
Abstract
This work demonstrates that natural rubber (NR) latex particles containing acetoacetoxy (AcAc) groups are able to undergo cross-linking upon film formation at ambient temperature by reaction with glutaraldehyde (GTA). Natural rubber latex grafted with poly(acetoacetoxyethyl methacrylate) (NR-g-PAAEM) was synthesized by seeded emulsion polymerization, using benzoyl peroxide (BPO) as an initiator in free radical polymerization. The degree of grafting of PAAEM in the graft copolymers was evaluated by 1H NMR technique. Transmission electron microscope (TEM) was used for investigation the particle morphology of the grafted NR latex. Since the AcAc groups are intentionally attached to the NR particles providing sites of cross-linking at ambient temperature, the cross-linking ability of these sites by reaction with GTA was then investigated. The results revealed that the latex film of NR-g-PAAEM with the addition of GTA had a much higher tensile strength in comparison with the film without GTA. The surface morphology of the NR-g-PAAEM latex film formed in the absence and presence of the GTA cross-linker was also investigated using atomic force microscopy (AFM). By GTA addition into the NR-g-PAAEM latex before film formation, an increase in the root-mean-square (RMS) roughness of the surface of the latex film was observed. Moreover, it was also observed that the NR-g-PAAEM films with the addition of GTA had higher activation energy for thermal degradation than that without the cross-linker. This confirms that the cross-linking reaction took place in the NR-g-PAAEM latex film as a result of its reaction with the GTA.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Accelerated-sulfur vulcanization is the most commonly used curing system for the natural rubber (NR) latex-based compounds. This is because sulfur vulcanization without accelerators and activators (most commonly zinc oxide, ZnO) is relatively inefficient and requires a longer period of curing time [1]. It is well accepted that zinc, in the forms of both zinc complexes (e.g., zinc dimethyl dithiocarbamate, ZDEC) and zinc oxide is essential to the process of sulfur vulcanization [2]. However, achieving reduction levels of ZnO in the rubber compounds has attracted much interest as it was officially classified as a dangerous material for the environment by the European Union in April 2004 [3]. Moreover, dithiocarbamate accelerators are considered as nitrosamine precursors, according to European law in Regulation TRGS 552 [4, 5]. Hence, a search is going on for nitrosamine precursor-free and zinc-free vulcanization systems, which not only benefits the environment but also lowers the operating costs of the curing process of the NR latex-based compounds.
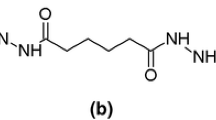
Acetoacetoxy (AcAc) is a chemically reactive group, due to its ability to undergo a keto–enol tautomerism, which is capable of reacting with various functional groups (i.e., isocyanates, diamines, and dialdehydes) at room and intermediate temperatures (<150°C) [6]. As a result, an AcAc functional group is frequently incorporated into the latex particles to prepare coatings and adhesives which are capable of cross-linking under ambient conditions [7–10]. Acetoacetoxyethyl methacrylate (AAEM) is a functional monomer that contains an AcAc group within its molecule (Scheme 1) and AcAc functional groups can be introduced onto the surface of latex particles by copolymerizing the base monomers with AAEM monomer during the process of the emulsion polymerization.

Chemical structures of a acetoacetoxyethyl methacrylate and b glutaraldehyde
Hence, in the present work, the AcAc groups were incorporated into the NR latex particles by grafting poly(acetoacetoxyethyl methacrylate) (PAAEM) onto the NR molecules in the latex stage. The morphology of the graft copolymers of NR and PAAEM (NR-g-PAAEM) particles was studied using TEM. The chemical structure of the synthesized graft copolymers was confirmed using 1H NMR. In this system, a chemical linkage between NR-g-PAAEM chains was achieved by cross-linking the AcAc groups in the grafted PAAEM chains with the suitable cross-linkers. A dialdehyde material, here glutaraldehyde (GTA, Scheme 1) was used to cross-link the AcAc groups in PAAEM repeated units. Attenuated total reflection Fourier transform infrared (ATR-FTIR) was employed to investigate changes in the functional groups of NR-g-PAAEM molecules after cross-linking with GTA which is a low-cost and commercially available dialdehyde cross-linking agent [11]. The major benefit of using GTA for cross-linking NR-g-PAAEM particles is that the reaction is feasible at ambient temperatures. Moreover, no dithiocarbamate accelerators or zinc activators are required for this cross-linking reaction to take place. This cross-linking reaction is, therefore, both a nitrosamine- and zinc-free system.
In this system, the cross-linking reactions can take place between the grafted AcAc groups onto the NR particles, and the GTA cross-linker. By GTA incorporation into the NR-g-PAAEM latex, two dfferent modes of cross-linking, i.e., the intra- and inter-particle cross-linkings can occur. When two grafted AcAc groups of the same NR-g-PAAEM particle, react with GTA, the cross-linking mode is called intra-particle cross-linking. For inter-particle cross-linking, the reaction occurs between neighboring NR-g-PAAEM particles that contain AcAc functional groups. Therefore, the term “cross-linking” as used herein represents both intra- and inter-particle cross-linkings between the AcAc groups. The formation of these cross-linked structures typically leads to a dramatic improvement in mechanical and thermal properties of the latex films.
