastasis. Cancer Res. 2013;73:172–83.
Mauro C, Crescenzi E, De Mattia R, Pacifico F, Mellone S, Salzano S, et al. Central role of the scaffold protein tumor necrosis factor receptor-associated factor 2 in regulating endoplasmic reticulum stress-induced apoptosis. J Biol Chem. 2006;281:2631–8.
Keestra-Gounder AM, Byndloss MX, Seyffert N, Young BM, Chavez-Arroyo A, Tsai AY, et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature. 2016;532:394–7.
Hu M, Luo Q, Alitongbieke G, Chong S, Xu C, Xie L, et al. Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol Cell. 2017;66:141–53. e6
Yang CH, Murti A, Pfeffer SR, Fan M, Du Z, Pfeffer LM. The role of TRAF2 binding to the type I interferon receptor in alternative NF kappaB activation and antiviral response. J Biol Chem. 2008;283:14309–16.
Schneider AT, Gautheron J, Feoktistova M, Roderburg C, Loosen SH, Roy S, et al. RIPK1 suppresses a TRAF2-dependent pathway to liver cancer. Cancer Cell. 2017;31:94–109.
Varfolomeev E, Goncharov T, Maecker H, Zobel K, Komuves LG, Deshayes K, et al. Cellular inhibitors of apoptosis are global regulators of NF-kappaB and MAPK activation by members of the TNF family of receptors. Sci Signal. 2012;5:ra22.
Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9:1371–8.
Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol. 2008;9:1364–70.
Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–8.
Sasai M, Tatematsu M, Oshiumi H, Funami K, Matsumoto M, Hatakeyama S, et al. Direct binding of TRAF2 and TRAF6 to TICAM-1/TRIF adaptor participates in activation of the Toll-like receptor 3/4 pathway. Mol Immunol. 2010;47:1283–91.
Ermolaeva MA, Michallet MC, Papadopoulou N, Utermohlen O, Kranidioti K, Kollias G, et al. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat Immunol. 2008;9:1037–46.
Collins AS, Ahmed S, Napoletano S, Schroeder M, Johnston JA, Hegarty JE, et al. Hepatitis C virus (HCV)-induced suppressor of cytokine signaling (SOCS) 3 regulates proinflammatory TNF-alpha responses. J Leukoc Biol. 2014;96:255–63.
Mikkelsen SS, Jensen SB, Chiliveru S, Melchjorsen J, Julkunen I, Gaestel M, et al. RIG-I-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: dependence on TRAF2 and TAK1. J Biol Chem. 2009;284:10774–82.
Hong S, Lim S, Li AG, Lee C, Lee YS, Lee EK, et al. Smad7 binds to the adaptors TAB2 and TAB3 to block recruitment of the kinase TAK1 to the adaptor TRAF2. Nat Immunol. 2007;8:504–13.
Bulek K, Liu C, Swaidani S, Wang L, Page RC, Gulen MF, et al. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol. 2011;12:844–52.
Shen RR, Zhou AY, Kim E, Lim E, Habelhah H, Hahn WC. IkappaB kinase {varepsilon} phosphorylates TRAF2 to promote mammary epithelial cell transformation. Mol Cell Biol. 2012;32:4756–68.
Zhang J, Stirling B, Temmerman ST, Ma CA, Fuss IJ, Derry JM, et al. Impaired regulation of NF-kappaB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest. 2006;116:3042–9.
Li L, Soetandyo N, Wang Q, Ye Y. The zinc finger protein A20 targets TRAF2 to the lysosomes for degradation. Biochim Biophys Acta. 2009;1793:346–53.
Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327:1135–9.
Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell. 2006;125:665–77.
Liang J, Saad Y, Lei T, Wang J, Qi D, Yang Q, et al. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J Exp Med. 2010;207:2959–73.
Chuang HC, Sheu WH, Lin YT, Tsai CY, Yang CY, Cheng YJ, et al. HGK/MAP4K4 deficiency induces TRAF2 stabilization and Th17 differentiation leading to insulin resistance. Nat Commun. 2014;5:4602.
Tobin D, van Hogerlinden M, Toftgard R. UVB-induced association of tumor necrosis factor (TNF) receptor 1/TNF receptor-associated factor-2 mediates activation of Rel proteins. Proc Natl Acad Sci U S A. 1998;95:565–9.
Etemadi N, Chopin M, Anderton H, Tanzer MC, Rickard JA, Abeysekera W, et al. TRAF2 regulates TNF and NF-kappaB signalling to suppress apoptosis and skin inflammation independently of Sphingosine kinase 1. Elife. 2015;4
Samak M, Fatullayev J, Sabashnikov A, Zeriouh M, Schmack B, Farag M, et al. Cardiac hypertrophy: an introduction to molecular and cellular basis. Med Sci Monit Basic Res. 2016;22:75–9.
Hilfiker-Klemer D, Landmesser U, Drexler H. Molecular mechanisms in heart failure—focus on cardiac hypertrophy, inflammation, angiogenesis, and apoptosis. J Am Coll Cardiol. 2006;48:A56–66.
Huang Y, Wu D, Zhang X, Jiang M, Hu C, Lin J, et al. Cardiac-specific Traf2 overexpression enhances cardiac hypertrophy through activating AKT/GSK3beta signaling. Gene. 2014;536:225–31.
Divakaran VG, Evans S, Topkara VK, Diwan A, Burchfield J, Gao F, et al. Tumor necrosis factor receptor-associated factor 2 signaling provokes adverse cardiac remodeling in the adult mammalian heart. Circ Heart Fail. 2013;6:535–43.
Qiao YQ, Shen J, Gu Y, Tong JL, Xu XT, Huang ML, et al. Gene expression of tumor necrosis factor receptor associated-factor (TRAF)-1 and TRAF-2 in inflammatory bowel disease. J Dig Dis. 2013;14:244–50.
Shen J, Qiao Y, Ran Z, Wang T, Xu J, Feng J. Intestinal protein expression profile identifies inflammatory bowel disease and predicts relapse. Int J Clin Exp Pathol. 2013;6:917–25.
Liu Y, Peng J, Sun T, Li N, Zhang L, Ren J, et al. Epithelial EZH2 serves as an epigenetic determinant in experimental colitis by inhibiting TNFalpha-mediated inflammation and apoptosis. Proc Natl Acad Sci U S A. 2017;114:E3796–E805.
