
Abstract
We examined expression and function of TLRs in enthesitis-related arthritis (ERA) patients. RNA levels of TLR1, TLR3, and TLRs 5–8 were measured in 24 ERA peripheral blood mononuclear cells (PBMC), 18 synovial fluid mononuclear cells (SFMC), and IRAK1, IRAK4, TRIF, TRAF3, and TRAF6 in 18 PBMC and 10 SFMC. IL-6 and IL-8 were measured in supernatants from ERA PBMC (n = 7), SFMC (n = 3), and healthy PBMC (n = 5) cultured with ligands for TLR1/2 (Pam 3-cys), TLR3 (poly I:C), TLR5 (flagellin), and TLR2/6 (zymosan). TLRs 1, 3, 5, and 6 were measured in whole blood (n = 20 ERA, seven healthy) and SFMC (n = 2) by flow cytometry. ERA PBMC compared to healthy PBMC and SFMC compared to ERA PBMC had higher RNA expression of TLR1, TLR3, TLR5, TLR6, IRAK1, IRAK4, TRIF, TRAF3, and TRAF6. TLR7 and TLR8 RNA expression was similar in all study groups. IL-6 and IL-8 levels were higher in stimulated ERA SFMC compared to ERA PBMC and in ERA PBMC compared to control PBMC. TLRs 1, 3, and 6 were also overexpressed at the protein level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Toll-like receptors (TLRs) are a key link between infection, injury, and inflammation. They recognize pathogen- and danger-associated molecular patterns (PAMPs and DAMPs), and subsequently trigger a pro-inflammatory cascade [1]. They also initiate the first step in adaptive immune response, by activating DCs and macrophages, which in turn prime T and B cells [2]. Although TLR-mediated inflammation is an important aspect of host defense, it is also linked to pathogenesis of several autoimmune diseases [3] like rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), inflammatory bowel disease (IBD), and type I diabetes. This is because excessive cytokine production and cell activation induced by TLR stimulation can damage tissues and organs.
TLRs are expressed in synovium of RA patients and were initially thought to play a role in exacerbation of disease activity in RA in response to infections [4]. However, it was subsequently discovered that TLRs are also able to up-regulate TNF-α production in response to endogenous ligands like HSPs [5] released after tissue damage, suggesting that TLRs can maintain an inflammatory response even in the absence of pathogen [6]. Necrotic cells, which are usually abundant in damaged tissue, e.g., in the arthritic joint, also activate TLRs [7]. It is possible that such mechanisms also operate in other arthritides.
Juvenile idiopathic arthritis (JIA) is a heterogeneous group of chronic, debilitating childhood arthritides and is grouped into seven categories as per the ILAR criteria [8]. JIA is supposed to arise in a genetically susceptible individual as a result of environmental triggers [9]. Among the various categories, enthesitis-related arthritis (ERA) is most prevalent in India [10, 11]. Microorganisms are one of the important environmental factors suspected to play a role in its pathogenesis, and this may be mediated via TLRs. Also, the synovial compartment contains a wide repertoire of endogenous TLR ligands. Thus, TLRs may contribute to increased inflammation in ERA patients.
We had earlier found that enteric bacteria implicated in reactive arthritis could be responsible for exacerbation of disease in ERA [12]. Further, fibroblast-like synoviocytes (FLS) from ERA patients were shown to produce pro-inflammatory cytokines like IL-6, IL-8 as well as matrix-degrading enzymes like MMP-1 and MMP-3 on stimulation by TLR ligands [13]. Later TLR 2 and TLR 4 expression was found to be increased both at protein and mRNA level in peripheral blood and synovial fluid mononuclear cells (PBMC and SFMC) of ERA patients; however, TLR 9 did not show any upregulation [14].
The present study was designed to further investigate the role of TLRs in ERA. We measured expression of TLRs 1, 3, 5–8, and their downstream adaptors (IRAK1, IRAK4, TRIF, TRAF3, and TRAF6) in mononuclear cells from ERA patients and found upregulation of membrane-bound TLRs, TLR3, and their signaling molecules in PBMC and SFMC. Further, we evaluated the response of PBMC and SFMC from ERA patients to stimulation with TLR ligands.