This paper is the first report on the chemical grafting of PAAEM onto NR molecules in the latex stage. It also highlights an ambient curing system for NR latex films, which does not require the presence of zinc activators and dithiocarbamate accelerators. The cross-linking of the produced NR-g-PAAEM latex films using GTA as a cross-linker was proved using tensile and thermogravimetric (TGA) analysis data. The changes in the morphology of NR-g-PAAEM latex film after cross-linking with GTA was also illustrated by atomic force microscopy (AFM) analysis. The ability to cross-link NR latex films under ambient conditions will be an advantage in the development of room temperature cure adhesives or coatings based on the modified NR latex.
Experimental
Materials
The high ammonia concentrated natural rubber (HANR) used for preparation of NR-g-PAAEM was procured from Yala Latex Co., Ltd (Yala, Thailand). The dry rubber content (DRC) of the HANR latex was 61.4%. The AAEM monomer was purchased from Sigma-Aldrich (Steinheim, Germany). Sodium dodecyl sulfate (SDS) as the anionic surfactant used for stabilizing the NR latex during graft copolymerization and benzoyl peroxide (BPO) as the free radical initiator to initiate the graft copolymerization were obtained from Sigma-Aldrich (Steinheim, Germany). The GTA, 25% (w/v) aqueous solution as the cross-linker for latex films and the 25% (v/v) ammonium hydroxide (NH4OH) used to adjust the total solids content of the grafting reaction were manufactured by Loba Chemie Pvt. Ltd. (Mumbai, India).
Preparation of graft copolymers of NR and PAAEM
Graft copolymerization of AAEM onto NR latex particles was carried out under a nitrogen flow in a thermostatic water bath. For the preparation of NR-g-PAAEM5 sample which contained 5 wt % of AAEM, NR latex (154.7 g), 2.5% (v/v) NH4OH (90.8 g), and SDS (1 g) were first put into the reaction vessel and stirred for 30 min. Then, a mixture of AAEM (5 g) and BPO (0.48 g) was added to the reaction vessel and the resulting mixture was stirred overnight at room temperature to allow the AAEM and BPO to penetrate into the NR seed particles. After that, the mixture was heated to 65 °C and the reaction was allowed to proceed for a further 6 h at 65 °C under stirring. Graft copolymers of NR-g-PAAEM with 10 and 15 wt% of AAEM (coded as NR-g-PAAEM10 and NR-g-PAAEM15, respectively) were prepared using the recipes presented in Table 1.
In general, the monomer conversion can be defined as the total weight of monomer polymerized divided by the initial weight of monomer in the grafting reaction. Thus, the percentage of AAEM monomer converted to polymer can be calculated using the following equation:
where w graft copolymers is the total weight of graft copolymers, w NR is the weight of the NR backbone before grafting and w AAEM monomer is the initial weight of AAEM monomer in the grafting reaction.
1H NMR characterization
1H NMR spectra were recorded on a Bruker spectrometer (Avance III, 400 MHz, Germany) in deuterated chloroform (CDCl3) using the solvent peak as the internal reference (i.e., 7.26 ppm).
Three main components including ungrafted natural rubber (free NR), ungrafted homopolymer (free PAAEM), and the obtained grafted copolymers are expected to exist in the crude graft copolymers samples. Therefore, the first and second components have to be removed from the crude graft copolymers before the levels of grafted PAAEM on the NR molecule can be determined.
At the end of the grafting reaction, about 20 mL of the graft copolymer latex was withdrawn from the reaction vessel, coagulated in methanol, and formed into a thin sheet. Then, the rubber sheet was leached with deionized water to remove any water-soluble impurities. After that, the sheet was dried to a constant weight in a vacuum oven at 40 °C. The free NR was first extracted from the crude graft copolymers with light petroleum ether at 80 °C for 24 h. The remaining product was then dried to constant weight in an oven at 40 °C under vacuum. The dried residue was further extracted with acetone at 60°C for 24 h to remove the free PAAEM. The residue was again dried to a constant weight in a vacuum oven at 40 °C before subjected to structural analysis.
Morphological study
A JEOL JEM-2010 transmission electron microscope, TEM, (Japan) was used to investigate the particles morphology of the crude graft copolymers. TEM samples were prepared by drying a drop of diluted latex (~0.05% total solid content) over a 400-mesh copper TEM grid in a dust-free environment. Thereafter, the dried latex film on the TEM grid was stained with osmium tetroxide vapor for 1 h prior to examination.
