
Abstract
Purpose of Review
Non-alcoholic fatty liver disease (NAFLD) is one of the most common causes of chronic liver disease with rising prevalence worldwide. Herein, we provide a comprehensive overview of the current knowledge supporting the role of ER stress and autophagy processes in NAFLD pathogenesis and progression. We also highlight the interrelation between these two pathways and the impact of ER stress and autophagy modulators on NAFLD treatment.
Recent Findings
The pathophysiological mechanisms involved in NAFLD progression are currently under investigation. The endoplasmic reticulum (ER) stress and the concomitant unfolded protein response (UPR) seem to contribute to its pathogenesis mainly due to high ER content in the liver which exerts significant metabolic functions and can be dysregulated. Furthermore, disruption of autophagy processes has also been identified in NAFLD. The crucial role of these two pathways in NAFLD is underlined by the fact that they have recently emerged as promising targets of therapeutic interventions.
Summary
There is a greater need for finding the natural/chemical compounds and drugs which can modulate the ER stress pathway and autophagy for the treatment of NAFLD. Clarifying the inter-relation between these two pathways and their interaction with inflammatory and apoptotic mechanisms will allow the development of additional therapeutic options which can better target and reprogram the underlying pathophysiological pathways, aiming to attenuate NAFLD progression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is characterized by accumulation of fat inside hepatocytes in the absence of excessive alcohol consumption [1]. NAFLD is strongly associated with metabolic syndrome features, such as obesity (particularly central), with increasing prevalence rates in both developed and developing countries which follow the corresponding rising prevalence of obesity and type 2 diabetes (T2DM) [1]. Currently, NAFLD constitutes the second most common cause for liver transplantation in the USA and is predicted to take the first place soon [1, 2]. Recently, a panel of international experts proposed that NAFLD should be renamed to metabolic-dysfunction- associated fatty liver disease (MAFLD) in order to better reflect the underlying pathophysiology [3]. NAFLD begins with simple steatosis, where triacylglycerol (TAG) accumulation is present in more than 5% of hepatocytes, while it evolves to fatty infiltration with inflammation, leading to the more severe form of NAFLD, namely nonalcoholic steatohepatitis (NASH) which can further progress into liver fibrosis and ultimately cirrhosis and hepatocellular carcinoma (HCC) [4].
The liver is a highly secretory organ and one of the major targets of insulin and glucagon [5•] and plays a fundamental role in the lipid and carbohydrate metabolism. Of note, the liver is characterized by the remarkable feature to proliferate upon damage [5•].
Hepatocytes have large amounts of endoplasmic reticulum (ER) that exert important liver metabolic functions such as protein and lipid synthesis, transmembrane protein folding and calcium homeostasis [6, 7]. Dysfunction of hepatic ER is implicated in the spectrum of the NAFLD pathology via activation of ER stress signaling [7, 8•].
Autophagy is another crucial process implicated in cellular homeostasis delivering cytoplasmic content to the lysosomes in order to degrade and/or recycle components to form macromolecules [9]. The potential role of autophagy in hepatic lipid metabolism has been recently recognized, while dysregulated autophagy has also been found to contribute to the pathogenesis of NAFLD [10•].
ER stress and autophagy co-operate to stimulate the degradation of intracellular lipid droplets (lipophagy) in liver. However, under increased and sustained ER stress, an aberrant inflammation, along with reduced autophagic process can lead to hepatocyte death [11, 12].
This review presents the current knowledge supporting the impact of ER stress signaling and autophagy and their interrelation on NAFLD, ranging from simple steatosis to NASH and HCC, while summarizing potential therapeutic interventions targeting these two processes.
Obesity and NAFLD
Obesity has been linked to the development of metabolic syndrome and its comorbidities, such as T2DM and NAFLD, and increases the risk of mortality in these individuals [13, 14]. Interestingly, obesity has emerged as an independent risk factor, increasing the risk of NAFLD incidence by 3.5-fold [15]. The global prevalence of NAFLD is estimated to be about 25.24% (over 2 billion people over the world) [16, 17], although prevalence can vary according to the diagnostic method used. Characteristically, when magnetic resonance spectroscopy—which is a highly sensitive method—was used to measure hepatic triglyceride content in a US study population, the prevalence of NAFLD was estimated to be 33.6% [18]. Of note, there is a higher prevalence of NAFLD among individuals with obesity compared to general population, although the exact percentages vary due to the different diagnostic methods and the characteristics of each studied population, such as nationality, age and predisposing factors, including T2DM [13]. In Italy, the Dionysos study identified that the prevalence of NAFLD was 75.8% in persons with obesity [19], while NAFLD prevalence was 27.1% among overweight Dutch and 81.7% among Dutch people with obesity [20], and 67.5% in US persons with obesity [21]. A study in a French population identified NAFLD with mild steatosis (5–33%) in 53% and moderate steatosis (33–66%) in 20% of patients with metabolically healthy obesity [22]. NAFLD has a prevalence between 15% and 80% in people with obesity in the Asia-Pacific region, with a prevalence ranged from 50 to 80% in Japanese people with obesity, between 70% and 80% in Chinese with obesity, from 10 to 50% in Korean individuals with obesity, between 15% and 20% in Indians with obesity and a prevalence of about 47% in Indonesian people with obesity [23]. Furthermore, the prevalence of obesity among global NAFLD patients is 51.3% [17]. Obesity can cause energy imbalance, insulin insensitivity and lipolysis, and alter metabolic functions such as insulin resistance and dyslipidemia, entities that can contribute to NAFLD occurrence [15, 16, 24]. Obesity can disturb the metabolism of the adipose tissue and when the excess energy cannot be stored in this tissue, then this role is taken over by other tissues, with the hepatic being one of them [13, 25]. In this case, circulating free fatty acids (FFAs) derived from increased hepatic de novo lipogenesis or from diminished uptake in subcutaneous adipose tissue can result in ectopic fat accumulation, in our case in the liver, and multiorgan insulin resistance, which is considered a major contributor towards NAFLD development [26, 27]. The high levels of lipids and carbohydrates that hepatocytes are exposed to due to obesity can lead to hepatocellular injury and consequently NAFLD due to ER stress, oxidative stress and mitochondrial dysfunction [11]. The accumulation of lipids in hepatocytes, if not appropriately managed, can provoke the infiltration of immune cells in the liver and the initiation of an inflammatory process [13, 28]. During this process the immune cells secrete inflammatory cytokines and immunomodulatory mediators that can worsen the hepatocyte dysfunction and lead to necrosis, hepatic steatosis and liver fibrosis, leading to NAFLD and NASH [15, 29]. Obesity can also affect the liver through hormones produced by the adipose tissue, the so called adipokines, which are implicated in the pathogenesis of NAFLD and its progression to NASH and HCC through their contribution to the aforementioned inflammation [30]. In particular, the adipokines are separated in pro- and anti-inflammatory ones and while in normal metabolic status there is a balance between the two categories, in obesity when the adipose tissue enlarges, the secreted adipokines shift towards a pro-inflammatory, steatogenic and fibrogenic profile [13, 31]. In addition, the interaction of the adipokines with the immune cell-derived cytokines can affect NAFLD pathogenesis and progression [13, 32].
ER Stress
The ER is responsible for proper folding of secreted and transmembrane proteins, and the accumulation of misfolded or unfolded proteins leads to ER stress and the activation of the unfolded protein response (UPR) to restore homeostasis [33, 34]. Activation of the UPR relies on the activation of three transmembrane ER stress sensors: the inositol-requiring enzyme 1 (IRE1), the PKR [double-stranded RNA-activated protein kinase]-like ER kinase (PERK) and the activating transcription factor 6 (ATF6) [33, 34]. These three sensors are maintained inactive through the binding of the ER chaperone GRP78/Binding immunoglobulin protein (BiP) on their cytosolic domains [8•, 35].
IRE1α, which catalyzes the first and most conserved branch of the ER stress response, is a transmembrane kinase/endoribonuclease (RNAse) [8•, 34]. Its cytosolic domain senses stress by binding unfolded proteins when GRP78/BiP is dislocated from the chaperone Hsp47, leading to autophosphorylation and dimerization of IRE1α and activation of its RNAse domain [8•, 34]. This domain cleaves the mRNA encoding the X-box binding protein 1 (XBP1), creating the spliced form XBP1s, that reinforces ER protein folding, secretion and degradation (ER-associated degradation-ERAD) [36]. Furthermore, the RNAse domain of IRE1α targets other ER-related mRNAs as well, through regulated IRE1α-dependent decay (RIDD) [37], which leads to pro-apoptotic signaling through destruction of specific microRNAs that block pro-apoptotic caspase-2 translation [38].
PERK is also transmembrane with an external domain that senses stress and a cytosolic kinase domain that preferentially phosphorylates the translation elongation factor elf2α which halts translation while at the same time it increases the amount of specific mRNAs, such as ATF4 [8•, 39]. ATF4 activates downstream genes, is implicated in restoring homeostasis and regulates the expression of the DNA damage-inducible transcript 3 (DDIT3, or growth arrest and DNA damage 153 (GADD153) or best known as CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP) [39], and subsequent growth arrest and DNA damage-inducible 34 (GADD34) [40].
ATF6 is a basic leucine zipper protein (bZIP) functioning as an ER stress response element, which promotes transcription of ER-resident chaperones [41]. Upon ER stress, ATF6α is translocated from the ER to the Golgi apparatus, where it is cleaved from the 90-kDA to the active 50-kDa protein by Site-1 protease (S1P) and Site-2 protease (S2P) [41, 42]. The active form is then transported to the nucleus [42] where it activates the transcription of ER chaperone genes, as well as of the transcription factors CHOP and XBP-1 [43].
Of note, apart from its role in protein processing, ER is also critically implicated in lipid synthesis in hepatocytes, thus it is implicated in lipid disorders, including hepatic steatosis [44]. Indeed, 59% of accumulated hepatic TAG causing NAFLD comes from non-esterified fatty acids, 26% from de novo lipogenesis, and only 15% from the diet [45], underlying the important role of lipogenesis in NAFLD pathophysiology.
ER Stress and NAFLD
The IRE1α/XBP1 Axis
The significance of IRE1α/XBP1 axis in the pathogenesis of NAFLD has been proven mostly by genetic ablation studies. When molecules of the three sensing pathways are deleted, the response to ER stress is compromised leading to hepatic steatosis [46]. Hepatocyte-specific deletion of IRE1α in mice led to defective protein transport from the ER to the Golgi apparatus, defective oxidative protein folding, and ER-associated degradation of misfolded proteins, factors that induce ER stress and activate the UPR [47]. These mice developed modest hepatosteatosis in the absence of ER stress, which was worsened after its induction [47]. IRE1α represses the expression of metabolic transcriptional regulators, including CCAAT/enhancer-binding protein (C/EBP)β, C/EBPδ, peroxisome proliferator-activated receptor γ (PPARγ) and enzymes involved in triglyceride biosynthesis, while is also necessary for apolipoprotein secretion when ER homeostasis is disrupted [47]. Furthermore, liver-specific XBP1 disruption is related to decreased hepatic lipogenesis [48]. Overexpression of Bax inhibitor-1 (BI-1) and concomitant inhibition of IRE1α endonuclease activity leads to downregulation of lipogenesis genes, such as stearoyl-CoA desaturase 1 (Scd-1) [48]. XBP-1 deficiency leads to a feedback activation of IRE1α and mRNA degradation of lipid metabolism genes, such as Dgat2, Acacb, Pcsk9, Angptl3, and Ces1 [49]. Apart from sensing the unfolded proteins, studies in yeast and mammalian cells have shown that the ER stress cytosolic (or transmembrane, in mammalian cells) sensing domain of IRE1α is sensitive to membrane lipid composition, suggesting a lipid sensing mechanism as well which can activate the UPR [50, 51]. Changes in membrane lipid composition can lead to IRE1α and PERK activation by enhanced dimerization via their transmembrane domain which proved to be the important domain for both—albeit especially for IRE1α—in terms of XBP1 splicing [51]. On the other hand, small heterodimer partner (SHP; also known as nuclear receptor subfamily 0, group B, member 2, NR0B2) deficiency downregulates XBP1s protein level and transcriptional activity, compromising the ER stress response [52]. Apart from the role of splicing in the formation of XBP1s which mediates key functions of the ER stress response, its post-translational modification plays an equally important role [53•]. In particular, deacetylation of XBP1s by sirtuin 6 (Sirt6) protects against hepatic steatosis, since in Sirt6 knockout and obese mice the acetylated XBP1s levels and hepatic steatosis were increased, while these phenomena were decreased after genetic overexpression and pharmacological activation of Sirt6 [53•]. The results linking acetylated XBP1s positively with NAFLD activity score (NAS) and negatively with Sirt6 level were also confirmed in liver biopsies from NAFLD patients [53•].
