
Abstract
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder caused by truncating mutations in the DMD gene. These result in the absence of the muscle fibre stabilizing dystrophin protein and progressive loss of muscle tissue and function. In-frame mutations with partially functional dystrophin generally lead to Becker muscular dystrophy (BMD) with a milder disease phenotype. This was the inspiration for the antisense-mediated exon-skipping approach that restores the dystrophin reading frame to allow production of a Becker-type dystrophin. This approach is mutation specific. Since exon 51 skipping is applicable to the largest group of DMD patients, two antisense compounds targeting exon 51 were developed first, i.e. drisapersen and eteplirsen. Ten years have passed since the first exon-skipping antisense compound was tested clinically in DMD patients. If objectively evaluated, initial trials were suboptimal with modest clinical success. Major hurdles were that, at the time of trial planning, natural history data and reliable outcome measures to detect clinical benefit were not available. Moreover, the levels of dystrophin that are restored in DMD patients are lower than those observed in BMD patients. This chapter looks back at the lessons that were learned during the development of DMD exon skipping so far, to allow for more optimal exon-skipping trials in the future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Duchenne muscular dystrophy
- Exon skipping
- Clinical trial
- Dystrophin level
- Natural history
- Outcome measure
- Disease heterogeneity
1 Exon Skipping for Duchenne Muscular Dystrophy
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder affecting around 1 in 5000 newborn males worldwide [1, 2]. Patients progressively lose muscle and generally become wheelchair-dependent by the age of 12, require assisted ventilation by the age of 20 and usually die in the third or fourth decade due to pulmonary or cardiac failure [3, 4].
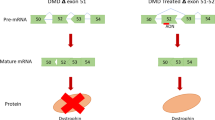
DMD is caused by out-of-frame mutations in the DMD gene that prevent the production of the muscle fibre stabilizing dystrophin protein [5, 6]. Lack of functional dystrophin makes muscle fibres more susceptible to damage resulting in chronic injury accompanied by inflammation and replacement of muscle fibres by adipose and fibrotic tissue [7]. Interestingly, the crucial functional parts of dystrophin are located at the beginning and at the end of the protein. Internal deletions or duplications in the DMD gene that maintain the reading frame give rise to partially functional dystrophins and generally lead to Becker muscular dystrophy (BMD) with a later onset and slower disease progression [4].
The finding that out-of-frame mutations generally lead to DMD while in-frame mutations generally lead to BMD was the inspiration for the antisense-mediated exon-skipping approach. Here, antisense oligonucleotides (AONs) are used as steric blockers that hide a targeted exon from the splicing machinery, causing it to be skipped so that the dystrophin reading frame is restored, allowing the production of a Becker-type dystrophin [8, 9]. The exon-skipping approach is mutation specific. DMD patients carry different types of mutations that vary in position and size within the DMD gene [5], and, as such, different exons need to be skipped to restore the reading frame for different mutations. In theory, the approach would be applicable to the majority of DMD gene mutations (55% of all patients and 80% of patients with deletions) [5, 10]. Moreover, the majority of the mutations are found at the ‘hot spot’ between exons 45 and 53; thus skipping of certain exons would apply to larger groups of patients, with exon 51 skipping being applicable to the largest group (13–14% of patients).
AONs are chemically modified DNA or RNA analogues. Early modifications involved phosphorothioate linkages to improve stability and pharmacokinetic properties and 2′-O-methyl RNA to render AONs RNase H resistant and making AONs suitable for splicing modulation [11]. These 2′-O-methyl RNA with a phosphorothioate backbone (2OMePS) AONs were the initial tool to modify splicing and skip one or more exons, thereby restoring dystrophin production in patient-derived cell models and animal models [9]. Phosphorodiamidate morpholino oligomers (PMOs) , containing a six-membered morpholine moiety instead of ribose and phosphorodiamidate linkages [12], have been explored as another chemistry for DMD exon skipping [13,14,15,16,17]. The mdx mouse model was helpful to explore exon-skipping efficiency for both chemistries. Interestingly, the AON uptake after systemic delivery in dystrophic muscles was found to be tenfold higher than in healthy muscles. This suggests that the dystrophic phenotype of the muscles lacking dystrophin facilitates AON uptake [9].
After encouraging preclinical results , both chemistries were tested in DMD patients who were amenable to exon 51 skipping. It has now been more than 10 years after the first exon-skipping AON was tested clinically in DMD patients. This chapter will give an overview of the decade-long clinical journey for AONs and will outline the lessons learned along the way.
2 Clinical Trials
Since exon 51 skipping is applicable to the largest number of DMD patients, two antisense compounds targeting exon 51 were developed first, i.e. drisapersen and eteplirsen. These components differ in their oligonucleotide backbone chemistries (i.e. 2OMePS and PMO, respectively). Drisapersen was developed by Prosensa/GSK/BioMarin, while eteplirsen was produced by AVI Biopharma/Sarepta. Since the exon-skipping approach is a mutation-specific genetic approach, it was not tested in healthy volunteers. First, safety data was available for both chemistries from trials for other indications. Secondly, exon 51 skipping would disrupt the reading frame in healthy volunteers and thus have the opposite effect compared to DMD patients.