Study on cross-linking reaction
The latex film samples were prepared by casting the crude prepared NR-g-PAAEM latex using 10 wt% of AAEM (NR-g-PAAEM10) (~40% DRC), with or without the addition of GTA, into a silicon mould (15 × 15 × 2 cm3). The amount of GTA required for reaction with AAEM groups was calculated based on the initial moles of AAEM present in the grafting reaction (Table 2). In the present study, two levels of GTA were added to the NR-g-PAAEM10 latex prior to film casting. The first level was corresponded to a stoichiometric ratio of GTA:AAEM of 0.5:1, while the second level was provided by a twofold stoichiometric excess of GTA (i.e., a GTA:AAEM molar ratio of 1:1). The cast films were dried in a hot air oven at 30 °C for 7 days before they were tested. The cross-linking reaction in the latex films was then investigated using ATR-FTIR, AFM, TGA and tensile tests.
ATR-FTIR analysis
Infrared spectra were recorded on a Bruker tensor 27 infrared spectrometer (Germany) fitted with an ATR accessory in the range 4000–400 cm−1 at a resolution of 4 cm−1.
Tensile testing
Tensile property of the latex films was examined according to ASTM D412 at a cross-head speed of 500 mm/min using a Hounsfield Tensometer model H 10 KS (UK). The testing was conducted using dog-bone-shaped specimens (51 mm × 254 mm) with a gauge of 38 mm wide and 51 mm long, and a minimum of 5 specimens were tested under each condition.
Equilibrium swelling measurements
Swelling studies were conducted on die-cut specimens of circular shape with a diameter of 20 mm. The dry weight of each specimen was determined before it was immersed in toluene in a glass chamber at room temperature. The specimen was periodically removed from the chamber to determine the weight gain after different immersion periods. This process was repeated until no further increase in the weight of the specimen was detected, indicating that swelling equilibrium had been reached. The volume fraction of rubber in the swollen network (V r) was then estimated from the swelling data, according to the method of Ellis and Welding as follows [12]:
where D is the de-swollen weight of the latex film, F is the weight fraction of insoluble components (i.e., other than the rubber) in the latex film, T is the initial weight of the latex film before swelling, A s is the weight of the solvent in the swollen film, and ρ r and ρ s are the densities of the rubber and solvent (toluene = 0.865 g/cm3), respectively.
Atomic force microscopy analysis
The samples of the latex films for AFM analyses were prepared by depositing NR-g-PAAEM latex onto a pre-cleaned mica substrate. The surface morphology of the cast films was investigated by an EasyScan 2 atomic force microscopy (Nanosurf AG, Liestal, Switzerland) in tapping mode using a Nano-sensor cantilever (i.e., the NCL-R type) with a typical static load of 10 nN at a dynamic frequency of 190 kHz operated in the air. AFM images were analyzed using analysis tools in the Nanosurf AFM software (v. 3.0) to determine the root-mean-square (RMS) roughness according to the following equation:
where Z(x) is the function that expresses the surface profile analyzed in terms of height (Z) and position (x) of the sample over the evaluation length (L).
Thermogravimetric analysis
Kinetics of thermal degradation of the NR-g-PAAEM films was investigated by TGA under non-isothermal conditions on a TGA Q100 (TA Instruments, USA) at four different heating rates (i.e., 5, 10, 20, and 40 °C/min) in nitrogen atmosphere with a flow rate of 20 mL/min. For each test, the initial mass of sample was about 5 mg.
Results and discussion
Structural characterization
In this work, graft copolymers of NR and PAAEM were prepared using 5, 10 and 15 wt% of AAEM (i.e., NR-g-PAAEM5, NR-g-PAAEM10, and NR-g-PAAEM15,) by seeded emulsion polymerization at 65 °C. The 1H NMR spectra for unmodified NR and NR-g-PAAEM10 are presented in Fig. 1. Three characteristic signals of cis-1,4-polyisoprene units were observed in both spectra at 5.13 ppm, 2.07 ppm and 1.67 ppm assigned to the olefinic proton (–C=CH–), methylene proton (–CH 2–C(CH3)=CH–CH 2–), and methyl proton (–C(CH 3)=CH–) in the repeating units of cis-1,4-polyisoprene, respectively [13, 14].

The 1HNMR spectra of unmodified NR and NR-g-PAAEM10
However, two signals at 2.18 ppm and 3.75 ppm were also observed in the spectrum of the NR-g-PAAEM10. The signal at 2.18 ppm was assigned to the methyl protons (CH 3–(C=O)–CH2–(C=O)) of the AcAc groups. The resonance peak at about 3.75 ppm was due to the methylene groups (–O–CH 2–CH 2–O–) connected to the oxygen atoms in the repeat units of the PAAEM. The signal of the methyl groups in the PAAEM repeat units also appeared in the spectra of the NR-g-PAAEM10 at 1.83 ppm.
The weight percentage of the grafted PAAEM in the graft copolymers was estimated from the integrated intensity ratio of the signals at 2.18 ppm and 5.13 ppm, as follows:
where I 2.18 is the integrated peak area of the methyl protons of the AcAc groups in the PAAEM repeat units at 2.18 ppm and the integer 3 in Eq (4) is the number of chemically equivalent protons that give rise to the corresponding peak. The term I 5.13 is the integrated peak area of the olefinic protons of NR at 5.13 ppm. The weight percentage of the grafted PAAEM in the NR-g-PAAEM was then determined as follows:
where mol %PAAEM is the mole percentage of PAAEM in the graft copolymers, mol %NR is the mole percentage of NR in the graft copolymers, M AAEM is the molar mass of repeating units for AAEM, and M NR is the molar mass of repeating units for NR.