Methods
Patients and Controls
ERA patients with active disease who satisfied ILAR criteria [8] and whose parents gave written informed consent were enrolled in the study. Peripheral blood and synovial fluid (where available) was obtained from patients. Peripheral blood was collected from similar gender non-related young adults to serve as healthy controls. Blood was drawn in sterile vials containing endotoxin-free lithium heparin. The study was approved by the institutional ethics committee.
Due to limitation of amount of blood sample available, all experiments could not be performed on all samples. The actual number of samples studied for each set of experiment is mentioned in the “Results” section.
Isolation of Mononuclear Cells
Peripheral blood and synovial fluid mononuclear cells (PBMC and SFMC) were isolated using density gradient centrifugation by layering on Histopaque 1077-1 (Sigma-Aldrich, St Louis, MO, USA). Cells were frozen in Trizol™ (Invitrogen, Carlsbad, CA, USA) at a concentration of not more than 7 × 106/ml and stored at −80°C.
RNA Isolation
RNA was isolated by columns (RNeasy kit; Qiagen, Valencia, CA, USA) using the manufacturer’s protocol with slight modification. After thawing the cells and vortexing to facilitate lysis, 200 μl of chloroform was added per milliliter of Trizol used. After centrifugation at 4°C for 10 min at 12,000 × g, the interface was separated and subsequent processing was done using columns. RNA was eluted in RNase-free water and estimated by spectrophotometer (Nanodrop, Wilmington, DE, USA). RNA integrity was checked using RNA 6000 Nano assay kit for Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA).
cDNA Conversion and Real-Time PCR
cDNA was prepared using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA) as per manufacturer’s instructions. Briefly, 2 μg RNA was added to master mix comprising RT buffer, dNTP mix, random hexamers, and reverse transcriptase. The mixture was incubated at 25°C for 10 min, 37°C for 120 min, and 85°C for 5 min. Subsequently, real-time PCR was performed using either SyBR Green or Taqman chemistry on Applied Biosystems 7500 Real-Time PCR system.
Assay for TLRs
Primers for SyBr Green assay were purchased from Sigma Aldrich, India. Details of primers used are given elsewhere [15]. Each reaction mixture of 20 μl comprised 100 ng cDNA, primers, SyBr Green (Applied Biosystems), and sterile water. 18S rRNA was used as housekeeping gene (HSG). Reaction conditions were initial denaturation of one cycle at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. A dissociation step was added in the end, comprising one cycle of 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s.
Assay for Adaptors
Taqman gene expression assay kits were purchased from Applied Biosystems, the IDs being Hs01018347_m1 (IRAK1), Hs00211610_m1 (IRAK4), Hs01090712_m1 (TRIF), Hs00237035_m1 (TRAF3), and Hs00377558_m1 (TRAF6). GAPDH (assay ID 4333764 F) was used as HSG, as assay for 18S rRNA was not commercially available. PCR was performed according to manufacturer’s instructions.
Data Analysis
Patient PBMC Ct (Ct = cycle threshold) was calibrated with PBMC of healthy control, while patient SFMC Ct was calibrated with corresponding SFMC. Relative fold change was determined using ΔΔCt method, where \( {\text{fold}}\,{\text{change}} = {2^{{ - \Delta \Delta {\text{Ct}}}}}\,{\text{and}}\,\Delta \Delta {\text{Ct}} = {\left[ {\Delta {\text{C}}{{\text{t}}_{\text{TLR}}} - \Delta {\text{C}}{{\text{t}}_{\text{HSG}}}} \right]_{{{\text{patient}}\,{\text{sample}}}}} - {\left[ {\Delta {\text{C}}{{\text{t}}_{\text{TLR}}} - \Delta {\text{C}}{{\text{t}}_{\text{HSG}}}} \right]_{{{\text{calibrator}}\,{\text{sample}}}}} \).
A relative fold change of 1 meant no difference in expression between patients and controls.