2.1 Drisapersen
2.1.1 Local Injections
Drisapersen was administrated intramuscularly into tibialis anterior muscles of four DMD patients. The injection was tolerated well, and no side effects were observed beyond some redness and swelling at the injection site. A biopsy taken from the injection site 28 days later showed that in all four patients, drisapersen induced specific skipping of exon 51 during pre-messenger RNA splicing of the dystrophin transcript and restored dystrophin locally [18]. Patients did not show any functional improvement, nor was this expected due to the localized nature of the treatment. Interestingly, the oldest patient, who had the most advanced stage of the disease as assessed by magnetic resonance imaging, showed dystrophin restoration in almost all muscle fibres. However, since he had only a limited number of fibres left, the absolute amount of dystrophin restored was much lower than those observed for the three younger patients. This result underlines that the therapeutic effect of exon-skipping treatment relies on the muscle quality at the time of treatment.
2.1.2 Systemic Phase 1–2a Trials
DMD is a disease that affects all skeletal muscles, and lifelong repeated AON treatment is required due to dystrophin mRNA transcript and protein turnover. This makes intramuscular injection of each muscle unfeasible. Therefore, subsequent trials involved systemic treatment, using subcutaneous injections as studies in the mdx mouse model had revealed that this resulted in lower kidney and liver exposure than intravenous delivery and speculating that this would be more patient-friendly than intravenous infusions.
First, drisapersen was tested for safety and efficacy in an open-label, dose-escalation phase 1–2a study, where 12 DMD patients were treated by weekly subcutaneous injections of drisapersen for 5 weeks, with groups of 3 patients receiving each of 4 possible doses (0.5, 2.0, 4.0 and 6.0 mg/kg) (PRO051-CLIN02, ClinicalTrials.gov Identifier NCT01910649) [19]. Treatment was tolerated well and resulted in detectable dystrophin production in patients treated with a dose of 2.0 mg/kg or higher. No functional effects were anticipated or observed after 5 weeks. After the dose-finding study, all 12 patients were enrolled in an open-label extension phase, during which they were treated subcutaneously weekly with a dose of 6.0 mg/kg (ClinicalTrials.gov identifier NCT01910649). Twelve weeks into this extension trial, drisapersen treatment was still well-tolerated without serious adverse events. Furthermore, the 10 ambulant patients showed a modest improvement in the distance walked in the 6-min walk test compared to the baseline at the initiation of the extension trial. Patients received weekly treatment for 72 weeks, followed by an 8-week treatment break and then cycles of 8 weekly treatments and 4-week treatment breaks of 6 mg/kg drisapersen for 188 weeks [20]. After 3.4 years, the most common observed adverse events were injection-site reactions and mild proteinuria and raised urinary α1-microglobulin levels. During the off-treatment periods, the proteinuria levels normalized. However, the injection-site reactions sometimes persisted.
Functionally, on average there was an improvement in 6-min walk test performances compared to the expected decline found in natural history studies of age-matched patients [21]. The distance walked in 6 min was stable for 8 of the 10 ambulant patients for the duration of the study, whereas 2 patients lost ambulation. While this finding was encouraging, it should be interpreted with caution, since it involved an open-label study and only a small number of patients.
2.1.3 Phase 2 Placebo-Controlled Trials
Prosensa had coordinated the local injection and the phase 2a dose-escalation trials. Following this, they in-licensed drisapersen to GlaxoSmithKline (GSK) . GSK then planned and coordinated three placebo-controlled trials. In the first phase 2 double-blind, three-arm, placebo-controlled study, different dosing regimens were compared in patients (DMD 114117, ClinicalTrials.gov Identifier NCT01153932). The study involved 53 DMD patients aged 5 years and older from 13 specialized centres in 9 countries. Patients were all in the early stage of the disease, since they had to be able to rise from the floor in less than 7 s [22]. All patients first received twice weekly doses of 6 mg/kg drisapersen or placebo during a 3-week period. After this period, patients were treated either continuously (once weekly) or intermittently (twice weekly at weeks 1, 3 and 5; once weekly at weeks 2, 4 and 6; and no active drug in weeks 7–10 of each 10-week cycle) for a total duration of 48 weeks. Patients from the drisapersen continuous group showed a significant increase in 6-min walk distance at 25 weeks (34 m; p = 0.01), while no significant differences were found for patients from the intermittent group. At week 49, the 6-min walk distance differed between drisapersen and placebo in 36 m and 27 m for the continuous and intermittent group, respectively (not statistically significant). Some decline towards baseline was observed in the continuous group between 25 and 49 weeks, whereas the intermittent group was relatively stable.
The second phase 2 placebo-controlled study compared different doses of drisapersen and involved 51 DMD patients in an early stage of the disease (6–8 years of age; time to rise from floor <15 s) (DMD114876, ClinicalTrials.gov Identifier NCT01462292). Patients were treated with placebo, 3 or 6 mg/kg drisapersen for 24 weeks. Patients treated with 6 mg/kg walked 27 m more than patients treated with placebo or 3 mg/kg; however this difference was not statistically significant [23].