The weight percentages of the grafted PAAEM calculated from the NMR spectra are given in Table 3. The calculated amounts of the grafted PAAEM on the NR molecules were 2.96, 5.87 and 7.59 wt%, when using 5, 10 and 15 wt% of AAEM in the grafting reactions, respectively. Moreover, the highest grafting efficiency of 73.92% was achieved when 10 wt% of AAEM was employed (Table 3). The grafting efficiency was estimated using a method described in details elsewhere [15, 16].
Morphological characterization
Morphologies of the unmodified and grafted NR particles (with 10 wt% of AAEM) are presented in Fig. 2. As osmium tetroxide (OsO4) was used to stain the C=C bonds in the NR molecules, the NR seed particle appears dark in the TEM micrographs. TEM analysis reveals that the particle morphology of NR modified by PAAEM grafting was different from that of the unmodified NR. A relatively smooth surface morphology was observed for the unmodified NR particle whereas the NR-g-PAAEM AEM particle exhibited a nodular morphology with PAAEM nodules on the surface of the NR particle. The presence of polar PAAEM nodules around the hydrophobic NR particle is confirmed by their comparatively bright appearance.

TEM micrographs of a unmodified NR and b NR-g-PAAEM10 latex particles
Benzoyl peroxide was employed as an oil-soluble initiator, in this study, and the formation of primary radicals (i.e., benzoyl radicals) was, therefore, expected to occur predominantly within the NR particles swollen with AAEM monomer. Therefore, it is assumed that the emulsion polymerization of AAEM takes place within the NR particles.
As the grafting reaction proceeded, the concentration of the PAAEM chains in the NR particles increased. This eventually leads to a phase separation due to incompatibility between the newly formed PAAEM and NR, as evidenced by the formation of the PAAEM clusters on the surface of NR particle (Fig. 2b).
Study of the cross-linking reaction
ATR-FTIR analysis
As noted earlier, the NR-g-PAAEM10 latex had the highest grafting efficiency (73.92%).
Therefore, this latex was chosen for the study of the cross-linking of NR-g-PAAEM latex film by GTA. The cross-linking in this system is expected to occur through the coupling reaction between the AcAc groups in the AAEM repeated units and the reactive aldehyde groups of the GTA. According to the proposed cross-linking reaction in Scheme 2 the stoichiometric molar ratio of GTA:AAEM is 0.5:1.

Proposed cross-linking reaction between the AcAc groups on the NR-g-PAAEM molecules with GTA
It can be seen from the proposed cross-link reaction in Scheme 2 that the GTA molecules react with the AcAc functional groups to yield the new double bond, which is in conjugation with the carbonyl groups of the AAEM repeated units. The FTIR spectra of the cast films formed from NR-g-PAAEM10 latex in the absence and presence of GTA are shown in Fig. 3. In all cases, the absorption peaks at about 2919, 1668 and 835 cm−1 are corresponded to –CH2 stretching, C=C stretching and =C–H out of plane bending vibrations of the cis-1,4-polyisoprene, respectively [13, 17].

FTIR spectra of NR-g-PAAEM10 latex films in the absence and presence of GTA (GTA:AAEM molar ratios of 0.5:1 and 1:1)
However, new peaks were appeared at about 1641 and 1558 cm−1 when GTA was incorporated into the latex prior to film casting. It was also observed that the intensity of the corresponding peaks increased with an increase in the molar ratio of GTA:AAEM from 0.5:1 to 1:1. The new peak centered at 1558 cm−1 is in the correct region for the stretching vibration of conjugated C = C double bonds. It is well accepted that conjugation shifts the IR absorbance to the lower frequencies (i.e., the lower wavenumbers) [18]. Thus, the new absorption peak at 1641 cm−1 is probably due to the C=O stretching vibration in the GTA molecule which is conjugated with the new double bond formed after incorporation of the GTA into the NR-g-PAAEM latex. Hence, these results support the hypothesis that the cross-linking in the latex films occurred through the reaction of the AcAc groups with the GTA during the film formation.
Tensile properties of the latex films
Figure 4 reveals that the incorporation of the GTA into the NR-g-PAAEM10 latex before film casting resulted in a marked increase in the tensile strength of the corresponding films. An increase of 341% in tensile strength was observed after adding an amount of GTA into the NR-g-PAAEM10 latex to achieve a 0.5:1 molar ratio prior to film formation. Moreover, the latex film formed in the presence of a two-fold excess of GTA (at 1:1 molar ratio of GTA:AAEM) exhibited an increase in tensile strength of 403% compared to its counterpart without GTA.

