
Abstract
Background
The transcription factor nuclear factor-E2-related factor-2 (Nrf2) is a key regulator for induction of hepatic antioxidative stress systems. We aimed to investigate whether activation of Nrf2 protects against steatohepatitis.
Method
Wild-type mice (WT), Nrf2 gene-null mice (Nrf2-null) and Keap1 gene-knockdown mice (Keap1-kd), which represent the sustained activation of Nrf2, were fed a methionine- and choline-deficient diet (MCDD) for 13 weeks and analyzed.
Results
In Keap1-kd fed an MCDD, steatohepatitis did not develop over the observation periods; however, in Nrf2-null fed an MCDD, the pathological state of the steatohepatitis was aggravated in terms of fatty change, inflammation, fibrosis and iron accumulation. In WT mice fed an MCDD, Nrf2 and antioxidative stress genes regulated by Nrf2 were potently activated in the livers, and in Keap1-kd, their basal levels were potently activated. Oxidative stress was significantly increased in the livers of the Nrf2-null and suppressed in the livers of the Keap1-kd compared to that of WT, based on the levels of 4-hydroxy-2-nonenal and malondialdehyde. Iron accumulation was greater in the livers of the Nrf2-null mice compared to those of the WT mice, and it was not observed in Keap1-kd. Further, the iron release from the isolated hepatocyte of Nrf2-null mice was significantly decreased. Sulforaphane, an activator of Nrf2, suppressed the pathological states and oxidative stress in the livers.
Conclusions
Nrf2 has protective roles against nutritional steatohepatitis through inhibition of hepatic iron accumulation and counteraction against oxidative stress-induced liver injury. Nrf2 activation by pharmaceutical intervention could be a new option for the prevention and treatment of steatohepatitis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Non-alcoholic steatohepatitis (NASH), as a component of metabolic syndrome, will represent an increasingly important global public health problem [1, 2]. In epidemiology, a follow-up study of NASH patients for 10 years has shown that the disease progresses to cirrhosis in ca. 20% of the patients and leads to death caused by liver cirrhosis in 8% [3]. Therefore, treatment strategies for NASH patients are urgently needed. The increased levels of free fatty acids, non-heme iron and inflammatory cytokines in the livers provide a perpetuating and propagating mechanism for oxidative stress via the process of reactive oxygen species (ROS) production [1, 2] and thereby lipid peroxidation [4]. The role of oxidative stress in the pathogenesis of NASH has been confirmed in the experimental models of NASH, as well as in human NASH [2, 5–8]. Under the pathological conditions of accumulation of oxidative stress, hepatic stellate cells (HSCs), the major sources of collagen and other extracellular matrix proteins, are activated and then transformed into proliferative fibrogenic cells [7, 9, 10]. Thus, NASH is developed, leading to liver cirrhosis.
The transcription factor termed nuclear factor-E2-related factor-2 (Nrf2) serves as a cellular sensor for oxidative stress. Nrf2 is sequestered in the cytosol by Kelch-like Ech-associated protein (Keap1). Upon an oxidative challenge, modification of Keap1 sulfhydryl groups results in the stabilization and nuclear translocation of Nrf2 [11]. Nrf2 plays crucial roles for antioxidant responsive element/electrophile-responsive element (ARE/EpRE)-mediated induction of antioxidative stress genes [12]. Our previous studies have shown that chemicals and drugs activating the Nrf2 regulatory pathway are shown to decrease oxidative stress, which in turn is implicated in the pathogenesis of numerous liver diseases [13, 14]. Moreover, our and other laboratories have shown that deletion of Nrf2 leads to severe progression of nutritional steatohepatitis [15–17]. However, the protective role of Nrf2 and the feasibility of drug therapy using Nrf2 activators against liver injuries in steatohepatitis have not yet been well elucidated.
Recently, Nrf2 has been reported to regulate iron efflux from macrophages through ferroportin-1 (Fpn1) gene transcription and control iron metabolism during inflammation [18]. Fpn1 is an iron exporter on the surface of absorptive intestinal enterocytes, macrophages, hepatocytes and placental cells, all of which release iron into plasma [19]. Iron excess in hepatocytes leads to generation of oxidative stress, cell toxicity and genotoxicity. Hepatic iron overload has been found in patients of NAFLD [5, 6], and moreover, is causatively associated with hepatic fibrosis in NASH patients [20].
