
Abstract
Graft copolymer of natural rubber and N-(4-hydroxyphenyl)maleimide (i.e., NR-g-HPM) was synthesized. It was found that the grafting yield increased upon increasing the grafting temperature and the highest grafted HPM content was obtained at 200 °C. Furthermore, increases in concentration of HPM led to drop in grafted HPM. Therefore, an optimum grafting temperature and dose of HPM were found to be 200 °C and 2 phr, respectively. Dynamically cured 60/40 NR-g-HPM/PP blends with various loading levels of HPM in graft copolymerization were then achieved by dynamic vulcanization. It was found that the blend with 2 phr of HPM exhibited the highest tensile strength, elongation-at-break, mixing torque during dynamic vulcanization, storage modulus and complex viscosity and the lowest tension set (i.e., the highest elasticity). This was attributed to the highest grafted HPM which created greater possibility to form linkage between NR-g-HPM and the phenolic modified PP compatibilizer molecules which promoted easier interactions between the blend components. TGA analysis found that the NR-g-HPM/PP blends exhibited two stages of weight loss while the pure PP exhibited a single stage. Furthermore, the NR-g-HPM/PP blend exhibited higher degradation temperature than that of the unmodified NR/PP blend which was the confirmation of higher heat resistance of NR-g-HPM.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Natural rubber molecules show low resistance to oxidation, UV irradiation, weathering, chemical and thermal stability. Therefore, chemical modification of NR molecules has been widely practiced to enhance those useful properties and to extend NR scope of applications. There are various NR products that have been chemically modified including maleated natural rubber (MNR) [1, 2], epoxidized natural rubber (ENR), hydrogenated natural rubber (HNR), chlorinated natural rubber (CNR) [3, 4], natural rubber grafted with various types of polymers, such as natural rubber-g-poly(methyl methacrylate) (NR-g-PMMA), natural rubber-g-polystyrene (NR-g-PS) [5, 6] and natural rubber-g-poly(dimethyl(methacryloyl oxymethyl)phosphonate) (NR-g-PDMMMP) [7].
Graft copolymers of natural rubber have the potential application to be used as compatibilizers for blending NR with other polymer components, which are identical to the grafted chains with similar polarity. These include blending of NR with poly(methyl methacrylate) with the aid of NR-g-PMMA [8] and similarly with polystyrene by using NR-g-PS [9] and blending with EVA by using NR-g-PDMMMP as compatibilizers [7]. In addition, natural rubber-g-maleic anhydride [1, 10] and NR-g-PMMA [5, 11] have been used as the main blend components to enhance some useful properties such as solvent resistance and anti-degradation.
N-(4-hydroxyphenyl)maleimide (HPM) is an interesting monomer, since it consists of pendant phenol group which could be introduced into the polymer backbone by copolymerization. This leads to formation of graft copolymer with high polarity and high heat resistance [12]. N-(4-hydroxyphenyl)maleimide (HPM) is an interesting monomer, since it consists of pendant phenol groups which could be introduced into the polymer backbone by copolymerization. This again leads to formation of graft copolymer with NR with high polarity and high heat resistance. Furthermore, pendant phenol groups in the HPM molecules might induce synergistic effect on the dynamic vulcanization of NR-g-HPM/PP blends by phenolic cured system. Also, the NR-g-HPM/PP blends can be used to produce material with resistance to non-polar solvent and high heat.
Nowadays, thermoplastic elastomers (TPEs) have been found to be the commercially interested class of polymeric materials. They are biphasic materials that possess the combined properties of glassy or semi-crystalline thermoplastics and soft elastomers, and enable rubbery materials to be processed as thermoplastics [13]. Thermoplastic vulcanizates (TPVs) are a special class of thermoplastic elastomer (TPE) with two-phase morphological properties, in which the rubber particles form the dispersed phase in the thermoplastic polymer matrix. TPVs offer many features of cross-linked rubbers, coupled with the processability of thermoplastic polymers. They exhibit outstanding low compression set and high extensibility in addition to solvent resistance related to the parent polymer pairs. Furthermore, application of TPV shows no property loss, which confirms the recyclability of these types of materials. TPVs can be filled with various types of fillers, can be used as impact modifiers, and can be colored to tailor their properties and appearance. One class of TPE based on blending natural rubber with thermoplastic has been known as thermoplastic natural rubber (TPNR) [14]. Most of the TPVs based on natural rubber have been employed on blending NR with polyethylene [15–17] and polypropylene [1, 18–20]. However, polypropylene is considered to be the best choice for blending with natural rubber due to its high softening temperature of about 150 °C and low glass transition temperature.
In this work, the graft copolymer of natural rubber and N-(4-hydroxyphenyl)maleimide (i.e., NR-g-HPM) was prepared via melt mixing process. Dynamically cured 60/40 NR-g-HPM/PP blends were also prepared by a phenolic dynamic vulcanization system. The effect of HPM content on mechanical, dynamic, rheological and morphological properties of the blends was investigated.
Experimental
Materials
Materials used in this work are listed in Table 1.
Synthesis of N-(4-hydroxyphenyl)maleimide (HPM)
HPM was prepared by gradually adding of p-aminophenol (0.22 mol) into a solution of maleic anhydride (0.22 mol) in dimethylformamide (DMF). Then, polyphosphoric acid (PPA) was added and the mixture was stirred for 2 h at 80 °C. The reaction mixture was then cooled down and transferred into the cold distilled water. The orange precipitate was separated by filtration and washed several times with distilled water, recrystallized with isopropanol, filtered and vacuum-dried for 2 h at 40 °C [21]. The molecular structures of the products were identified by FTIR and 1H NMR techniques.