Sress–strain curves of NR-g-PAAEM10 latex films in the absence and presence of GTA (GTA:AAEM molar ratios of 0.5:1 and 1:1)
However, the elongation-at-break of the latex film was found to decrease slightly with an increase in the molar ratio of GTA:AAEM. This observation is probably the result of an increase in the degree of cross-linking in the latex film with increasing levels of GTA cross-linker. When NR-g-PAAEM chains are linked together extensively by cross-linking to form a three-dimensional network, the toughness of the latex films increases. This was also consistent with the observation that the stress–strain behavior of the latex film changed from rubbery to plastic (i.e., into a solid and tough material) when the GTA cross-linker was added prior to film casting (Fig. 4).
It is widely known that the degree of swelling is inversely related to the cross-link density of the vulcanized rubber. A lower swelling value indicates a higher cross-link density of the rubber network. Estimation of cross-link densities (ν) in the NR-g-PAAEM films with the two levels of GTA (i.e., GTA:AAEM molar ratios of 0.5:1 and 1:1) can typically be obtained from equilibrium swelling measurements in toluene, using the Flory-Rehner equation [19] as follows:
where V r is the volume fraction of the rubber in the swollen network, V s is the molar volume of the solvent (toluene = 106.28 cm3/mol) and χ is the Flory–Huggins interaction parameter for a specific polymer–solvent pair.
This equation relates the volume fraction of the rubber in the swollen state (V r) to the cross-link density in the film. However, the value of the Flory–Huggins interaction parameter (χ) for the NR-g-PAAEM-toluene system is not known. In addition, χ is not constant and it depends mainly on both temperature and composition [20]. The dependence of χ on the concentration of vulcanizing agent was also demonstrated by Westlinning et al. [21] on the vulcanizates of NR. A decrease in the value of χ for the NR-benzene system was observed as the concentration of the sulfur vulcanizing agents (i.e., benzothiazole disulfide/sulfur system) increased. McKenna and co-workers [22] reported that the value of χ for the peroxide vulcanization (i.e., cumyl peroxide) of NR was dependent on the cross-link density of the vulcanizates. Variation in the values of χ with the cross-link density has also been reported on other rubbers (i.e., polybutadiene rubber (BR) [23] and styrene butadiene rubber (SBR) [24]).
Therefore, in the present work, the cross-linking level in the latex films was expressed by values of V r rather than cross-link density because of the uncertainty in the value of χ.
It is well accepted that the value of V r changes with the cross-link density in rubber vulcanizates [23] and, for any given value of the χ, the cross-link density always increases as V r increases [25]. The NR-g-PAAEM films were first immersed in toluene at 25 °C in a dark environment until equilibrium swelling was reached. The values of V r in the swollen films of the NR-g-PAAEM were then estimated from the swelling data according to Eq. (2), as given in Table 4. The results indicated that the latex film with the addition of a twofold excess of GTA (V r = 0.146) had a higher level of cross-linking than the film with an amount of GTA necessary to achieve a molar ratio of 0.5:1 (V r = 0.135). This corroborates the observed higher tensile strength for the former type of the latex film. It is also suggested that when a twofold excess of GTA was employed, there was sufficient GTA present to create cross-linking between all of the available AAEM groups. Hence, these results provided irrefutable evidence that cross-linking between the AcAc functional groups occurred during film formation due to the reaction with GTA.
Surface morphology of the latex films
The surface morphology of the NR-g-PAAEM10 latex films with or without the addition of GTA was investigated by AFM. The films were dried at 30 °C for 7 days before being subjected to AFM analyses. In Fig. 5, in all cases, no spherical contours of the NR-g-PAAEM10 latex particles are seen on the film surfaces. The result also reveals that when the NR-g-PAAEM10 film was formed in the absence of a cross-linker, the latex particles coalesced reasonably well at ambient temperature to form a continuous film with only a few surface indentations. However, many indentations were observed on the surfaces of the latex films formed in the presence of the GTA cross-linker. The appearance of these indentation structures is probably attributable to the collapse of the NR-g-PAAEM latex particles during film formation.

AFM micrographs of NR-g-PAAEM10 latex films: without GTA (a) and in the presence of it with GTA:AAEM molar ratios of b 0.5:1 and c 1:1
Since GTA is a hydrophilic cross-linker, it stays predominantly in the aqueous phase after being added to NR-g-PAAEM latex. Cross-linking in this system occurs when GTA molecules come into physical contact with the AcAc groups in the NR-g-PAAEM molecules. This leads both to the formation of intra-particle cross-linking within the same particle, and to inter-particle cross-linking between different particles. When inter-diffusion of the NR-g-PAAEM molecules between particles takes place during film formation, a continuous film is expected to form as the glass transition temperature (T g) value of the NR-g-PAAEM10 (−54 °C) was significantly below the ambient temperature as determined by DMTA.