In this study, we studied the protective roles of Nrf2 against the development of nutritional steatohepatitis, with special reference to iron metabolism in the livers. Wild-type mice (WT), Nrf2 gene-null mice (Nrf2-null) and Keap1 gene-knockdown mice (Keap1-kd), which represent the sustained activation of Nrf2, were fed a methionine- and choline-deficient diet (MCDD), which is widely used for research on steatohepatitis [7, 8]. We also studied the effect of sulforaphane (SFN), a compound in broccoli sprouts that is known to be a potent Nrf2 activator [21], on the development of steatohepatitis. We first demonstrated that Nrf2 inhibits hepatic iron accumulation and counteracts against oxidative stress-induced liver injury in steatohepatitis patients. Moreover, the treatment with SFN reduced the iron accumulation and oxidative stress in the livers by an upregulation of Nrf2, which in turn led to the decreased fibrosis. Nrf2 activation by pharmaceutical intervention could be a new option for the prevention and treatment of steatohepatitis.
Materials and methods
Animals
Male 10–12-week-old WT, Nrf2-null and Keap1-kd mice [13–15] on a C57BL/6 background were fed an MCDD or control diet (Oriental Yeast, Tokyo, Japan) for 13 weeks. At the end of the experiment, serum and liver tissue specimens were collected for analysis. SFN was mixed at 0.05% (wt/wt) in MCDD (MPBio, CA, USA) and administered. All experiments were performed under protocols approved by the Institutional Animal Care and Use Committees of the University of Tsukuba.
Biochemical and histological analysis
Serum concentrations of aspartate aminotransferase (AST), alanine aminotransferase (ALT) and alkaline phosphatase (ALP), were measured by SRL Inc. (Tokyo, Japan). The serum concentration of hepcidin 1 was measured in the laboratories of MCProt Biotechnology (Kanazawa, Japan). Liver tissues were fixed in 10% paraformaldehyde, embedded in paraffin and stained with hematoxylin–eosin (HE), a Masson trichrome, Sirius red and Berlin blue solution.
Triglyceride concentrations, GSH levels and MDA concentrations of liver tissues
Triglyceride concentrations, glutathione (GSH) levels and malondialdehyde (MDA) concentrations of liver tissues specimens were measured as previously described [15].
Immunoblot analysis
Immunoblot analysis was performed by using liver total homogenates and nuclear fraction as previously described [13, 15, 18].
Immunohistochemistry
For immunostainings of 4-hydroxy-2-nonenal (4-HNE), 2-μm-thick tissue sections were stained using the indirect immunoperoxidase method with anti-4HNE mAb (JaiCA, Shizuoka, Japan) as previously described [15].
Real-time quantitative polymerase chain reaction
Steady-state mRNA levels in the specimens were determined by real-time quantitative PCR using recently detailed methodology [13]. Primers and probes used for this study have been described previously [13, 15, 18]. Data were normalized to the amounts of GAPDH present in each specimen and then averaged.
Non-heme iron contents in cells and liver tissues
Non-heme iron contents of cells and liver tissue specimens were measured as previously described [22].
Isolation and culture of primary mouse hepatocytes
Primary hepatocytes were isolated from 10- to 15-week-old C57BL6 male mice by collagenase perfusion according to the method described previously [21] with slight modification. Briefly, the liver parenchymal hepatocytes were seeded at a density of 1 × 105 cells/cm2 with D-MEM medium supplemented with 10% FBS. From the 5th day cells were then exposed for 24 h either to control medium (D-MEM) or identical medium that was manufactured to be completely deficient in methionine and choline (MCD medium) purchased from Invitrogen. All experiments were performed at least three times using different cell culture preparations from other animals.
Iron release experiment
Iron release was measured using 59FeCl as previously described [18]. The percent of iron release was calculated according to the following equation: percent 59Fe release = [(cpm in medium)/(cpm in medium + cpm in cells)] × 100.