Piao JH, Yagita H, Okumura K, Nakano H. Aberrant accumulation of interleukin-10-secreting neutrophils in TRAF2-deficient mice. Immunol Cell Biol. 2012;90:881–8.
Ignatius Irudayam J, Contreras D, Spurka L, Ren S, Kanagavel V, Ramaiah A, et al. Profile of inflammation-associated genes during hepatic differentiation of human pluripotent stem cells. Data Brief. 2015;5:871–8.
Ryu HM, Park SG, Yea SS, Jang WH, Yang YI, Jung G. Gene expression analysis of primary normal human hepatocytes infected with human hepatitis B virus. World J Gastroenterol. 2006;12:4986–95.
Gonzalvez F, Lawrence D, Yang B, Yee S, Pitti R, Marsters S, et al. TRAF2 sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol Cell. 2012;48:888–99.
Chen Z, Sheng L, Shen H, Zhao Y, Wang S, Brink R, et al. Hepatic TRAF2 regulates glucose metabolism through enhancing glucagon responses. Diabetes. 2012;61:566–73.
Culpan D, Cram D, Chalmers K, Cornish A, Palmer L, Palmer J, et al. TNFR-associated factor-2 (TRAF-2) in Alzheimer’s disease. Neurobiol Aging. 2009;30:1052–60.
Chung JY, Park HR, Lee SJ, Lee SH, Kim JS, Jung YS, et al. Elevated TRAF2/6 expression in Parkinson’s disease is caused by the loss of Parkin E3 ligase activity. Lab Invest. 2013;93:663–76.
Liu G, Guo H, Guo C, Zhao S, Gong D, Zhao Y. Involvement of IRE1alpha signaling in the hippocampus in patients with mesial temporal lobe epilepsy. Brain Res Bull. 2011;84:94–102.
Rajabi P, Alaee M, Mousavizadeh K, Samadikuchaksaraei A. Altered expression of TNFSF4 and TRAF2 mRNAs in peripheral blood mononuclear cells in patients with systemic lupus erythematosus: association with atherosclerotic symptoms and lupus nephritis. Inflamm Res. 2012;61:1347–54.
Zhu LJ, Landolt-Marticorena C, Li T, Yang X, Yu XQ, Gladman DD, et al. Altered expression of TNF-alpha signaling pathway proteins in systemic lupus erythematosus. J Rheumatol. 2010;37:1658–66.
Raghav SK, Gupta B, Agrawal C, Chaturvedi VP, Das HR. Expression of TNF-alpha and related signaling molecules in the peripheral blood mononuclear cells of rheumatoid arthritis patients. Mediators Inflamm. 2006;2006:12682.
Reuss R, Mirau A, Mistarz M, Kraus J, Bodeker RH, Oschmann P. TRAF2 is upregulated in relapsing-remitting multiple sclerosis. Neuroimmunomodulation. 2013;20:177–83.
Lin WJ, Su YW, Lu YC, Hao Z, Chio II, Chen NJ, et al. Crucial role for TNF receptor-associated factor 2 (TRAF2) in regulating NFkappaB2 signaling that contributes to autoimmunity. Proc Natl Acad Sci U S A. 2011;108:18354–9.
Lalani AI, Luo C, Han Y, Xie P. TRAF3: a novel tumor suppressor gene in macrophages. Macrophage (Houst). 2015;2:e1009.
• Lalani AI, Moore CR, Luo C, Kreider BZ, Liu Y, Morse HC 3rd, et al. Myeloid cell TRAF3 regulates immune responses and inhibits inflammation and tumor development in mice. J Immunol. 2015;194:334–48. This article reports the direct in vivo evidence of myeloid cell TRAF3 in inhibiting inflammatory responses, chronic inflammation, and tumor development
Yi Z, Wallis AM, Bishop GA. Roles of TRAF3 in T cells: many surprises. Cell Cycle. 2015;14:1156–63.
Bishop GA. TRAF3 as a powerful and multitalented regulator of lymphocyte functions. J Leukoc Biol. 2016;100:919–26.
Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity. 2007;27:253–67.
Xie P, Poovassery J, Stunz LL, Smith SM, Schultz ML, Carlin LE, et al. Enhanced Toll-like receptor (TLR) responses of TNFR-associated factor 3 (TRAF3)-deficient B lymphocytes. J Leukoc Biol. 2011;90:1149–57.
Chen Z, Shen H, Sun C, Yin L, Tang F, Zheng P, et al. Myeloid cell TRAF3 promotes metabolic inflammation, insulin resistance, and hepatic steatosis in obesity. Am J Physiol Endocrinol Metab. 2015;308:E460–9.
Chen Z, Canet MJ, Sheng L, Jiang L, Xiong Y, Yin L, et al. Hepatocyte TRAF3 promotes insulin resistance and type 2 diabetes in mice with obesity. Mol Metab. 2015;4:951–60.
Zapata JM, Llobet D, Krajewska M, Lefebvre S, Kress CL, Reed JC. Lymphocyte-specific TRAF3-transgenic mice have enhanced humoral responses and develop plasmacytosis, autoimmunity, inflammation, and cancer. Blood. 2009;113:4595–603.
Hu J, Zhu XH, Zhang XJ, Wang PX, Zhang R, Zhang P, et al. Targeting TRAF3 signaling protects against hepatic ischemia/reperfusions injury. J Hepatol. 2016;64:146–59.
Jiang X, Deng KQ, Luo Y, Jiang DS, Gao L, Zhang XF, et al. Tumor necrosis factor receptor-associated factor 3 is a positive regulator of pathological cardiac hypertrophy. Hypertension. 2015;66:356–67.
Yao Z, Xing L, Boyce BF. NF-kappaB p100 limits TNF-induced bone resorption in mice by a TRAF3-dependent mechanism. J Clin Invest. 2009;119:3024–34.
Xiu Y, Xu H, Zhao C, Li J, Morita Y, Yao Z, et al. Chloroquine reduces osteoclastogenesis in murine osteoporosis by preventing TRAF3 degradation. J Clin Invest. 2014;124:297–310.
Gong J, Li ZZ, Guo S, Zhang XJ, Zhang P, Zhao GN, et al. Neuron-specific tumor necrosis factor receptor-associated factor 3 is a central regulator of neuronal death in acute ischemic stroke. Hypertension. 2015;66:604–16.
Hacker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol. 2011;11:457–68.
Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity. 2012;36:742–54.