However, when cross-linking reaction occurs mostly near the surface and interfaces, a highly cross-linked shell is formed (Fig. 6). This cross-linked shell seems to act as a physical barrier to hinder inter-diffusion of the rubber chains across contacting surfaces during the film formation process [26]. Based on the AFM results in Fig. 5, it appears that the cross-linking reaction partially occurred before the entire inter-diffusion of the rubber chains could be achieved. Consequently, the coalescence of NR-g-PAAEM latex particles in the latex film was retarded. Moreover, the density of cross-linking at or near the interface is expected to be higher than the interior of the latex particles. After the water evaporated during the drying process, the denser cross-linked shell of the latex particles collapsed because part of the interior of the latex particles remained soft and partially unvulcanized. The collapse of the hard shell-soft core latex particles led to the formation of indentation structures on the film surface [26].

Proposed mechanism of film formation for NR-g-PAAEM latex in the presence of GTA cross-linker, adapted from [26]
The RMS roughness values for the surface of the NR-g-PAAEM films formed in the absence and presence of GTA are presented in Table 4. The results show that the surface roughness values for the NR-g-PAAEM films with the addition of GTA were much higher than those obtained for the NR-g-PAAEM films without GTA. Moreover, a slight increase in the RMS roughness value was observed at the higher molar ratio of GTA:AAEM. These results also provided solid evidence that cross-linking occurred in the NR-g-PAAEM latex film by reacting with GTA, during film formation.
Thermal stability of the latex films
TGA analysis was conducted to examine the effect of cross-linking on the thermal stability of the NR-g-PAAEM films. The residual mass percentage as a function of temperature for NR-g-PAAEM films, with or without addition of GTA, is shown in Fig. 7.

TGA thermograms of NR-g-PAAEM10 films in the absence and presence of GTA (GTA:AAEM molar ratios of 0.5:1 and 1:1) at heating rate of 10°C.min−1
It can be seen in Fig. 7 that the TGA curves for both types of NR-g-PAAEM films (with or without GTA) appeared to have two degradation steps. The first step (i.e., about 7% weight loss), occurring in the temperature ranges of 220–300 °C, is probably attributable to the initial scission of the vinylidene end groups (–C=CH2), formed because of the disproportionation in the termination reaction of AAEM. Termination processes in radical polymerizations of most vinyl monomers (i.e., styrene, acrylonitrile, and methyl acrylate) occur mainly due to the combination of factors. However, it has been extensively reported that disproportionation is the main mode of termination of polymerizations of α-methylvinyl monomers (e.g., methyl methacrylate) [27]. The AAEM is one of the α-methylvinyl monomers, which contains readily abstractable β-hydrogen in its molecule (Scheme 3). Additionally, the radicals at the ends of the growing chains of PAAEM are sterically hindered due to the presence of bulky substituents in the close vicinity of the radical center. Thus, it can be postulated that disproportionation is the preponderant termination mechanism in the polymerization of AAEM.

Postulated mechanism for disproportionation between growing chains of PAAEM, adapted from [27]
The main decomposition of the NR-g-PAAEM films (a weight loss of about 91%) occured in the second degradation step from 364 to 440 °C, which was mainly attributed to the degradation of polymer backbone. In general, a random scission process is a primary degradation pathway for the most polymers. This degradation process involves the breaking of the polymer backbone at random points into shorter fragments. The resulting fragments will eventually evaporate when their size are small enough.
However, it was also observed that the mass loss for the NR-g-PAAEM10 films with GTA in the temperature range of 400–450 °C was lower than that of the film without GTA (Table 5). At 450 °C, about 91–92% weight loss was observed for the NR-g-PAAEM10 films with GTA while the film without GTA showed about 96% weight loss. The lower weight loss of the former film at 450 °C indicates an increase in thermal stability up on the addition of GTA to the NR-g-PAAEM10 latex prior to film casting.
Thermal degradation kinetics
The activation energy (E a) for thermal degradation of NR-g-PAAEM10 films was estimated using the Flynn–Wall–Ozawa (FWO) method [28–30] as the following equation:
where β is the heating rate (°C/min), A is the pre-exponential factor (min−1), g(α) is the integral function of conversion, R is the universal gas constant (J/K.mol), and T is the absolute temperature (K). According to Eq (7), if logβ is plotted against 1/T, a straight line graph should be obtained. Then, the E a can be estimated from the slope of this plot, which is equal to –0.4567E a/R, at a given conversion. The degree of conversion (α) represents a fraction of compound decomposed which can be calculated from mass loss measurements using the Eq (8) [31, 32]:
where m o and m f are the initial and final masses of the mass loss region of interest, respectively, and m T is the mass of the sample at temperature T.
It is well known that the heating rate is one of the most important factors in TGA analysis. This is because the thermal degradation depends on both temperature and time [33]. When a higher heating rate is employed, a shorter time is required to heat a sample to a certain temperature. This means that, at a higher heating rate, the sample stays at each temperature shorter than that at a lower heating rate. As a result, the thermal degradation of the sample at a higher heating rate will occur slower than that at a lower heating rate due to the shorter residence time.