Statistics
Values are given as mean ± standard error of the mean (SEM). When two groups were compared, unpaired t test was used for data analysis. Multiple group comparisons were performed by two-way ANOVA. A P value of <0.05 was defined as statistically significant.
Results
Suppression of MCDD-induced steatohepatitis progression by Nrf2
Treating mice with MCDD for 13 weeks resulted in signs of steatohepatitis in the WT mouse livers, namely fat droplet deposition, inflammatory cell infiltration and fibrosis (Fig. 1a). These changes were more apparent in the Nrf2-null mouse livers than in the WT mouse livers, while the Keap1-kd mouse livers did not show many pathological changes in fat deposition, inflammation or fibrosis. Among other effects, the fibrosis noted in the Nrf2-null mouse livers was bridging fibrosis. Berlin blue staining of the liver tissue showed that iron deposition was particularly evident near blood vessels. This change reflected the intensity of inflammation and fibrosis, and was more evident in Nrf2-null mouse livers than in WT mouse livers. However, the change was not evident in the Keap1-kd mouse livers. Immunostaining with 4-HNE was performed to determine the presence of lipid peroxides, and the chromatic response resembled the known pattern of iron deposition. More intense and extensive chromatic responses were noted in Nrf2-null mouse livers than in WT mouse livers, while deposition was poor in Keap1-kd mouse livers (Fig. 1a).

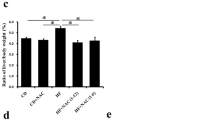
Sustained activation of Nrf2 protects against progression of steatohepatitis induced by an MCDD. a H&E-, Sirius red-, Masson trichrome-, Berlin blue- and 4-HNE-stained sections of representative liver specimens from the WT, the Nrf2-null and Keap1-kd mice fed an MCDD for 13 weeks (bars 100 μm). b NAFLD activity score (NAS) in liver samples fed an MCDD for 13 weeks. Values are mean ± SE. *P < 0.05, **P < 0.01, significantly different between the two groups. The numbers in parentheses represent the number of animals examined in each group. c Analysis of blood biochemistry (AST, ALT and ALP) in the WT, Nrf2-null and Keap1-kd fed an MCDD for 6 or 13 weeks. The numbers in parentheses represent the number of animals examined in each group. d Survival curve in the WT, Nrf2-null and Keap1-kd mice
Pathological changes in the liver were quantified using the NAFLD activity score (NAS). The Nrf2-null mouse livers showed 1.5- to 2-fold greater changes in the scores for fat deposition, inflammation, fibrosis and iron deposition compared to the WT mouse livers, while these changes were approximately half as intense in the Keap1-kd mouse livers (Fig. 1b). Signs of steatohepatitis were more intense in the Nrf2-null mouse livers than in the WT mouse livers, while they were minimal in the Keap1-kd mouse livers, thus indicating that the activity of steatohepatitis is associated with the intensity of Nrf2 expression. Evaluation of hepatopathy on the basis of blood biochemical data showed that AST, ALT and ALP levels at 6 weeks after the start of MCDD treatment were higher in the Nrf2-null mouse than in WT and Keap1-kd mouse (Fig. 1c). At 13 weeks after beginning MCDD treatment, AST and ALT levels were higher in the WT mice than in the other mice, and elevated AST and ALT levels were suppressed in the Keap1-kd mice (Fig. 1c). This result may be attributable to a difference in the timing of the peak in hepatic dysfunction—peak of dysfunction in the WT mouse livers occurred later than that in the Nrf2-null mouse livers. The ALP level was significantly lower in Keap1-kd mice than in WT and Nrf2-null mice. Figure 1d shows the mouse survival curve. Death of Nrf2-null mice began 8 weeks after the start of MCDD treatment. By the end of the 13-week MCDD treatment, 15 of 36 Nrf2-null mice had died. Only a few WT and Keap1-kd mice had died. These results indicate that deletion of Nrf2 is fatal during prolonged MCDD treatment.