Functional Assay
A total of 105 PBMC from seven ERA patients and five healthy controls and SFMC from three paired ERA samples were cultured in 10% cRPMI (Sigma-Aldrich) and stimulated with N-palmitoyl-S-[2,3-bis(palmitoyloxy-(2RS)-propyl]-(R)-cysteine (Pam3cys, TLR-1 ligand; 10 μg/ml; EMC Microcollection GmbH, Germany), polyinosinic:polycytidylic acid (poly I:C, TLR3 ligand; 20 ng/ml; Invivogen, San Diego, CA, USA), flagellin (TLR 5 ligand; 5 μg/ml; Invivogen), or zymosan (TLR-6 ligand; 10 μg/ml; Sigma-Aldrich). TNF-α (eBiosciences, CA, USA; 20 ng/ml) was used as positive control and unstimulated cells served as negative control. Supernatants were harvested after 48 h and IL-6 (BD OptEIA Kit, San Jose, CA, USA) and IL-8 (R&D Systems, Minneapolis, MN, USA) levels were measured by ELISA.
Flow Cytometry
Based on the results of experiments 4 and 5, we also decided to measure expression of TLRs1, 3, 5, and 6 at the protein level. To minimize the amount of samples to be taken from patients, this set of experiments was performed on whole blood (collected in EDTA vials). SFMC were isolated from SF (if available). Anti-TLR primary and PE-labeled secondary antibodies were purchased from Imgenex (Orissa, India) and FITC-conjugated anti-CD14 antibody from (BD Biosciences, San Diego, CA, USA). Isotypes were rabbit IgG for TLR1 and TLR5, and mouse IgG1 kappa for TLR3 and TLR6. Secondary antibodies to rabbit or mouse primary antibodies were raised in goat.
Anti-CD14 and anti-TLRs 1, 5, and 6 primary antibodies were added to 100 μl whole blood and incubated in the dark for 30 min. Two milliliters of 1× RBC lysing solution (BD Biosciences) was added to all tubes followed by incubation for 15 min in the dark. Samples were washed with 2 ml PBS for 5 min at 470 × g. PE-labeled secondary antibody was added to all tubes, and incubation and washing was done as above.
For TLR3 staining, samples were fixed with 20 μl fixation buffer (AB Serotec, UK) and incubated in the dark for 20 min. Following washing, the cells were incubated with 20 μl of permeabilization buffer (AB Serotec) and washed as above. Primary antibody was added, followed by incubation and washing. Lastly, cells were stained with PE-labeled secondary antibody.
To see the effect of Fc blockade, blood from two samples (one healthy control and one patient) was treated with 10% human serum (heat inactivated at 56°C for 1 h or used as such) for 15 min at room temperature before addition of antibodies.
A total of 105 cells were acquired on BD FACSCalibur and data analysis using CellQuest software (BD Biosciences). Lymphocytes and monocytes were gated from total population, then (median fluorescence intensity) MFI was calculated for CD14-positive cells expressing TLRs of interest.
Statistical Analysis
Data were analyzed using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA). Wilcoxon signed rank test was employed for comparison of PBMC–SFMC pairs, while Mann–Whitney U test was used to compare data of healthy controls and ERA patients. A p value less than 0.05 was considered significant.
Results
Twenty-four patients were included in the study. Paired synovial fluid was available from 10 patients. Patient details are given in Table I. Median age of seven healthy controls was 24 years.
RNA Levels of TLRs
Twenty-four patients and six healthy controls were studied for determination of mRNA expression of TLRs and 18 patients and five healthy controls for expression of adaptors. Comparison of normalized Cts revealed that levels of TLRs 3 and 6 in ERA PBMC were significantly higher than healthy PBMC while the levels of TLRs 1, 5, 7, and 8 were not significantly different (Fig. 1a).

Expression of TLR mRNA: comparison of normalized Cts (Delta Ct=CtTLR − Ct18s) between healthy PBMC, ERA PBMC, and ERA SFMC
Relative to healthy controls (Supplementary Fig. S1a), patient PBMC had higher expression of TLR1 (median = 2.5, range = 1.5–3.6), TLR3 (median = 3, range = 2.2–3.6), TLR5 (median = 2.2, range = 1.7–3.1), and TLR6 (median = 2.5, range = 1.3–2.3). No difference was observed in levels of TLR7 (median = 1.3, range = 0.8–1.7) and TLR8 (median = 1.3, range = 0.9–1.8).