Upon ER stress and concomitant UPR activation, activated IRE1α can lead to subsequent activation of several downstream molecules, such as the pro-inflammatory and pro-apoptotic c-Jun-NH2-terminal kinases (JNKs) which lead to inflammation and cell death, as well as the nuclear factor-κB (NF-κB) [8•]. NF-κB in the liver acts as a link between hepatic injury and fibrosis—and even progress to hepatocellular carcinoma—with a double role, mediating both pro-inflammatory and anti-apoptotic responses to protect cells from death while inflammatory or immune responses are initiated [54]. This is necessary since these responses are responsible for protecting against pathogens/infections, but should also be tightly regulated to prevent liver injury from the toxic pathogen products [54]. Indeed, NF-κB can act as a “two-edged sword” in NAFLD, since balance between its two roles is needed: normal or slightly elevated NF-κB activity can protect hepatocytes from death, but exaggerated activity can lead to fibrosis and NASH through facilitating excessive inflammation [8•, 54]. Data from cell culture experiments suggest that free fatty acids induce the UPR which when prolonged leads to cell death through CHOP-mediated activation of NF-κB signaling [55]. Furthermore, in mice deficient for BI-1, the unrestrained action of IRE1α RNAse leads to NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation, increased thioredoxin-interacting protein (TXNIP) levels and enhanced NASH [56]. Indeed, hyper-activated IRE1α increases TNXIP mRNA stability and protein expression leading to programmed cell death through NLRP3 inflammasome induction [57], and possibly to NASH occurrence. However, it seems that, not only IRE1α hyper-activation, but also its downregulation can exacerbate hepatic steatosis progression. Data from cultured hepatocytes, which were also confirmed in high-fat diet (HFD) fed mice and human liver biopsies, suggest that the inactivation of the RNAse activity of IRE1α caused by a HFD disrupts the RIDD-mediated degradation of its microRNA targets [58]. The latter includes the miR-200 and miR-34 families, and, in turn, was shown to lead to decreased abundance of their targets, peroxisome proliferator-activated receptor α (PPARα) and deacetylase sirtuin 1 (SIRT1), that are implicated in lipid homeostasis [58]. As a result, lipids accumulate and progression towards hepatic steatosis is noted [58]. Furthermore, studies on liver biopsies from patients with metabolic syndrome with or without NAFLD or NASH found that NASH is associated with failure to splice XBP-1 and generate XBP-1s [59]. Palmitate which contains palmitic acid -a saturated fatty acid- has been found to activate the IRE1α pathway, which activates CHOP and JNK proteins leading to enhanced expression of the BH3-only proteins PUMA and Bim which activate the apoptosis regulator BAX and lead to hepatocyte death [60].
In HCC, IRE1α signaling has been associated with tumor initiation [61]. XBP-1s expression levels have been found elevated in HCC human biopsies [62]. Studies in mice revealed that primary tumors display increased XBP-1 expression which correlated with heightened aggressiveness, namely faster doubling times of the tumor [63].
The PERK-eIF2a-ATF4 Axis
The PERK-eIF2a-ATF4 axis has been found to be primarily implicated in lipogenesis and steatosis regulation [8•]. Enforced overexpression of the GADD34 phosphatase in the liver, which selectively dephosphorylates elf2α, leads to downregulation of phosphorylated elf2α and subsequent attenuation of the adipogenic nuclear receptor PPARγ and the metabolic transcriptional regulators C/EBPα and C/EBPβ [64]. This sequence of events has been linked to enhanced glucose tolerance and insulin sensitivity, as well as with lower incidence of hepatic steatosis in HFD fed mice compared to normal diet fed counterparts [64]. However, complete dephosphorylation of elf2α leads to hepatic steatosis in mice, underpinning the importance of a basal elf2α phosphorylation level in order to avoid fat accumulation when ER stress is challenged [46]. Liver biopsies from nondiabetic patients with obesity revealed that elf2α phosphorylation increased with worsening insulin resistance [65]. Similar to IRE1α, PERK lacking its luminal unfolded protein sensing domain, can sense aberrations in lipid composition through its transmembrane domain and activate the UPR [51]. Moreover, ATF4 was found to play a significant role in regulating hepatic lipid metabolism since its deficiency protected mice from hypertriglyceridemia and steatosis in response to high fructose diet, by attenuating hepatic lipogenesis [66]. ATF4-null mice are lean, protected from age-induced and diet-induced obesity and they are also protected from diet-induced diabetes, hyperlipidemia and hepatosteatosis [67]. CD154 (CD40 ligand) is a key player in inflammatory processes and CD154 knock-out mice fed a diet rich in olive oil were found to develop hepatic steatosis [68]. These mice also displayed reduced apolipoprotein B100 (apoB100) expression and secretion of very low-density lipoproteins, associated with modified UPR, reduced XBP-1s and phosphorylated elf2α, thereby providing another link between inflammation and the UPR [68]. ATF4 was also found to functionally promote lipid biosynthesis, while overexpressed ATF4 stimulated the UPR and led to liver steatosis in zebrafish model system as well [69]. Interestingly, ATF4 is suppressed by microRNA-214 (miR-214) and this suppression is reversed by the long noncoding RNA maternally expressed gene 3 (MEG3) [70]. Studies in primary hepatocytes, as well as in HFD fed mice and ob/ob mice, revealed this interaction and highlighted the role of MEG3 in hepatic insulin resistance promotion [70], a role that may be also implicated in NAFLD. As mentioned above, excessive NF-κB activation can cause NASH development. Indeed, data from cultured mouse fibroblasts show that the phosphorylation of elf2α by PERK inhibits the synthesis of IκB, an inhibitor of NF-κB, leading consequently to NF-κB activation [71]. PERK activation phosphorylates and, in turn, activates the transcription factor NF-E2-related factor-2 (Nrf2), which controls the amount of reactive oxygen species (ROS) through regulating glutathione levels during UPR [72]. Thus, PERK-mediated Nrf2 activation during ER stress alleviates the oxidative stress, thereby increasing cell survival [72]. The significant protective role of Nrf2 against steatohepatitis has also been proven in animal models. Specifically, Nrf2 null mice fed a methionine- and choline-deficient diet for 13 weeks displayed deteriorating NASH compared to wild-type mice, in terms of increased oxidative stress, iron accumulation, fatty change, inflammation and fibrosis [73]. On the other hand, although activation of Nrf2 had no effect in fat accumulation, oxidative stress and iron deposition, it significantly decreased inflammation and fibrosis [73]. Moreover, Nrf2 activation via PERK can lead to downregulation of TNXIP transcription, which links oxidative stress to inflammasome activation, and, thereby, protects hepatic cells from pyroptosis [74]. In addition, the farnesoid X receptor (FXR) inhibits the ER stress-induced NLRP3 inflammasome activation that leads to hepatocyte death, inflammation and progression of liver fibrosis by inhibiting TXNIP activity [75]. FXR exerts these protective effects against NASH progression through the PERK pathway, and specifically through the downstream transcriptional regulator CHOP [75].
On the contrary, PERK activation as well as IRE1α activation induced CHOP overexpression that leads to hepatocyte death through pyroptosis and apoptosis [76]. The PERK-CHOP branch of ER stress, in particular, has been found to upregulate dual-specificity phosphatase 5 (DUSP5) expression in liver fibrosis—both in human and animal models—which in turn contributes to hepatocyte death [77]. Interestingly, liver biopsies from patients with metabolic syndrome with or without NAFLD or NASH revealed that phosphorylated elf2α was increased in NAFLD and NASH patients with metabolic syndrome compared to those with normal liver histology, although levels of the downstream effectors ATF4, CHOP and GADD34 remained unchanged [59].
Regarding the role of PERK-eIF2a-ATF4 axis in HCC, the PERK pathway is activated during tumor progression [61]. Experiments with transformed mouse embryonic fibroblasts and their tumorigenic effects in mice revealed that PERK plays a role in tumor adaptation to hypoxic conditions by regulating the translation of proangiogenic genes implicated in cell-cell matrix remodeling, adhesion and extracellular matrix proteolysis [78].
The ATF6α Axis
There are data indicative of a protective role of ATF6α pathway in hepatic steatosis [8•]. ATF6α knockout mice when challenged with tunicamycin, an ER stress inducing agent, display CHOP upregulation, liver dysfunction and steatosis due to lipid accumulation [46, 79]. ATF6 has been found to bind and suppress the sterol regulatory element-binding protein 2 (SREBP2) and diminishes its lipogenic effect [80]. Additionally, ATF6 physically interacts with PPARα in hepatocytes, upregulates its transcriptional activity, promotes fatty acid oxidation and protects against hepatic steatosis [81]. ATF6α reduces hepatic glucose output [82] and improves insulin signaling, reducing hyperglycemia and hyperinsulinemia when overexpressed in livers of obese mice [83], pointing towards a positive effect of ATF6α in regulating insulin signaling. However, more recent data have implicated the activation of ATF6 signaling pathway in NAFLD progression [84]. Indeed, the downregulation of ATF6 pathway attenuated NAFLD progression by reducing the ER stress-induced inflammation and apoptosis in liver cells of HFD-fed NAFLD mice [84]. Regarding NASH, empagliflozin found to attenuate steatosis, hepatocellular ballooning and lobular inflammation by—among others—reducing ATF6 expression, in HFD-fed Apo(-/-) mice [85••].
Moreover, the ATF6α pathway seems to be the last to operate in HCC compared to IRE1α and PERK pathways, and it is modestly activated only after tumor initiation [61]. ATF6α has been also identified as a regulator in HCC. Indeed, ATF6 mRNA levels have been found increased and the activated ATF6 product has displayed high nuclear localization in HCC biopsies [62]. Furthermore, ATF6α upregulated 18 genes specifically expressed in an ATF6α-transfected HCC cell line and HCC tissues in comparison to a vector transfected cell line and non-cancerous liver tissues [86]. Finally, ATF6α is necessary for the adaptation of dormant cancer cells to chemotherapy, nutrient starvation, and changes in the in vivo microenvironment, promoting their survival [87]. ATF6 has been recognized as a potential activator of CHOP in the tumor environment with both molecules being elevated in HCC induced by the carcinogen N-diethylnitrosamine (DEN) [88]. CHOP overexpression in HCC tumors did not lead to apoptosis, suggesting a change in CHOP function from pro-apoptotic to tumorigenic [88]. CHOP’s role as a promoting factor for HCC has been identified from other studies as well. More specifically, CHOP null mice display reduced apoptosis, cellular proliferation and fibrosis, key factors for HCC development [89]. In human liver biopsies, CHOP expression increased in parallel to NAFLD progression, from steatosis to NASH to HCC [89]. The ATF6 and IRE1/XBP-1 pathways lead to the transformation-associated expression of the GRP78 gene in HCC [62]. Interestingly, autoantibodies against GRP78 were significantly higher in the serum of patients with HCC compared to control groups and could possibly serve as potential diagnostic markers [90].
Autophagy
Autophagy is an evolutionary conserved process in which cellular components are enclosed in membrane vesicles and transported to lysosomes for degradation [91]. However, the purpose of autophagy is not only the degradation of unnecessary material, but mainly the recycling of simple components, like amino acids or monosaccharides, which can then be reused in the formation of their more complex macromolecules, and thereby maintain cellular homeostasis and survival in periods of starvation [91]. Macro-autophagy, the major type of autophagy, involves the enclosure of cytosolic material, including proteins and degraded organelles, into an isolation membrane named phagophore, which forms the autophagosome [92]. The latter then fuses with lysosomes (with or without previously fusing with endosomes) to form the autolysosome and degrade its contents [92]. The mammalian target of rapamycin (mTOR) functions as a nutrient sensor that can activate or not the autophagy mechanism through regulation of the ULK1 complex consisting of unc-51-like kinase 1/2 (ULK1/2), Atg13 and focal adhesion kinase family interacting protein of 200 kDa (FIP200) [92, 93•]. The ULK1 complex is suppressed under nutrient-rich conditions and is activated translocating to the ER under starvation and autophagy-inducing conditions [92, 93•]. Apart from the mTOR complex which is responsible mainly for short-term regulation of autophagy [94], long-term regulation can be accomplished by the transcription factors class O of forkhead box transcription factors (FoxO) and transcription factor EB (TFEB), which under starvation and in the absence of insulin signaling translocate to the nucleus and transactivate their target autophagic genes [95]. Beclin 1 is also a central player in autophagy functioning more downstream, and is starvation-sensitive (interacting with Bcl-2 under nutrient-rich conditions and dissociating from Bcl-2 under nutrient limitations) [92, 93•]. Light chain 3 (LC3), an autophagy-related protein 8 (Atg8) ortholog, is another major player in autophagy, with its membrane-bound LC3II form being required for autophagosome formation, in contrast to the cytosolic LC3I form [93•, 96]. Finally, important players in autophagosome formation are the autophagy-related (Atg) proteins. Although 31 Atg genes have been identified, only 15 have been characterized as core Atg genes necessary for the biogenesis of autophagy-related membranes, namely Atg1-10, 12-14, 16 and 18 [97]. A characteristic autophagy substrate whose levels are commonly used as an indicator of autophagy activation and autophagic flux is p62/sequestosome 1 (SQSTM1), an autophagy adaptor protein which controls the formation of protein aggregates and inclusion bodies, links ubiquitinated proteins to LC3 and transfers them to autophagosomes for autophagic turnover [98]. Autophagy is necessary for the degradation of p62/SQSTM1, which is an activator of NF-κB and of the oxidative-stress responsive transcription factor Nrf2, which as previously mentioned is implicated in UPR as well [93•, 99, 100].