The TGA curves of the NR-g-PAAEM10 film with addition of a 1:1 molar ratio of GTA:AAEM obtained at four different heating rates of 5, 10, 20, and 40 °C/min are presented in Fig. 8. It is clearly seen that the TGA curves were shifted towards higher temperatures as the heating rate was increased. Therefore, a particular level of conversion was obtained for a higher temperature at a higher heating rate. On this basis, the E a at different conversions for the NR-g-PAAEM10 films can be estimated from the plot of ln β versus 1/T according to Eq (7). In this study, a range of conversion from 0.2 to 0.8 was considered for calculating the E a based on the FWO method. Figure 9 shows FWO plots of the NR-g-PAAEM10 film with a twofold excess of GTA (GTA:AAEM molar ratio of 1:1) at different values of conversion. The dependence of the E a on the extent of conversion for this corresponding film is given in Fig. 10. It can be seen that the E a values for the thermal degradation of the NR-g-PAAEM films showed a strong dependence on the extent of conversion. The E a increased from about 264.25 kJ/mol at the conversion of 0.2 to 412.74 kJ/mol at the conversion of 0.8.

TGA curves at four different heating rates for thermal decomposition of NR-g-PAAEM films under nitrogen

Plots of lnβ vs. 1/T at different degrees of conversion (α = 0.2–0.8) for NR-g-PAAEM10 films with the GTA:AAEM molar ratio of 1:1

Plots of of E a vs. degree of conversion observed in the thermal degradation of the NR-g-PAAEM10 latex films with and without GTA
Different types of bonds are generally present in the polymer chains. However, the weakest bond in the chains has the highest probability of breaking, and typically takes place at the low temperatures. In contrast, the cleavage of relative strong bonds occurs at high temperatures. Thus, less E a is required to induce the thermal decomposition of the NR-g-PAAEM10 films at low conversions. Additionally, the E a for the NR-g-PAAEM10 films with GTA was also observed to be much higher than that of the film without GTA (Fig. 10). This proves that the thermal stability of the NR-g-PAAEM10 film was significantly improved by the cross-linking of the AcAc groups through addition of GTA during film formation.
Conclusion
Grafting PAAEM onto NR latex particles was carried out by emulsion polymerization, using benzoyl peroxide as the initiator. The successful synthesis of NR-g-PAAEM was confirmed by 1H NMR spectroscopy. TEM analysis revealed that the NR-g-PAAEM latex particles had a nodular morphology with PAAEM nodules on the surface of the NR particles. The feasibility of the creating cross-linking between NR-g-PAAEM particles during film formation under ambient conditions was then investigated using GTA as the cross-linker. The FTIR spectra indicate that the cross-linking between the rubber chains occurred through the reaction between the AcAc groups with GTA cross-linker. Additionally, a considerable increase in the tensile strength of the cast film was observed when GTA was added into the NR-g-PAAEM latex prior to film casting. The surfaces of the NR-g-PAAEM latex films were also found to become much rougher after the addition of GTA. These results conclusively proved that cross-linking was formed between NR-g-PAAEM particles during film formation by reaction with GTA. The determination of E a for the thermal degradation of NR-g-PAAEM films, using the Flynn–Wall–Ozawa method, provided information about the effects of cross-linking on the thermal stability of the films. It was observed that the cast films with GTA had much higher E a values than the films without it, reflecting an increase in the thermal stability by the addition of GTA. Hence, these results provided irrefutable evidence that NR-g-PAAEM particles can undergo cross-linking during film formation by reaction with the GTA. The ability to cross-link films based on the modified NR latex under ambient temperatures is the major advantage of this curing system. Moreover, it is a nitrosamine-free and zinc-free curing system, which requires only one component (i.e., GTA) to conduct the vulcanization. In addition, the incorporation of AcAc functional groups onto the NR molecules should improve its polarity and wettability on polar surfaces such as wood, paper, and leather. Thus, this system offers the potential of developing coatings and adhesives based on modified NR latex, which is capable of curing under ambient temperature.
