
Abstract
Eteplirsen (Exondys 51) is an antisense oligonucleotide designed to induce exon 51 skipping that is developed by Sarepta Therapeutics. Intravenous eteplirsen has received accelerated approval from the US FDA for the treatment of Duchenne muscular dystrophy (DMD) in patients with a confirmed mutation of the DMD gene amenable to exon 51 skipping. Eteplirsen has orphan drug designation in the USA and EU, and rare paediatric disease designation in the USA for use in DMD. In the phase III PROMOVI trial, eteplirsen significantly increased dystrophin levels from baseline in muscle tissues of 12 evaluable patients with DMD after 48 weeks of treatment. This finding is supported by data from phase II trials. Long-term treatment with eteplirsen was associated with a decrease in the rate of decline in ambulation and pulmonary function in an open-label extension of a phase II trial. Eteplirsen was generally well tolerated in clinical trials. This article summarizes the milestones in the development of eteplirsen leading to this first approval for DMD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Duchenne muscular dystrophy (DMD) is caused by mutations in the dystrophin gene (also known as DMD gene) that encodes dystrophin, a structural protein that protects muscles from strain-induced injury [1, 2]. DMD mutations abrogate the mRNA open reading frame, thus preventing the expression of functional dystrophin [1, 2]. The standard-of-care treatment for DMD include glucocorticoids, which, however, do not modify the underlying disease [2]. Thus, there is a high unmet medical need for DMD treatments. A potential therapeutic approach for treating this disease is to convert an out-of-frame DMD mutation to an in-frame mutation, which would theoretically preserve the mRNA reading frame and produce a truncated, but functional dystrophin [2]. This conversion can be achieved by antisense-mediated ‘exon skipping’ [1, 2].
Eteplirsen (Exondys 51) is an antisense oligonucleotide that targets dystrophin pre-mRNA and induces exon 51 skipping [2]. It was developed by Sarepta Therapeutics for the treatment of DMD [2]. Intravenous eteplirsen received accelerated approval from the US FDA in September 2016 for the treatment of DMD in patients with a confirmed mutation of the DMD gene amenable to exon 51 skipping [3]. The approval was based on increased dystrophin levels seen in skeletal muscles (a surrogate endpoint for DMD) of eteplirsen-treated patients with DMD in clinical trials. A clinical benefit (such as improved motor function) of eteplirsen in DMD has not been clearly established. Continued approval of eteplirsen in DMD may be contingent upon verification of clinical benefit in confirmatory trials. Eteplirsen is available in the USA as single-dose vials containing 100 or 500 mg eteplirsen (50 mg/mL). The recommended dosage is 30 mg/kg once weekly administered as a 35–60 min infusion. Eteplirsen has not been evaluated in patients with renal or hepatic impairment [3].

Clinical development of eteplirsen for the treatment of DMD in patients with a confirmed mutation of the DMD gene amenable to exon 51 skipping
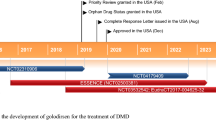
Eteplirsen has been granted orphan drug designation in the USA and EU [4] and rare paediatric disease designation in the USA [5] for use in DMD. Eteplirsen is protected by multiple US patents and an EU patent [4]. The primary patent for the drug (U.S. 9,018,368) is presumed to be valid and enforceable up to June 2025 [6]. Eteplirsen was undergoing phase II clinical development in the UK; however no recent development has been reported.
1.1 Company Agreements
In November 2014, Sarepta Therapeutics announced a multi-product, multi-year agreement with Flagship Biosciences for developing digitally automatic, fast and consistent methods of measuring dystrophin, a key biomarker of therapeutic efficacy in muscular dystrophy [7].
Eteplirsen was developed under an exclusive license agreement between Sarepta Therapeutics and the University of Western Australia [4]. The agreement was initially signed in November 2008 and was amended and restated in April 2013. Under the terms of the agreement, the university received an upfront payment from Sarepta in 2013, and it may receive additional milestone payments and royalties on sales [4].