Preparation of natural rubber grafted N-(4-hydroxyphenyl)maleimide (NR-g-HPM)
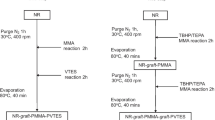
NR-g-HPM was synthesized by melt mixing process. Natural rubber (i.e., STR 5L) was first masticated in an internal mixer, Brabender Plasticorder (PLE-330; Duisburg, Germany) at 170 °C at a rotor speed of 60 rpm for 2 min. HPM (2 phr) was then added into the mixing chamber and continued mixing for 10 min. In this work, NR-g-HPMs with various grafting temperatures (170, 180, 190, 200 and 210 °C) and concentrations of HPM (2, 4, 6, 8 and 10 phr) were prepared. The molecular structure of NR-g-HPM was then identified by FTIR and 1H NMR techniques.
Preparation of dynamically cured NR-g-HPM/PP blends
The NR-g-HPM was first compounded using a formulation, as shown in Table 2. A Brabender Plasticorder (PLE-330, Duisburg, Germany) with a rotor speed of 60 rpm at 40 °C was exploited by a mixing schedule shown in Table 2. The dynamically cured NR-g-HPM/PP blends at a fixed blend ratio of 60/40 with PhHRJ-PP compatibilizer were then prepared via dynamic vulcanization during melt mixing at 180 °C at a rotor speed of 60 rpm by the mixing schedule shown in Table 3. The blend products were received from the mixer and cooled down to room temperature and then pelletized. The dumb-bell shaped specimens were fabricated by using a thermoplastic injection molding machine (Welltec Machinery, Hong Kong). In this work, NR-g-HPM with various quantities of HPM at 2, 4, 6, 8 and 10 phr was used to prepare TPVs. Furthermore, the dynamically cured NR-g-HPM/PP blends were introduced to study the mechanical, morphological, solvent resistance and thermal properties.
Characterization and testing methods
FTIR
HPM sample was grinded and mixed with KBr powder and then pressed into a thin disc which was then characterized by a Nicolet Magna FTIR (560 Omnic ESP, Nicolet Instruments, USA) spectrometer. On the other hand, each NR-g-HPM sample was dissolved in chloroform and was cast on KBr disc to form a thin film. After the evaporation of chloroform, FTIR was used to determine the functional groups in NR-g-HPM molecules.
1H NMR
HPM sample was dissolved in deuterium chloroform and then characterized by proton nuclear magnetic resonance (Unity Inova 500 Hz, Varian Co., Germany). The NR-g-HPM samples were first dissolved in toluene and then precipitated in ethanol. The samples were dried in vacuum oven at 40 °C for 48 h and then dissolved in deuterium chloroform before characterizing by 1H NMR.
Thermal properties
Thermo gravimetric analysis (TGA) and dynamic mechanical analysis (DMA) were used to study the thermal behavior of the samples. TGA analysis of NR-g-HPM was performed by a thermo gravimetric analyzer TGA, STA 6000 thermal analyzer (Perkin Elmer Co., USA) with a constant heating rate of 20 °C/min under nitrogen atmosphere in the temperature range of 30 to 800 °C. On the other hand, DMA was performed by a Perkin Elmer DMA 8000 in a dual cantilever mode at a frequency of 1 Hz. The temperature range of −100 to 100 °C was set with a heating rate of 5 °C.
Mechanical properties
Dumb-bell shaped specimens were prepared by a thermoplastic injection-molding machine with a clamping force of 90 tons (Welltec Machinery, Hong Kong). Tensile properties of the dynamically cured NR-g-HPM/PP blends were measured at 23 °C using 2 mm thick injection-molded samples according to ASTM D412. The Hounsfield tensile testing machine (model H 10KS, Hounsfield Test Equipment Co., England) was used at a speed of 500 mm/min. Tensile properties in terms of modulus at 100 % elongation, tensile strength and elongation-at-break were determined. Tension set was also determined at room temperature according to ISO 2285. That is, the samples were kept under tension at a fixed elongation and time interval (i.e., 100 % and 10 min, respectively). The test specimens were then released and conditioned for another 10 min. The dimensions were eventually determined and compared with the original shapes. The hardness was also measured according to ISO 7619 by employing a Shore A Durometer.
Dynamic properties
Dynamic properties of the NR-g-HPM/PP blends were characterized using a rotor less oscillating shear rheometer (RheoTech MDPT, Cuyahoya Falls, USA) at 180 °C. The oscillation frequency was set in the range of 1–15 Hz at a constant strain of 3 %. This was to assure that the test was in the range of linear viscoelasticity. The storage (G′) and loss shear (G″) moduli, loss factor, tan δ = G″/G″ as well as the complex viscosity (i.e., η* = 3G*/ω = η″ + iη′) of the TPVs were characterized.
Morphological characterization
Morphological characterization of dynamically cured NR-g-HPM/PP blends was performed using a Jeol scanning electron microscope (JSM-5200, Jeol, Japan). The blends were first cryogenically cracked in liquid nitrogen to avoid any morphological changes. The polypropylene phase was then extracted by dissolving the samples in hot xylene. The extracted samples were dried in vacuum oven at 40 °C for 24 h. The dried surfaces were then gold-coated and examined by scanning electron microscope.
Results and discussion
Synthesis and characterization of HPM and NR-g-HPM
Figure 1 shows infrared spectrum of N-(4-hydroxyphenyl)maleimide (HPM). It can be seen that the absorption peaks at 1,709 and 1,516 cm−l indicate the stretching vibrations of carbonyl groups (–C=O) of imide ring and C=C stretching vibration of aromatic rings, respectively. Furthermore, the absorption peak at 3,109 cm−l indicates the asymmetric stretching vibration of C–H bonding of olefinic groups. Also, a peak at 3,483 cm−l is observed, which is assigned to the symmetric stretching vibration of O–H bond. This proves the occurrence of HPM from a reaction of maleic anhydride and p-aminophenol, as shown in Scheme 1 [22]. It is seen that maleic anhydride reacts with p-aminophenol in a medium of polyphosphoric acid and dimethyl formamide. Ring opening of maleic anhydride was followed by addition reaction at the nitrogen atom of p-aminophenol. Therefore, HPM was formed which was a combination of maleic anhydride and p-aminophenol.