Cai X, Du J, Liu Y, Xia W, Liu J, Zou M, et al. Identification and characterization of receptor-interacting protein 2 as a TNFR-associated factor 3 binding partner. Gene. 2013;517:205–11.
Chen X, Yang X, Zheng Y, Yang Y, Xing Y, Chen Z. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell. 2014;5:369–81.
Guan K, Wei C, Zheng Z, Song T, Wu F, Zhang Y, et al. MAVS promotes inflammasome activation by targeting ASC for K63-linked ubiquitination via the E3 ligase TRAF3. J Immunol. 2015;194:4880–90.
Hildebrand JM, Yi Z, Buchta CM, Poovassery J, Stunz LL, Bishop GA. Roles of tumor necrosis factor receptor associated factor 3 (TRAF3) and TRAF5 in immune cell functions. Immunol Rev. 2011;244:55–74.
Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol. 2010;11:70–5.
Pedros C, Zhang Y, JK H, Choi YS, Canonigo-Balancio AJ, Yates JR 3rd, et al. TRAF-like motif of the inducible costimulator ICOS controls development of germinal center TFH cells via the kinase TBK1. Nat Immunol. 2016;17:825–33.
Hu H, Brittain GC, Chang JH, Puebla-Osorio N, Jin J, Zal A, et al. OTUD7B controls non-canonical NF-kappaB activation through deubiquitination of TRAF3. Nature. 2013;494:371–4.
Xiao Y, Jin J, Chang M, Chang JH, Hu H, Zhou X, et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat Med. 2013;19:595–602.
Li P, Liu H, Zhang Y, Liao R, He K, Ruan X, et al. Endotoxin tolerance inhibits degradation of tumor necrosis factor receptor-associated factor 3 by suppressing Pellino 1 expression and the K48 ubiquitin ligase activity of cellular inhibitor of apoptosis protein 2. J Infect Dis. 2016;214:906–15.
Zhong B, Liu X, Wang X, Li H, Darnay BG, Lin X, et al. Ubiquitin-specific protease 25 regulates TLR4-dependent innate immune responses through deubiquitination of the adaptor protein TRAF3. Sci Signal. 2013;6:ra35.
Lin D, Zhang M, Zhang MX, Ren Y, Jin J, Zhao Q, et al. Induction of USP25 by viral infection promotes innate antiviral responses by mediating the stabilization of TRAF3 and TRAF6. Proc Natl Acad Sci U S A. 2015;112:11324–9.
Jiang B, Shen H, Chen Z, Yin L, Zan L, Rui L. Carboxyl terminus of HSC70-interacting protein (CHIP) down-regulates NF-kappaB-inducing kinase (NIK) and suppresses NIK-induced liver injury. J Biol Chem. 2015;290:11704–14.
Panda S, Nilsson JA, Gekara NO. Deubiquitinase MYSM1 regulates innate immunity through inactivation of TRAF3 and TRAF6 complexes. Immunity. 2015;43:647–59.
Wang Y, Shaked I, Stanford SM, Zhou W, Curtsinger JM, Mikulski Z, et al. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity. 2013;39:111–22.
Yi Z, Lin WW, Stunz LL, Bishop GA. The adaptor TRAF3 restrains the lineage determination of thymic regulatory T cells by modulating signaling via the receptor for IL-2. Nat Immunol. 2014;15:866–74.
Lin YC, Huang DY, Chu CL, Lin YL, Lin WW. The tyrosine kinase Syk differentially regulates Toll-like receptor signaling downstream of the adaptor molecules TRAF6 and TRAF3. Sci Signal. 2013;6:ra71.
Zhou Y, He C, Yan D, Liu F, Liu H, Chen J, et al. The kinase CK1varepsilon controls the antiviral immune response by phosphorylating the signaling adaptor TRAF3. Nat Immunol. 2016;17:397–405.
Ma C, Lin W, Liu Z, Tang W, Gautam R, Li H, et al. NDR1 protein kinase promotes IL-17- and TNF-alpha-mediated inflammation by competitively binding TRAF3. EMBO Rep. 2017;18:586–602.
Ma C, Spies NP, Gong T, Jones CX, Chu WM. Involvement of DNA-PKcs in the type I IFN response to CpG-ODNs in conventional dendritic cells in TLR9-dependent or -independent manners. PLoS One. 2015;10:e0121371.
Cullell N, Muino E, Carrera C, Torres N, Krupinski J, Fernandez-Cadenas I. Role of TRAF3 in neurological and cardiovascular diseases: an overview of recent studies. Biomol Concepts. 2017;
Ooi JY, Tuano NK, Rafehi H, Gao XM, Ziemann M, XJ D, et al. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics. 2015;10:418–30.
Shen J, Qiao YQ, Ran ZH, Wang TR. Up-regulation and pre-activation of TRAF3 and TRAF5 in inflammatory bowel disease. Int J Med Sci. 2013;10:156–63.
Momeni M, Zainodini N, Bidaki R, Hassanshahi G, Daneshvar H, Khaleghinia M, et al. Decreased expression of toll like receptor signaling molecules in chronic HBV infected patients. Hum Immunol. 2014;75:15–9.
Wang PX, Zhang XJ, Luo P, Jiang X, Zhang P, Guo J, et al. Hepatocyte TRAF3 promotes liver steatosis and systemic insulin resistance through targeting TAK1-dependent signalling. Nat Commun. 2016;7:10592.
Yao Z, Lei W, Duan R, Li Y, Luo L, Boyce BF. RANKL cytokine enhances TNF-induced osteoclastogenesis independently of TNF receptor associated factor (TRAF) 6 by degrading TRAF3 in osteoclast precursors. J Biol Chem. 2017;292:10169–79.
Yang C, McCoy K, Davis JL, Schmidt-Supprian M, Sasaki Y, Faccio R, et al. NIK stabilization in osteoclasts results in osteoporosis and enhanced inflammatory osteolysis. PLoS One. 2010;5:e15383.
Perez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. 2010;33:400–11.
Gallego-Fabrega C, Carrera C, Reny JL, Fontana P, Slowik A, Pera J, et al. TRAF3 epigenetic regulation is associated with vascular recurrence in patients with ischemic stroke. Stroke. 2016;47:1180–6.
Zhu S, Pan W, Shi P, Gao H, Zhao F, Song X, et al. Modulation of experimental autoimmune encephalomyelitis through TRAF3-mediated suppression of interleukin 17 receptor signaling. J Exp Med. 2010;207:2647–62.