Statistical analysis of normalized Cts revealed that compared to ERA PBMC, ERA SFMC had significantly higher RNA levels of TLRs 1, 3, 5, and 6, but not TLR7 and TLR8 (Fig. 1b).
Expression in SFMC relative to PBMC (Supplementary Fig. S1b) was found to be higher for TLR1 [median (range) relative fold change = 2.5 (2.3–3.7)], TLR3 [median (range) relative fold change = 3.0 (2.4–3.3)], TLR5 [median (range) relative fold change = 2.3 (1.5–3.0)], and TLR6 [median (range) relative fold change = 2.8 (2.3–3.5)] while TLR7 [median (range) relative fold change = 1.6 (1.5–2.3)] and TLR8 [median (range) relative fold change = 1.7 (1.4–1.8)] exhibited no change in expression.
RNA Levels of Adaptors
Compared to healthy controls, patient PBMC had higher expression of IRAK1 (median = 2.3, range = 2–2.5), IRAK4 (median = 2, range = 1.3–2.4), TRIF (median = 2.2, range = 1.8–2.6), TRAF3 (median = 2.1, range = 1.9–3), and TRAF6 (median = 2.6, range = 1.9–2.9). Normalized Cts were significantly higher in case of ERA PBMC for all adaptors (Fig. 2a and Supplementary Fig. S2a).

Expression of adaptor mRNA: comparison of normalized Cts (Delta Ct=Ctadaptors − CtGAPDH) between healthy PBMC, ERA PBMC, and ERA SFMC
In paired samples, SFMC once again had higher expression of IRAK1 (median = 2.4, range = 1.5–2.8), IRAK4 (median = 2.3, range = 2–2.7), TRIF (median = 2.1, range = 1.5–2.4), TRAF3 (median = 2.1, range = 1.7–2.5), and TRAF6 (median = 2.2, range = 2–2.7). However, none of the normalized Ct values were statistically different between ERA PBMC and ERA SFMC (Fig. 2b and Supplementary Fig. S2b).
Functional Assay
Functional assay was done with seven ERA PBMC, five healthy PBMC, and three ERA SFMC.
Patient PBMC (Fig. 3) produced more IL-6 and IL-8 compared to healthy PBMC on stimulation with Pam 3 Cys, poly I:C, flagellin, and zymosan. There was no significant difference in production of IL-6 and IL-8 on stimulation with TNF-α. In paired samples, SFMC showed a trend towards higher IL-6 and IL-8 production compared to PBMC. However, no statistical analysis could be done because of small numbers of SFMC samples.

Production of IL-6 and IL-8 by ERA PBMC (n = 7), ERA SFMC (n = 3), and healthy PBMC (n = 5) in response to TLR ligand stimulation. Error bars represent interquartile range. ns non-significant
Flow Cytometry for TLRs 1, 3, 5, and 6
Whole blood from 20 JIA–ERA patients and seven healthy controls was analyzed. Paired SF could be obtained from only two patients. Fc receptor blockade did not affect the staining with specific antibodies (Supplementary Fig. S3).
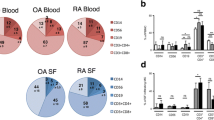
Patients’ CD14 cells had significantly higher expression of TLRs 1, 3, and 6 while TLR5 showed no difference (Fig. 4). Expression of all TLRs was higher on SFMC (MFI TLR1 = 1,176; TLR3 = 1,560; TLR5 = 737.8; TLR6 = 1,849); however, no statistical test could be applied as we had only two SF samples. Representative flow cytometry plots are shown in Fig. 4.

Expression of TLRs 1, 3, 5, and 6 in JIA–ERA blood (n = 20) compared to healthy blood (n = 7). Panel (a) shows median fluorescence intensity. Representative flow cytometry dot plots of blood from patient (b) and healthy control (c). Iso isotype control, FL1 channel for reading FITC fluorescence (CD14), FL2 channel for reading PE fluorescence (TLR)
Discussion
The data presented in this paper shows that peripheral blood and synovial fluid mononuclear cells of ERA patients have increased levels of RNA of TLRs 1, TLR3, TLR5, and TLR6 and their adaptors IRAK1, IRAK4, TRIF, TRAF3, and TRAF6. When stimulated with ligands for these TLRs, PBMC and SFMC produced a higher amount of pro-inflammatory cytokines IL-6 and IL-8 as compared to cells from healthy controls. At the protein level, ERA PBMC had higher expression of TLRs 1, 3, and 6 compared to healthy PBMC while SFMC had higher expression of TLRs 1, 3, 5, and 6. Intracellular receptors TLR7 and 8 showed no dysregulation in ERA patients at the mRNA level.