Autophagy and NAFLD
In the context of NAFLD, autophagy is related to lipid metabolism, insulin resistance, hepatocellular injury and inflammation [93•]. Overall, autophagy has been generally found reduced in NAFLD [101••]. Lipid droplets, where excess free fatty acids and glucose are converted to triglycerides and stored, need a conjugation system of LC3 and Atg7 for their formation [102]. Atg7 deficiency, which leads to suppression of Atg5, results in ER stress induction and severe insulin resistance [103]. Furthermore, pharmacological induction of autophagy by carbamazepine and rapamycin mitigated hepatic steatosis and improved insulin sensitivity in a HFD-induced mouse model of NAFLD [104]. Aggregation of the autophagic substrate p62 (SQSTM1) in hepatocytes was found in 68% or 88% of patients with NAFLD according to two clinical studies, while absence of p62 expression was identified in the control groups consisting of patients with normal liver function or with liver metastatic tumors without markers of infection for hepatitis or HIV [105, 106]. Moreover, liver-specific overexpression of p62 induced steatosis in regular chow fed mice [107]. Hepatic autophagy was suppressed in HFD-fed mice which displayed insulin resistance and hyperinsulinemia, while it has been shown that insulin inhibits the expression of key autophagy genes in a FoxO1-dependent manner [108]. Furthermore, mTOR mRNA and protein expression levels were significantly increased in rats fed a HFD which exhibited mild and severe steatosis compared to rats fed a standard diet, indicating that mTOR contributes to NAFLD development and progression [109]. Wang et al. exploring the underlying mechanisms found that mTOR regulates insulin resistance and chronic liver inflammation [109], while in vitro and animal studies showed that mTOR activation accompanied with inflammation aggravated NAFLD through disrupting low density lipoprotein receptor (LDLR) expression [110].
Autophagy impairment has also been linked to NASH development. The expression levels of Beclin-1 were found significantly higher in patients with NASH compared to patients with simple steatosis [111]. Protein levels of p62 were also elevated in patients with NASH compared to patients with steatosis and individuals with normal liver, along with the ratio of LC3II/L3-I proteins which was significantly increased in patients with NASH and steatosis compared to normal liver subjects [111]. These data indicate decreased autophagy in patients with hepatic steatosis and NASH [111]. However, a study performing Western blot analysis in liver tissues identified no difference in p62 protein levels between NAFLD, NASH patients and normal controls [112]. In line with the clinical data, increased phosphorylated (activated) mTOR and its downstream target S6K1 proteins were observed in mice with steatosis and NASH compared to control mice fed a regular chow diet, indicating autophagy suppression [111]. Indeed, p62 protein and LC3II/LC3I ratio were increased in steatosis and NASH-model mice compared to control, and correlated with prolonged and terminal ER stress [111]. Liver-specific overexpression of p62 in mice exacerbated NASH-induced fibrosis in the methionine-choline deficient diet model of NASH [107]. LC3B has also been found elevated in another NASH mouse model [113]. However, as mentioned elsewhere, evaluation of autophagic flux via p62 levels and LC3II/LC3I ratio, although initially interpreted as a reliable index of macro-autophagy [114], should always be conducted carefully, since the LC3I levels can display naturally occurring fluctuations and p62 levels can be elevated due to other factors, independently of autophagy [94].
Regarding HCC, the role of autophagy remains controversial and seems to be dual [115]. As such, autophagy can limit chromosomal instability and suppress tumor initiation by eliminating senescent and “defected” cells, whereas can also promote cancer cell survival by providing energy and milestones for them through recycling damaged organelles, DNA and proteins [94]. Emerging evidence points towards a tumor suppressive role of autophagy. Mice with heterozygous disruption of Beclin-1 gene display reduced autophagy and increased cellular proliferation, and develop HCC among other types of tumors [116]. In terms of mRNA expression [117] and protein expression assessed by immunohistochemistry [118, 119] and Western blot analysis [119], Beclin-1 was also found lower in human HCC tissues in comparison to non-cancerous tissues. Interestingly, lower Beclin-1 protein expression was associated with poorer survival in patients with HCC [118], and could serve as a marker for HCC prognosis [119]. However, a discrepancy exists here, since an older study had identified increased Beclin-1 mRNA and protein expression in HCC [120]. LC3 protein can also serve as a prognostic marker [121] for predicting survival and recurrence after HCC surgical removal; low levels or absence of LC3 from adjacent non-tumor tissues were found to be predictive of immediate mortality [121, 122]. Furthermore, low LC3 expression in tumor and adjacent non-tumor tissues was associated with repeated recurrence after surgical resection of HCC [121, 123]. By contrast, divergent findings come from other studies indicating that high expression of LC3B was correlated with vascular invasion, metastasis and poor prognosis and survival in HCC patients [124]. In line with the aforementioned, LC3 was highly expressed in HCC compared to non-cancerous tissues, while, in correlation to hypoxia-induced factor 1α (HIF1α), it was significantly associated with tumor size, serving as a predictor of HCC recurrence after surgery, albeit only in large tumors [125]. Several tumor suppressors, such as protein tyrosine phosphatase receptor type O (PTPRO) [126], and TGFβ-activated kinase 1 (TAK1) [127], induce autophagy by increasing LC3 expression [126] and decreasing p62 expression [126] and mTOR complex 1 (mTORC1) formation [127] and thus suppressing proliferation of human HCC cell lines [126] or hepatic carcinogenesis in mice [127]. According to the role of mTORC1 as a consistent autophagy inhibitor [128], activation of mTOR signaling has been identified in patients with HCC, while inhibition of mTOR decreased tumor growth and extended survival in experimental HCC xenograft models [129]. Interestingly, mTOR activation has been found to occur selectively in human and mice HCCs with a background of metabolic syndrome and NASH, unlike tumors derived from other etiologies, such as those related to hepatitis viral infection [130]. Additionally, activation of mTORC1 specifically in mice livers leads to spontaneous HCC development [131]. Deletion of Atg5 and Atg7 in the liver leads to development of benign liver adenomas, originating from autophagy-deficient hepatocytes as shown by p62 autophagy substrate accumulation [132]. Expression levels of Atg5, Beclin 1, and Atg7 mRNA were downregulated in HCC cell lines compared to normal cell lines, while Beclin 1 gene expression was also decreased in HCC tissue samples compared to non-tumor samples [133]. Accumulation of the autophagy substrate p62 is also linked to hepatic carcinogenesis, since p62 was originally discovered in HCC cell inclusions, while it was undetectable in non-neoplastic liver tissue [134,135,136]. Further studies revealed that high p62 expression predisposes to rapid recurrence after surgical treatment of HCC [137]. Moreover, p62 is needed for activation of NRF2 and mTORC1 and protects HCC-initiating cells from cell death caused by oxidative stress [137]. Overexpression of p62 in human hepatoma cell lines caused anti-apoptotic effects [135]. In addition, young transgenic mice that overexpress p62 display a steatotic phenotype in the absence of inflammation or liver damage [138], suggesting that p62 promotes the progression of NAFLD towards HCC [139].
Notably, there is also evidence that autophagy promotes cancer survival. In a study by Lage et al., immunohistochemical analysis of p62 revealed reduced content in HCC specimens compared to non-cancerous liver tissue, with p62 levels being decreased as HCC cells were differentiated further [140]. Contradicting data exist also for mTOR, since pharmacological mTOR inhibition promoted HCC development, at least in mice with hepatocyte-specific ablation of the specific mTORC1 subunit Raptor [141]. It should be highlighted that, autophagy seems to inhibit tumor suppressors further promoting HCC development once hepatic carcinogenesis has initiated. Indeed, it has been demonstrated that autophagy inhibition by Atg5 deletion in mice led to the appearance of only benign tumors instead of hepatic carcinogenesis, even after treatment with HCC-inducing agents [142]. Of interest, inhibition of autophagy by pharmacological inhibitors or siRNA against Atg5 and Atg7 was found to sensitize HCC cells to the multikinase inhibitor linifanib [143]. Furthermore, reduced Atg7 expression by the micro-RNA miR-375 was shown to reduce viability of cancer cells under hypoxic conditions [144].
Recent data suggest that the balance of activation of autophagy-related molecules is crucial to preserve the normal physiology of the liver, while an imbalance in either direction can result in harmful effects. For instance, in autophagy-deficient mice both hyper- and hypo-activation of mTOR molecules was found to be linked to the development of hepatic tumors, including HCC [145••]. This underlines the potential detrimental effects that can occur by the chronic use of mTOR inhibitors due to disruption of the mTOR balance [145••].
ER Stress and Autophagy Interaction
Summarizing the current knowledge, it is evident that ER stress and autophagy play an important role in NAFLD/NASH initiation and progression.
Hyperglycemia, insulin resistance, and lipid accumulation (from dietary lipid influx and de novo lipogenesis) can induce proteostasis and trigger ER stress in hepatic cells [11] so that the UPR adaptive signaling pathway is activated to restore it, by promoting autophagy. Autophagy stimulates also the degradation of intracellular lipid droplets (lipophagy).
Of interest, there is an interplay between ER stress and autophagy, as ER stress is a well-established positive regulator of autophagy via mainly ATF4 and Xbp1 [12], while autophagy can reduce ER stress, so that it can act protectively by inhibiting apoptosis through caspase inactivation [146].
Nevertheless, under chronic stress, the UPR turns from adaptive to terminal, reduces autophagic process, and activates pro-apoptotic pathways, leading thus to hepatocyte death [12].
AMPK is a known positive regulator of autophagy. Zhou et al. demonstrated that the inhibition of a key enzyme in lipid metabolism (Scd-1) increased AMPK activity and autophagy, attenuating thus hepatic steatosis, in HFD-fed mice [147]. Moreover, in line with the aforementioned, we showed that a sodium-glucose cotransporter-2 (SGLT-2) inhibitor (SGLT-2i) can increase autophagy through AMPK phosphorylation, and reduce the HFD-induced ER stress and hepatic cell apoptosis in ApoE(-/-) mice, alleviating the progression of NAFLD [85••].
However, it should be noted that in HCC, the ER stress and autophagy are differentially regulated and interact, probably under the influence of other critical oncogenic signaling pathways.
Treatment
Both genetic and lifestyle factors appear to contribute to the pathogenesis of NAFLD, and, at least at the early stages of the disease, lifestyle changes—namely improved diet, weight management, and increased physical activity—can be an effective strategy to manage NAFLD [148]. Although further research is clearly required, this beneficial effect appears to involve autophagy-related mechanisms. Indeed, a study on rats showed that exercise can reduce NASH effects on mitochondrial permeability transition pore and promote autophagy and mitochondrial fusion towards a protective direction [149]. Furthermore, exercise can be beneficial in NAFLD by promoting autophagy in the liver which could protect from hepatic fat accumulation, as shown in HFD-fed mice [150, 151]. Recent data also confirmed that physical activity, even without dietary changes, can diminish the progression of NAFLD to NASH and tumorigenesis by activating liver autophagy [152•]. However, apart from the changes in lifestyle, in many cases pharmacological intervention is further needed.
Current guidelines on the management of NAFLD do not recommend any drug treatment; however, given the strong association between T2DM, obesity and NAFLD, various anti-diabetic and anti-obesity drugs have been assessed regarding their usefulness in the treatment of NAFLD (Table 1).
Among the main categories of the anti-diabetic drugs that have been tested in clinical trials are insulin sensitizers (metformin and glitazones), glucagon-like peptide-1 (GLP-1)analogues, dipeptidyl-peptidase-4 (DPP-4) inhibitors, and SGLT-2i.
Jalali et al. conducted a meta-analysis of clinical trials evaluating the role of metformin in NAFLD. They included six randomized controlled studies with 307 individuals and their results emphasized the importance of metformin administration for improving liver function in non-diabetic NAFLD patients [157].
A recent meta-analysis assessed the efficacy of pioglitazone, DPP-4 inhibitors, GLP-1 analogues and SGLT-2i agents. Of note, according to their findings, pioglitazone demonstrates significant improvements in liver enzymes and in liver steatosis, fibrosis and parenchymal inflammation in NAFLD/NASH patients with diabetes as well as without diabetes [158]. Regarding the effects of the other three drug classes, there are findings suggesting possible improvement in liver enzymes in NAFLD subjects without diabetes; however, current data in this area are still very limited, especially for DPP-4i and SGLT2-i in NAFLD patients without diabetes and in NASH patients.
Anti-obesity treatment with orlistat has also been evaluated for its effectiveness to improve NAFLD. Two small prospective studies have yielded contradictory results, showing either improvement or no effect of orlistat in decreasing liver enzymes and liver fat as estimated by ultrasound [161, 165, 168]. Of note, a recent open-label, 24-week, randomized clinical trial identified orlistat treatment as an independent predictor of steatosis improvement [166].
Of great interest, several compounds have been identified to modulate ER stress or autophagy levels as a therapeutic strategy to attenuate the progression of NAFLD. To this aim, several natural compounds, hormones, and drugs have been evaluated in NAFLD animal models and human studies regarding attenuating NAFLD progression through modulating ER stress and autophagy. Tables 2 and 3 summarize selected key modulators of the ER stress pathway and autophagy, respectively, and their corresponding targets/mechanism(s) of action for the treatment of NAFLD.
Table 2 shows that focusing on the implication of ER stress as a mediating mechanism, most of the chemical compounds that exert favourable effects on NAFLD/NASH have been tested in animal models. Only selonsertib, an ASK1 inhibitor, was assessed in patients with nonalcoholic steatohepatitis and demonstrated reduction of liver fibrosis [174].
Moreover, the effect of natural compounds on ER stress pathway has been evaluated mainly in hepatocarcinoma cell lines.
It is of great interest that although almost all the categories of anti-diabetic drugs have been assessed for their usefulness in NAFLD in clinical trials, the ER-stress as target mechanism have been studied only for SGLT2-i, and only in animal models [85, 181].