In December 2006, AVI BioPharma (now Sarepta Therapeutics) and Ercole Biotech entered into a cross-license and drug discovery collaboration agreement for investigating the potential therapeutic effects of shifting splicing of specific gene targets [8]. AVI and Ercole refer to their technology/drug discovery platform as Exon Skipping Pre-RNA Interference Technology (ESPRIT) and Splice Switching Oligonucleotide (SSO), respectively. Under the terms of the agreement, Ercole also acquired an exclusive license to AVI’s third-generation antisense chemistry [Neugene® molecules, also referred to as phosphorodiamidate morpholino oligomers (PMOs)] for the specific targets selected by Ercole [8]. In May 2007, the two companies entered into a license and drug development agreement for DMD and beta thalassemia programmes, led by AVI and Ercole, respectively [9]. AVI acquired Ercole in March 2008 [10].
2 Scientific Summary
Eteplirsen is a PMO designed to bind to exon 51 of dystrophin pre-mRNA, resulting in skipping of this exon during mRNA processing. This allows for production of an internally truncated dystrophin in patients with DMD mutations that are amenable to exon 51 skipping [3].
2.1 Pharmacodynamics
Intramuscular injection of eteplirsen 0.9 mg in 900 µL saline into the extensor digitorum brevis muscles induced the expression of dystrophin locally in five patients with DMD in a proof-of-concept phase I/II trial (NCT00159250) [11]. Immunostaining revealed that the mean intensity of dystrophin expression in randomly chosen sections of eteplirsen-treated muscles was 22–32% of that seen in healthy muscles and was 17% (p = 0.002) greater than in the saline-treated contralateral muscles. When only dystrophin-positive fibres were assessed, the intensity in the eteplirsen-treated muscles was up to 42% of that in healthy muscle. The mean number of dystrophin-positive fibres was also significantly (p = 0.02) higher in the eteplirsen-treated muscles versus saline control (269.8 vs. 6.6). Furthermore, a Western blot assay showed distinct bands of dystrophin at the expected molecular weight in the eteplirsen-treated muscles. Skipping of exon 51 in these muscles was confirmed by the reverse-transcription polymerase chain reaction (RT-PCR). Sarcolemmal colocalization of dystrophin glycoprotein complex suggested that the dystrophin produced in the eteplirsen-treated muscles was functional. A lower dose of eteplirsen (0.09 mg in 900 µL saline) was also tested in two patients, but was found to be ineffective. This was a single-blind, placebo-controlled, dose-escalation study conducted in the UK in boys aged 3–8 years with a classic clinical diagnosis of DMD who had exon deletions that were responsive to exon 51 skipping [11].
In patients with DMD, eteplirsen treatment did not significantly change levels of dystromirs (miR-1, -206, -31, -133a, -133b), which are considered as potential serum biomarkers for monitoring the disease severity; however, there was a trend towards normalization of these dystromirs post treatment [12]. Eteplirsen up to 5 mg/mL showed no mutagenic potential in a bacterial reverse mutation assay, an in vitro mammalian chromosome aberration test or a mouse bone marrow micronucleus test [13].
2.2 Pharmacokinetics
The pharmacokinetics of eteplirsen was approximately dose-proportional and linear after multiple weekly intravenous administrations across a dosage range of 0.5–50 mg/kg/week in male paediatric patients with DMD [3, 14, 15]. The plasma concentration-time profiles were largely similar after single or multiple administrations [3].
The peak plasma concentration (Cmax) of eteplirsen was reached near the end of infusions (i.e. 1.1–1.2 h across the dose range studied) and showed multi-phasic decline thereafter, reaching 0.07% of Cmax 24 h after the end of infusion [3]. The inter-subject variability of eteplirsen Cmax and the area under the concentration-time curve ranged from 20 to 55%. Following weekly intravenous infusions of eteplirsen 30 mg/kg, its mean apparent volume of distribution was 600 mL/kg. In vitro data suggested that plasma protein binding of eteplirsen was 6–17% in humans. Eteplirsen did not accumulate significantly after multiple weekly administrations across a dosage range of 0.5–50 mg/kg/week [3].