Most of the previous studies [16–18] in inflammatory arthritides have studied the role of TLRs 2 and 4, but this is the first study to that has looked at a wide array of TLRs in ERA. Similar to our data, a study by Ospelt et al. [19] found increased expression of RNA of TLRs 1–6, but not 7–10 in RA synovial fibroblasts. However, no data on overexpression of these TLRs in PBMC of patients with inflammatory arthritis is available. Even though the differences in median dCt between the groups (patient PBMC versus control PBMC and patient PBMC versus SFMC) are in the range of 1–2, suggesting a 2–4-fold increase in mRNA level, there was a significant difference in protein expression and upon stimulation, patient’s cells produced higher levels of pro-inflammatory cytokines indicating that this TLR overexpression is of functional significance. Overexpression of surface TLRs, especially those recognizing exogenous ligands like cell wall components and flagellin, suggests that the disease may be initiated or worsened by microbial triggers.
TLR1 and TLR5 have been implicated in inflammation, but not in arthritis. TLR1 and 6 form heterodimers with TLR2 [20, 21], and this could be one mechanism by which they may play a role in arthritis, as TLR2 has been shown to be overexpressed both in PBMC, SFMC, synovial tissue, and synovial fibroblasts in patients with inflammatory arthritis. Further, TLR2 knockout mice have less severe arthritis in Streptococcal cell wall arthritis model [22]. TLR5 has been found to promote an inflammatory response in dextran-induced injury to gut epithelial cells [23].
TLR3 was the only intracellular receptor overexpressed in the present study. Activation of TLR3 results in the production of type I interferons, which in turn upregulates the expression of TLR3. Thus, a positive feedback loop is formed which amplifies inflammation. TLR3 promotes osteoclast differentiation of human monocytes and induces RANKL expression in rheumatoid arthritis-derived fibroblast-like synoviocytes [24]. TLR3 signaling plays a pivotal role in pristine-induced arthritis, a rat model for RA [25]. In a recent paper, Agarwal et al. [26] suggested that extracellular matrix of dermal fibroblasts may contain TLR3 ligands and cause exacerbation of scleroderma. Similarly, ligands present in synovial compartment may enhance TLR3 signaling. TRIF, the adaptor for signaling downstream of TLR3, was also upregulated in ERA patients, suggesting a role for TLR3 in arthritis.
Most of the adaptors downstream of TLR were found to be upregulated, suggesting that there may be increased TLR signaling in these cells. While the role of these proteins has not been extensively investigated in humans, there are several animal models to show that knocking out these adaptors results in protection from disease. KBxN mice having inactive IRAK4 [27] were completely protected against arthritis. IRAK1-deficient mice have reduced IL-17 production and increased Foxp3 expression [28]. Another study demonstrated reduction of MMP-13 production on knocking down TRAF6 and IRAK1 in human chondrocytes [29]. However, the cause for this upregulation is still uncertain. It may be due to feedback regulation due to inflammatory milieu, promoter region polymorphisms, or regulation by microRNAs. Indeed, polymorphism in TRAF5 gene shows a strong association with RA [30].
Increased production of pro-inflammatory cytokines IL-6 and IL-8 by patients’ cells on stimulation with TLR ligands suggests that the cells also had higher expression of TLRs at protein level, and this may be one of the mechanism through which TLRs may be perpetuating inflammation. We had earlier reported increased production of IL-6 and MMP3 on stimulation with these ligands in patients with JIA [14]. Other studies in RA have also shown increased production of IL-6 upon stimulation of fibroblast-like synoviocytes with TLR3 and TLR5 [19] and by PBMC in response to LPS stimulation [31]. Stimulation by ligands for TLR2, TLR4, and TLR6 resulted in a synergistic increase in IL-1β production in a murine model of arthritis [32]. Another group has shown the ability of TLRs to collaborate with other pattern recognition receptors and cause secretion of TNF α, IL-1β, and IL-8 [33].