Myles A, Rahman MT, Aggarwal A. Membrane-bound toll-like receptors are overexpressed in peripheral blood and synovial fluid mononuclear cells of enthesitis-related arthritis category of juvenile idiopathic arthritis (JIA-ERA) patients and lead to secretion of inflammatory mediators. J Clin Immunol. 2012;32:488–96.
Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls. Am J Hum Genet. 2013; 92:854-65.
Chang JH, Hu H, Jin J, Puebla-Osorio N, Xiao Y, Gilbert BE, et al. TRAF3 regulates the effector function of regulatory T cells and humoral immune responses. J Exp Med. 2014;211:137–51.
Yi Z, Stunz LL, Bishop GA. TNF receptor associated factor 3 plays a key role in development and function of invariant natural killer T cells. J Exp Med. 2013;210:1079–86.
Xie P, Kraus ZJ, Stunz LL, Liu Y, Bishop GATNF. Receptor-associated factor 3 is required for T cell-mediated immunity and TCR/CD28 signaling. J Immunol. 2011;186:143–55.
Jenkinson SR, Williams JA, Jeon H, Zhang J, Nitta T, Ohigashi I, et al. TRAF3 enforces the requirement for T cell cross-talk in thymic medullary epithelial development. Proc Natl Acad Sci U S A. 2013;110:21107–12.
Kedinger V, Rio MC. TRAF4, the unique family member. Adv Exp Med Biol. 2007;597:60–71.
Rousseau A, Rio MC, Alpy F. TRAF4, at the crossroad between morphogenesis and cancer. Cancers (Basel). 2011;3:2734–49.
Shiels H, Li X, Schumacker PT, Maltepe E, Padrid PA, Sperling A, et al. TRAF4 deficiency leads to tracheal malformation with resulting alterations in air flow to the lungs. Am J Pathol. 2000;157:679–88.
Regnier CH, Masson R, Kedinger V, Textoris J, Stoll I, Chenard MP, et al. Impaired neural tube closure, axial skeleton malformations, and tracheal ring disruption in TRAF4-deficient mice. Proc Natl Acad Sci U S A. 2002;99:5585–90.
Rousseau A, Wilhelm LP, Tomasetto C, Alpy F. The phosphoinositide-binding protein TRAF4 modulates tight junction stability and migration of cancer cells. Tissue Barriers. 2014;2:e975597.
Wu L, Chen X, Zhao J, Martin B, Zepp JA, Ko JS, et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J Exp Med. 2015;212:1571–87.
Li JM, Fan LM, Christie MR, Shah AM. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: role of p47phox phosphorylation and binding to TRAF4. Mol Cell Biol. 2005;25:2320–30.
Esparza EM, Arch RH. TRAF4 functions as an intermediate of GITR-induced NF-kappaB activation. Cell Mol Life Sci. 2004;61:3087–92.
Teng L, Fan LM, Meijles D, Li JM. Divergent effects of p47(phox) phosphorylation at S303-4 or S379 on tumor necrosis factor-alpha signaling via TRAF4 and MAPK in endothelial cells. Arterioscler Thromb Vasc Biol. 2012;32:1488–96.
Takeshita F, Ishii KJ, Kobiyama K, Kojima Y, Coban C, Sasaki S, et al. TRAF4 acts as a silencer in TLR-mediated signaling through the association with TRAF6 and TRIF. Eur J Immunol. 2005;35:2477–85.
Marinis JM, Homer CR, McDonald C, Abbott DW. A novel motif in the Crohn’s disease susceptibility protein, NOD2, allows TRAF4 to down-regulate innate immune responses. J Biol Chem. 2011;286:1938–50.
Marinis JM, Hutti JE, Homer CR, Cobb BA, Cantley LC, McDonald C, et al. IkappaB kinase alpha phosphorylation of TRAF4 downregulates innate immune signaling. Mol Cell Biol. 2012;32:2479–89.
Zepp JA, Liu C, Qian W, Wu L, Gulen MF, Kang Z, et al. Cutting edge: TNF receptor-associated factor 4 restricts IL-17-mediated pathology and signaling processes. J Immunol. 2012;189:33–7.
Zepp JA, Wu L, Qian W, Ouyang W, Aronica M, Erzurum S, et al. TRAF4-SMURF2-mediated DAZAP2 degradation is critical for IL-25 signaling and allergic airway inflammation. J Immunol. 2015;194:2826–37.
Abell AN, Johnson GL. MEKK4 is an effector of the embryonic TRAF4 for JNK activation. J Biol Chem. 2005;280:35793–6.
Shen J, Qiao Y, Ran Z, Wang T. Different activation of TRAF4 and TRAF6 in inflammatory bowel disease. Mediators Inflamm. 2013;2013:647936.
Aston C, Jiang L, Sokolov BP. Microarray analysis of postmortem temporal cortex from patients with schizophrenia. J Neurosci Res. 2004;77:858–66.
Muller N, Weidinger E, Leitner B, Schwarz MJ. The role of inflammation in schizophrenia. Front Neurosci. 2015;9:372.
Blaise S, Kneib M, Rousseau A, Gambino F, Chenard MP, Messadeq N, et al. In vivo evidence that TRAF4 is required for central nervous system myelin homeostasis. PLoS One. 2012;7:e30917.
Au PY, Yeh WC. Physiological roles and mechanisms of signaling by TRAF2 and TRAF5. Adv Exp Med Biol. 2007;597:32–47.
Doyle MS, Collins ES, FitzGerald OM, Pennington SR. New insight into the functions of the interleukin-17 receptor adaptor protein Act1 in psoriatic arthritis. Arthritis Res Ther. 2012;14:226.
Buchta CM, Bishop GA. TRAF5 negatively regulates TLR signaling in B lymphocytes. J Immunol. 2014;192:145–50.
Liu S, Chen J, Cai X, Wu J, Chen X, Wu YT, et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife. 2013;2:e00785.
Shang J, Li L, Wang X, Pan H, Liu S, He R, et al. Disruption of tumor necrosis factor receptor-associated factor 5 exacerbates murine experimental colitis via regulating T helper cell-mediated inflammation. Mediators Inflamm. 2016;2016:9453745.