Although ambulation improvements in this young population with early stage of the disease appear very encouraging, both phase 2 studies were exploratory and contained small numbers of patients in each treatment group. Moreover, both studies were not sufficiently powered to be able to detect significant differences and clinical benefits.
In all phase 2 trials and the following open-label studies using subcutaneous injections of drisapersen, injection-site reactions and proteinuria were more frequently reported in drisapersen-treated patients. Similar injection-site reactions have also been reported for mipomersen, an AON of comparable chemistry that was approved by the Food and Drug Administration (FDA, USA) for the treatment of familial hypercholesterolaemia [24]. These injection-site reactions do not occur after intravenous delivery, which has been explored in clinical trials for AONs targeting exons 44, 45 and 53.
2.1.4 Phase 3 Placebo-Controlled Trial
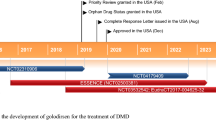
In parallel with the two phase 2 trials, the safety and effectiveness of treatment with drisapersen were tested in a large phase 3 trial involving 186 ambulant patients between 5 and 16 years (DMD114044, ClinicalTrials.gov Identifier NCT01254019). Patients were treated with placebo (n = 61) or 6 mg/kg drisapersen (n = 125) for 48 weeks, and the primary outcome measure was the 6-min walk test. At the end of the trial, drisapersen-treated patients walked 10.3 m more than the placebo group, which was not clinically relevant or statistically significant [25]. Consequently, GSK stopped the clinical development of drisapersen, and all rights returned to Prosensa. In early 2015, BioMarin acquired Prosensa and reanalysed the clinical data. Post hoc analysis of the data from the phase 2 and 3 trials revealed that patients in the phase 3 trial were on average older and had a more advanced disease stage than patients in the phase 2 trials. Therefore, analysis was performed on the subset of patients who would have met the selection criteria for phase 2 trials, revealing that for this group the treatment difference in 6-min walk test was 21.5 m (p = 0.131) [25]. Given that all studies had open-label extension arms, for a substantial number of patients, 96-week treatment data were available. Analysis of this data revealed that when compared to natural history data, longer-term drisapersen treatment appears to slow down disease progression in younger patients but also in older patients [26].
Based on these findings, drug registration applications were filed with the FDA and the European Medicines Agency (EMA) . FDA declined approval for drisapersen, saying the ‘standard of substantial evidence of effectiveness has not been met’. In May 2016, BioMarin announced they had withdrawn the application with EMA [27] and that they would stop the clinical developments of their current exon-skipping components, to focus on investing in research of next-generation oligonucleotides [28].
2.2 Eteplirsen
2.2.1 Local Injection Study
Like drisapersen, eteplirsen was also first tested in a local injection study. Here, the extensor digitorum brevis (EDB) muscles of seven DMD patients were injected with eteplirsen at doses of 0.09 mg (n = 2) and 0.9 mg (n = 5) (ClinicalTrials.gov Identifier NCT00159250). The contralateral EDB served as a control and received only saline injection [15]. EDB muscles were selected based on their preservation observed with magnetic resonance and the responsiveness to exon 51 skipping in cultured fibroblasts obtained from skin biopsies. Muscle biopsies taken between 3 and 4 weeks after injections showed dystrophin restoration in all 5 patients treated with the higher dose. Intramuscular administration of eteplirsen appeared to be safe and on average intensity of dystrophin staining was 17% higher in treated muscles than the intensity in the contralateral control muscles. This proof-of-concept study led to systemic clinical trials in DMD patients.
2.2.2 Dose-Funding and Efficacy Phase 2 Trials
Following proof-of-concept after the local injection study, systemic trials were performed for eteplirsen. The studies used intravenous infusion as a delivery route. Due to poorer solubility of the PMO compound, subcutaneous injections were not feasible.
The safety and biochemical efficacy of eteplirsen was first examined in an open-label, dose-escalation phase 2 study involving 19 ambulant patients with DMD aged 5–15 years (ClinicalTrials.gov Identifier NCT00844597). Several doses of eteplirsen were tested (0.5, 1.0, 2.0, 4.0, 10.0 and 20.0 mg/kg body weight), and muscle biopsies were taken from the biceps at the start and from the contralateral biceps after 12 weeks of weekly intravenous treatment [29]. Overall, eteplirsen was well-tolerated with no serious drug-related adverse effects. Seven patients responded to treatment showing exon 51 skipping and dystrophin restoration. Three patients showed a clear response to treatment with 21%, 15% and 55% of dystrophin-positive fibres, while the other four patients demonstrated only increases between 6 and 8%. Notably, newly produced dystrophin was functional, as the dystrophin-associated glycoprotein complex (DGC) was restored at the sarcolemma. However, since even in the 20 mg/kg dose group, there were patients in whom no increase in dystrophin expression was observed; the conclusion was that probably a higher dose was needed.