In contrast, autophagy has been found to be involved in the favorable effects of four anti-diabetic drug classes (GLP-1, glitazones, metformin and SGLT2-i) on NAFLD/NASH progression albeit again only in animal studies [85, 182, 220,221,222, 225, 226].
As in the case of ER stress, Table 3 shows that an important number of natural and chemical compounds have been tested for their efficacy to modulate the autophagy process only in hepatocarcnoma cell lines. Interestingly, not even one clinical study has investigated the implication of autophagy as target mechanism that mediates the favorable effect of any compound or drug on NAFLD.
Conclusion and Perspectives
The incidence of NAFLD spectrum is rising and is estimated to affect more than 130 million patients worldwide by 2030 [231•].
The involvement of ER stress and autophagy in the pathogenesis and progression of NAFLD is gaining increasing interest.
Herein, we reviewed in vitro, animal and human studies on the deterioration of the aforementioned mechanisms in hepatic cells leading to NAFLD, NASH and HCC.
It is evident from the existing evidence that the complex etiology and pathophysiology of NAFLD with the implication of genetic and environmental factors make it difficult to extrapolate from in vitro and animal research on humans, and this is reflected in the divergent results that emerged from various studies.
Undoubtedly, the plethora of genetically engineered mouse models, diets or synthetic compounds that are now available can further facilitate the research of NAFLD.
However, it still remains a challenge to create ideal animal models reproducing the whole spectrum of the NAFLD, in order to fully clarify the disease pathophysiology and ascertain effective treatments. Realizing that the “humanizing” of NAFLD animal models can become possible to a limited extent, humanizing of computational models derived from animal experiments could further aid the attempts of translating relevant research findings from animal to humans [232].
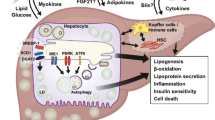
Most studies—especially focusing on diet-induced NAFLD progression—indicate that there is a counterbalance between ER stress and autophagy, where ER stress induces autophagy which sequentially attenuates ER stress and apoptosis of hepatic cells (Fig. 1). It appears that a shift of this balance to the side of ER stress, due to either sustained terminal ER stress or diminished autophagy, can result to hepatic cell apoptosis and liver injury.

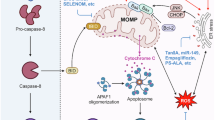
Cross-talk between ER stress and autophagy. The ER stress pathway is induced by hypeglycemia, the ER stress response is initiated upon accumulation of misfolded and unfolded proteins in the ER lumen. Dissociation of Bip from the ER stress sensors (PERK, IRE1, and ATF6) leads to subsequent activation of 3-arms of UPR signaling. Phosphorylation of PERK results in activation of the downstream translation factor eIF2α, leading to the activation of ATF4 transcription factor and over-expression of CHOP. Both ATF-4 and CHOP can regulate the expression of a number of Bcl-2 family proteins and autophagy regulatory proteins such Atg5 and p62. The ATF6α, upon UPR activation, is transported to the Golgi apparatus where is processed by Site-1 protease (S1P) and S2P and becomes activated. Activation of IRE1 induces XBP1 splicing to generate active XBP1s. XBP1s enters the nucleus to transcribe proteins involved in ER associated degradation (ERAD). Activation of IRE1 also activates the JNK signaling cascade which in turn increases both phosphorylation of Bcl-2 as well as the formation of LC3II. Phosphorylation of Bcl-2 disrupts the Bcl-2/Beclin1 interaction and therefore induces activation of autophagy. All three transcription factors (XBP1s, ATF4, and ATF6) are translocated to the nucleus where they induce the expression of target genes including genes related with autophagosome formation. The autophagy pathway is mainly controlled by the AMPK/mTOR-signaling axis. AMPK inhibits mTORC1 leading to initiation of autophagy. AMPK activation induces the phosphorylation of FoxO transcription factor and its translocation into the nucleus where it can bind to the promoter of autophagy genes and elevate their expression. AMPK activation is also the essential step towards activation of ULK1 complex which in turn leads to the activation of the class III PI3K complex (Atg14/Beclin 1/Vps34/p150) by phosphorylating Beclin 1. These steps are required for the formation of autophagosome which is then transformed into a double-membranous vesicle by recruitment of the Atg (12, 5, 16) complex and the lipidation of LC3I into phosphatidylethanolamine-conjugated(PE) LC3II. The LC3II participates in the formation and expansion of phagophore along with Atg8 and Atg7 and ubiquitinated proteins such as p62. The mature autophagosome undergoes fusion with a lysosome to form autolysosome. Activation of autophagy can induce cell survival by clearing damaged organelles and inhibit apoptosis though inhibition of caspase-8 activation
Taking into account that other than hepatic cells can also contribute to the pathogenesis of steatohepatitis and tumorigenesis such as Kupffer, endothelial, epithelial and immune cells, studies on the role of ER stress and autophagy should be expanded to these cells which also shape the milieu in the spectrum of NAFLD. To this direction, a recent study showed that a myeloid-specific IRE1α deletion results in an altered transcriptional profile of hepatic macrophages and diminishes the diabetes-induced NASH, as well as HCC development [233]. Moreover, recent data suggest that cell-type-specific reprogramming of the liver cell transcriptomes is linked to NASH pathogenesis [234•]. Multi-omics approaches could offer a new strategy to investigate the interactive processes of ER stress and autophagy from the perspective of NAFLD and its progression.
Better understanding of the interrelations between ER stress and autophagy, and how both these pathways can interact with inflammation and apoptotic mechanisms is expected to further aid the development of new therapeutic options which can better target and reprogram these underlying pathways. Indeed, both these processes are now intensively explored as targets of various compounds and drugs aiming to attenuate NAFLD progression.
Data Availability
Not applicable
Code Availability
Not applicable
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Gadiparthi C, Spatz M, Greenberg S, Iqbal U, Kanna S, Satapathy SK, et al. NAFLD Epidemiology, Emerging Pharmacotherapy, Liver Transplantation Implications and the Trends in the United States. J Clin Transl Hepatol. 2020;8(2):215–21. https://doi.org/10.14218/JCTH.2020.00014.
Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62(1 Suppl):S47–64. https://doi.org/10.1016/j.jhep.2014.12.012.
Fouad Y, Waked I, Bollipo S, Gomaa A, Ajlouni Y, Attia D. What's in a name? Renaming 'NAFLD' to 'MAFLD'. Liver Int. 2020;40(6):1254–61. https://doi.org/10.1111/liv.14478.
Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu Rev Pathol. 2016;11:451–96. https://doi.org/10.1146/annurev-pathol-012615-044224.
• Rutkowski DT. Liver function and dysfunction - a unique window into the physiological reach of ER stress and the unfolded protein response. FEBS J. 2019;286(2):356–78. https://doi.org/10.1111/febs.14389The liver due to its unique properties as a highly secretory tissue which regulates peripheral metabolism and can proliferate upon damage underlined the fact that the UPR extends beyond the improvement of ER protein folding to control of metabolism and inflammation in terms of regulating the cycles of cell death and immune cell recruitment.
Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473(7348):528–31. https://doi.org/10.1038/nature09968.
Ashraf NU, Sheikh TA. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic Res. 2015;49(12):1405–18. https://doi.org/10.3109/10715762.2015.1078461.
• Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2018;69(4):927–47. https://doi.org/10.1016/j.jhep.2018.06.008A comprehensive review summarizing evidence about the role of ER stress in several NAFLD manifestations, such as insulin resistance, lipid and calcium homeostasis, autophagic flux and inflammation and also presenting ER stress-modulating therapies for the treatment of the NAFLD entitites.
Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105–37. https://doi.org/10.1146/annurev-pathol-020712-163918.
• Khawar MB, Gao H, Li W. Autophagy and Lipid Metabolism. Adv Exp Med Biol. 2019;1206:359–74. https://doi.org/10.1007/978-981-15-0602-4_17A book chapter which summarizes the molecular mechanisms of the autophagic degradation of lipids and their implication in various metabolic disorders, including NAFLD.
Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism. 2016;65(8):1049–61. https://doi.org/10.1016/j.metabol.2016.02.014.
Guo B, Li Z. Endoplasmic reticulum stress in hepatic steatosis and inflammatory bowel diseases. Front Genet. 2014;5:242. https://doi.org/10.3389/fgene.2014.00242.
Polyzos SA, Kountouras J, Mantzoros CS. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism. 2019;92:82–97. https://doi.org/10.1016/j.metabol.2018.11.014.
Global BMIMC, Di Angelantonio E, Bhupathiraju Sh N, Wormser D, Gao P, Kaptoge S, et al. Body-mass index and all-cause mortality: individual-participant-data meta-analysis of 239 prospective studies in four continents. Lancet. 2016;388(10046):776–86. https://doi.org/10.1016/S0140-6736(16)30175-1.
Li L, Liu DW, Yan HY, Wang ZY, Zhao SH, Wang B. Obesity is an independent risk factor for non-alcoholic fatty liver disease: evidence from a meta-analysis of 21 cohort studies. Obes Rev. 2016;17(6):510–9. https://doi.org/10.1111/obr.12407.
Thandra KC, Barsouk A, Saginala K, Aluru JS, Rawla P, Barsouk A. Epidemiology of non-alcoholic fatty liver disease and risk of hepatocellular carcinoma progression. Clin Exp Hepatol. 2020;6(4):289–94. https://doi.org/10.5114/ceh.2020.102153.
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. https://doi.org/10.1002/hep.28431.
Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005;288(2):E462–8. https://doi.org/10.1152/ajpendo.00064.2004.
Bellentani S, Saccoccio G, Masutti F, Croce LS, Brandi G, Sasso F, et al. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann Intern Med. 2000;132(2):112–7. https://doi.org/10.7326/0003-4819-132-2-200001180-00004.
van den Berg EH, Amini M, Schreuder TC, Dullaart RP, Faber KN, Alizadeh BZ, et al. Prevalence and determinants of non-alcoholic fatty liver disease in lifelines: A large Dutch population cohort. PLoS One. 2017;12(2):e0171502. https://doi.org/10.1371/journal.pone.0171502.
Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140(1):124–31. https://doi.org/10.1053/j.gastro.2010.09.038.
Frey S, Patouraux S, Debs T, Gugenheim J, Anty R, Iannelli A. Prevalence of NASH/NAFLD in people with obesity who are currently classified as metabolically healthy. Surg Obes Relat Dis. 2020;16(12):2050–7. https://doi.org/10.1016/j.soard.2020.07.009.
Amarapurkar DN, Hashimoto E, Lesmana LA, Sollano JD, Chen PJ, Goh KL, et al. How common is non-alcoholic fatty liver disease in the Asia-Pacific region and are there local differences? J Gastroenterol Hepatol. 2007;22(6):788–93. https://doi.org/10.1111/j.1440-1746.2007.05042.x.
Samocha-Bonet D, Dixit VD, Kahn CR, Leibel RL, Lin X, Nieuwdorp M, et al. Metabolically healthy and unhealthy obese--the 2013 Stock Conference report. Obes Rev. 2014;15(9):697–708. https://doi.org/10.1111/obr.12199.
Polyzos SA, Mantzoros CS. Leptin in health and disease: facts and expectations at its twentieth anniversary. Metabolism. 2015;64(1):5–12. https://doi.org/10.1016/j.metabol.2014.10.017.
Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. The Lancet. 2010;375(9733):2267–77. https://doi.org/10.1016/S0140-6736(10)60408-4.
Magkos F, Mantzoros CS. Body fat redistribution and metabolic abnormalities in HIV-infected patients on highly active antiretroviral therapy: novel insights into pathophysiology and emerging opportunities for treatment. Metabolism. 2011;60(6):749–53. https://doi.org/10.1016/j.metabol.2010.09.011.
Makri E, Goulas A, Polyzos SA. Epidemiology, Pathogenesis, Diagnosis and Emerging Treatment of Nonalcoholic Fatty Liver Disease. Arch Med Res. 2021;52(1):25–37. https://doi.org/10.1016/j.arcmed.2020.11.010.
Tilg H, Moschen AR, Szabo G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2016;64(3):955–65. https://doi.org/10.1002/hep.28456.
Boutari C, Perakakis N, Mantzoros CS. Association of Adipokines with Development and Progression of Nonalcoholic Fatty Liver Disease. Endocrinol Metab (Seoul). 2018;33(1):33–43. https://doi.org/10.3803/EnM.2018.33.1.33.
Polyzos SA, Kountouras J, Mantzoros CS. Adipose tissue, obesity and non-alcoholic fatty liver disease. Minerva Endocrinol. 2017;42(2):92–108. https://doi.org/10.23736/S0391-1977.16.02563-3.
Polyzos SA, Kountouras J, Mantzoros CS. Adipokines in nonalcoholic fatty liver disease. Metabolism. 2016;65(8):1062–79. https://doi.org/10.1016/j.metabol.2015.11.006.
Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529(7586):326–35. https://doi.org/10.1038/nature17041.
Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–6. https://doi.org/10.1126/science.1209038.
Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal. 2009;11(9):2307–16. https://doi.org/10.1089/ARS.2009.2485.
Hetz C, Martinon F, Rodriguez D, Glimcher LH. The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol Rev. 2011;91(4):1219–43. https://doi.org/10.1152/physrev.00001.2011.
Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186(3):323–31. https://doi.org/10.1083/jcb.200903014.
Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. 2012;338(6108):818–22. https://doi.org/10.1126/science.1226191.
Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–4. https://doi.org/10.1038/16729.
Ma Y, Hendershot LM. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem. 2003;278(37):34864–73. https://doi.org/10.1074/jbc.M301107200.
Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273(50):33741–9. https://doi.org/10.1074/jbc.273.50.33741.
Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6(6):1355–64. https://doi.org/10.1016/s1097-2765(00)00133-7.
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–91. https://doi.org/10.1016/s0092-8674(01)00611-0.
Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119(5):1201–15. https://doi.org/10.1172/JCI37007.
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–51. https://doi.org/10.1172/JCI23621.
Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell. 2008;15(6):829–40. https://doi.org/10.1016/j.devcel.2008.10.015.
Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011;30(7):1357–75. https://doi.org/10.1038/emboj.2011.52.
Bailly-Maitre B, Belgardt BF, Jordan SD, Coornaert B, von Freyend MJ, Kleinridders A, et al. Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance. J Biol Chem. 2010;285(9):6198–207. https://doi.org/10.1074/jbc.M109.056648.
So JS, Hur KY, Tarrio M, Ruda V, Frank-Kamenetsky M, Fitzgerald K, et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012;16(4):487–99. https://doi.org/10.1016/j.cmet.2012.09.004.
Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, Kimata Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol Biol Cell. 2011;22(18):3520–32. https://doi.org/10.1091/mbc.E11-04-0295.
Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci U S A. 2013;110(12):4628–33. https://doi.org/10.1073/pnas.1217611110.
Sun S, Kelekar S, Kliewer SA, Mangelsdorf DJ. The orphan nuclear receptor SHP regulates ER stress response by inhibiting XBP1s degradation. Genes Dev. 2019;33(15-16):1083–94. https://doi.org/10.1101/gad.326868.119.
• Bang IH, Kwon OK, Hao L, Park D, Chung MJ, Oh BC, et al. Deacetylation of XBP1s by sirtuin 6 confers resistance to ER stress-induced hepatic steatosis. Exp Mol Med. 2019;51(9):1–11. https://doi.org/10.1038/s12276-019-0309-0Post-translational modification of XBP1s, apart from the obvious post-transcriptional modification of splicing, is implicated in the protection against ER stress-induced hepatic steatosis. Sirtuin 6 created the deacetylated form of XBP1s which protects from ER stress-induced hepatic steatosis.
Luedde T, Schwabe RF. NF-kappaB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8(2):108–18. https://doi.org/10.1038/nrgastro.2010.213.
Willy JA, Young SK, Stevens JL, Masuoka HC, Wek RC. CHOP links endoplasmic reticulum stress to NF-kappaB activation in the pathogenesis of nonalcoholic steatohepatitis. Mol Biol Cell. 2015;26(12):2190–204. https://doi.org/10.1091/mbc.E15-01-0036.
Lebeaupin C, Vallee D, Rousseau D, Patouraux S, Bonnafous S, Adam G, et al. Bax inhibitor-1 protects from nonalcoholic steatohepatitis by limiting inositol-requiring enzyme 1 alpha signaling in mice. Hepatology. 2018;68(2):515–32. https://doi.org/10.1002/hep.29847.
Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16(2):250–64. https://doi.org/10.1016/j.cmet.2012.07.007.
Wang JM, Qiu Y, Yang Z, Kim H, Qian Q, Sun Q, et al. IRE1alpha prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci Signal. 2018;11(530). https://doi.org/10.1126/scisignal.aao4617.
Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134(2):568–76. https://doi.org/10.1053/j.gastro.2007.10.039.
Akazawa Y, Cazanave S, Mott JL, Elmi N, Bronk SF, Kohno S, et al. Palmitoleate attenuates palmitate-induced Bim and PUMA up-regulation and hepatocyte lipoapoptosis. J Hepatol. 2010;52(4):586–93. https://doi.org/10.1016/j.jhep.2010.01.003.
Vandewynckel YP, Laukens D, Bogaerts E, Paridaens A, Van den Bussche A, Verhelst X, et al. Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: a PERK for hepatocellular carcinoma therapy. Hepatol Int. 2015;9(1):93–104. https://doi.org/10.1007/s12072-014-9582-0.
Shuda M, Kondoh N, Imazeki N, Tanaka K, Okada T, Mori K, et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol. 2003;38(5):605–14. https://doi.org/10.1016/s0168-8278(03)00029-1.
Spiotto MT, Banh A, Papandreou I, Cao H, Galvez MG, Gurtner GC, et al. Imaging the unfolded protein response in primary tumors reveals microenvironments with metabolic variations that predict tumor growth. Cancer Res. 2010;70(1):78–88. https://doi.org/10.1158/0008-5472.CAN-09-2747.
Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7(6):520–32. https://doi.org/10.1016/j.cmet.2008.04.011.
Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A. 2011;108(39):16381–5. https://doi.org/10.1073/pnas.1113359108.
Xiao G, Zhang T, Yu S, Lee S, Calabuig-Navarro V, Yamauchi J, et al. ATF4 protein deficiency protects against high fructose-induced hypertriglyceridemia in mice. J Biol Chem. 2013;288(35):25350–61. https://doi.org/10.1074/jbc.M113.470526.
Seo J, Fortuno ES 3rd, Suh JM, Stenesen D, Tang W, Parks EJ, et al. Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes. 2009;58(11):2565–73. https://doi.org/10.2337/db09-0335.
Villeneuve J, Lepreux S, Mulot A, Berard AM, Higa-Nishiyama A, Costet P, et al. A protective role for CD154 in hepatic steatosis in mice. Hepatology. 2010;52(6):1968–79. https://doi.org/10.1002/hep.23935.
Yeh KY, Lai CY, Lin CY, Hsu CC, Lo CP, Her GM. ATF4 overexpression induces early onset of hyperlipidaemia and hepatic steatosis and enhances adipogenesis in zebrafish. Sci Rep. 2017;7(1):16362. https://doi.org/10.1038/s41598-017-16587-9.
Zhu X, Li H, Wu Y, Zhou J, Yang G, Wang W. lncRNA MEG3 promotes hepatic insulin resistance by serving as a competing endogenous RNA of miR-214 to regulate ATF4 expression. Int J Mol Med. 2019;43(1):345–57. https://doi.org/10.3892/ijmm.2018.3975.
Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, et al. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol. 2003;23(16):5651–63. https://doi.org/10.1128/mcb.23.16.5651-5663.2003.
Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279(19):20108–17. https://doi.org/10.1074/jbc.M314219200.
Okada K, Warabi E, Sugimoto H, Horie M, Tokushige K, Ueda T, et al. Nrf2 inhibits hepatic iron accumulation and counteracts oxidative stress-induced liver injury in nutritional steatohepatitis. J Gastroenterol. 2012;47(8):924–35. https://doi.org/10.1007/s00535-012-0552-9.
Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26. https://doi.org/10.1146/annurev-pharmtox-011112-140320.
Han CY, Rho HS, Kim A, Kim TH, Jang K, Jun DW, et al. FXR Inhibits Endoplasmic Reticulum Stress-Induced NLRP3 Inflammasome in Hepatocytes and Ameliorates Liver Injury. Cell Rep. 2018;24(11):2985–99. https://doi.org/10.1016/j.celrep.2018.07.068.
Lebeaupin C, Proics E, de Bieville CH, Rousseau D, Bonnafous S, Patouraux S, et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015;6:e1879. https://doi.org/10.1038/cddis.2015.248.
Jo HJ, Yang JW, Park JH, Choi ES, Lim CS, Lee S, et al. Endoplasmic Reticulum Stress Increases DUSP5 Expression via PERK-CHOP Pathway, Leading to Hepatocyte Death. Int J Mol Sci. 2019;20(18). https://doi.org/10.3390/ijms20184369.
Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol. 2006;26(24):9517–32. https://doi.org/10.1128/MCB.01145-06.
Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, et al. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell. 2010;21(17):2975–86. https://doi.org/10.1091/mbc.E09-02-0133.
Zeng L, Lu M, Mori K, Luo S, Lee AS, Zhu Y, et al. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004;23(4):950–8. https://doi.org/10.1038/sj.emboj.7600106.
Chen X, Zhang F, Gong Q, Cui A, Zhuo S, Hu Z, et al. Hepatic ATF6 Increases Fatty Acid Oxidation to Attenuate Hepatic Steatosis in Mice Through Peroxisome Proliferator-Activated Receptor alpha. Diabetes. 2016;65(7):1904–15. https://doi.org/10.2337/db15-1637.
Wang Y, Vera L, Fischer WH, Montminy M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460(7254):534–7. https://doi.org/10.1038/nature08111.
Ozcan L, Ghorpade DS, Zheng Z, de Souza JC, Chen K, Bessler M, et al. Hepatocyte DACH1 Is Increased in Obesity via Nuclear Exclusion of HDAC4 and Promotes Hepatic Insulin Resistance. Cell Rep. 2016;15(10):2214–25. https://doi.org/10.1016/j.celrep.2016.05.006.
Chen Z, Liu Y, Yang L, Liu P, Zhang Y, Wang X. MiR-149 attenuates endoplasmic reticulum stress-induced inflammation and apoptosis in nonalcoholic fatty liver disease by negatively targeting ATF6 pathway. Immunol Lett. 2020;222:40–8. https://doi.org/10.1016/j.imlet.2020.03.003.
•• Nasiri-Ansari N, Nikolopoulou C, Papoutsi K, Kyrou I, Mantzoros CS, Kyriakopoulos G, et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(-/-) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. IJMS. 2021;22(2):818 ApoE (-/-) mice treated with empagliflozin for five weeks exhibited reduced fasting glucose, total cholesterol and triglyceride serum levels, as well as decreased NAFLD progression, NAFLD activity score and reduced expression of lipogenic enzymes and inflammatory molecules, compared to the control group. Empagliflozin significantly reduced ER stress while activating autophagy via increased AMPK phosphorylation, decreased mTOR and increased LC3B expression.
Arai M, Kondoh N, Imazeki N, Hada A, Hatsuse K, Kimura F, et al. Transformation-associated gene regulation by ATF6alpha during hepatocarcinogenesis. FEBS Lett. 2006;580(1):184–90. https://doi.org/10.1016/j.febslet.2005.11.072.
Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci U S A. 2008;105(30):10519–24. https://doi.org/10.1073/pnas.0800939105.
Scaiewicz V, Nahmias A, Chung RT, Mueller T, Tirosh B, Shibolet O. CCAAT/enhancer-binding protein homologous (CHOP) protein promotes carcinogenesis in the DEN-induced hepatocellular carcinoma model. PLoS One. 2013;8(12):e81065. https://doi.org/10.1371/journal.pone.0081065.
Toriguchi K, Hatano E, Tanabe K, Takemoto K, Nakamura K, Koyama Y, et al. Attenuation of steatohepatitis, fibrosis, and carcinogenesis in mice fed a methionine-choline deficient diet by CCAAT/enhancer-binding protein homologous protein deficiency. J Gastroenterol Hepatol. 2014;29(5):1109–18. https://doi.org/10.1111/jgh.12481.
Shao Q, Ren P, Li Y, Peng B, Dai L, Lei N, et al. Autoantibodies against glucose-regulated protein 78 as serological diagnostic biomarkers in hepatocellular carcinoma. Int J Oncol. 2012;41(3):1061–7. https://doi.org/10.3892/ijo.2012.1515.
Sheng R, Qin ZH. History and Current Status of Autophagy Research. Adv Exp Med Biol. 2019;1206:3–37. https://doi.org/10.1007/978-981-15-0602-4_1.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–41. https://doi.org/10.1016/j.cell.2011.10.026.
• Wu WKK, Zhang L, Chan MTV. Autophagy, NAFLD and NAFLD-Related HCC. Adv Exp Med Biol. 2018;1061:127–38. https://doi.org/10.1007/978-981-10-8684-7_10A book chapter which presents evidence that defective autophagy is implicated in the pathogenesis of NAFLD through regulating lipid metabolism, insulin resistance, hepatocellular injury and inflammation and presents autophagy modulators that could treat NAFLD or block its progression towards HCC.
Lavallard VJ, Gual P. Autophagy and non-alcoholic fatty liver disease. Biomed Res Int. 2014;2014:120179. https://doi.org/10.1155/2014/120179.
Lavallard VJ, Meijer AJ, Codogno P, Gual P. Autophagy, signaling and obesity. Pharmacol Res. 2012;66(6):513–25. https://doi.org/10.1016/j.phrs.2012.09.003.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–8. https://doi.org/10.1093/emboj/19.21.5720.
Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10(7):458–67. https://doi.org/10.1038/nrm2708.
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–63. https://doi.org/10.1016/j.cell.2007.10.035.
Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 1999;18(11):3044–53. https://doi.org/10.1093/emboj/18.11.3044.
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–23. https://doi.org/10.1038/ncb2021.
•• Kim YS, Kim SG. Endoplasmic reticulum stress and autophagy dysregulation in alcoholic and non-alcoholic liver diseases. Clin Mol Hepatol. 2020;26(4):715–27. https://doi.org/10.3350/cmh.2020.0173A comprehensive review providing evidence that ER stress and autophagy are closely linked to each other and their interrelation contributes to the occurrence of alcoholic and non-alcoholic liver diseases, including NAFLD and its entities.
Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, et al. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun. 2009;382(2):419–23. https://doi.org/10.1016/j.bbrc.2009.03.039.
Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11(6):467–78. https://doi.org/10.1016/j.cmet.2010.04.005.
Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58(5):993–9. https://doi.org/10.1016/j.jhep.2013.01.011.
Fukuo Y, Yamashina S, Sonoue H, Arakawa A, Nakadera E, Aoyama T, et al. Abnormality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease. Hepatol Res. 2014;44(9):1026–36. https://doi.org/10.1111/hepr.12282.
Fukushima H, Yamashina S, Arakawa A, Taniguchi G, Aoyama T, Uchiyama A, et al. Formation of p62-positive inclusion body is associated with macrophage polarization in non-alcoholic fatty liver disease. Hepatol Res. 2018;48(9):757–67. https://doi.org/10.1111/hepr.13071.
Simon Y, Kessler SM, Gemperlein K, Bohle RM, Muller R, Haybaeck J, et al. Elevated free cholesterol in a p62 overexpression model of non-alcoholic steatohepatitis. World J Gastroenterol. 2014;20(47):17839–50. https://doi.org/10.3748/wjg.v20.i47.17839.
Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284(45):31484–92. https://doi.org/10.1074/jbc.M109.033936.
Wang Y, Shi M, Fu H, Xu H, Wei J, Wang T, et al. Mammalian target of the rapamycin pathway is involved in non-alcoholic fatty liver disease. Mol Med Rep. 2010;3(6):909–15. https://doi.org/10.3892/mmr.2010.365.
Liu J, Ma KL, Zhang Y, Wu Y, Hu ZB, Lv LL, et al. Activation of mTORC1 disrupted LDL receptor pathway: a potential new mechanism for the progression of non-alcoholic fatty liver disease. Int J Biochem Cell Biol. 2015;61:8–19. https://doi.org/10.1016/j.biocel.2015.01.011.
Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5:e1179. https://doi.org/10.1038/cddis.2014.162.
Lee S, Kim S, Hwang S, Cherrington NJ, Ryu DY. Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease. Oncotarget. 2017;8(38):63370–81. https://doi.org/10.18632/oncotarget.18812.
Adkins Y, Schie IW, Fedor D, Reddy A, Nguyen S, Zhou P, et al. A novel mouse model of nonalcoholic steatohepatitis with significant insulin resistance. Lab Invest. 2013;93(12):1313–22. https://doi.org/10.1038/labinvest.2013.123.
Karim MR, Kanazawa T, Daigaku Y, Fujimura S, Miotto G, Kadowaki M. Cytosolic LC3 ratio as a sensitive index of macroautophagy in isolated rat hepatocytes and H4-II-E cells. Autophagy. 2007;3(6):553–60. https://doi.org/10.4161/auto.4615.
Wang Z, Han W, Sui X, Fang Y, Pan H. Autophagy: A novel therapeutic target for hepatocarcinoma (Review). Oncol Lett. 2014;7(5):1345–51. https://doi.org/10.3892/ol.2014.1916.
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–20. https://doi.org/10.1172/JCI20039.
Kotsafti A, Farinati F, Cardin R, Cillo U, Nitti D, Bortolami M. Autophagy and apoptosis-related genes in chronic liver disease and hepatocellular carcinoma. BMC Gastroenterol. 2012;12:118. https://doi.org/10.1186/1471-230X-12-118.
Qiu DM, Wang GL, Chen L, Xu YY, He S, Cao XL, et al. The expression of beclin-1, an autophagic gene, in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer. 2014;14:327. https://doi.org/10.1186/1471-2407-14-327.
Sun H, Yu J, Wen Z, Wang M, Chen W. Decreased expression of Beclin-1 in patients with hepatocellular carcinoma. J BUON. 2019;24(2):634–41.
Kang KF, Wang XW, Chen XW, Kang ZJ, Zhang X, Wilbur RR, et al. Beclin 1 and nuclear factor-kappaBp65 are upregulated in hepatocellular carcinoma. Oncol Lett. 2013;5(6):1813–8. https://doi.org/10.3892/ol.2013.1307.
Lee YJ, Hah YJ, Kang YN, Kang KJ, Hwang JS, Chung WJ, et al. The autophagy-related marker LC3 can predict prognosis in human hepatocellular carcinoma. PLoS One. 2013;8(11):e81540. https://doi.org/10.1371/journal.pone.0081540.
Lin CW, Lin CC, Lee PH, Lo GH, Hsieh PM, Koh KW, et al. The autophagy marker LC3 strongly predicts immediate mortality after surgical resection for hepatocellular carcinoma. Oncotarget. 2017;8(54):91902–13. https://doi.org/10.18632/oncotarget.19763.
Lin CW, Chen YS, Lin CC, Lee PH, Lo GH, Hsu CC, et al. Autophagy-related gene LC3 expression in tumor and liver microenvironments significantly predicts recurrence of hepatocellular carcinoma after surgical resection. Clin Transl Gastroenterol. 2018;9(6):166. https://doi.org/10.1038/s41424-018-0033-4.
Wu DH, Jia CC, Chen J, Lin ZX, Ruan DY, Li X, et al. Autophagic LC3B overexpression correlates with malignant progression and predicts a poor prognosis in hepatocellular carcinoma. Tumour Biol. 2014;35(12):12225–33. https://doi.org/10.1007/s13277-014-2531-7.
Toshima T, Shirabe K, Matsumoto Y, Yoshiya S, Ikegami T, Yoshizumi T, et al. Autophagy enhances hepatocellular carcinoma progression by activation of mitochondrial beta-oxidation. J Gastroenterol. 2014;49(5):907–16. https://doi.org/10.1007/s00535-013-0835-9.
Zhang W, Hou J, Wang X, Jiang R, Yin Y, Ji J, et al. PTPRO-mediated autophagy prevents hepatosteatosis and tumorigenesis. Oncotarget. 2015;6(11):9420–33. https://doi.org/10.18632/oncotarget.3353.
Inokuchi-Shimizu S, Park EJ, Roh YS, Yang L, Zhang B, Song J, et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J Clin Invest. 2014;124(8):3566–78. https://doi.org/10.1172/JCI74068.
Dossou AS, Basu A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers (Basel). 2019;11(10). https://doi.org/10.3390/cancers11101422.
Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135(6):1972-83, 83 e1-11. https://doi.org/10.1053/j.gastro.2008.08.008.
Okuno T, Kakehashi A, Ishii N, Fujioka M, Gi M, Wanibuchi H. mTOR Activation in Liver Tumors Is Associated with Metabolic Syndrome and Non-Alcoholic Steatohepatitis in Both Mouse Models and Humans. Cancers (Basel). 2018;10(12). https://doi.org/10.3390/cancers10120465.
Li T, Weng J, Zhang Y, Liang K, Fu G, Li Y, et al. mTOR direct crosstalk with STAT5 promotes de novo lipid synthesis and induces hepatocellular carcinoma. Cell Death Dis. 2019;10(8):619. https://doi.org/10.1038/s41419-019-1828-2.
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. https://doi.org/10.1101/gad.2016211.
Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008;68(22):9167–75. https://doi.org/10.1158/0008-5472.CAN-08-1573.
Zhang J, Chan EKL. Autoantibodies to IGF-II mRNA binding protein p62 and overexpression of p62 in human hepatocellular carcinoma. Autoimmunity Reviews. 2002;1(3):146–53. https://doi.org/10.1016/S1568-9972(02)00030-7.
Kessler SM, Pokorny J, Zimmer V, Laggai S, Lammert F, Bohle RM, et al. IGF2 mRNA binding protein p62/IMP2-2 in hepatocellular carcinoma: antiapoptotic action is independent of IGF2/PI3K signaling. Am J Physiol Gastrointest Liver Physiol. 2013;304(4):G328–36. https://doi.org/10.1152/ajpgi.00005.2012.
Bao L, Chandra PK, Moroz K, Zhang X, Thung SN, Wu T, et al. Impaired autophagy response in human hepatocellular carcinoma. Exp Mol Pathol. 2014;96(2):149–54. https://doi.org/10.1016/j.yexmp.2013.12.002.
Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell. 2016;29(6):935–48. https://doi.org/10.1016/j.ccell.2016.04.006.
Tybl E, Shi FD, Kessler SM, Tierling S, Walter J, Bohle RM, et al. Overexpression of the IGF2-mRNA binding protein p62 in transgenic mice induces a steatotic phenotype. J Hepatol. 2011;54(5):994–1001. https://doi.org/10.1016/j.jhep.2010.08.034.
Simon Y, Kessler SM, Bohle RM, Haybaeck J, Kiemer AK. The insulin-like growth factor 2 (IGF2) mRNA-binding protein p62/IGF2BP2-2 as a promoter of NAFLD and HCC? Gut. 2014;63(5):861–3. https://doi.org/10.1136/gutjnl-2013-305736.
Lage H, Kellner U, Tannapfel A, Dietel M. Expression of a glypican-related 62-kDa antigen is decreased in hepatocellular carcinoma in correspondence to the grade of tumor differentiation. Virchows Arch. 2001;438(6):567–73. https://doi.org/10.1007/s004280000377.
Umemura A, Park EJ, Taniguchi K, Lee JH, Shalapour S, Valasek MA, et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab. 2014;20(1):133–44. https://doi.org/10.1016/j.cmet.2014.05.001.
Tian Y, Kuo CF, Sir D, Wang L, Govindarajan S, Petrovic LM, et al. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ. 2015;22(6):1025–34. https://doi.org/10.1038/cdd.2014.201.
Pan H, Wang Z, Jiang L, Sui X, You L, Shou J, et al. Autophagy inhibition sensitizes hepatocellular carcinoma to the multikinase inhibitor linifanib. Sci Rep. 2014;4:6683. https://doi.org/10.1038/srep06683.
Chang Y, Yan W, He X, Zhang L, Li C, Huang H, et al. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology. 2012;143(1):177–87 e8. https://doi.org/10.1053/j.gastro.2012.04.009.
•• Yang H, Ni HM, Ding WX. The double-edged sword of MTOR in autophagy deficiency induced-liver injury and tumorigenesis. Autophagy. 2019;15(9):1671–3. https://doi.org/10.1080/15548627.2019.1634445Both hyper- and hypoactivation of mTOR have detrimental effects on liver function and can lead to HCC occurrence. The chonic use of mTOR inhibitors which disrupt its balance can augment these effects and generally, it is underlined that the balance of activation of autophagy-related molecules is necessary to preserve liver’s normal physiology.
Song S, Tan J, Miao Y, Li M, Zhang Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J Cell Physiol. 2017;232(11):2977–84. https://doi.org/10.1002/jcp.25785.
Zhou Y, Zhong L, Yu S, Shen W, Cai C, Yu H. Inhibition of stearoyl-coenzyme A desaturase 1 ameliorates hepatic steatosis by inducing AMPK-mediated lipophagy. Aging (Albany NY). 2020;12(8):7350–62. https://doi.org/10.18632/aging.103082.
Zelber-Sagi S, Godos J, Salomone F. Lifestyle changes for the treatment of nonalcoholic fatty liver disease: a review of observational studies and intervention trials. Therap Adv Gastroenterol. 2016;9(3):392–407. https://doi.org/10.1177/1756283X16638830.
Goncalves IO, Passos E, Diogo CV, Rocha-Rodrigues S, Santos-Alves E, Oliveira PJ, et al. Exercise mitigates mitochondrial permeability transition pore and quality control mechanisms alterations in nonalcoholic steatohepatitis. Appl Physiol Nutr Metab. 2016;41(3):298–306. https://doi.org/10.1139/apnm-2015-0470.
Wang B, Zeng J, Gu Q. Exercise restores bioavailability of hydrogen sulfide and promotes autophagy influx in livers of mice fed with high-fat diet. Can J Physiol Pharmacol. 2017;95(6):667–74. https://doi.org/10.1139/cjpp-2016-0611.
Rosa-Caldwell ME, Lee DE, Brown JL, Brown LA, Perry RA Jr, Greene ES, et al. Moderate physical activity promotes basal hepatic autophagy in diet-induced obese mice. Appl Physiol Nutr Metab. 2017;42(2):148–56. https://doi.org/10.1139/apnm-2016-0280.
• Guarino M, Kumar P, Felser A, Terracciano LM, Guixe-Muntet S, Humar B, et al. Exercise Attenuates the Transition from Fatty Liver to Steatohepatitis and Reduces Tumor Formation in Mice. Cancers (Basel). 2020;12(6). https://doi.org/10.3390/cancers12061407Exercise, indepedently of dietary changes, attenuated the progression from NAFLD to NASH, improved biochemical and histological parameters of NAFLD, impeded the progression of fibrosis and tumorigenesis associated with enhanced activation of AMPK signaling and favored liver autophagy in a choline-deficient, HFD-fed mouse model.
Marchesini G, Brizi M, Bianchi G, Tomassetti S, Zoli M, Melchionda N. Metformin in non-alcoholic steatohepatitis. Lancet. 2001;358(9285):893–4. https://doi.org/10.1016/s0140-6736(01)06042-1.
Li Y, Liu L, Wang B, Wang J, Chen D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed Rep. 2013;1(1):57–64. https://doi.org/10.3892/br.2012.18.
Cunha V, Cotrim HP, Rocha R, Carvalho K, Lins-Kusterer L. Metformin in the prevention of hepatocellular carcinoma in diabetic patients: A systematic review. Ann Hepatol. 2020;19(3):232–7. https://doi.org/10.1016/j.aohep.2019.10.005.