In vitro, eteplirsen did not appear to be metabolized by hepatic microsomes in human [3]. Most of the administered eteplirsen was eliminated within 24 h, with an elimination half-life of 3–4 h. [3, 14, 15]. The total clearance of eteplirsen was 339 mL/h/kg after administration of 30 mg/kg/week for 12 weeks [3]. Renal clearance accounted for approximately two-thirds of the administered intravenous dose within 24 h [3, 14, 15].
In vitro, eteplirsen did not significantly inhibit cytochrome P450 (CYP) enzymes CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP3A4/5, and did not induce CYP2B6 or CYP3A4 [3, 16]. Induction of CYP1A2 by eteplirsen was considerably less than by omeprazole, a prototypical inducer of this enzyme. Eteplirsen was not a substrate or a significant inhibitor of the main human transporters (OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, P-gp, BCRP, MRP2 and BSEP). The drug-drug interaction potential of eteplirsen is expected to be low because of its low plasma protein binding, no microsomal metabolism and minimal interactions with CYP enzymes or drug transporters [3].
Features and properties of eteplirsen
Alternative names | AVI 4658; AVI-4658 PMO; Exondys 51 |
Class | Antisense oligonucleotides; morpholines |
Mechanism of action | Induces exon 51 skipping |
Route of administration | Intravenous |
Pharmacodynamics | Increases dystrophin levels in muscle tissues of patients with Duchenne muscular dystrophy |
Pharmacokinetics | No significant accumulation; majority of the administered dose is eliminated within 24 h; elimination half-life 3–4 h; renal clearance accounts for approximately two-thirds of total clearance; low drug-drug interaction potential |
Most frequent (incidence ≥25%) adverse events | Balance disorder, vomiting and contact dermatitis |
ATC codes | |
WHO ATC code | M09A-X (other drugs for disorders of the musculo-skeletal system) |
EphMRA ATC code | M5X (all other musculoskeletal products) |
Chemical name | RNA, [P-deoxy-P-(dimethylamino)](2′,3′-dideoxy-2′,3′-imino-2′,3′-seco)(2′a→5′)(C-m5U- C-C-A-A-C-A-m5U-C-A-A-G-G-A-A-G-A-m5U-G-G-C-A-m5U-m5U-m5U-C-m5U-A-G), 5′-[P-[4-[[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]carbonyl]-1-piperazinyl]-N,N- dimethylphosphonamidate] |
2.3 Therapeutic Trials
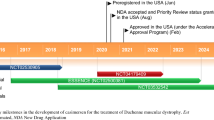
Interim data from the phase III PROMOVI trial (4658-301; NCT02255552) showed that eteplirsen increased dystrophin levels in muscle tissues of patients with DMD [3]. In 12 evaluable patients, the mean dystrophin level assessed by Western blot increased significantly (p = 0.008) from baseline at 48 weeks (0.16 vs. 0.44% of that in a healthy subject). In PROMOVI (n = 160 planned), evaluable patients (mean age 8.9 years) who were on a stable dose of corticosteroids for ≥6 months received open-label eteplirsen 30 mg/kg once weekly for 48 weeks [3].
This finding was supported by data from phase II trials [14, 15, 17]. Eteplirsen increased dystrophin production in skeletal muscle of boys with confirmed out-of-frame DMD deletions amenable to exon 51 skipping in a 24-week, double-blind, placebo-controlled trial (4658-US-201; NCT01396239) [15] and its open-label extension (4658-US-202; NCT01540409) [15, 17], and in a 12-week, open-label, dose-escalation trial (AVI-4658-28; NCT00844597) [14]. Induction of exon 51 skipping by eteplirsen was confirmed by RT-PCR [14, 15, 17]. The restored dystrophin was functional, as confirmed by localisation of dystrophin-associated proteins (such as α-sarcoglycan and neuronal nitric oxide synthase) at the sarcolemma [14, 15, 17]. Long-term treatment with eteplirsen appeared to decrease the rate of decline in ambulation and pulmonary function [17].