A subsequent trial involved 12 patients with DMD aged 7–13 years. The trial started as a placebo-controlled, double-blind trial. Patients were randomized to weekly intravenous infusions of 30 or 50 mg/kg/weeks eteplirsen or placebo (n = 4/group) for 24 weeks [30]. At week 25, the study became an open-label trial, and placebo patients switched to 30 or 50 mg/kg eteplirsen (n = 2/group), and all patients have been receiving weekly intravenous infusions now for over 4 years (ClinicalTrials.gov Identifier NCT01396239). An increase in dystrophin production was the primary endpoint, but function was also assessed by the 6-min walk test. No increase in dystrophin was observed after 12 weeks of treatment with 50 mg/kg eteplirsen. In biopsies taken at week 24, however, the percentage of dystrophin-positive fibres was increased to 23% in patients treated with 30 mg/kg of eteplirsen, while no increase was found in placebo-treated patients. After longer treatment (48 weeks), even greater increases of dystrophin-positive fibres (52% and 43% in the 30 and 50 mg/kg cohorts, respectively) were observed. Furthermore, restored dystrophin appeared to be functional, since sarcoglycans and neuronal nitric oxide synthase were localized at the sarcolemma [31].
Two of the patients in the 30 mg/kg group lost ambulation within the first 3 months of the study. During the 3 years of follow-up, the 10 remaining ambulant patients showed a lower degree of decline in their 6-min walk distance than would be expected from the natural history. Namely, the eteplirsen-treated patients declined 100 m, while the cohort of 13 untreated Belgium and Italian DMD patients declined 250 m in a 3-year time frame [31]. As mentioned before, comparisons of small groups of patients should be interpreted with caution. Nevertheless, Sarepta filed for accelerated approval with the FDA. However, the FDA was hesitant to approve eteplirsen based on such a small number of patients and also questioned the robustness of the dystrophin quantification method, which involved manual counting of dystrophin-positive fibres by a pathologist, while information on the quantity of dystrophin was lacking. A fourth biopsy was taken from patients after 188 weeks of treatment. Western blot analysis quantification revealed an increase of dystrophin of 0.9% [32, 33].
2.2.3 Open-Labelled Confirmatory Phase 3 Trial
In September 2014, Sarepta initiated an open-labelled phase 3 trial to provide confirmatory evidence of eteplirsen efficacy (ClinicalTrials.gov Identifier: NCT02255552). The trial involved 80 ambulant DMD patients amenable to exon 51 skipping, who received weekly intravenous dosing of 30 mg/kg eteplirsen for up to 96 weeks, while 80 matched DMD patients with mutations not amenable to exon 51 skipping served as controls for safety and functional outcome measures.
FDA requested Sarepta to confirm increased dystrophin expression by western blot analysis from biopsies taken from these patients before and after 48 weeks of eteplirsen treatment [34]. Western blot analysis of 13 patients showed an increase in dystrophin in some patients, ranging from 0.22% to 0.32% of normal [35]. Notably about half of the patients had no or minimal apparent increases in dystrophin expression. Although the levels of dystrophin restoration were lower than anticipated, eteplirsen was granted accelerated approval under provisions that Sarepta will confirm the drug’s clinical benefit before 2021 [36].
3 Lesson Learned
There have been several lessons learnt from the exon-skipping studies. Sometimes things could not have been foreseen, e.g. the injection-site reactions after subcutaneous injections of drisapersen were never observed in mice. In retrospect, intravenous delivery would have been preferred and probably should be considered for future trials using high doses of PS-modified AONs.
However, some of the lessons learned relate to the field being unprepared for clinical trials.
At the onset of the clinical trials , neither natural history data of the disease were available nor did functional outcome measures exist. This realization inspired multiple stakeholder collaboration meetings involving academics, regulators and representatives from industry and patient advocacy groups to identify gaps, collect additional data and develop new outcome measures [3, 37, 38]. However, this is an effort that is still ongoing, while the first systemic trials were initiated in 2008.
The 6-min walk test was used in these trials, but this test was not developed for DMD but borrowed from the cardiovascular field to measure muscle function in ambulant patients. Since the test had not been performed by DMD patients and no natural history data for this test existed, the heterogeneity of the disease had not been fully appreciated. With the onset of therapy trials, the field started collecting natural history data for the 6-min walk test [39,40,41]. This revealed that generally the 6-min walk distance declines nonlinearly and younger patients (≤7 years of age) are stable or can even increase in their walk distance within 1 year [39]. Later the 6-min walk distance stabilizes, followed by a slow decline and finally a rapid decline just before losing ambulation [42]. Given that the exon-skipping approach aims to slow down disease progression and prolong the ambulation period, ideally patients in the decline phase are selected for future clinical trials (it is not possible to measure a slower decline in stable patients) [32, 42]. However, once the rapid decline has started, it may be too late to achieve a therapeutic effect on walking function. Thus, currently a specific subset of patients is selected in clinical trials using the 6-min walk test , i.e. the patients where one expects to be able to detect a slower disease progression in a 1-year trial. This is generally assumed to be patients with a baseline 6-min walk distance near 350 m [3].
Looking back on past trials with the current knowledge, it is clear to see how initial trials may have been suboptimal. For instance, the phase 2 drisapersen trials involved only very young patients in a relatively stable phase of the disease [20], while the phase 3 trial involved patients between 5 and 16 years of age [25], resulting in high variability. The unexpected heterogeneity can also give rise to uncertainty within trials, e.g. in the phase 2 drisapersen trial testing different dosing regimens, the continuous treatment regime appeared to contain higher number of younger and more functional patients compared to the intermitted regime explaining improved 6-min walk distance in the continuous but not in the intermitted group at 25 weeks [25].