Infrared spectrum of N-(4-hydroxyphenyl)maleimide (HPM)

Reaction of maleic anhydride and p-aminophenol at 80 °C
Figure 2 shows 1H NMR spectrum of N-(4-hydroxyphenyl)maleimide. It can be seen that the peaks are observed at the chemical shifts of 7.15 (2) and 6.89 (3) ppm assigned to the protons of aromatic rings. Furthermore, the peaks with the chemical shift of 6.82 (1) and 5.08 (4) ppm are observed. These two peaks are attributed to the protons of imide rings and hydroxyl groups, respectively. This confirms the formation of N-(4-hydroxyphenyl)-maleimide, as the reaction shown in Scheme 1.

1H NMR spectrum of N-(4-hydroxyphenyl)maleimide
Figure 3 shows the infrared spectra of natural rubber-g-N-(4-hydroxyphenyl)maleimide (i.e., NR-g-HPM) compared with pure NR and pure HPM. In the spectrum of NR-g-HPM, the peaks can be seen at 1,709 and 1,516 cm−l, which are assigned to the stretching vibrations of carbonyl groups in imide rings and C=C stretching vibration of aromatic rings in HPM, respectively. This may confirm that the HPM moiety has been successfully grafted onto natural rubber molecules. Furthermore, the absorption peak at 835 cm−l of = C–H out-of-plane bending of isoprene units in natural rubber molecules is observed in the spectrum of NR-g-HPM. Therefore, the grafting reaction of HPM onto the natural rubber molecules may occur according to the proposed reaction mechanism shown in Scheme 2 [21].

Infrared spectra of natural rubber grafted N-(4-hydroxyphenyl)maleimide (NR-g-HPM) compared with infrared spectrum of natural rubber and HPM

Reaction mechanism for grafting of N-(4-hydroxyphenyl)maleimide onto natural rubber molecules
Figure 4 shows 1H NMR spectra of natural rubber-g-N-(4-hydroxyphenyl)maleimide (NR-g-HPM). An intense peak is observed at a chemical shift of 5.1 ppm, which is assigned to the protons in [(CH3)–C=C–H] of unsaturated unit of natural rubber backbone. Furthermore, two signals are observed at chemical shifts of 7.15 (A) and 6.89 (B) ppm. These are assigned to the protons of aromatic and imide rings in HPM molecules, respectively. This result confirms the formation of graft copolymer of N-(4-hydroxyphenyl)maleimide onto natural rubber molecules (i.e., NR-g-HPM). The reaction mechanism is shown in Scheme 2.

Proton NMR of natural rubber-g-N-(4-hydroxyphenyl)maleimide (NR-g-HPM)
Effect of grafting temperature and monomer concentration on grafted HPM content in NR-g-HPM
The NR-g-HPM is synthesized by a fixed concentration of HPM at 2 phr at various mixing temperatures of 170, 180, 190, 200 and 210 °C. After selecting the optimum mixing temperature, various concentrations of HPM at 2, 4, 6, 8, and 10 phr are used to prepare the graft copolymer. In this work, the grafting level is determined by quantifying the peak height ratio of infrared spectra peak at 1,709 cm−1 (stretching vibration of carbonyl groups in imide rings) to 835 cm−1 (=C–H out-of-plane bending of natural rubber molecules). This is assembled to the quantification of grafted maleic anhydride onto EPDM backbone using the peak height ratio of 1,707 cm−1 (–C=O stretching vibration of maleic anhydride) and 1,460 cm−1 (–CH out-of-plane bending in EPDM) [23].
Figure 5 shows the infrared spectra of natural rubber-g-N-(4-hydroxyphenyl)maleimide prepared with various grafting temperatures. Also, Fig. 6 shows the peak height ratio of the peaks at 1,709 to 835 cm−1 based on the FTIR spectra in Fig. 5. It can be seen that the peak height ratio increased with increasing grafting temperature with the maximum grafting content at approximately 200 °C. This is attributed to reaction rate increased with increasing temperature, according to the well-known Arrhenius equation (i.e., k = Ae−Ea/RT). A decreasing trend of the peak height ratio is observed in the reaction at temperature higher than 200 °C. This is attributed to competitive reactions among homopolymerization, oxidative degradation and chain scission. Therefore, the grafting temperature at 200 °C is the optimum condition that could be used to prepare the NR-g-HPM.

Infrared spectra of natural rubber grafted N-(4-hydroxyphenyl)maleimide (NR-g-HPM) prepared at various temperatures

Influence of grafting temperature on peak height ratio of 1,709 to 835 cm−1
Figure 7 shows the infrared spectra of natural rubber-g-N-(4-hydroxyphenyl)maleimide prepared with various quantities of HPM. Also, the peak height ratios of the peaks at the wavenumber of 1,709 to 835 cm−1 of the infrared spectra in Fig. 7 are calculated and the results are shown in Fig. 8. It can be seen that the peak height ratio is fallen with increasing quantities of HPM. This might be attributed to the incompatibility between the non-polar molecules of natural rubber and polar molecules of HPM, which induces the agglomeration of HPM and its separation from rubber phase [24]. Therefore, the concentration of HPM at 2 phr is an optimum concentration used throughout this work.