Kim KS, Lee BW. Beneficial effect of anti-diabetic drugs for nonalcoholic fatty liver disease. Clin Mol Hepatol. 2020;26(4):430–43. https://doi.org/10.3350/cmh.2020.0137.
Jalali M, Rahimlou M, Mahmoodi M, Moosavian SP, Symonds ME, Jalali R, et al. The effects of metformin administration on liver enzymes and body composition in non-diabetic patients with non-alcoholic fatty liver disease and/or non-alcoholic steatohepatitis: An up-to date systematic review and meta-analysis of randomized controlled trials. Pharmacol Res. 2020;159:104799. https://doi.org/10.1016/j.phrs.2020.104799.
Kumar J, Memon RS, Shahid I, Rizwan T, Zaman M, Menezes RG, et al. Antidiabetic drugs and non-alcoholic fatty liver disease: A systematic review, meta-analysis and evidence map. Dig Liver Dis. 2021;53(1):44–51. https://doi.org/10.1016/j.dld.2020.08.021.
Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135(4):1176–84. https://doi.org/10.1053/j.gastro.2008.06.047.
Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362(18):1675–85. https://doi.org/10.1056/NEJMoa0907929.
Younossi ZM, Reyes MJ, Mishra A, Mehta R, Henry L. Systematic review with meta-analysis: non-alcoholic steatohepatitis - a case for personalised treatment based on pathogenic targets. Aliment Pharmacol Ther. 2014;39(1):3–14. https://doi.org/10.1111/apt.12543.
Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology. 2003;38(4):1008–17. https://doi.org/10.1053/jhep.2003.50420.
Ratziu V, Giral P, Jacqueminet S, Charlotte F, Hartemann-Heurtier A, Serfaty L, et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135(1):100–10. https://doi.org/10.1053/j.gastro.2008.03.078.
Ratziu V, Charlotte F, Bernhardt C, Giral P, Halbron M, Lenaour G, et al. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51(2):445–53. https://doi.org/10.1002/hep.23270.
Zelber-Sagi S, Kessler A, Brazowsky E, Webb M, Lurie Y, Santo M, et al. A double-blind randomized placebo-controlled trial of orlistat for the treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2006;4(5):639–44. https://doi.org/10.1016/j.cgh.2006.02.004.
Ye J, Wu Y, Li F, Wu T, Shao C, Lin Y, et al. Effect of orlistat on liver fat content in patients with nonalcoholic fatty liver disease with obesity: assessment using magnetic resonance imaging-derived proton density fat fraction. Therap Adv Gastroenterol. 2019;12:1756284819879047. https://doi.org/10.1177/1756284819879047.
Khoo J, Hsiang JC, Taneja R, Koo SH, Soon GH, Kam CJ, et al. Randomized trial comparing effects of weight loss by liraglutide with lifestyle modification in non-alcoholic fatty liver disease. Liver Int. 2019;39(5):941–9. https://doi.org/10.1111/liv.14065.
Harrison SA, Fecht W, Brunt EM, Neuschwander-Tetri BA. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology. 2009;49(1):80–6. https://doi.org/10.1002/hep.22575.
Shen B, Feng H, Cheng J, Li Z, Jin M, Zhao L, et al. Geniposide alleviates non-alcohol fatty liver disease via regulating Nrf2/AMPK/mTOR signalling pathways. J Cell Mol Med. 2020;24(9):5097–108. https://doi.org/10.1111/jcmm.15139.
Rojas C, Pan-Castillo B, Valls C, Pujadas G, Garcia-Vallve S, Arola L, et al. Resveratrol enhances palmitate-induced ER stress and apoptosis in cancer cells. PLoS One. 2014;9(12):e113929. https://doi.org/10.1371/journal.pone.0113929.
Hu F, Han J, Zhai B, Ming X, Zhuang L, Liu Y, et al. Blocking autophagy enhances the apoptosis effect of bufalin on human hepatocellular carcinoma cells through endoplasmic reticulum stress and JNK activation. Apoptosis. 2014;19(1):210–23. https://doi.org/10.1007/s10495-013-0914-7.
Miao Q, Bi LL, Li X, Miao S, Zhang J, Zhang S, et al. Anticancer effects of bufalin on human hepatocellular carcinoma HepG2 cells: roles of apoptosis and autophagy. Int J Mol Sci. 2013;14(1):1370–82. https://doi.org/10.3390/ijms14011370.
Tufanli O, Telkoparan Akillilar P, Acosta-Alvear D, Kocaturk B, Onat UI, Hamid SM, et al. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc Natl Acad Sci U S A. 2017;114(8):E1395–E404. https://doi.org/10.1073/pnas.1621188114.
Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology. 2018;67(2):549–59. https://doi.org/10.1002/hep.29514.
Lee DH, Han DH, Nam KT, Park JS, Kim SH, Lee M, et al. Ezetimibe, an NPC1L1 inhibitor, is a potent Nrf2 activator that protects mice from diet-induced nonalcoholic steatohepatitis. Free Radic Biol Med. 2016;99:520–32. https://doi.org/10.1016/j.freeradbiomed.2016.09.009.
• Sharma RS, Harrison DJ, Kisielewski D, Cassidy DM, McNeilly AD, Gallagher JR, et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell Mol Gastroenterol Hepatol. 2018;5(3):367–98. https://doi.org/10.1016/j.jcmgh.2017.11.016Pharmacologic activation of Nrf2 in insulin resistant mice with obesity reversed insulin resistance suppressed hepatic steatosis and mitigated NASH and liver fibrosis, underlining the potential of the use of Nrf2 activators to treat NASH.
Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011;332(6025):91–4. https://doi.org/10.1126/science.1201396.
Ben Mosbah I, Alfany-Fernandez I, Martel C, Zaouali MA, Bintanel-Morcillo M, Rimola A, et al. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010;1:e52. https://doi.org/10.1038/cddis.2010.29.
Moreira AJ, Ordonez R, Cerski CT, Picada JN, Garcia-Palomo A, Marroni NP, et al. Melatonin Activates Endoplasmic Reticulum Stress and Apoptosis in Rats with Diethylnitrosamine-Induced Hepatocarcinogenesis. PLoS One. 2015;10(12):e0144517. https://doi.org/10.1371/journal.pone.0144517.
Kim HJ, Joe Y, Kim SK, Park SU, Park J, Chen Y, et al. Carbon monoxide protects against hepatic steatosis in mice by inducing sestrin-2 via the PERK-eIF2alpha-ATF4 pathway. Free Radic Biol Med. 2017;110:81–91. https://doi.org/10.1016/j.freeradbiomed.2017.05.026.
•• Petito-da-Silva TI, Souza-Mello V, Barbosa-da-Silva S. Empaglifozin mitigates NAFLD in high-fat-fed mice by alleviating insulin resistance, lipogenesis and ER stress. Mol Cell Endocrinol. 2019;498:110539. https://doi.org/10.1016/j.mce.2019.110539Empaglifozin mitigated the development of NAFLD in HFD-fed mice by attenuating insulin resistance, lipogenesis and ER stress as shown by the reduced expression of genes involved in hepatic lipogenesis and genes involved in ER stress. Thus, empagliflozin could be used to treat the progression of hepatic steatosis.
Sharma S, Mells JE, Fu PP, Saxena NK, Anania FA. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS One. 2011;6(9):e25269. https://doi.org/10.1371/journal.pone.0025269.
Zhang H, Li K, Lin Y, Xing F, Xiao X, Cai J, et al. Targeting VCP enhances anticancer activity of oncolytic virus M1 in hepatocellular carcinoma. Sci Transl Med. 2017;9(404). https://doi.org/10.1126/scitranslmed.aam7996.
Huang Q, Wang T, Yang L, Wang HY. Ginsenoside Rb2 Alleviates Hepatic Lipid Accumulation by Restoring Autophagy via Induction of Sirt1 and Activation of AMPK. Int J Mol Sci. 2017;18(5). https://doi.org/10.3390/ijms18051063.
Ren H, Wang D, Zhang L, Kang X, Li Y, Zhou X, et al. Catalpol induces autophagy and attenuates liver steatosis in ob/ob and high-fat diet-induced obese mice. Aging (Albany NY). 2019;11(21):9461–77. https://doi.org/10.18632/aging.102396.
Gong LL, Li GR, Zhang W, Liu H, Lv YL, Han FF, et al. Akebia Saponin D Decreases Hepatic Steatosis through Autophagy Modulation. J Pharmacol Exp Ther. 2016;359(3):392–400. https://doi.org/10.1124/jpet.116.236562.
Shen C, Dou X, Ma Y, Ma W, Li S, Song Z. Nicotinamide protects hepatocytes against palmitate-induced lipotoxicity via SIRT1-dependent autophagy induction. Nutr Res. 2017;40:40–7. https://doi.org/10.1016/j.nutres.2017.03.005.
Li X, Gong H, Yang S, Yang L, Fan Y, Zhou Y. Pectic Bee Pollen Polysaccharide from Rosa rugosa Alleviates Diet-Induced Hepatic Steatosis and Insulin Resistance via Induction of AMPK/mTOR-Mediated Autophagy. Molecules. 2017;22(5). https://doi.org/10.3390/molecules22050699.
Sinha RA, Farah BL, Singh BK, Siddique MM, Li Y, Wu Y, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology. 2014;59(4):1366–80. https://doi.org/10.1002/hep.26667.
Zheng X, Dai W, Chen X, Wang K, Zhang W, Liu L, et al. Caffeine reduces hepatic lipid accumulation through regulation of lipogenesis and ER stress in zebrafish larvae. J Biomed Sci. 2015;22:105. https://doi.org/10.1186/s12929-015-0206-3.
Zhou J, Farah BL, Sinha RA, Wu Y, Singh BK, Bay BH, et al. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, stimulates hepatic autophagy and lipid clearance. PLoS One. 2014;9(1):e87161. https://doi.org/10.1371/journal.pone.0087161.
Porcu C, Sideri S, Martini M, Cocomazzi A, Galli A, Tarantino G, et al. Oleuropein Induces AMPK-Dependent Autophagy in NAFLD Mice, Regardless of the Gender. Int J Mol Sci. 2018;19(12). https://doi.org/10.3390/ijms19123948.
Veskovic M, Mladenovic D, Milenkovic M, Tosic J, Borozan S, Gopcevic K, et al. Betaine modulates oxidative stress, inflammation, apoptosis, autophagy, and Akt/mTOR signaling in methionine-choline deficiency-induced fatty liver disease. Eur J Pharmacol. 2019;848:39–48. https://doi.org/10.1016/j.ejphar.2019.01.043.
Ohashi T, Nakade Y, Ibusuki M, Kitano R, Yamauchi T, Kimoto S, et al. Conophylline inhibits high fat diet-induced non-alcoholic fatty liver disease in mice. PLoS One. 2019;14(1):e0210068. https://doi.org/10.1371/journal.pone.0210068.
Zhong J, Gong W, Chen J, Qing Y, Wu S, Li H, et al. Micheliolide alleviates hepatic steatosis in db/db mice by inhibiting inflammation and promoting autophagy via PPAR-gamma-mediated NF-small ka. CyrillicB and AMPK/mTOR signaling. Int Immunopharmacol. 2018;59:197–208. https://doi.org/10.1016/j.intimp.2018.03.036.
Zhang R, Chu K, Zhao N, Wu J, Ma L, Zhu C, et al. Corilagin Alleviates Nonalcoholic Fatty Liver Disease in High-Fat Diet-Induced C57BL/6 Mice by Ameliorating Oxidative Stress and Restoring Autophagic Flux. Front Pharmacol. 2019;10:1693. https://doi.org/10.3389/fphar.2019.01693.
Chen X, Chan H, Zhang L, Liu X, Ho IHT, Zhang X, et al. The phytochemical polydatin ameliorates non-alcoholic steatohepatitis by restoring lysosomal function and autophagic flux. J Cell Mol Med. 2019;23(6):4290–300. https://doi.org/10.1111/jcmm.14320.
Zhou T, Ye L, Bai Y, Sun A, Cox B, Liu D, et al. Autophagy and apoptosis in hepatocellular carcinoma induced by EF25-(GSH)2: a novel curcumin analog. PLoS One. 2014;9(9):e107876. https://doi.org/10.1371/journal.pone.0107876.
Cao Z, Zhang H, Cai X, Fang W, Chai D, Wen Y, et al. Luteolin Promotes Cell Apoptosis by Inducing Autophagy in Hepatocellular Carcinoma. Cell Physiol Biochem. 2017;43(5):1803–12. https://doi.org/10.1159/000484066.
Luo Y, Song L, Wang X, Huang Y, Liu Y, Wang Q, et al. Uncovering the Mechanisms of Cryptotanshinone as a Therapeutic Agent Against Hepatocellular Carcinoma. Front Pharmacol. 2020;11:1264. https://doi.org/10.3389/fphar.2020.01264.
•• Yao C, Liu BB, Qian XD, Li LQ, Cao HB, Guo QS, et al. Crocin induces autophagic apoptosis in hepatocellular carcinoma by inhibiting Akt/mTOR activity. Onco Targets Ther. 2018;11:2017–28. https://doi.org/10.2147/OTT.S154586Crocin, a glucosidic derivative from saffron, the dried dark-red stigma of Crocus sativus L., can induce autophagic apoptosis in HCC cells by inhibiting Akt/mTOR signaling pathway as shown by the increased Beclin-1, decreased p62 and accumulated LC3-II protein levels.