As mentioned, DMD progresses slowly when measured with the 6-min walk test, and the exon-skipping compounds aim to slow down disease progression. This has an impact on trial duration. The EMA guidance recommends DMD trials to be placebo-controlled and lasting at least 1 year [43], while the draft FDA guidance suggests 18–24 months [44]. The phase 2 trial design for eteplirsen was originally not set up for drug registration, as underlined by the small number of patients and the fact that there was no placebo group beyond the first 24 weeks. Therefore, results of the 6-min walk test had to be compared between eteplirsen-treated patients and historical controls selected from natural history data of baseline-matched patients from Belgium and Italy [31, 32]. This is a challenging exercise, because variation in care in different countries will influence disease progression. As such it is not surprising that FDA was not convinced by this data and requested Sarepta to provide compelling functional data in future study as a condition of the accelerated approval [32].
Currently, the 6-min walk is often selected as the primary functional endpoint in phase 2 and 3 trials for DMD. However, it has several disadvantages. First, it was not developed for DMD. As such, a lot of effort was needed to define the clinically meaningful difference for patients as 30 m [41]. Being able to walk 30 m more in 6 min may not appear clinically relevant. However, it has become clear that the distance walked in 6 min is predictive for when patients will lose ambulation, which clearly is clinically relevant. Alternative outcome measures are now developed as well, such as the North Star Ambulatory Assessment, which captures multiple items that are relevant to patients, such as the ability to climb stairs (and therefore traverse thresholds) and get up from the floor. The performance upper limb (PUL) functional outcome measure was established in collaboration with patients and can also be used in non-ambulant patients. However, these outcome measures have been newly developed, and natural history data is only now being collected. If there is one lesson from this all, it is that ideally outcome measures should be available at the time first trials are initiated.
Another thing that has become clear is that it is unlikely that exon skipping will convert a DMD patient into a BMD patient. First, the levels of dystrophin that are restored in patients after exon skipping are a lot lower than those expressed in BMD patients . However, preclinical studies in mouse models revealed that very low levels (less than 4%) of dystrophin are beneficial for survival [45]. Furthermore, patients amenable to exon 44 skipping show higher baseline levels of dystrophin due to spontaneous exon 44 skipping, which result in clinical benefits such as prolonged ambulation and slower disease progression [46, 47]. However, the higher dystrophin levels are present from birth, while dystrophin expression will only be induced at the time of intervention in DMD patients. At that time muscle damage will already have accumulated. It is currently not known how much dystrophin is required to slow down disease progression in DMD patients, but it is likely that the levels may vary for young patients with relatively good muscle quality and for older patients with progressive muscle tissue loss.
4 Future Perspectives
New oligonucleotide chemistries are currently in development aiming to achieve more widespread restoration of dystrophin throughout the whole body’s muscle including the heart.
Heart failure is one of the main causes of death in DMD patients, and targeting heart remains one of the most significant challenges [14]. Among oligonucleotide chemistries tested in preclinical trials, cell-penetrating peptide-conjugated PMO (PPMO) efficiently induced dystrophin expression in whole body muscles and the heart and improved heart function [48, 49]. Although preclinical testing of PPMOs in mdx mice appeared to be safe, monkeys are more sensitive to dose-dependent PPMO-related toxicity, which can lead to kidney degeneration [50]. If it is possible to lower the toxicity, e.g. through structural modifications, PPMOs could be a promising therapeutic compound for DMD.
Another next-generation exon 51 DMD compound was recently developed by Wave Life Sciences Ltd. Wave has presented that their stereopure component induces higher exon-skipping levels and results in better uptake in skeletal muscle and the heart. Wave is planning to initiate their first clinical trial involving ambulatory and non-ambulatory DMD patients in 2017 [51].
A major hurdle of the exon-skipping approach is that the DMD mutations are very heterogeneous, while the exon-skipping approach is highly mutation specific. Each AON is considered as a new drug by the regulators. To address this, multi-exon skipping has been proposed as a method that is applicable for larger groups of patients. For example, skipping exons 45–55 would apply to 40% of all patients [10], and Becker patients with a deletion of exons 45–55 show a mild disease phenotype [52]. However, this approach needs 11 AONs targeting 11 exons, which is challenging [53]. Multi-exon skipping is currently at a preclinical stage, and several hurdles need to be addressed including low efficacy and potentially high toxicity. A better understanding of the DMD intron splicing order and usage of new-generation antisense oligonucleotides may reduce the number of AONs required to skip exons 45–55 and reduce the therapeutic of individual AONs [54].
Exon-skipping therapy development for DMD is very dynamic. New AON chemistries and modifications are tested in cell and animal models, and outcome measures have been developed and natural history collected. While initial trials perhaps were not optimal, it is hoped that future AON trials will benefit from the work that has been done so far.