Infrared spectra of natural rubber grafted N-(4-hydroxyphenyl)maleimide (NR-g-HPM) prepared with various quantities of HPM

Influence of HPM quantity on peak height ratio of the peaks at 1,709 to 835 cm−1
Mechanical properties of dynamically cured 60/40 NR-g-HPM/PP blends
Figure 9 shows the mixing torques of dynamic vulcanization based on blending of 60/40 NR-g-HPM/PP blends with various quantities of HPM used in the graft copolymerization. The first peaks of the mixing torque are related to the incorporation of PP into the mixing chamber until complete melting of PP at a mixing time of approximately 3 min. A low amount of PhHRJ-PP blend compatibilizer (i.e., 5 wt% of PP) is then added and mixing is continued for 2 min. The second peaks at a mixing time of approximately 5.5 min arise due to the viscosity of the blends after incorporation of the rubber compounds into the mixing chamber. The mixing torque is then dropped owing to the lowering viscosity of the rubber compounds. The third peaks of mixing torques can be seen at approximately 7.5 min and it is due to the dynamic cross-linking reaction of the rubber phase.

Mixing torques and time of dynamically cured 60/40 NR-g-HPM/PP blends with various quantities of HPM
During the cross-linking reaction, an abrupt increase in mixing torque in the mixing chamber is observed due to onset of dynamic vulcanization. This causes increasing trends of shear and extensional viscosities of the rubber phase. Therefore, the vulcanizing rubber phase is forced to break into smaller rubber particles at this stage. The mixing is further performed to ensure the complete vulcanization of the rubber phase or until the plateau mixing torque reaches a total mixing time of approximately 12 min. In Fig. 9, it is also seen that the mixing torque at the end of mixing decreased with increasing quantities of HPM used in the graft copolymerization. This might be attributed to the higher loading level of HPM which results in higher lubricating effect in the blend system due to the higher content of un-grafted HPM (Fig. 8).
In this work, phenolic modified polypropylene (i.e., PhHRJ-PP) is added in the blend formulation as a blend compatibilizer with a loading level of 5 wt% of PP. It has been proved that the incorporation of PhHRJ-PP in NR-g-HPM/PP blends results in grafting reaction between the phenolic modified PP molecules and NR-g-HPM. Furthermore, PP moiety in the PhHRJ-PP has the capability to mix with PP molecules in the blend component. This results in improvements in mechanical, rheological and morphological properties of the dynamically cured NR-g-HPM/PP blends [25].
Figure 10 shows the stress–strain curves of dynamically cured 60/40 NR-g-HPM/PP blends with various quantities of HPM used in graft copolymerization. It can be seen that the blends exhibit close values of Young’s moduli (i.e., slope at an initial part of the curve). However, the blend with 2 phr of HPM shows the highest toughness (i.e., area underneath the curve). Table 4 summarizes the tensile strength and elongation-at-break of dynamically cured NR-g-HPM/PP blends with various quantities of HPM based on the failed locations in Fig. 10. The results of tension set and hardness measurements are also given in Table 4. It can be seen that the tensile strength and elongation-at-break are decreased with increasing quantities of HPM used in the graft copolymerization. This might be due to the blends with higher concentration of HPM producing higher free or un-grafted HPM (Fig. 8), which is capable of reacting with phenolic modified PP compatibilizer and also with phenolic curing agent. This influences the retardation of the compatibilizing effect (i.e., Scheme 3) and dynamic vulcanization reaction which thereafter lead to inferior mechanical properties. In Table 4, the lowest tension set value is observed in the blend with 2 phr of HPM, which indicates the highest rubber elasticity of the material. Other types of blends show higher similar values of tension set which imply lower flexibility. Furthermore, it is also seen that the NR-g-HPM/PP blends with different levels of HPM used in the graft copolymerization show similar hardness indices. Therefore, different types of NR-g-HPM showed significant reduction effects on the hardness.