In the 4658-US-201 trial, 12 patients were randomized to eteplirsen 30 or 50 mg/kg, or placebo once weekly for 24 weeks [15]. At 12 weeks, the mean change from baseline in dystrophin-positive fibres was not significant with the 50 mg/kg dose (coprimary endpoint). However, there was a significant (p ≤ 0.002) increase after 24 weeks of treatment with the 30 mg/kg dose versus placebo (mean change from baseline +22.9 vs. −4.0%; coprimary endpoint). These data suggest that the effect of eteplirsen on dystrophin production was time-dependent. With respect to muscle function, the adjusted mean change from baseline at 24 weeks in the six-minute walk test (6MWT) distance in the eteplirsen 30 and 50 mg, and placebo groups was −128.2, −0.3 and −25.8 m, respectively. The large decline in the eteplirsen 30 mg group was attributed to two patients who showed rapid disease progression after enrolment into the study. This trial included boys aged 7−13 years who were on stable glucocorticoids and were able to walk 200–400 m (mean 381.9 m at baseline) on the 6MWT [15].
Patients completing the 4658-US-201 trial received open-label eteplirsen for additional 212 weeks in the 4658-US-202 trial [eteplirsen recipients continued same dosage (continuous eteplirsen group) and placebo recipients were switched to eteplirsen 30 (n = 2) or 50 (n = 2) mg/kg at week 25 (delayed eteplirsen group)] [15, 17]. At 48 weeks, the mean change from baseline in dystrophin-positive fibres was significant in both the continuous (47.3%; p ≤ 0.001) and the delayed (37.7%; p ≤ 0.009) eteplirsen groups [15]. These findings were supported by immunostaining assay, which showed a significant (p ≤ 0.01) increase in the intensity of dystrophin expression in both groups at 48 weeks [15]. At 180 weeks, the mean dystrophin level in eteplirsen recipients was 0.93% of that in healthy subjects [3].
In the 4658-US-202 trial, eteplirsen recipients had some clinical benefit [1, 15, 17, 18]. At 48 weeks, the adjusted mean change from baseline in 6MWT distance in the eteplirsen 30 and 50 mg/kg, and the delayed eteplirsen group was −153.4, +21 and −68.4 m, respectively; when the two patients in the eteplirsen 30 mg/kg group who had rapid disease progression were excluded, the remaining six patients in the continuous eteplirsen group had a significant (p < 0.001) benefit of 67.3 m versus the delayed eteplirsen group [15]. At 168 weeks, the continuous eteplirsen group (n = 6 evaluable) had a significant (p ≤ 0.017) benefit of 65.4 m on the 6MWT, compared with the delayed eteplirsen group (n = 4) [18]. After 3 years of treatment, eteplirsen recipients had a statistically significant (p < 0.01) and clinically meaningful reduction in the decline of 6MWT distance when compared with age- and genotype-matched untreated historical control subjects (a difference of 148 m) [2, 17]. At 4 years, two of the 12 eteplirsen-treated boys lost ambulation, compared with 10 of 11 in the historical control group [1].
Eteplirsen treatment was also associated with a smaller decline in pulmonary function tests relative to published natural history of patients with DMD [17]. After 3 years of treatment, mean percentages of predicted maximum inspiratory pressure, maximum expiratory pressure and forced vital capacity declined by 2.2, 5.0 and 9.4%, respectively, in patients who received eteplirsen in the 4658-US-202 trial; the corresponding expected rates might be 11.5, 11.5 and 14.3% in patients not receiving eteplirsen [17].