Suppression of hepatic fat accumulation and oxidative stress by Nrf2
Neutral fat levels in the liver tissue after 6-week control diet ingestion were significantly higher in Nrf2-null mice than in WT mice. Neutral fat levels in the Keap1-kd mouse livers were significantly lower than those in the WT mouse livers at both 6 and 13 weeks after the start of control diet ingestion (Fig. 2a). Six-week MCDD treatment resulted in significantly elevated neutral fat levels in WT and Nrf2-null mouse livers, while levels in Keap1-kd mouse livers were not elevated; neutral fat levels were significantly lower in Keap1-kd mouse livers than in WT and Nrf2-null mouse livers (Fig. 2a). Treatment with MCDD for 13 weeks resulted in elevated neutral fat levels in the Keap1-kd mouse liver, but similar to those with 6-week MCDD treatment. These levels remained significantly lower than in WT and Nrf2-null mouse livers (Fig. 2a).

Sustained activation of Nrf2 inhibits hepatic oxidative stress, inflammatory cytokines and fibrosis stimulation factors in steatohepatitis induced by an MCDD. a Triglyceride, malondialdehyde (MDA) and glutathione (GSH) of liver tissue in the WT, Nrf2-null and Keap1-kd mice fed an MCDD for 6 or 13 weeks. Data are given as mean ± SE (n = 8–15/group). # P < 0.05, ## P < 0.01, significantly different from the WT with control feeding; a P < 0.05, aa P < 0.01, significantly different from the Nrf2-null with control feeding; † P < 0.05, ++ P < 0.01, significantly different from the WT with MCD feeding; b P < 0.05, bb P < 0.01, significantly different from the Nrf2-null with MCD feeding; brackets *P < 0.05, **P < 0.01, significantly different between the two groups. b Immunoblot analysis of α-Sma proteins in livers of the WT, Nrf2-null and Keap1-kd fed a control diet or an MCDD for 13 weeks. Bar graph shows quantitation of optical density of the immunoblots. Data are given as mean ± SE (n = 8/group). c Steady-state mRNA levels of the factors involved in the inflammatory cytokines in livers of the WT, Nrf2-null and Keap1-kd mice fed a control diet or MCDD for 6 or 13 weeks. Data are given as mean ± SE (n = 6–7/group). d Steady-state mRNA levels of the factors involved in the fibrogenesis in livers of the WT, Nrf2-null and Keap1-kd mice fed a control diet or MCDD for 6 or 13 weeks. Data are given as mean ± SE (n = 6–7/group)
We measured the levels of MDA, a lipid peroxide, to evaluate the intensity of oxidative stress in the liver tissue (Fig. 2a). Treatment with MCDD resulted in significantly elevated MDA levels in WT and Nrf2-null mouse livers. MDA levels in Keap1-kd mouse livers were unexpectedly slightly higher than those in WT mouse livers at both 6 and 13 weeks after the start of control diet ingestion (Fig. 2a). Following MCDD treatment, MDA levels remained significantly lower in Keap1-kd mouse livers than in WT and Nrf2-null mouse livers (Fig. 2a).
Basal levels of GSH, an endogenous antioxidant found in the liver tissue, did not differ significantly among the WT, Nrf2-null and Keap1-kd mouse livers (Fig. 2a). At 6 and 13 weeks after the start of MCDD treatment, GSH levels were significantly lower than the pretreatment levels in WT, Nrf2-null and Keap1-kd mouse livers. Furthermore, the GSH level was significantly lower in Nrf2-null and Keap1-kd mouse livers than in the WT mouse livers. There was no significant difference in this parameter between Nrf2-null and Keap1-kd mouse livers.
Figure 2b shows the results of immunoblot analysis of the alpha smooth muscle actin (α-Sma) protein expression level, an indicator of HSC activation and hepatic fibrosis. At 13 weeks after the start of control diet ingestion, the α-Sma expression level did not differ among WT, Nrf2-null and Keap1-kd mouse livers, while treatment with MCDD increased the expression of α-Sma in WT and Nrf2-null mouse livers. No increase was observed in the Keap1-kd mouse livers; the level was significantly lower than that in WT and Nrf2-null mouse livers (Fig. 2b).