•• Nagashima H, Okuyama Y, Asao A, Kawabe T, Yamaki S, Nakano H, et al. The adaptor TRAF5 limits the differentiation of inflammatory CD4(+) T cells by antagonizing signaling via the receptor for IL-6. Nat Immunol. 2014;15:449–56. This article reports the direct in vivo evidence of TRAF5 in IL-6 receptor/gp130 signaling, Th17 differentiation, and Th17-mediated EAE
Nagashima H, Okuyama Y, Hayashi T, Ishii N, So T. TNFR-associated factors 2 and 5 differentially regulate the instructive IL-6 receptor signaling required for Th17 development. J Immunol. 2016;196:4082–9.
So T, Salek-Ardakani S, Nakano H, Ware CF, Croft M. TNF receptor-associated factor 5 limits the induction of Th2 immune responses. J Immunol. 2004;172:4292–7.
Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat Immunol. 2011;12:853–60.
Wang X, Yang J, Han L, Zhao K, Wu Q, Bao L, et al. TRAF5-mediated Lys-63-linked polyubiquitination plays an essential role in positive regulation of RORgammat in promoting IL-17A expression. J Biol Chem. 2015;290:29086–94.
Zhong B, Liu X, Wang X, Chang SH, Wang A, Reynolds JM, et al. Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat Immunol. 2012;13:1110–7.
Missiou A, Rudolf P, Stachon P, Wolf D, Varo N, Aichele P, et al. TRAF5 deficiency accelerates atherogenesis in mice by increasing inflammatory cell recruitment and foam cell formation. Circ Res. 2010;107:757–66.
Bian Z, Dai J, Hiroyasu N, Guan H, Yuan Y, Gan L, et al. Disruption of tumor necrosis factor receptor associated factor 5 exacerbates pressure overload cardiac hypertrophy and fibrosis. J Cell Biochem. 2014;115:349–58.
Gao L, Wang PX, Zhang Y, CJ Y, Ji Y, Wang X, et al. Tumor necrosis factor receptor-associated factor 5 (Traf5) acts as an essential negative regulator of hepatic steatosis. J Hepatol. 2016;65:125–36.
Wang L, Lu Y, Guan H, Jiang D, Guan Y, Zhang X, et al. Tumor necrosis factor receptor-associated factor 5 is an essential mediator of ischemic brain infarction. J Neurochem. 2013;126:400–14.
Gaffen SL. Recent advances in the IL-17 cytokine family. Curr Opin Immunol. 2011;23:613–9.
Xiang Q, Chen L, Hou S, Fang J, Zhou Y, Bai L, et al. TRAF5 and TRAF3IP2 gene polymorphisms are associated with Behcet’s disease and Vogt-Koyanagi-Harada syndrome: a case-control study. PLoS One. 2014;9:e84214.
Xiang Q, Chen L, Fang J, Hou S, Wei L, Bai L, et al. TNF receptor-associated factor 5 gene confers genetic predisposition to acute anterior uveitis and pediatric uveitis. Arthritis Res Ther. 2013;15:R113.
Wu H, Arron JR. TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. Bioessays. 2003;25:1096–105.
Walsh MC, Lee J, Choi Y. Tumor necrosis factor receptor-associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol Rev. 2015;266:72–92.
Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408:600–5.
Zhang H, Wu C, Matesic LE, Li X, Wang Z, Boyce BF, et al. Ubiquitin E3 ligase Itch negatively regulates osteoclast formation by promoting deubiquitination of tumor necrosis factor (TNF) receptor-associated factor 6. J Biol Chem. 2013;288:22359–68.
Yang YJ, Chen W, Carrigan SO, Chen WM, Roth K, Akiyama T, et al. TRAF6 specifically contributes to FcepsilonRI-mediated cytokine production but not mast cell degranulation. J Biol Chem. 2008;283:32110–8.
Bonito AJ, Aloman C, Fiel MI, Danzl NM, Cha S, Weinstein EG, et al. Medullary thymic epithelial cell depletion leads to autoimmune hepatitis. J Clin Invest. 2013;123:3510–24.
Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94:192–203.
Zucchelli S, Codrich M, Marcuzzi F, Pinto M, Vilotti S, Biagioli M, et al. TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum Mol Genet. 2010;19:3759–70.
Zucchelli S, Marcuzzi F, Codrich M, Agostoni E, Vilotti S, Biagioli M, et al. Tumor necrosis factor receptor-associated factor 6 (TRAF6) associates with huntingtin protein and promotes its atypical ubiquitination to enhance aggregate formation. J Biol Chem. 2011;286:25108–17.
Xiao X, Balasubramanian S, Liu W, Chu X, Wang H, Taparowsky EJ, et al. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nat Immunol. 2012;13:981–90.
Volpi C, Fallarino F, Pallotta MT, Bianchi R, Vacca C, Belladonna ML, et al. High doses of CpG oligodeoxynucleotides stimulate a tolerogenic TLR9-TRIF pathway. Nat Commun. 2013;4:1852.
Watanabe T, Asano N, Meng G, Yamashita K, Arai Y, Sakurai T, et al. NOD2 downregulates colonic inflammation by IRF4-mediated inhibition of K63-linked polyubiquitination of RICK and TRAF6. Mucosal Immunol. 2014;7:1312–25.
Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity. 2011;34:854–65.
Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity. 2011;34:843–53.
Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat Immunol. 2012;13:823–31.
Mellett M, Atzei P, Horgan A, Hams E, Floss T, Wurst W, et al. Orphan receptor IL-17RD tunes IL-17A signalling and is required for neutrophilia. Nat Commun. 2012;3:1119.
Lim S, Bae E, Kim HS, Kim TA, Byun K, Kim B, et al. TRAF6 mediates IL-1beta/LPS-induced suppression of TGF-beta signaling through its interaction with the type III TGF-beta receptor. PLoS One. 2012;7:e32705.
Adamson SE, Griffiths R, Moravec R, Senthivinayagam S, Montgomery G, Chen W, et al. Disabled homolog 2 controls macrophage phenotypic polarization and adipose tissue inflammation. J Clin Invest. 2016;126:1311–22.
Xie JJ, Liang JQ, Diao LH, Altman A, Li Y. TNFR-associated factor 6 regulates TCR signaling via interaction with and modification of LAT adapter. J Immunol. 2013;190:4027–36.
Lin FT, Lin VY, Lin VT, Lin WC. TRIP6 antagonizes the recruitment of A20 and CYLD to TRAF6 to promote the LPA2 receptor-mediated TRAF6 activation. Cell Discov. 2016;2
Wi SM, Moon G, Kim J, Kim ST, Shim JH, Chun E, et al. TAK1-ECSIT-TRAF6 complex plays a key role in the TLR4 signal to activate NF-kappaB. J Biol Chem. 2014;289:35205–14.