In the AVI-4658-28 trial, six cohorts of patients (n = 19) received eteplirsen 0.5–20 mg/kg once weekly for 12 weeks [14]. At 12 weeks, seven of the 19 patients (including six receiving 10 or 20 mg/kg) showed a significant (p = 0.0203), dose-dependent, linear increase in dystrophin expression, as assessed by at least two of three methods of dystrophin quantification (number of dystrophin-positive fibres, immunostaining and Western blot). In these patients, mean dystrophin fluorescence intensity increased significantly (p = 0.0287) from pre- to post-treatment (8.9 vs. 16.4% of that in normal muscle). Three patients (one each in the 2, 10 and 20 mg/kg cohort) showed high dystrophin expression, as assessed by all three methods. Muscle biopsies from patients receiving the 10 or 20 mg/kg dosages showed a reduction in inflammatory infiltrate, a well-known inflammatory response in DMD, with the seven dystrophin responders having a significant (p < 0.05), dose-dependent reduction in cluster of differentiation 3 (CD3) cells. There were no dose-dependent changes in muscle function tests (North Star ambulatory assessment, myometry, step activity monitoring and 6MWT) [14]. At baseline, patient age was 6–13 years, 6MWT distance was 138–515 m, and all but one patient were on corticosteroids.
Key clinical trials of eteplirsen
Drug(s) | Indication | Phase | Status | Location(s) | Identifier | Sponsor |
|---|---|---|---|---|---|---|
Eteplirsen | DMD | III | Recruiting | USA | NCT02255552; 4658-301 | Sarepta Therapeutics |
Eteplirsen | DMD | II | Recruiting | USA | NCT02420379, 4658-203 | Sarepta Therapeutics |
Eteplirsen | DMD | II | Ongoing | USA | NCT02286947, 4658-204 | Sarepta Therapeutics |
Eteplirsen | DMD | II | Ongoing | USA | NCT01540409, 4658-us-202 | Sarepta Therapeutics |
Eteplirsen, placebo | DMD | II | Completed | USA | NCT01396239, 4658-us-201 | Sarepta Therapeutics |
Eteplirsen | DMD | I/II | Completed | UK | NCT00844597, AVI-4658-28 | Sarepta Therapeutics, British Medical Research Council |
Eteplirsen | DMD | I/II | Completed | UK | NCT00159250, 05/MRE12/32 | Imperial College London |
2.4 Adverse Events
Eteplirsen was general well tolerated in patients with DMD participating in clinical trials, with no systemic reactions or treatment-related serious adverse events [3, 14, 15, 17]. In the 4658-US-201 trial, the common treatment-emergent adverse events (TEAEs) in eteplirsen 30 or 50 mg/kg/week recipients (n = 8) with incidence ≥25% more than in placebo recipients (n = 4) over 24 weeks included balance disorder (38 vs. 0%), vomiting (38 vs. 0%) and contact dermatitis (25 vs. 0%) [17]. In patients who received eteplirsen ≥30 mg/kg/week for up to 208 weeks, adverse events occurring with an incidence of ≥10% (and occurring more frequently than with the same dose in the 4658-US-201 trial) included vomiting, contusion, excoriation, arthralgia, rash, catheter site pain and upper respiratory tract infection [3]. Transient erythema, facial flushing, and elevated temperature have been reported on days of eteplirsen administration [3].
The most common TEAEs reported over 168 weeks in the 4658-US-202 trial (n = 8) included procedural pain (75%), proteinuria (62%), vomiting (50%), hypokalemia (50%), back pain (50%), headache (50%) and balance disorder (50%) [17]. Six eteplirsen recipients reported seven adverse events that were deemed to be possibly or probably related to study drug by the investigator. These included thrombosed tunnelled port catheters (two events), mild erythema (two), mild and transient proteinuria (two), and white blood cell (WBC) count decreased to 3.70 × 109/L in one patient who had a history of low WBC count (one). There were no treatment interruption or dose modification because of an adverse event, and there were no treatment discontinuations or deaths. Protein was found in 19 of 609 urine samples from eteplirsen recipients tested over ≈3 years; however, the levels were low in most cases, and all increases were transient and resolved spontaneously with no evidence of renal toxicity. Eteplirsen was not associated with hepatic toxicity. Indeed, DMD-related elevations in creatine kinase, aspartate aminotransferase and alanine aminotransferase levels decreased over the course of treatment with eteplirsen [17].
Where reported, there were no clinically relevant effects of eteplirsen on vital signs, physical examination (including injection-site reaction), electrocardiograms, echocardiograms, haematology, coagulation, or pulmonary, renal, liver or bone-marrow functions [14, 15]. Anti-dystrophin antibodies [11, 14] or dystrophin-induced T-cell responses [15] were not observed after eteplirsen treatment.