References
Mendell JR, Shilling C, Leslie ND, Flanigan KM, al-Dahhak R, Gastier-Foster J, Kneile K, Dunn DM, Duval B, Aoyagi A, Hamil C, Mahmoud M, Roush K, Bird L, Rankin C, Lilly H, Street N, Chandrasekar R, Weiss RB (2012) Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol 71(3):304–313. https://doi.org/10.1002/ana.23528
Moat SJ, Bradley DM, Salmon R, Clarke A, Hartley L (2013) Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur J Hum Genet 21(10):1049–1053. https://doi.org/10.1038/ejhg.2012.301
Straub V, Balabanov P, Bushby K, Ensini M, Goemans N, De Luca A, Pereda A, Hemmings R, Campion G, Kaye E, Arechavala-Gomeza V, Goyenvalle A, Niks E, Veldhuizen O, Furlong P, Stoyanova-Beninska V, Wood MJ, Johnson A, Mercuri E, Muntoni F, Sepodes B, Haas M, Vroom E, Aartsma-Rus A (2016) Stakeholder cooperation to overcome challenges in orphan medicine development: the example of Duchenne muscular dystrophy. Lancet Neurol 15(8):882–890. https://doi.org/10.1016/s1474-4422(16)30035-7
Emery AE (2002) The muscular dystrophies. Lancet 359(9307):687–695. https://doi.org/10.1016/s0140-6736(02)07815-7
Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, Dawkins H, Lamont L, Roy AJ, Chamova T, Guergueltcheva V, Chan S, Korngut L, Campbell C, Dai Y, Wang J, Barisic N, Brabec P, Lahdetie J, Walter MC, Schreiber-Katz O, Karcagi V, Garami M, Viswanathan V, Bayat F, Buccella F, Kimura E, Koeks Z, van den Bergen JC, Rodrigues M, Roxburgh R, Lusakowska A, Kostera-Pruszczyk A, Zimowski J, Santos R, Neagu E, Artemieva S, Rasic VM, Vojinovic D, Posada M, Bloetzer C, Jeannet PY, Joncourt F, Diaz-Manera J, Gallardo E, Karaduman AA, Topaloglu H, El Sherif R, Stringer A, Shatillo AV, Martin AS, Peay HL, Bellgard MI, Kirschner J, Flanigan KM, Straub V, Bushby K, Verschuuren J, Aartsma-Rus A, Beroud C, Lochmuller H (2015) The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat 36(4):395–402. https://doi.org/10.1002/humu.22758
Hoffman EP, Brown RH Jr, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51(6):919–928
Blake DJ, Weir A, Newey SE, Davies KE (2002) Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev 82(2):291–329. https://doi.org/10.1152/physrev.00028.2001
Aartsma-Rus A, van Ommen GJ (2007) Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA 13(10):1609–1624. https://doi.org/10.1261/rna.653607
Aartsma-Rus A (2010) Antisense-mediated modulation of splicing: therapeutic implications for Duchenne muscular dystrophy. RNA Biol 7(4):453–461
Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen GJ, den Dunnen JT (2009) Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat 30(3):293–299. https://doi.org/10.1002/humu.20918
Jarver P, O’Donovan L, Gait MJ (2014) A chemical view of oligonucleotides for exon skipping and related drug applications. Nucleic Acid Ther 24(1):37–47. https://doi.org/10.1089/nat.2013.0454
Summerton J, Weller D (1997) Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev 7(3):187–195. https://doi.org/10.1089/oli.1.1997.7.187
Fletcher S, Honeyman K, Fall AM, Harding PL, Johnsen RD, Wilton SD (2006) Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J Gene Med 8(2):207–216. https://doi.org/10.1002/jgm.838
Heemskerk HA, de Winter CL, de Kimpe SJ, van Kuik-Romeijn P, Heuvelmans N, Platenburg GJ, van Ommen GJ, van Deutekom JC, Aartsma-Rus A (2009) In vivo comparison of 2′-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping. J Gene Med 11(3):257–266. https://doi.org/10.1002/jgm.1288
Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, Guglieri M, Ashton E, Abbs S, Nihoyannopoulos P, Garralda ME, Rutherford M, McCulley C, Popplewell L, Graham IR, Dickson G, Wood MJ, Wells DJ, Wilton SD, Kole R, Straub V, Bushby K, Sewry C, Morgan JE, Muntoni F (2009) Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol 8(10):918–928. https://doi.org/10.1016/s1474-4422(09)70211-x
Gebski BL, Mann CJ, Fletcher S, Wilton SD (2003) Morpholino antisense oligonucleotide induced dystrophin exon 23 skipping in mdx mouse muscle. Hum Mol Genet 12(15):1801–1811
Alter J, Lou F, Rabinowitz A, Yin H, Rosenfeld J, Wilton SD, Partridge TA, Lu QL (2006) Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med 12(2):175–177. https://doi.org/10.1038/nm1345
van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, den Dunnen JT, Koop K, van der Kooi AJ, Goemans NM, de Kimpe SJ, Ekhart PF, Venneker EH, Platenburg GJ, Verschuuren JJ, van Ommen GJ (2007) Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med 357(26):2677–2686. https://doi.org/10.1056/NEJMoa073108
Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, Holling T, Janson AA, Platenburg GJ, Sipkens JA, Sitsen JM, Aartsma-Rus A, van Ommen GJ, Buyse G, Darin N, Verschuuren JJ, Campion GV, de Kimpe SJ, van Deutekom JC (2011) Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med 364(16):1513–1522. https://doi.org/10.1056/NEJMoa1011367