Quantitative PCR was used to analyze hepatic expression of Tnf-α and Mcp-1 (inflammatory cytokines, Fig. 2c) and α1-procollagen and Tgf-β1 (fibrosis stimulation factors, Fig. 2d). At 13 weeks after the start of control diet ingestion, Tnf-α expression was significantly higher in Nrf2-null mouse livers than in WT mouse livers. At 6 and 13 weeks after the start of MCDD treatment, Tnf-α expression was significantly higher than its pretreatment level in both WT and Nrf2-null mouse livers, with the magnitude of increase significantly greater in the Nrf2-null mouse livers than in the WT mouse livers. In the Keap1-kd mouse livers, Tnf-α expression did not increase and was significantly lower than that in WT and Nrf2-null mouse livers. Changes similar to those in Tnf-α expression in the liver were noted in the expression of Mcp-1, α1-procollagen and Tgf-β1. Expression levels of these factors were significantly higher in MCDD-treated Nrf2-null mouse livers. These results appear to reflect the presence of inflammatory cell infiltration and fibrosis in the histopathologic findings (Fig. 1a).
Changes in the expression of Nrf2 and antioxidative stress genes regulated by Nrf2
Immunoblot analysis was conducted to evaluate changes in the expression of Nrf2 and oxidative stress response genes regulated by Nrf2 following MCDD treatment. Following control diet ingestion, Nrf2 expression was not detected in the Nrf2-null mouse livers, while the Keap1-kd mouse livers showed approximately fourfold higher Nrf2 expression than the WT mouse livers, thereby indicating constitutive Nrf2 expression in the mouse livers (Fig. 3a). Nrf2 expression in the WT mouse livers was approximately threefold higher following 1-week MCDD treatment than following control diet ingestion. Interestingly, the increase in Nrf2 expression in the WT mouse livers attenuated over time and was not observed at 13 weeks after the start of MCDD treatment. In the Keap1-kd mouse livers, Nrf2 expression was high at baseline, and MCDD treatment did not induce a marked increase in Nrf2 expression. However, the Keap1-kd mouse livers showed a greater increase in Nrf2 expression following MCDD treatment than the WT mouse livers. This increase was also observed at 13 weeks after the start of MCDD treatment (Fig. 3a).

Immunoblot analysis of a Nrf2 proteins in nuclear fraction of livers and b Nqo1, Gsta1 and γ-Gcs proteins in livers of the WT, Nrf2-null and Keap1-kd mice fed a control diet or an MCDD for 1 or 6 or 13 weeks. Bar graph shows quantitation of optical density of the immunoblots. Data are given as mean ± SE (n = 8/group). # P < 0.05, ## P < 0.01, significantly different from the WT with control feeding; a P < 0.05, aa P < 0.01, significantly different from the Nrf2-null with control feeding; † P < 0.05, ++ P < 0.01, significantly different from the WT with MCD feeding; b P < 0.05, bb P < 0.01, significantly different from the Nrf2-null with MCD feeding; brackets *P < 0.05, **P < 0.01, significantly different between the two groups
The expression levels of γ-glutamylcysteine synthetase γ-Gcs), a rate-limiting enzyme of GSH, NAD(P)H: quinone oxidoreductase 1 (Nqo1) and glutathione S-transferase a1 (Gsta1), which are detoxifying enzymes regulated by Nrf2, were low in Nrf2-null mouse livers and markedly high in Keap1-kd mouse livers following control diet ingestion, and this trend was similar to that of Nrf2 expression (Fig. 3b). Following treatment with MCDD, WT mouse livers showed an increase in the expression levels of γ-GCS, Nqo1 and Gsta1, and these increases were similar but slower than the increase in Nrf2 expression; their expressions were minimal or did not occur in Nrf2-null mouse livers. In Keap1-kd mouse livers, the basal expression level was high, and MCDD treatment did not markedly increase expression. However, Nqo1, Gsta1 and γ-Gcs expression levels following MCDD treatment were markedly higher in Keap1-kd mouse livers than in WT and Nrf2-null mouse livers.