Stress-strain curves of dynamically cured 60/40 NR-g-HPM/PP blends with various quantities of HPM

Possible reaction mechanism of grafting reaction between phenolic modified polypropylene (PhHRJ-PP) and NR-g-HPM molecules
Morphological properties of dynamically cured 60/40 NR-g-HPM/PP blends
Figure 11 shows SEM micrographs of the fractured surface of the dynamically cured 60/40 NR-g-HPM/PP blends with various quantities of HPM used in the graft copolymerization. It is seen that the size of vulcanized rubber domains increased with increasing quantities of HPM. This is attributed to the higher level of free HPM molecules which has led to stronger lubricating effect in the blend system. Also, some amounts of HPM molecules react with PhHRJ-PP compatibilizer and HRJ-10518 phenolic curing agent which lower the compatibilization and vulcanization reactions. Therefore, during dynamic vulcanization, the vulcanizing NR-g-HPM phase exhibits lower flow resistance, as observed in terms of mixing torque shown in Fig. 9 where the mixing torque of the blend system with 10 phr of HPM has exhibited the lowest value. Therefore, lower shear and extensional viscosities are the two main parameters influenced on breaking up the vulcanizing rubber domains into smaller particles. Hence, the size of vulcanized rubber particles is increased with increasing quantities of HPM used in the graft copolymerization. This result correlates well with the mixing torque (Fig. 10), tensile strength and elongation-at-break (Table 4). It is noted that larger vulcanized rubber domains dispersed in the PP matrix reduce the interfacial adhesion between rubber particles and PP phase, and hence lower tensile strength and elongation-at-break properties together with poor set properties. This result agrees with the findings of Coran et al. [26], who suggested that the reduction in size of rubber particles is responsible for the increase in stress- and strain-at-break.

SEM micrographs of dynamically cured 60/40 NR-g-HPM/PP blends with various quantities of HPM used in the graft copolymerization: a 2 phr, b 4 phr, c 6 phr, d 8 phr and e 10 phr
Dynamic properties of dynamically cured 60/40 NR-g-HPM/PP blends
Figure 12 shows the storage modulus as a function of frequency for various 60/40 NR-g-HPM/PP blends. As expected, the storage modulus or elastic response of the material increased with frequency and it is related to the shorter time for molecular relaxation. At a given frequency, the storage modulus increased with decreasing the loading amount of HPM in the graft copolymerization of NR and HPM. The maximum value of storage modulus is observed at a loading amount of 2 wt% of HPM. Higher loading amounts show a decreasing trend of the storage modulus. Therefore, the dynamically cured 60/40 NR-g-HPM/PP blends with 2 wt% HPM provided the highest elasticity to the vulcanized rubber phase. These results corresponded to the mechanical properties in Table 4, where the lowest tension set and highest tensile strength and elongation-at-break are observed in the dynamically cured NR-g-HPM/PP blends with 2 wt% HPM.

Storage modulus as a function of frequency of dynamically cured 60/40 NR-g-HPM/PP blends with various quantities of HPM
Figure 13 shows the complex viscosity as a function of frequency for various 60/40 NR-g-HPM/PP blends. A shear-thinning behavior (i.e., decreasing trend of complex viscosity with increasing magnitude of oscillating frequency) is observed for all the blends. At a given frequency, the complex viscosity results are in good agreement with the trend of storage modulus in Fig. 12. That is the dynamically cured NR-g-HPM/PP blends with 2 wt% HPM exhibited the highest viscosity. Higher amount of HPM used in the graft copolymerization resulted in free or more ungrafted HPM (Figs. 7, 8). Therefore, some excess amount of HPM might form small micelles which produce a lubricating effect to the system, and hence a lower flow resistance or lower viscosity is observed.