Geng J, Sun X, Wang P, Zhang S, Wang X, Wu H, et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat Immunol. 2015;16:1142–52.
Zhou X, Liu Z, Cheng X, Zheng Y, Zeng F, He Y. Socs1 and Socs3 degrades Traf6 via polyubiquitination in LPS-induced acute necrotizing pancreatitis. Cell Death Dis. 2015;6:e2012.
McBerry C, Gonzalez RM, Shryock N, Dias A, Aliberti J. SOCS2-induced proteasome-dependent TRAF6 degradation: a common anti-inflammatory pathway for control of innate immune responses. PLoS One. 2012;7:e38384.
Iha H, Peloponese JM, Verstrepen L, Zapart G, Ikeda F, Smith CD, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J. 2008;27:629–41.
Wei J, Wei C, Wang M, Qiu X, Li Y, Yuan Y, et al. The GTPase-activating protein GIT2 protects against colitis by negatively regulating Toll-like receptor signaling. Proc Natl Acad Sci U S A. 2014;111:8883–8.
Dong XM, Liu ED, Meng YX, Liu C, Bi YL, Wu HW, et al. Keratin 8 limits TLR-triggered inflammatory responses through inhibiting TRAF6 polyubiquitination. Sci Rep. 2016;6:32710.
Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–56.
Lin XW, Xu WC, Luo JG, Guo XJ, Sun T, Zhao XL, et al. WW domain containing E3 ubiquitin protein ligase 1 (WWP1) negatively regulates TLR4-mediated TNF-alpha and IL-6 production by proteasomal degradation of TNF receptor associated factor 6 (TRAF6). PLoS One. 2013;8:e67633.
Qiu Q, Zheng Z, Chang L, Zhao YS, Tan C, Dandekar A, et al. Toll-like receptor-mediated IRE1alpha activation as a therapeutic target for inflammatory arthritis. EMBO J. 2013;32:2477–90.
Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–60.
Garg AV, Ahmed M, Vallejo AN, Ma A, Gaffen SL. The deubiquitinase A20 mediates feedback inhibition of interleukin-17 receptor signaling. Sci Signal. 2013;6:ra44.
Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. 2016;48:67–73.
Jean-Charles PY, Zhang L, Wu JH, Han SO, Brian L, Freedman NJ, et al. Ubiquitin-specific protease 20 regulates the reciprocal functions of beta-arrestin2 in toll-like receptor 4-promoted nuclear factor kappaB (NFkappaB) activation. J Biol Chem. 2016;291:7450–64.
Yang S, Wang B, Humphries F, Hogan AE, O’Shea D, Moynagh PN. The E3 ubiquitin ligase Pellino3 protects against obesity-induced inflammation and insulin resistance. Immunity. 2014;41:973–87.
Yuk JM, Shin DM, Lee HM, Kim JJ, Kim SW, Jin HS, et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat Immunol. 2011;12:742–51.
Jiao S, Zhang Z, Li C, Huang M, Shi Z, Wang Y, et al. The kinase MST4 limits inflammatory responses through direct phosphorylation of the adaptor TRAF6. Nat Immunol. 2015;16:246–57.
Kim E, Beon J, Lee S, Park SJ, Ahn H, Kim MG, et al. Inositol polyphosphate multikinase promotes Toll-like receptor-induced inflammation by stabilizing TRAF6. Sci Adv. 2017;3:e1602296.
Lee HJ, Jang SH, Kim H, Yoon JH, Chung KC. PINK1 stimulates interleukin-1beta-mediated inflammatory signaling via the positive regulation of TRAF6 and TAK1. Cell Mol Life Sci. 2012;69:3301–15.
Hedl M, Yan J, Abraham C. IRF5 and IRF5 disease-risk variants increase glycolysis and human M1 macrophage polarization by regulating proximal signaling and Akt2 activation. Cell Rep. 2016;16:2442–55.
Siednienko J, Jackson R, Mellett M, Delagic N, Yang S, Wang B, et al. Pellino3 targets the IRF7 pathway and facilitates autoregulation of TLR3- and viral-induced expression of type I interferons. Nat Immunol. 2012;13:1055–62.
Luu K, Greenhill CJ, Majoros A, Decker T, Jenkins BJ, Mansell A. STAT1 plays a role in TLR signal transduction and inflammatory responses. Immunol Cell Biol. 2014;92:761–9.
Wei J, Yuan Y, Jin C, Chen H, Leng L, He F, et al. The ubiquitin ligase TRAF6 negatively regulates the JAK-STAT signaling pathway by binding to STAT3 and mediating its ubiquitination. PLoS One. 2012;7:e49567.
Jung SM, Lee JH, Park J, Oh YS, Lee SK, Park JS, et al. Smad6 inhibits non-canonical TGF-beta1 signalling by recruiting the deubiquitinase A20 to TRAF6. Nat Commun. 2013;4:2562.
Wu H, Li XM, Wang JR, Gan WJ, Jiang FQ, Liu Y, et al. NUR77 exerts a protective effect against inflammatory bowel disease by negatively regulating the TRAF6/TLR-IL-1R signalling axis. J Pathol. 2016;238:457–69.
Li XM, Zhang S, He XS, Guo PD, Lu XX, Wang JR, et al. Nur77-mediated TRAF6 signalling protects against LPS-induced sepsis in mice. J Inflamm (Lond). 2016;13:4.
Tenekeci U, Poppe M, Beuerlein K, Buro C, Muller H, Weiser H, et al. K63-ubiquitylation and TRAF6 pathways regulate mammalian P-body formation and mRNA decapping. Mol Cell. 2016;62:943–57.
Slotwinski R, Sarnecka A, Dabrowska A, Kosalka K, Wachowska E, Balan BJ, et al. Innate immunity gene expression changes in critically ill patients with sepsis and disease-related malnutrition. Cent Eur J Immunol. 2015;40:311–24.
Song Z, Yao C, Yin J, Tong C, Zhu D, Sun Z, et al. Genetic variation in the TNF receptor-associated factor 6 gene is associated with susceptibility to sepsis-induced acute lung injury. J Transl Med. 2012;10:166.
Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–24.
Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999;4:353–62.
Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–9.
Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–92.