Regulation of iron excretion from hepatocytes and suppression of liver tissue iron deposition by Nrf2
Histopathological examination showed that MCDD treatment resulted in a significant increase in iron deposition in Nrf2-null mouse livers, while it suppressed iron deposition in Keap1-kd mouse livers (Fig. 1a, b). Non-heme iron levels in the liver tissue were measured to directly evaluate liver tissue iron levels. Liver tissue non-heme iron levels after 13 weeks of control diet ingestion were significantly higher in Nrf2-null mouse livers than in WT mouse livers (Fig. 4a). MCDD treatment increased liver tissue non-heme iron levels in WT, Nrf2-null and Keap1-kd mice. Following 13-week MCDD treatment, non-heme iron levels were significantly higher in Nrf2-null mouse livers than in WT mouse livers and were significantly lower in Keap1-kd mouse livers. These results are consistent with the results of tissue staining using Berlin blue (Fig. 1a, b).

The role of Nrf2 on iron metabolism in liver and isolated hepatocyte. a Non-heme iron contents of liver tissue in the WT, Nrf2-null and Keap1-kd mice fed an MCDD for 6 or 13 weeks. Data are given as mean ± SE (n = 8–15/group). # P < 0.05, ## P < 0.01, significantly different from the WT with control feeding; a P < 0.05, aa P < 0.01, significantly different from the Nrf2-null with control feeding; † P < 0.05, ++ P < 0.01, significantly different from the WT with MCD feeding; b P < 0.05, bb P < 0.01, significantly different from the Nrf2-null with MCD feeding; brackets *P < 0.05, **P < 0.01, significantly different between the two groups. b Serum hepcidin 1 in the WT, Nrf2-null and Keap1-kd mice fed an MCDD for 6 or 13 weeks. Data are given as mean ± SE (n = 7–8/group). c Steady-state Fpn1 mRNA levels of the WT, Nrf2-null and Keap1-kd mice fed a control or MCDD for 6 or 13 weeks. Data are given as mean ± SE (n = 6–7/group). d Immunoblot analysis of Fpn1 and hepcidin gene (Hamp) proteins in livers of the WT, Nrf2-null and Keap1-kd mice fed a control diet or an MCDD for 6 or 13 weeks. Bar graph shows quantitation of optical density of the immunoblots. Data are given as mean ± SE (n = 8/group). e Non-heme iron contents and f iron release of isolated hepatocyte from the WT, Nrf2-null and Keap1-kd mice with or without an MCD medium for 24 h. The means of three independent experiments are shown with the SEM
Iron uptake by hepatocytes takes place primarily via the transferrin receptor (TfR), and iron excretion from hepatocytes is primarily mediated by Fpn1. Moreover, hepcidin 1 binds to Fpn1 and induces its internalization and degradation. This restricts iron release from liver iron stores. The liver Fpn1 expression level was analyzed using quantitative PCR and immunoblot analysis (Fig. 4c, d). Fpn1 mRNA levels rose significantly in WT and Keap1-kd mouse livers following MCDD treatment, while elevation was not observed in Nrf2-null mouse livers following MCDD treatment. The elevation in Fpn1 mRNA level following MCDD treatment was generally greater in Keap1-kd mouse livers than in WT mouse livers (Fig. 4c). Fpn1 protein expression level following control diet ingestion showed no significant difference among WT, Nrf2-null and Keap1-kd mouse livers. There was also no significant difference in this parameter after 6-week MCDD treatment. However, following 13-week MCDD treatment, Fpn1 expression increased significantly in WT and Keap1-kd mouse livers (Fig. 4d). The increase of Fpn1 expression was more significant in the Keap1-kd mouse livers than in the WT mouse livers. These results suggest that MCDD treatment induces activation of iron excretion from the liver in Keap1-kd mice but not in Nrf2-null mice.