Tao R, Sun WY, Yu DH, Qiu W, Yan WQ, Ding YH, et al. Sodium cantharidinate induces HepG2 cell apoptosis through LC3 autophagy pathway. Oncol Rep. 2017;38(2):1233–9. https://doi.org/10.3892/or.2017.5779.
Zheng J, Shao Y, Jiang Y, Chen F, Liu S, Yu N, et al. Tangeretin inhibits hepatocellular carcinoma proliferation and migration by promoting autophagy-related BECLIN1. Cancer Manag Res. 2019;11:5231–42. https://doi.org/10.2147/CMAR.S200974.
Leu YS, Chen YJ, Chen CC, Huang HL. Induction of Autophagic Death of Human Hepatocellular Carcinoma Cells by Armillaridin from Armillaria mellea. Am J Chin Med. 2019;47(6):1365–80. https://doi.org/10.1142/S0192415X19500708.
•• Zhang B, Yin X, Sui S. Resveratrol inhibited the progression of human hepatocellular carcinoma by inducing autophagy via regulating p53 and the phosphoinositide 3kinase/protein kinase B pathway. Oncol Rep. 2018;40(5):2758–65. https://doi.org/10.3892/or.2018.6648Resveratrol (trans-3,4′,5-trihydroxystilbene), which is a polyphenolic compound derived from grapes and originally isolated from the roots of white hellebore, exerted antitumor effects through autophagy activation as shown by the upregulated autophagosome formation, LC3-II/I ratio and Beclin-1 protein levels and the reduced p62 expression in a HCC cell line.
Mao Z, Han X, Chen D, Xu Y, Xu L, Yin L, et al. Potent effects of dioscin against hepatocellular carcinoma through regulating TP53-induced glycolysis and apoptosis regulator (TIGAR)-mediated apoptosis, autophagy, and DNA damage. Br J Pharmacol. 2019;176(7):919–37. https://doi.org/10.1111/bph.14594.
Hu P, Ke C, Guo X, Ren P, Tong Y, Luo S, et al. Both glypican-3/Wnt/beta-catenin signaling pathway and autophagy contributed to the inhibitory effect of curcumin on hepatocellular carcinoma. Dig Liver Dis. 2019;51(1):120–6. https://doi.org/10.1016/j.dld.2018.06.012.
Ji Y, Li L, Ma YX, Li WT, Li L, Zhu HZ, et al. Quercetin inhibits growth of hepatocellular carcinoma by apoptosis induction in part via autophagy stimulation in mice. J Nutr Biochem. 2019;69:108–19. https://doi.org/10.1016/j.jnutbio.2019.03.018.
Iannucci LF, Cioffi F, Senese R, Goglia F, Lanni A, Yen PM, et al. Metabolomic analysis shows differential hepatic effects of T2 and T3 in rats after short-term feeding with high fat diet. Sci Rep. 2017;7(1):2023. https://doi.org/10.1038/s41598-017-02205-1.
Zhu S, Wu Y, Ye X, Ma L, Qi J, Yu D, et al. FGF21 ameliorates nonalcoholic fatty liver disease by inducing autophagy. Mol Cell Biochem. 2016;420(1-2):107–19. https://doi.org/10.1007/s11010-016-2774-2.
Wu P, Zhao J, Guo Y, Yu Y, Wu X, Xiao H. Ursodeoxycholic acid alleviates nonalcoholic fatty liver disease by inhibiting apoptosis and improving autophagy via activating AMPK. Biochem Biophys Res Commun. 2020;529(3):834–8. https://doi.org/10.1016/j.bbrc.2020.05.128.
Xue W, Wang J, Jiang W, Shi C, Wang X, Huang Y, et al. Caveolin-1 alleviates lipid accumulation in NAFLD associated with promoting autophagy by inhibiting the Akt/mTOR pathway. Eur J Pharmacol. 2020;871:172910. https://doi.org/10.1016/j.ejphar.2020.172910.
Okada H, Takabatake R, Honda M, Takegoshi K, Yamashita T, Nakamura M, et al. Peretinoin, an acyclic retinoid, suppresses steatohepatitis and tumorigenesis by activating autophagy in mice fed an atherogenic high-fat diet. Oncotarget. 2017;8(25):39978–93. https://doi.org/10.18632/oncotarget.18116.
Liu YL, Yang PM, Shun CT, Wu MS, Weng JR, Chen CC. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy. 2010;6(8):1057–65. https://doi.org/10.4161/auto.6.8.13365.
Wei PL, Huang CY, Chang YJ. Propyl gallate inhibits hepatocellular carcinoma cell growth through the induction of ROS and the activation of autophagy. PLoS One. 2019;14(1):e0210513. https://doi.org/10.1371/journal.pone.0210513.
Wang J, Sun P, Chen Y, Yao H, Wang S. Novel 2-phenyloxypyrimidine derivative induces apoptosis and autophagy via inhibiting PI3K pathway and activating MAPK/ERK signaling in hepatocellular carcinoma cells. Sci Rep. 2018;8(1):10923. https://doi.org/10.1038/s41598-018-29199-8.
Zhu L, Wu Q, Quan B, Yang J, Yang J, Hou W, et al. Autophagy inhibition by reversine and its suppressive effects on human hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2020;528(1):105–11. https://doi.org/10.1016/j.bbrc.2020.05.054.
Yan Y, Jiang K, Liu P, Zhang X, Dong X, Gao J, et al. Bafilomycin A1 induces caspase-independent cell death in hepatocellular carcinoma cells via targeting of autophagy and MAPK pathways. Sci Rep. 2016;6:37052. https://doi.org/10.1038/srep37052.
Liu C, Liu L, Zhu HD, Sheng JQ, Wu XL, He XX, et al. Celecoxib alleviates nonalcoholic fatty liver disease by restoring autophagic flux. Sci Rep. 2018;8(1):4108. https://doi.org/10.1038/s41598-018-22339-0.
Hsiao PJ, Chiou HC, Jiang HJ, Lee MY, Hsieh TJ, Kuo KK. Pioglitazone Enhances Cytosolic Lipolysis, beta-oxidation and Autophagy to Ameliorate Hepatic Steatosis. Sci Rep. 2017;7(1):9030. https://doi.org/10.1038/s41598-017-09702-3.
Tong W, Ju L, Qiu M, Xie Q, Chen Y, Shen W, et al. Liraglutide ameliorates non-alcoholic fatty liver disease by enhancing mitochondrial architecture and promoting autophagy through the SIRT1/SIRT3-FOXO3a pathway. Hepatol Res. 2016;46(9):933–43. https://doi.org/10.1111/hepr.12634.
He Q, Sha S, Sun L, Zhang J, Dong M. GLP-1 analogue improves hepatic lipid accumulation by inducing autophagy via AMPK/mTOR pathway. Biochem Biophys Res Commun. 2016;476(4):196–203. https://doi.org/10.1016/j.bbrc.2016.05.086.
• Huang Z, Fang W, Liu W, Wang L, Liu B, Liu S, et al. Aspirin induces Beclin-1-dependent autophagy of human hepatocellular carcinoma cell. Eur J Pharmacol. 2018;823:58–64. https://doi.org/10.1016/j.ejphar.2018.01.031A widely used medication, aspirin, inhibited HCC development by inducing autophagy as shown by the increased LC3II/LC3I ratio, autophagic flux and decreased p62 expression in HCC cell lines. Interestingly these effects were dependent on Beclin-1 activation.
Chen YJ, Chi CW, Su WC, Huang HL. Lapatinib induces autophagic cell death and inhibits growth of human hepatocellular carcinoma. Oncotarget. 2014;5(13):4845–54. https://doi.org/10.18632/oncotarget.2045.
•• Gao C, Fang L, Zhang H, Zhang WS, Li XO, Du SY. Metformin Induces Autophagy via the AMPK-mTOR Signaling Pathway in Human Hepatocellular Carcinoma Cells. Cancer Manag Res. 2020;12:5803–11. https://doi.org/10.2147/CMAR.S257966Metformin (1,1-dimethylbiguanide hydrochloride), which is a biguanide derivative and one of the most used first-line anti-diabetic drugs, inhibited hepatocellular carcinoma HepG2 cell viability via activating autophagy as shown by the downregulation of p62 and LC3-II, with the decrease in LC3-II being attributed to activation of autophagic flux and not formation of autophagosomes in these cells. Furthermore, metformin activated AMPK and inhibited mTOR phosphorylation, showing that the effect of metformin on autophagy is exerted by the AMPK/mTOR signaling pathway.
Linden MA, Lopez KT, Fletcher JA, Morris EM, Meers GM, Siddique S, et al. Combining metformin therapy with caloric restriction for the management of type 2 diabetes and nonalcoholic fatty liver disease in obese rats. Appl Physiol Nutr Metab. 2015;40(10):1038–47. https://doi.org/10.1139/apnm-2015-0236.
• Xie B, He X, Guo G, Zhang X, Li J, Liu J, et al. High-throughput screening identified mitoxantrone to induce death of hepatocellular carcinoma cells with autophagy involvement. Biochem Biophys Res Commun. 2020;521(1):232–7. https://doi.org/10.1016/j.bbrc.2019.10.114Mitoxantrone, an antineoplastic agent that inhibits the type II topoisomerase, was found to inhibit the growth and proliferation of HCC cells by inducing mTOR-dependent autophagy activation as shown by the increased expression of LC3-II and Beclin-1 proteins and the downregulation of p62 protein.
Liang YC, Chang CC, Sheu MT, Lin SY, Chung CC, Teng CT, et al. The Antihistamine Deptropine Induces Hepatoma Cell Death through Blocking Autophagosome-Lysosome Fusion. Cancers (Basel). 2020;12(6). https://doi.org/10.3390/cancers12061610.
•• Wang Y, Wang L. Effect of Combined Sorafenib/Cisplatinum Treatment on the Autophagy and Proliferation of Hepatocellular Carcinoma hepG2 Cells in Vitro. Asian Pac J Cancer Prev. 2020;21(10):2853–7. https://doi.org/10.31557/APJCP.2020.21.10.2853A combination treatment of Sorafenib (a multi-kinase inhibitor) and Cisplatinum (a chemotherapy medication) inhibited human hepatocellular carcinoma HepG2 cell proliferation by inducing autophagy as shown by the downregulated mTOR levels and the increased LC3II expression.
Shen L, Yang Y, Ou T, Key CC, Tong SH, Sequeira RC, et al. Dietary PUFAs attenuate NLRP3 inflammasome activation via enhancing macrophage autophagy. J Lipid Res. 2017;58(9):1808–21. https://doi.org/10.1194/jlr.M075879.
• Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67(1):123–33. https://doi.org/10.1002/hep.29466NAFLD-related liver diseases and mortality are predicted to increase in the United States along with continued high rates of adult obesity, type 2 diabetes mellitus and an aging population. Thus, the need to discover therapeutic interventions and strategies to slow the growth of NAFLD is urgent in order to mitigate disease burden.
•• Brubaker DK, Lauffenburger DA. Translating preclinical models to humans. Science. 2020;367(6479):742–3. https://doi.org/10.1126/science.aay8086The problem of generalizing results from animal models to human patients is responsible for the large proportion of failures encountered in moving therapeutics from preclinical studies to clinical trials and impedes drug development. Instead of attempting to “humanize” animal experimental models, which is possible to a very limited extent, greater success may be obtained by humanizing computational models derived from animal experiments.
Van Campenhout S, Tilleman L, Lefere S, Vandierendonck A, Raevens S, Verhelst X, et al. Myeloid-specific IRE1alpha deletion reduces tumour development in a diabetic, non-alcoholic steatohepatitis-induced hepatocellular carcinoma mouse model. Metabolism. 2020;107:154220. https://doi.org/10.1016/j.metabol.2020.154220.
• Ægidius HM, Veidal SS, Feigh M, Hallenborg P, Puglia M, Pers TH, et al. Multi-omics characterization of a diet-induced obese model of non-alcoholic steatohepatitis. Sci Rep. 2020;10(1):1148. https://doi.org/10.1038/s41598-020-58059-7A diet-induced mouse model of NASH was used to perform multi-omics analysis in order to improve the understanding of the complex biological processes underlying NASH occurrence. Bulk RNA-sequencing and proteomics showed a clear distinction between phenotypes and a good correspondence between mRNA and protein level regulations, apart from specific regulatory events discovered by each technology. Transcriptomics-based gene set enrichment analysis revealed changes associated with key clinical manifestations of NASH, including impaired lipid metabolism, increased extracellular matrix formation/remodeling and pro-inflammatory responses, whereas proteomics-based gene set enrichment analysis pinpointed metabolic pathway perturbations. Integration with single-cell RNA-sequencing data identified key regulated cell types involved in development of NASH demonstrating the cellular heterogeneity and complexity of NASH pathogenesis.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of Interest
The authors declared no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Eva Kassi and Harpal S. Randeva are joint last authors and corresponding authors.
Ioannis Kyrou and Narjes Nasiri-Ansari are joint second authors.
This article is part of the Topical Collection on Metabolism
Rights and permissions
About this article

Cite this article
Flessa, CM., Kyrou, I., Nasiri-Ansari, N. et al. Endoplasmic Reticulum Stress and Autophagy in the Pathogenesis of Non-alcoholic Fatty Liver Disease (NAFLD): Current Evidence and Perspectives. Curr Obes Rep 10, 134–161 (2021). https://doi.org/10.1007/s13679-021-00431-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13679-021-00431-3