References
Kuriakose AP (1996) Vulcanization (overview). In: Salamone JC (ed) Polymeric materials encyclopedia. CRC Press, New York, pp 8625–8630
Nieuwenhuizen PJ (2001) Zinc accelerator complexes: versatile homogeneous catalysts in sulfur vulcanization. Appl Catal A-Gen 207:55–68
Chapman A, Johnson T (2005) The role of zinc in the vulcanisation of styrene butadiene rubbers. Kautsch Gummi Kunstst 58:358–361
Chauvin B, Mangere JL (2002) Rubber composition which is free of carcinogenic nitrosamine precursor and serves as connecting rubber. US Patent 20020180078 A1
Higashira T, Yokota A, Matsumoto N (2010) Rubber-metal laminate. US Patent 20100261004 A1
Tillet G, Boutevin B, Ameduri B (2011) Chemical reactions of polymer cross-linking and post-cross-linking at room and medium temperature. Prog Polym Sci 36:191–217
Feng J, Pham H, Macdonald P, Winnik MA, Geurts JM, Zirkzee H, Van Es S, German AL (1998) Formation and cross-linking of latex films through the reaction of acetoacetoxy groups with diamines under ambient conditions. J Coating Technol 70:57–68
González I, Asua JM, Leiza JR (2006) Cross-linking in acetoacetoxy functional waterborne cross-linkable latexes. Macromol Symp 243:53–62
Zhou W, Qu Q, Yu W, An Z (2014) Single monomer for multiple tasks: polymerization induced self-assembly, functionalization and cross-linking, and nanoparticle loading. ACS Macro Lett 3:1220–1224
Li H, Newell K, Cheek J (2015) Controlled cross-linking of latex polymers with polyfunctional amines. US Patent 20150259562 A1
Johns A, Aan MPS, Johns J, Bhagyashekar MS, Nakason C, Kalkornsurapranee E (2015) Optimization study of ammonia and glutaraldehyde contents on vulcanization of natural rubber latex. Iran Polym J 24:901–909
Ellis B, Welding GN (1964) Estimation, from swelling, of the structural contribution of chemical reactions to the vulcanization of natural rubber Part I. General method. Rubber Chem Technol 37:563–570
Kookarinrat C, Paoprasert P (2015) Versatile one-pot synthesis of grafted-hydrogenated natural rubber. Iran Polym J 24:123–133
Thuong NT, Yamamoto Y, Nghia PT, Kawahara S (2016) Analysis of damage in commercial natural rubber through NMR. Polym Degrad Stabil 123:155–161
Thongnuanchan B, Ninjan R, Kaesaman A, Nakason C (2015) A novel method to crosslink natural rubber latex adhesive at ambient temperature. Polym Bull 72:135–155
Yusof NH, Kosugi K, Song TK, Kawahara S (2016) Preparation and characterization of poly(stearyl methacrylate) grafted natural rubber in latex stage. Polymer 88:43–51
Nakason C, Sasdipan K, Kaesaman A (2014) Novel natural rubber-g-N-(4-hydroxyphenyl) maleimide: synthesis and its preliminary blending products with polypropylene. Iran Polym J 23:1–12
Smith MB (2015) Organic chemistry: an acid-base approach, 2nd edn. CRC Press, Boca Raton
Flory PJ, Rehener J (1943) Statistical mechanics of cross-linked polymer networks. II. Swelling. J Chem Phys 11:521–526
Orwoll RA (1977) The polymer-solvent interaction parameter χ. Rubber Chem Technol 50:451–479
Westlinning H, Wolff S (1968) Considerations on structure of natural rubber vulcanizates. Rubber Chem Tchnol 41:659–668
McKenna GB, Flynn KM, Chen Y (1990) Swelling in crosslinked natural rubber: experimental evidence of the crosslink density dependence of χ. Polymer 31:1937–1945
Marzocca AJ, Garraza ALR, Mansilla MA (2010) Evaluation of the polymer–solvent interaction parameter χ for the system cured polybutadiene rubber and toluene. Polym Test 29:119–126
Marzocca AJ (2007) Evaluation of the polymer–solvent interaction parameter χ for the system cured styrene butadiene rubber and toluene. Eur Polym J 43:2682–2689
Hergenrother WL, Hilton AS (2003) Use of χ as a function of volume fraction of rubber to determine crosslink density by swelling. Rubber Chem Technol 76:832–845
Ho CC, Khew MC (1999) Surface morphology of prevulcanized natural rubber latex films by atomic force microscopy: new insight into the prevulcanization mechanism. Langmuir 15:6208–6219
Yamada B, Zetterlund PB (2002) General chemistry of radical polymerization. In: Matyjaszewski K, Davis TP (eds) Handbook of radical polymerization. John Wiley and Sons, New York, pp 117–186
Ozawa T (1965) A new method of analyzing thermogravimetric data. Bull Chem Soc Jpn 38:1881–1886
Popescu C (1996) Integral method to analyze the kinetics of heterogeneous reactions under non-isothermal conditions a variant on the Ozawa-Flynn-Wall method. Thermochim Acta 285:309–323
Kim W, Kim SD, Lee SB, Hong IN (2000) Kinetic characterization of thermal degradation process for commercial rubbers. J Ind Eng Chem 6:348–355
Park JW, Oh SC, Lee HP, Kim HT, Yoo KO (2000) A kinetic analysis of thermal degradation of polymers using a dynamic method. Polym Degrad Stabil 67:535–540
Ceamanos J, Mastral JF, Millera A, Aldea ME (2002) Kinetics of pyrolysis of high density polyethylene. Comparison of isothermal and dynamic experiments. J Anal Appl Pyrol 65:93–110
Zhang S, Li L, Kumar A (2008) Materials characterization techniques. CRC Press, Boca Raton
Acknowledgments
This work was supported by the Research Fund of Prince of Songkla University, SAT581267S. The authors would like to thank the Research and Development Office (RDO) for editing this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Thongnuanchan, B., Ninjan, R. & Nakason, C. Acetoacetoxy functionalized natural rubber latex capable of forming cross-linkable film under ambient conditions. Iran Polym J 26, 41–53 (2017). https://doi.org/10.1007/s13726-016-0497-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13726-016-0497-6