TLR ligands—which may be endogenous, like HSPs, fibronection, necrotic cells produced during tissue damage in the synovial compartment and exogenous, like microbes—if present in circulation or synovium can induce secretion of pro-inflammatory cytokines through TLR pathway thus contributing to ERA pathogenesis. Microbial trigger like enteric bacteria have been implicated in pathogenesis of ERA [34] and RA [35], but so far no specific organism has been associated with pathogenesis. The current hypothesis is that, following infection, PAMPs activate TLRs and result in uncontrolled inflammation. Synovitis may be due to homing of primed immune cells and/or because of presence of TLR ligands in the synovium. Activation of TLRs can also occur by endogenous ligands, which are abundant in the synovial compartment. Although it still remains to be elucidated whether TLR overexpression is a cause of arthritis or a result of some feedback loop, yet our results show that their increased activity likely contributes to maintenance of the inflammatory milieu in ERA patients.
A potential therapeutic avenue would be to block TLRs or their adaptors. Opal and Huber [36] propose the use of soluble TLRs, which could bind to cognate ligands leading to downstream signaling. Trials are underway to evaluate efficacy of TLR antagonists in curbing sepsis, autoimmunity, and inflammation disorders. NI-0101, a TLR4 antagonist, delayed disease development in a mouse model of colitis [37]. A TLR2 blocker OPN-305 has shown good results in animal models of inflammation and myocardial ischemia [38]. Data is also available regarding blocking of downstream adaptors. A MyD88 inhibitor ST2825 inhibited secretion of IL-1 in animal models of lupus, inflammatory bowel disease, and multiple sclerosis [39]. Song et al. have shown that IRAK1 and IRAK4 have to be inhibited together to ameliorate inflammation [40]. These researches, together with our findings, suggest that impairing the TLR signaling axis may have possible therapeutic benefits. However, complete/prolonged suppression may increase susceptibility to microbial infections.
Our study has some limitations. We have not estimated levels of phosphorylated adaptors in ERA patients, which would have shed better light on TLR signaling in disease pathogenesis. However, no antibodies are available for phosphorylated adaptors. Secondly, we were able to get only a small number of samples for our functional assay and, finally, all our patients were on either methotrexate or NSAIDS, or both, which may have had some influence on expression of TLR though no effect has been shown so far. Nonetheless, our study does highlight the role of TLRs in inflammation in ERA patients. Thus, the TLR pathway can be studied as a therapeutic target in ERA.
Conclusion
To conclude, our observations suggest involvement of Toll-like receptors in the pathogenesis of JIA-ERA. Upregulation of TLRs and their adaptors (probably in response to microbial trigger) leads to uncontrolled inflammation and tissue injury. Thus, blocking the TLR signaling pathway may aid in disease remission.
References
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2004;124:783–801.
Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–5.
Drexler SK, Foxwell BM. The role of toll-like receptors in chronic inflammation. Int J Biochem Cell Biol. 2009;42:506–18.
van der Heijden IM, Wilbrink B, Tchetverikov I, Schrijver IA, Schouls LM, Hazenberg MP, et al. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum. 2000;43:593–8.
Huang Q, Sobkoviak R, Jockheck-Clark AR, et al. Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol. 2009;182:4965–73.
Andreakos E, Foxwell B, Feldmann M. Is targeting toll-like receptors and their signaling pathway a useful therapeutic approach to modulating cytokine-driven inflammation? Immunol Rev. 2004;202:250–65.
Li M, Carpio DF, Zheng Y, Bruzzo P, Singh V, Ouaaz F, Medzhitov RM, Beg AA. An essential role of NF-κB/toll like receptor pathway in induction of inflammatory and tissue repair gene expression by necrotic cells. J Immunol. 2001;3:375–80.
Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390–2.
Ellis JA, Munro JE, Ponsonby AE. Possible environmental determinants of juvenile idiopathic arthritis. Rheumatology. 2010;49:411–25.