Zhang Y, Xu X, Ceylan-Isik AF, Dong M, Pei Z, Li Y, et al. Ablation of Akt2 protects against lipopolysaccharide-induced cardiac dysfunction: role of Akt ubiquitination E3 ligase TRAF6. J Mol Cell Cardiol. 2014;74:76–87.
Lv S, Qiu X, Li J, Li W, Zhang C, Zhang ZN, et al. Suppression of CRTC2-mediated hepatic gluconeogenesis by TRAF6 contributes to hypoglycemia in septic shock. Cell Discov. 2016;2:16046.
Liu G, Tsuruta Y, Gao Z, Park YJ, Abraham E. Variant IL-1 receptor-associated kinase-1 mediates increased NF-kappa B activity. J Immunol. 2007;179:4125–34.
Chandra R, Federici S, Bishwas T, Nemeth ZH, Deitch EA, Thomas JA, et al. IRAK1-dependent signaling mediates mortality in polymicrobial sepsis. Inflammation. 2013;36:1503–12.
Wan Y, Xiao H, Affolter J, Kim TW, Bulek K, Chaudhuri S, et al. Interleukin-1 receptor-associated kinase 2 is critical for lipopolysaccharide-mediated post-transcriptional control. J Biol Chem. 2009;284:10367–75.
Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202.
Yu X, Yi H, Guo C, Zuo D, Wang Y, Kim HL, et al. Pattern recognition scavenger receptor CD204 attenuates Toll-like receptor 4-induced NF-kappaB activation by directly inhibiting ubiquitination of tumor necrosis factor (TNF) receptor-associated factor 6. J Biol Chem. 2011;286:18795–806.
Wang Y, Tang Y, Teng L, Wu Y, Zhao X, Pei G. Association of beta-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2006;7:139–47.
Karimi K, Sarir H, Mortaz E, Smit JJ, Hosseini H, De Kimpe SJ, et al. Toll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophages. Respir Res. 2006;7:66.
Liu X, Cao H, Li J, Wang B, Zhang P, Dong Zhang X, et al. Autophagy induced by DAMPs facilitates the inflammation response in lungs undergoing ischemia-reperfusion injury through promoting TRAF6 ubiquitination. Cell Death Differ. 2017;24:683–93.
Han D, Walsh MC, Kim KS, Hong SW, Lee J, Yi J, et al. Dendritic cell expression of the signaling molecule TRAF6 is required for immune tolerance in the lung. Int Immunol. 2017;29:71–8.
Maezawa Y, Nakajima H, Suzuki K, Tamachi T, Ikeda K, Inoue J, et al. Involvement of TNF receptor-associated factor 6 in IL-25 receptor signaling. J Immunol. 2006;176:1013–8.
Ji YX, Zhang P, Zhang XJ, Zhao YC, Deng KQ, Jiang X, et al. The ubiquitin E3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via TAK1-dependent signalling. Nat Commun. 2016;7:11267.
Zhang D, Liu X, Chen X, Gu J, Li F, Zhang W, et al. Role of the MAPKs/TGF-beta1/TRAF6 signaling pathway in atrial fibrosis of patients with chronic atrial fibrillation and rheumatic mitral valve disease. Cardiology. 2014;129:216–23.
Zhang D, Chen X, Wang Q, Wu S, Zheng Y, Liu X. Role of the MAPKs/TGF-beta1/TRAF6 signaling pathway in postoperative atrial fibrillation. PLoS One. 2017;12:e0173759.
Polykratis A, van Loo G, Xanthoulea S, Hellmich M, Pasparakis M. Conditional targeting of tumor necrosis factor receptor-associated factor 6 reveals opposing functions of Toll-like receptor signaling in endothelial and myeloid cells in a mouse model of atherosclerosis. Circulation. 2012;126:1739–51.
McDermott E, Ryan EJ, Tosetto M, Gibson D, Burrage J, Keegan D, et al. DNA methylation profiling in inflammatory bowel disease provides new insights into disease pathogenesis. J Crohns Colitis. 2016;10:77–86.
• Han D, Walsh MC, Cejas PJ, Dang NN, Kim YF, Kim J, et al. Dendritic cell expression of the signaling molecule TRAF6 is critical for gut microbiota-dependent immune tolerance. Immunity. 2013;38:1211–22. This article provides the direct in vivo evidence of the importance of DC TRAF6 in controlling immune tolerance and preventing enteritis
Han D, Walsh MC, Kim KS, Hong SW, Lee J, Yi J, et al. Microbiota-independent ameliorative effects of antibiotics on spontaneous Th2-associated pathology of the small intestine. PLoS One. 2015;10:e0118795.
Cejas PJ, Walsh MC, Pearce EL, Han D, Harms GM, Artis D, et al. TRAF6 inhibits Th17 differentiation and TGF-beta-mediated suppression of IL-2. Blood. 2010;115:4750–7.
Vlantis K, Polykratis A, Welz PS, van Loo G, Pasparakis M, Wullaert A. TLR-independent anti-inflammatory function of intestinal epithelial TRAF6 signalling prevents DSS-induced colitis in mice. Gut. 2016;65:935–43.
Sajadi SM, Mirzaei V, Hassanshahi G, Khorramdelazad H, Daredor HY, Hosseini SM, et al. Decreased expressions of Toll-like receptor 9 and its signaling molecules in chronic hepatitis B virus-infected patients. Arch Pathol Lab Med. 2013;137:1674–9.
Balasubramanyam M, Aravind S, Gokulakrishnan K, Prabu P, Sathishkumar C, Ranjani H, et al. Impaired miR-146a expression links subclinical inflammation and insulin resistance in type 2 diabetes. Mol Cell Biochem. 2011;351:197–205.
Lenin R, Sankaramoorthy A, Mohan V, Balasubramanyam M. Altered immunometabolism at the interface of increased endoplasmic reticulum (ER) stress in patients with type 2 diabetes. J Leukoc Biol. 2015;98:615–22.
Guo C, Zhang L, Nie L, Zhang N, Xiao D, Ye X, et al. Association of polymorphisms in the MyD88, IRAK4 and TRAF6 genes and susceptibility to type 2 diabetes mellitus and diabetic nephropathy in a southern Han Chinese population. Mol Cell Endocrinol. 2016;429:114–9.
Lan P, Fan Y, Zhao Y, Lou X, Monsour HP, Zhang X, et al. TNF superfamily receptor OX40 triggers invariant NKT cell pyroptosis and liver injury. J Clin Invest. 2017;127:2222–34.