2.5 Ongoing Clinical Trials
The PROMOVI trial is currently ongoing with an estimated completion date of May 2019. Two phase II studies are evaluating eteplirsen in patients with early (NCT02420379) or advanced (NCT02286947) stage DMD.
3 Current Status
Intravenous eteplirsen received its first global approval in the USA on 19 September 2016 for the treatment of DMD in patients with a confirmed mutation of the DMD gene amenable to exon 51 skipping.
References
Miceli MC, Nelson SF. The case for eteplirsen: paving the way for precision medicine. Mol Genet Metab. 2016;118(2):70–1.
Sarepta Therapeutics Inc. Eteplirsen briefing document (NDA 206488). 2016. http://www.fda.gov. Accessed 5 Oct 2016.
Sarepta Therapeutics Inc. Exondys 51(eteplirsen) injection, for intravenous use: US prescribing information. 2016. http://www.fda.gov. Accessed 6 Oct 2016.
Sarepta Therapeutics Inc. United States Securities And Exchange Commission filing: form 10-K for the fiscal year ended December 31, 2015. 2016. http://www.sarepta.com. Accessed 5 Oct 2016.
Sarepta Therapeutics Inc. Sarepta Therapeutics receives rare pediatric disease designation from FDA for eteplirsen for the potential treatment of Duchenne muscular dystrophy [media release]. 21 Aug 2015. http://www.sarepta.com.
Sarepta Therapeutics Inc. Sarepta Therapeutics announces USPTO decision in patent interference case with BioMarin Pharmaceutical [media release]. 30 Sep 2015. http://www.sarepta.com.
Sarepta Therapeutics Inc. Sarepta Therapeutics announces third quarter 2014 financial results and recent corporate developments [media release]. 6 Nov 2014. http://www.sarepta.com.
AVI BioPharma Inc. AVI BioPharma and Ercole Biotech announce cross-license and drug discovery collaboration for alternative splicing therapeutics [media release]. 21 Dec 2006. http://www.avibio.com.
AVI BioPharma Inc, Ercole Biotech Inc. AVI BioPharma and Ercole Biotech announce license and drug development agreement for Duchenne muscular dystrophy and beta thalassemia [media release]. 3 May 2007. http://www.avibio.com.
AVI BioPharma Inc. AVI BioPharma announces close of Ercole Biotech acquisition. [media release]. 25 Mar 2008. http://www.avibio.com.
Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8(10):918–28.
Zaharieva IT, Calissano M, Scoto M, et al. Dystromirs as serum biomarkers for monitoring the disease severity in Duchenne muscular dystrophy. PLoS One. 2013;8(11):e80263.
Sazani P, Weller DL, Shrewsbury SB. Safety pharmacology and genotoxicity evaluation of AVI-4658. Int J Toxicol. 2010;29(2):143–56.
Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378(9791):595–605.
Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74(5):637–47.
Sazani P, Magee T, Charleston JS, et al. In vitro pharmacokinetic evaluation of eteplirsen, SRP4045, and SRP4053; three phosphorodiamidate morpholino oligomers (PMO) for the treatment of patients with duchenne muscular dystrophy (DMD) [abstract no. P5.061]. Neurology. 2015;84(14 Suppl).
Mendell JR, Goemans N, Lowes LP, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. 2016;79(2):257–71.
Mendell JR, Rodino-Klapac L, Sahenk Z, et al. Eteplirsen, a phosphorodiamidate morpholino oligomer (PMO) for duchenne muscular dystrophy (DMD): clinical update [abstract no. 130]. Ann Neurol. 2015;78(Suppl 19):S212.
Disclosure
The preparation of this review was not supported by any external funding. During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the author on the basis of scientific completeness and accuracy. Yahiya Y. Syed is a salaried employee of Adis, Springer SBM.
Author information
Authors and Affiliations
Corresponding author
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Rights and permissions
About this article

Cite this article
Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 76, 1699–1704 (2016). https://doi.org/10.1007/s40265-016-0657-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-016-0657-1