Kunjir V, Venugopalan A, Chopra A. Profile of Indian patients with juvenile onset chronic inflammatory joint disease using the ILAR classification criteria for JIA: a community-based cohort study. J Rheumatol. 2010;37:1756–62.
Aggarwal A, Misra RN. Juvenile rheumatoid arthritis in India: rarity of antinuclear antibody and uveitis. Indian J Pediatr. 1996;63:301–4.
Saxena N, Aggarwal A, Misra RN. Is the enthesitis-related arthritis subtype of juvenile idiopathic arthritis a form of chronic reactive arthritis? Rheumatology. 2006;45:1129–32.
Agarwal S, Misra R, Aggarwal A. TLR ligands induce metalloproteinases expression in human fibroblast like synoviocytes from patients with juvenile idiopathic arthritis. Ind J Med Res. 2010;131:771–9.
Myles A, Aggarwal A. Expression of Toll-like receptors 2 and 4 is increased in peripheral blood and synovial fluid monocytes of patients with enthesitis-related arthritis subtype of juvenile idiopathic arthritis. Rheumatology. 2011;50:481–8.
Fitzner N, Clauberg S, Essman F, Liebmann J, Kolb-Bachofen V. Human skin endothelial cells can express all 10 TLR genes and respond to respective ligands. Clin Vaccine Immunol. 2008;15:138–46.
Iwshashi M, Yamamura M, Aita T, et al. Expression of toll-like receptor 2 on CD16+ blood monocytes and synovial tissue macrophages in rheumatoid arthritis. Arthritis Rheum. 2004;50:1457–67.
Radstake TR, Roelofs MF, Jenniskens YM, et al. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthritis Rheum. 2004;50:3856–65.
Kim KW, Cho ML, Lee SH, et al. Human rheumatoid synovial fibroblasts promote osteoclastogenic activity by activating RANKL via TLR-2 and TLR-4 activation. Immunol Lett. 2007;110:54–64.
Ospelt C, Brentano F, Rengel Y, Stanczyk J, Kolling C, Tak PP, et al. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum. 2008;58:3684–92.
Abreu MT, Arditi M. Innate immunity and toll-like receptors: clinical implications of basic science research. J Pediatr. 2004;144:421–9.
Janssens S, Beyaert R. Role of Toll-like receptors in pathogen recognition. Clin Microbiol Rev. 2003;16:637–46.
Joosten LA, Koenders MI, Smeets RL, et al. Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88. J Immunol. 2003;171:6145–53.
Rhee SH, Im E, Riegler M, Kokkotou E, O’Brien M, Pothoulakis C. Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc Natl Acad Sci USA. 2005;102:13610–5.
Kim KW, Cho ML, Oh HJ, Kim HR, Kang CM, Heo YM, Lee SH, Kim HY. TLR-3 enhances osteoclastogenesis through upregulation of RANKL expression from fibroblast-like synoviocytes in patients with rheumatoid arthritis. Immunol Lett. 2009;124:9–17.
Meng L, Zhu W, Jiang C, He X, Hou W, Zheng F, Holmdahl R, Lu S. Toll-like receptor 3 upregulation in macrophages participates in the initiation and maintenance of pristane-induced arthritis in rats. Arthritis Res Ther. 2010;12:R103.
Agarwal SK, Wu M, Livingston CK, Parks DH, Mayes MD, Arnett FC, Tan FK. Toll-like receptor 3 upregulation by type I interferon in healthy and scleroderma dermal fibroblasts. Arthritis Res Ther. 2011;13:R3.
Koziczak-Holbro M, Littlewood-Evans A, Pollinger B, Kovarik J, Dawson J, Zenke G, et al. The critical role of kinase activity of interleukin-1 receptor-associated kinase 4 in animal models of joint inflammation. Arthritis Rheum. 2009;60:1661–71.
Maitra U, Davis S, Reilly CM, Li L. Differential regulation of Foxp3 and IL-17 expression in CD4 T helper cells by IRAK-1. J Immunol. 2009;182:5763–9.
Ahmad R, Sylvester J, Zafarullah M. MyD88, IRAK1 and TRAF6 knockdown in human chondrocytes inhibits interleukin-1-induced matrix metalloproteinase-13 gene expression and promoter activity by impairing MAP kinase activation. Cell Sig. 2007;19:2549–57.