Goemans NM, Tulinius M, van den Hauwe M, Kroksmark AK, Buyse G, Wilson RJ, van Deutekom JC, de Kimpe SJ, Lourbakos A, Campion G (2016) Long-term efficacy, safety, and pharmacokinetics of drisapersen in Duchenne muscular dystrophy: results from an open-label extension study. PLoS One 11(9):e0161955. https://doi.org/10.1371/journal.pone.0161955
Goemans N, Tulinius M, Kroksmark AK, Wilson R, van den Hauwe M, Campion G (2016) Comparison of ambulatory capacity and disease progression of Duchenne muscular dystrophy subjects enrolled in the drisapersen DMD114673 study with a matched natural history cohort of subjects on daily corticosteroids. Neuromuscul Disord 27:203. https://doi.org/10.1016/j.nmd.2016.11.013
Voit T, Topaloglu H, Straub V, Muntoni F, Deconinck N, Campion G, De Kimpe SJ, Eagle M, Guglieri M, Hood S, Liefaard L, Lourbakos A, Morgan A, Nakielny J, Quarcoo N, Ricotti V, Rolfe K, Servais L, Wardell C, Wilson R, Wright P, Kraus JE (2014) Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol 13(10):987–996. https://doi.org/10.1016/s1474-4422(14)70195-4
Economides C (March 17, 2014) Prosensa announces 48-week data from a U.S. phase II placebo-controlled study of drisapersen in 51 DMD boys. https://globenewswire.com/news-release/2014/03/17/618957/10072933/en/Prosensa-Announces-48-Week-Data-from-a-U-S-Phase-II-Placebo-Controlled-Study-of-Drisapersen-in-51-DMD-Boys.html
Toth PP (2013) Emerging LDL therapies: mipomersen-antisense oligonucleotide therapy in the management of hypercholesterolemia. J Clin Lipidol 7(3 Suppl):S6–S10. https://doi.org/10.1016/j.jacl.2013.02.004
FDA briefing document peripheral and central nervous system drugs advisory committee meeting (November 24, 2015)
BioMarin announces data analysis demonstrating consistent efficacy of Kyndrisa™ (drisapersen) in comparable patients across three randomized studies (2015). http://investors.bmrn.com/releasedetail.cfm?ReleaseID=943823
Withdrawal assessment report published at EMA. http://www.ema.europa.eu/docs/en_GB/document_library/Application_withdrawal_assessment_report/2016/09/WC500212619.pdf
BioMarin announces withdrawal of market authorization application for Kyndrisa™ (drisapersen) in Europe (2016). http://investors.bmrn.com/releasedetail.cfm?ReleaseID=973536
Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F (2011) Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378(9791):595–605. https://doi.org/10.1016/s0140-6736(11)60756-3
Mendell JR, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, Alfano L, Gomez AM, Lewis S, Kota J, Malik V, Shontz K, Walker CM, Flanigan KM, Corridore M, Kean JR, Allen HD, Shilling C, Melia KR, Sazani P, Saoud JB, Kaye EM (2013) Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 74(5):637–647. https://doi.org/10.1002/ana.23982
Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, Kaye EM, Mercuri E (2016) Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 79(2):257–271. https://doi.org/10.1002/ana.24555
Aartsma-Rus A, Krieg AM (2017) FDA approves eteplirsen for Duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Ther 27:1. https://doi.org/10.1089/nat.2016.0657
Eteplirsen FDA briefing document (2016). http://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/peripheralandcentralnervoussystemdrugsadvisorycommittee/ucm497064.pdf
Sarepta therapeutics announces FDA request for dystrophin data prior to making a decision on eteplirsen NDA (2016). http://investorrelations.sarepta.com/phoenix.zhtml?c=64231&p=irol-newsArticle&ID=2175522
Center for drug evaluation and research (2016). http://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/206488_summary%20review_Redacted.pdf
FDA grants accelerated approval to first drug for Duchenne muscular dystrophy (2016). http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm521263.htm
Muntoni F (2010) The development of antisense oligonucleotide therapies for Duchenne muscular dystrophy: report on a TREAT-NMD workshop hosted by the European Medicines Agency (EMA), on September 25th 2009. Neuromuscul Disord 20(5):355–362. https://doi.org/10.1016/j.nmd.2010.03.005
Aartsma-Rus A, Ferlini A, Goemans N, Pasmooij AM, Wells DJ, Bushby K, Vroom E, Balabanov P (2014) Translational and regulatory challenges for exon skipping therapies. Hum Gene Ther 25(10):885–892. https://doi.org/10.1089/hum.2014.086
Goemans N, Klingels K, van den Hauwe M, Boons S, Verstraete L, Peeters C, Feys H, Buyse G (2013) Six-minute walk test: reference values and prediction equation in healthy boys aged 5 to 12 years. PLoS One 8(12):e84120. https://doi.org/10.1371/journal.pone.0084120