Complex viscosity as a function of frequency of dynamically cured 60/40 NR-g-HPM/PP blends with various quantities of HPM
TGA analysis of dynamically cured 60/40 NR-g-HPM/PP blends
Figure 14 shows the thermo gravimetric and derivative of thermo gravimetric curves of pure PP and dynamically cured 60/40 NR-g-HPM/PP blend with 2 phr of HPM used in the graft copolymerization. The TGA and derivative of TGA (i.e., DTG) for dynamically cured unmodified NR/PP blend is also carried out for a comparison purpose. Furthermore, Table 5 summarizes the decomposition temperature (T d) of dynamically cured NR-g-HPM/PP blend compared with unmodified NR/PP blend and pure PP. It can be seen that pure PP shows a single stage of weight loss at the T d of approximately 452 °C. However, the dynamically cured NR-g-HPM/PP and unmodified NR/PP blends show two-steps of weight loss. That is, the unmodified NR/PP blends show the T d of approximately 389 and 458 °C at the first and second stage, respectively. However, the dynamically cured NR-g-HPM/PP blend exhibits higher Td of approximately 411 and 461 °C at the first and second stages, respectively. The first decomposition stage is related to the degradation of rubber component while the second stage is related to the degradation of PP component. It is clear that NR-g-HPM enhances the thermal stability of the blend by rising degradation temperature of 22 and 3 °C in the first and second degradation stages. This is indicated by higher heat resistance of the dynamically cured NR-g-HPM/PP blend compared with the dynamically cured unmodified NR/PP blend.