Potter C, Eyre S, Cope A, Worthington J, Barton A. Investigation of association between the TRAF family genes and RA susceptibility. Ann Rheum Dis. 2007;66:1322–6.
Kowalski ML, Wolska A, Grzegorczyk J, et al. Increased responsiveness to toll-like receptor 4 stimulation in peripheral blood mononuclear cells from patients with recent onset rheumatoid arthritis. Mediators Inflamm. 2008;2008:132732.
Jung YO, Cho ML, Lee SY, et al. Synergism of toll-like receptor 2 (TLR2), TLR4, and TLR6 ligation on the production of tumor necrosis factor (TNF)-alpha in a spontaneous arthritis animal model of interleukin (IL)-1 receptor antagonist-deficient mice. Immunol Lett. 2009;123:138–43.
van Heel DA, Ghosh S, Butler M, Hunt K, Foxwell BM, Mengin-Lecreulx D, et al. Synergistic enhancement of Toll-like receptor responses by NOD1 activation. Eur J Immunol. 2005;35:2471–6.
Saxena N, Misra R, Aggarwal A. Is the enthesitis-related arthritis subtype of juvenile idiopathic arthritis a form of chronic reactive arthritis? Rheumatology. 2006;45:1129.
Edwards CJ. Commensal gut bacteria and the etiopathogenesis of rheumatoid arthritis. J Rheumatol. 2008;35:1477–14797.
Opal SM, Huber CE. Bench-to-bedside review: toll-like receptors and their role in septic shock. Crit Care. 2002;6:125–36.
Ungaro R, Fukata M, Hsu D, Hernandez Y, Breglio K, Chen A, et al. A novel toll-like receptor 4 (TLR4) antagonist antibody ameliorates inflammation but impairs mucosal healing in murine colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1167–79.
Arslan F, de Kleijn DP, Timmers L, Doevendans PA, Pasterkamp G. Bridging innate immunity and myocardial ischemia/reperfusion injury: the search for therapeutic targets. Curr Pharm Des. 2008;14:1205–16.
Loiarro M, Capolunghi F, Fantò N, Gallo G, Campo S, Arseni B, et al. Pivotal advance: inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J Leukoc Biol. 2007;82:801–10.
Song KW, Talamas FX, Suttmann RT, Olson PS, Barnett JW, Lee SW, et al. The kinase activities of interleukin-1 receptor associated kinase (IRAK)-1 and 4 are redundant in the control of inflammatory cytokine expression in human cells. Mol Immunol. 2009;46:1458–66.
Acknowledgments
This study was supported by a grant from the Indian Council of Medical Research and Department of Biotechnology, Government of India to A.A. A.M. is supported by Council of Scientific and Industrial Research–Senior Research Fellowship.
Conflict of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Fig. S1
{kind=link}
RNA levels of TLRs normalized to 18S rRNA. a Fold change in patient PBMC (n = 24) relative to healthy PBMC. b Fold change in patient (n = 7) SFMC relative to corresponding PBMC (JPEG 58 kb)
Supplementary Fig. S2
{kind=link}
RNA levels of adaptors normalized to GAPDH. a Fold change in patient PBMC (n = 18) relative to healthy PBMC. b Fold change in patient (n = 10) SFMC relative to corresponding PBMC (JPEG 60 kb)
Supplementary Fig. S3
{kind=link}
Flow cytometry histograms comparing expression of various toll-like receptors in whole blood upon blocking Fc receptors with 10% human serum. Samples were treated with either HI Serum heat inactivated serum, UT Serum untreated serum (used without activation), or left untreated (no serum) (JPEG 214 kb)
Rights and permissions
About this article
Cite this article
Myles, A., Rahman, M.T. & Aggarwal, A. Membrane-Bound Toll-Like Receptors are Overexpressed in Peripheral Blood and Synovial Fluid Mononuclear Cells of Enthesitis-Related Arthritis Category of Juvenile Idiopathic Arthritis (JIA–ERA) Patients and Lead to Secretion of Inflammatory Mediators. J Clin Immunol 32, 488–496 (2012). https://doi.org/10.1007/s10875-011-9640-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-011-9640-5