Cheng KK, Lam KS, Wang Y, Wu D, Zhang M, Wang B, et al. TRAF6-mediated ubiquitination of APPL1 enhances hepatic actions of insulin by promoting the membrane translocation of Akt. Biochem J. 2013;455:207–16.
Nishida A, Inatomi O, Fujimoto T, Imaeda H, Tani M, Andoh A. Interleukin-36alpha induces inflammatory mediators from human pancreatic myofibroblasts via a MyD88 dependent pathway. Pancreas. 2017;46:539–48.
Zhou XY, Zhou ZG, Ding JL, Wang L, Wang R, Zhou B, et al. TRAF6 as the key adaptor of TLR4 signaling pathway is involved in acute pancreatitis. Pancreas. 2010;39:359–66.
Su L, Chen Z, Yan Y, Liang B, Xie J, Chen Q, et al. Between TRAF6 gene polymorphisms and susceptibility of ischemic stroke in southern Chinese Han population. J Mol Neurosci. 2015;57:386–92.
Wu D, Lee YC, Liu HC, Yuan RY, Chiou HY, Hung CH, et al. Identification of TLR downstream pathways in stroke patients. Clin Biochem. 2013;46:1058–64.
Zheng CZ, Shu YB, Luo YL, Luo J. The role of miR-146a in modulating TRAF6-induced inflammation during lupus nephritis. Eur Rev Med Pharmacol Sci. 2017;21:1041–8.
Zhu Y, Xue Z, Di L. Regulation of MiR-146a and TRAF6 in the diagnose of lupus nephritis. Med Sci Monit. 2017;23:2550–7.
Zhu LJ, Yang TC, Wu Q, Yuan LP, Chen ZW, Luo MH, et al. Tumor necrosis factor receptor-associated factor (TRAF) 6 inhibition mitigates the pro-inflammatory roles and proliferation of rheumatoid arthritis fibroblast-like synoviocytes. Cytokine. 2017;93:26–33.
Namjou B, Choi CB, Harley IT, Alarcon-Riquelme ME, Kelly JA, Glenn SB, et al. Evaluation of TRAF6 in a large multiancestral lupus cohort. Arthritis Rheum. 2012;64:1960–9.
Chiffoleau E, Kobayashi T, Walsh MC, King CG, Walsh PT, Hancock WW, et al. TNF receptor-associated factor 6 deficiency during hemopoiesis induces Th2-polarized inflammatory disease. J Immunol. 2003;171:5751–9.
King CG, Kobayashi T, Cejas PJ, Kim T, Yoon K, Kim GK, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Med. 2006;12:1088–92.
King CG, Buckler JL, Kobayashi T, Hannah JR, Bassett G, Kim T, et al. Cutting edge: requirement for TRAF6 in the induction of T cell anergy. J Immunol. 2008;180:34–8.
Shimo Y, Yanai H, Ohshima D, Qin J, Motegi H, Maruyama Y, et al. TRAF6 directs commitment to regulatory T cells in thymocytes. Genes Cells. 2011;16:437–47.
Muto G, Kotani H, Kondo T, Morita R, Tsuruta S, Kobayashi T, et al. TRAF6 is essential for maintenance of regulatory T cells that suppress Th2 type autoimmunity. PLoS One. 2013;8:e74639.
Lee YH, Song GG. Associations between TNFSF4 and TRAF1-C5 gene polymorphisms and systemic lupus erythematosus: a meta-analysis. Hum Immunol. 2012;73:1050–4.
Kurreeman FA, Goulielmos GN, Alizadeh BZ, Rueda B, Houwing-Duistermaat J, Sanchez E, et al. The TRAF1-C5 region on chromosome 9q33 is associated with multiple autoimmune diseases. Ann Rheum Dis. 2010;69:696–9.
Potter C, Eyre S, Cope A, Worthington J, Barton A. Investigation of association between the TRAF family genes and RA susceptibility. Ann Rheum Dis. 2007;66:1322–6.
Zervou MI, Sidiropoulos P, Petraki E, Vazgiourakis V, Krasoudaki E, Raptopoulou A, et al. Association of a TRAF1 and a STAT4 gene polymorphism with increased risk for rheumatoid arthritis in a genetically homogeneous population. Hum Immunol. 2008;69:567–71.
Wang H, Chen W, Wang L, Li F, Zhang C, Xu L. Tumor necrosis factor receptor-associated factor 6 promotes migration of rheumatoid arthritis fibroblast-like synoviocytes. Mol Med Rep. 2015;11:2761–6.
Zarzycka B, Seijkens T, Nabuurs SB, Ritschel T, Grommes J, Soehnlein O, et al. Discovery of small molecule CD40-TRAF6 inhibitors. J Chem Inf Model. 2015;55:294–307.
Moriya J, Takeuchi K, Tai K, Arai K, Kobayashi N, Yoneda N, et al. Structure-based development of a protein-protein interaction inhibitor targeting tumor necrosis factor receptor-associated factor 6. J Med Chem. 2015;58:5674–83.
Aarts S, Seijkens TTP, Kusters PJH, van der Pol SMA, Zarzycka B, Heijnen P, et al. Inhibition of CD40-TRAF6 interactions by the small molecule inhibitor 6877002 reduces neuroinflammation. J Neuroinflammation. 2017;14:105.
van den Berg SM, Seijkens TT, Kusters PJ, Zarzycka B, Beckers L, den Toom M, et al. Blocking CD40-TRAF6 interactions by small-molecule inhibitor 6860766 ameliorates the complications of diet-induced obesity in mice. Int J Obes (Lond). 2015;39:782–90.
Acknowledgements
This study was supported by the National Institutes of Health grants R01 CA158402 and R21 AI128264 (P. Xie), the Department of Defense grant W81XWH-13-1-0242 (P. Xie), a Pilot Award from the Cancer Institute of New Jersey through Grant Number P30CA072720 from the National Cancer Institute (P. Xie), and a Busch Biomedical Grant (P. Xie).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no competing financial interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Immunology and Inflammation
Rights and permissions
About this article

Cite this article
Lalani, A.I., Zhu, S., Gokhale, S. et al. TRAF Molecules in Inflammation and Inflammatory Diseases. Curr Pharmacol Rep 4, 64–90 (2018). https://doi.org/10.1007/s40495-017-0117-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-017-0117-y