TGA thermograms a and derivative of TGA thermograms b of dynamically cured 60/40 NR-g-HPM/PP blends with 2 phr of HPM compared with unmodified NR/PP blend
Conclusion
Natural rubber-g-N-(4-hydroxyphenyl)maleimide (NR-g-HPM) was successfully prepared via melt processing method. This was confirmed by the infrared absorption peaks at 1,709 and 1,516 cm−1 which indicated the presence of C=O of imide ring and C=C of aromatic ring of HPM grafted onto the natural rubber molecules. In addition, the chemical shift in 1H NMR spectrum at 7.15 and 6.89 ppm was observed from the proton of aromatic ring of HPM and the signal at 6.82 ppm which was attributed to the proton of imide ring of HPM. These results confirmed the formation of NR-g-HPM along with natural rubber chain. The effects of grafting temperature and quantities of HPM on grafted HPM content were also studied. An increasing trend of grafted HPM was observed upon increasing the grafting temperature by reaching its maximum at 200 °C. In addition, the grafted HPM content decreased with increasing HPM content in the graft copolymerization. The effect of HPM quantity on the properties of the dynamically cured 60/40 NR-g-HPM/PP blends was also investigated. It was found that tensile strength, elongation-at-break, mixing torque, storage modulus and complex viscosity of the blends decreased with increasing quantities of HPM used in the graft copolymerization. Moreover, the size of vulcanized rubber particles of the blends and tension set increased with increasing quantities of HPM. Thermal stability of dynamically cured NR-g-HPM/PP blend was also characterized by TGA. It was found that the blends showed two stages of weight loss while the pure PP exhibited a single stage. Furthermore, the NR-g-HPM/PP blend exhibited higher degradation temperature than that of the unmodified NR/PP blend. This indicated the higher heat resistance of NR-g-HPM, and it was possible to prepare thermoplastic elastomers with higher heat resistance suitable for industrial applications.
References
Nakason C, Saiwari S, Kaesaman A (2006) Rheological properties of maleated natural rubber/polypropylene blends with phenolic modified polypropylene and polypropylene-g-maleic anhydride compatibilizers. Polym Test 25:413–423
Nakason C, Kaesaman A, Supasanthitikul P (2004) The grafting of maleic anhydride onto natural rubber. Polym Test 23:35–41
Bradbury JH, Perera MCS (1985) Epoxidation of natural rubber studied by NMR spectroscopy. J Appl Polym Sci 30:3347–3364
Perera MCS (1987) Surface modification of natural rubber laces. J Appl Polym Sci 34:2591–2600
Thiraphattaraphun L, Kiatkamjornwong S, Prasassarakich P, Damronglerd SJ (2001) Natural rubber-g-methyl methacrylate/poly(methyl methacrylate) blends. Appl Polym Sci 81:428–439
Arayapranee W, Prasassarakich P, Rempel GLJ (2003) Process variables and their effects on grafting reactions of styrene and methyl methacrylate onto natural rubber. Appl Polym Sci 89:63–74
Intharapat P, Derouet D, Nakason C (2010) Dynamically cured natural rubber/EVA blends: influence of NR-g-poly(dimethyl (methacryloyloxymethyl)phosphonate) compatibilizer. Polym Adv Technol 21:310–321
Oommen Z, Thomas S, Premalatha CK, Kuriakose B (1997) Melt rheological behaviour of natural rubber/poly(methyl methacrylate)/natural rubber-g-poly(methyl methacrylate) blends. Polymer 38:5611–5621
Asaletha R, Kumaran MG, Thomas S (1998) Thermal behaviour of natural rubber/polystyrene blends: thermo gravimetric and differential scanning calorimetric analysis. Polym Degrad Stab 61:431–439