McDonald CM, Henricson EK, Abresch RT, Florence JM, Eagle M, Gappmaier E, Glanzman AM, Spiegel R, Barth J, Elfring G, Reha A, Peltz S (2013) The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve 48(3):343–356. https://doi.org/10.1002/mus.23902
Mercuri E, Signorovitch JE, Swallow E, Song J, Ward SJ (2016) Categorizing natural history trajectories of ambulatory function measured by the 6-minute walk distance in patients with Duchenne muscular dystrophy. Neuromuscul Disord 26(9):576–583. https://doi.org/10.1016/j.nmd.2016.05.016
Mazzone ES, Pane M, Sormani MP, Scalise R, Berardinelli A, Messina S, Torrente Y, D’Amico A, Doglio L, Viggiano E, D’Ambrosio P, Cavallaro F, Frosini S, Bello L, Bonfiglio S, De Sanctis R, Rolle E, Bianco F, Magri F, Rossi F, Vasco G, Vita G, Motta MC, Donati MA, Sacchini M, Mongini T, Pini A, Battini R, Pegoraro E, Previtali S, Napolitano S, Bruno C, Politano L, Comi GP, Bertini E, Mercuri E (2013) 24 month longitudinal data in ambulant boys with Duchenne muscular dystrophy. PLoS One 8(1):e52512. https://doi.org/10.1371/journal.pone.0052512
Guideline on the clinical investigation of medicinal products for the treatment of Duchenne and Becker muscular dystrophy (2015). http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/12/WC500199239.pdf
Duchenne muscular dystrophy and related dystrophinopathies: developing drugs for treatment guidance for industry (2015). http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/UCM450229.pdf
van Putten M, Hulsker M, Nadarajah VD, van Heiningen SH, van Huizen E, van Iterson M, Admiraal P, Messemaker T, den Dunnen JT, ’t Hoen PA, Aartsma-Rus A (2012) The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS One 7(2):e31937. https://doi.org/10.1371/journal.pone.0031937
van den Bergen JC, Ginjaar HB, Niks EH, Aartsma-Rus A, Verschuuren JJ (2014) Prolonged ambulation in Duchenne patients with a mutation amenable to exon 44 skipping. J Neuromuscul Dis 1(1):91–94
Pane M, Mazzone ES, Sormani MP, Messina S, Vita GL, Fanelli L, Berardinelli A, Torrente Y, D’Amico A, Lanzillotta V, Viggiano E, D’Ambrosio P, Cavallaro F, Frosini S, Bello L, Bonfiglio S, Scalise R, De Sanctis R, Rolle E, Bianco F, Van der Haawue M, Magri F, Palermo C, Rossi F, Donati MA, Alfonsi C, Sacchini M, Arnoldi MT, Baranello G, Mongini T, Pini A, Battini R, Pegoraro E, Previtali SC, Napolitano S, Bruno C, Politano L, Comi GP, Bertini E, Morandi L, Gualandi F, Ferlini A, Goemans N, Mercuri E (2014) 6 minute walk test in Duchenne MD patients with different mutations: 12 month changes. PLoS One 9(1):e83400. https://doi.org/10.1371/journal.pone.0083400
Jearawiriyapaisarn N, Moulton HM, Buckley B, Roberts J, Sazani P, Fucharoen S, Iversen PL, Kole R (2008) Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol Ther 16(9):1624–1629. https://doi.org/10.1038/mt.2008.120
Jearawiriyapaisarn N, Moulton HM, Sazani P, Kole R, Willis MS (2010) Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc Res 85(3):444–453. https://doi.org/10.1093/cvr/cvp335
Moulton HM, Moulton JD (2010) Morpholinos and their peptide conjugates: therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim Biophys Acta 1798(12):2296–2303. https://doi.org/10.1016/j.bbamem.2010.02.012
WAVE life sciences to advance next-generation nucleic acid therapies to address unmet need in Duchenne muscular dystrophy (2016). http://ir.wavelifesciences.com/phoenix.zhtml?c=254233&p=irol-newsArticle&ID=2166326
Beroud C, Tuffery-Giraud S, Matsuo M, Hamroun D, Humbertclaude V, Monnier N, Moizard MP, Voelckel MA, Calemard LM, Boisseau P, Blayau M, Philippe C, Cossee M, Pages M, Rivier F, Danos O, Garcia L, Claustres M (2007) Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum Mutat 28(2):196–202. https://doi.org/10.1002/humu.20428
Aoki Y, Yokota T, Wood MJ (2013) Development of multiexon skipping antisense oligonucleotide therapy for Duchenne muscular dystrophy. Biomed Res Int 2013:402369. https://doi.org/10.1155/2013/402369
Gazzoli I, Pulyakhina I, Verwey NE, Ariyurek Y, Laros JF, ’t Hoen PA, Aartsma-Rus A (2016) Non-sequential and multi-step splicing of the dystrophin transcript. RNA Biol 13(3):290–305. https://doi.org/10.1080/15476286.2015.1125074
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Pasteuning-Vuhman, S., Aartsma-Rus, A. (2019). What We Have Learned from 10 Years of DMD Exon-Skipping Trials. In: Duan, D., Mendell, J. (eds) Muscle Gene Therapy. Springer, Cham. https://doi.org/10.1007/978-3-030-03095-7_43
Download citation
DOI: https://doi.org/10.1007/978-3-030-03095-7_43
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-03094-0
Online ISBN: 978-3-030-03095-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)