Pichaiyut S, Nakason C, Kaesaman A, Kiatkamjornwong S (2008) Influences of blend compatibilizers on dynamic, mechanical, and morphological properties of dynamically cured maleated natural rubber and high-density polyethylene blends. Polym Test 27:566–580
Pechurai W (2005) Preparation of thermoplastic elastomer from NR-g-PMMA and PMMA blends by dynamic vulcanization. In: MSc Thesis, Faculty of Science and Technology, Prince of Songkla University, Pattani, Thailand
Reghunadhan Nair CP, Mathew D, Ninan KN (1999) Free radical copolymerisation of N-(4-hydroxy phenyl) maleimide with vinyl monomers: solvent and penultimate-unit effects. Eur Polym J 35:1829–1840
Holden G (2000) Understanding thermoplastic elastomer. Hanser Publicher, Munich
Ibrahim A, Dahlan M (1998) Thermoplastic natural rubber blends. Prog Polym Sci 23:665–706
Machado AV, van Duin M (2005) Dynamic vulcanisation of EPDM/PE-based thermoplastic vulcanisates studied along the extruder axis. Polymer 46:6575–6586
Nakason C, Nuansomsri K, Kaesaman A, Kiatkamjornwong S (2006) Dynamic vulcanization of natural rubber/high-density polyethylene blends: effect of compatibilization, blend ratio and curing system. Polym Test 25:782–796
Pechurai W, Nakason C, Sahakaro K (2008) Thermoplastic natural rubber based on oil extended NR and HDPE blends: blend compatibilizer, phase inversion composition and mechanical properties. Polym Test 27:621–631
Ismail H, Suryadiansyah S (2002) Thermoplastic elastomers based on polypropylene/natural rubber and polypropylene/recycle rubber blends. Polym Test 21:389–395
Oh JS, Isayev AI, Rogunova MA (2003) Continuous ultrasonic process for in situ compatibilization of polypropylene/natural rubber blends. Polymer 44:2337–2349
Thitithammawong A, Nakason C, Sahakaro K, Noordermeer JWM (2007) Thermoplastic vulcanizates based on epoxidized natural rubber/polypropylene blends: selection of optimal peroxide type and concentration in relation to mixing conditions. Eur Polym J 43:4008–4018
Somkieowan S (2009) Rubber bound antioxidants: natural rubber grafted with maleimide derivatives. In: MSc Thesis, Faculty of Science and Technology, Prince of Songkla University, Pattani, Thailand
Park JO, Jang SH (1992) Synthesis and characterization of bismaleimides from epoxy resins. J Polym Sci Part A Polym Chem 30:723–729
Barra GMO, Bertolino JR, Crespo JS, Pires ATN, Soldi V (1999) Maleic anhydride grafting on EPDM: qualitative and quantitative determination. Braz Chem Soc 10:31–34
Grigoryeva OP, Karger-Kocsis J (2000) Melt grafting of maleic anhydride onto an ethylene–propylene–diene terpolymer (EPDM). Eur Polym J 36:1419–1429
Sasdipan K (2009) High heat resistance thermoplastic vulcanizate based on natural rubber-g-N-(4-hydroxy phenyl) maleimide. In: MSc Thesis, Faculty of Science and Technology, Prince of Songkla University, Pattani, Thailand
Coran AY, Patel RP (1996) Thermoplastic elastomers based on dynamically vulcanized elastomer-thermoplastic blends. In: Holden G, Legge NR, Quirk R, Schroeder HE (eds) Thermoplastic elastomers. Hanser Publisher, New York
Acknowledgments
The authors would like to thank the Center of Excellence in Natural Rubber Technology (CoE-NR) and the funding from the government budget for financial support. This work was also supported by the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nakason, C., Sasdipan, K. & Kaesaman, A. Novel natural rubber-g-N-(4-hydroxyphenyl)maleimide: synthesis and its preliminary blending products with polypropylene. Iran Polym J 23, 1–12 (2014). https://doi.org/10.1007/s13726-013-0194-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13726-013-0194-7