Regarding the serum hepcidin 1 levels (Fig. 4b), at 6 weeks after the start of either the control or MCD diet, the levels were not significantly different among the WT, Nrf2-null and Keap1-kd mice. At 13 weeks after the start of a control diet, the levels were lower in the Keap1-kd mice compared with WT and Nrf2-null mice. At 13 weeks after the start of MCDD, the levels were decreased significantly in WT mice but not in Nrf2-null and Keap1-kd mice. The hepatic Hamp levels (Fig. 4d) were significantly lower in the Keap1-kd mice compared with WT and Nrf2-null mice at 6 and 13 weeks after the start of a control diet. After MCDD treatment for 6 and 13 weeks, the levels were significantly decreased in the livers of WT and Nrf2-null mice but not in Keap1-kd mice (Fig. 4d). Taken together, the expression levels of hepcidin 1 and Hamp were not increased in the serum and livers of Nrf2-null mice fed MCDD. Therefore, in this study, the results of the increased hepatic iron contents associated with the loss of Fpn1 induction in Nrf2-null mice fed MCDD would not be attributed to an increase in the expression levels of hepcidin 1 and Hamp.
For a more detailed analysis of iron metabolism in hepatocytes, we isolated hepatocytes from WT, Nrf2-null and Keap1-kd mouse livers, and changes in iron metabolism depending on Nrf2 expression level were analyzed using primary hepatocyte cultures (Fig. 4e, f). To reproduce in vivo environments, hepatocytes from WT, Nrf2-null, and Keap1-kd mice were divided into an ordinary medium culture group and an MCD medium culture group. Analysis of intracellular iron levels revealed significantly higher iron levels in Nrf2-null mouse hepatocytes in the MCD medium culture group (Fig. 4e). An analysis of iron kinetics using radioisotope-labeled iron showed that Nrf2-null mice excrete significantly less iron from hepatocytes compared to WT and Keap1-kd mice (Fig. 4f). However, no increase in RI excretion was noted in Keap1-kd mouse hepatocytes as compared to that in WT mouse hepatocytes (Fig. 4f).
Effects of sulforaphane (an Nrf2 activator) in alleviating MCDD-induced steatohepatitis
Whether or not activation of Nrf2 can suppress steatohepatitis progression was examined from a therapeutic perspective. SFN, an Nrf2 activator, was administered together with MCDD to evaluate the effects of this treatment in WT mice. Histopathological changes of the liver were compared between WT mice treated with MCDD alone and WT mice treated with MCDD + SFN for 13 weeks (Fig. 5a). Figure 5b shows pathological changes resulting from this treatment, with NAS serving as an indicator. HE staining revealed no significant change in fat droplet deposition. Inflammatory cell infiltration decreased in the SFN-treated group. Evaluation of hepatic fibrosis using Sirius red and MT staining showed that fiber extension (stretching) was less evident in the SFN-treated group, thus indicating suppression of fibrosis. Immunostaining with 4-HNE also revealed suppressed iron deposition and suppressed chromatic response to 4-HNE in the SFN-treated group, while no marked change was noted in the Keap1-kd mouse livers (Fig. 5a). Analysis of NAS also revealed no effect of SFN in alleviating fat deposition, although inflammation and fibrosis were significantly alleviated in the SFN-treated group. Analysis of NAS revealed no significant differences in iron deposition.

Sulforaphane inhibits hepatic oxidative stress and counteracts inflammation and fibrosis in steatohepatitis induced by an MCDD. a H&E-, Sirius red-, Masson trichrome-, Berlin blue- and 4-HNE-stained sections of representative liver specimens from the WT mice fed an MCDD with or without SFN for 13 weeks (bars 100 μm). b NAFLD activity score (NAS) in liver samples fed a control diet or an MCDD with or without SFN for 13 weeks. Values are mean ± SE (n = 10–20/group). *P < 0.05, **P < 0.01, significantly different between the two groups. c Triglyceride, malondialdehyde (MDA) and non-heme iron contents of liver tissue in the WT mice fed a control diet or an MCDD with or without SFN for 6 or 13 weeks. Data are given as mean ± SE (n = 8–15/group). d Immunoblot analysis of α-Sma proteins in livers of the WT mice fed a control diet or an MCDD with or without SFN for 13 weeks. Bar graph shows quantitation of optical density of the immunoblots. Data are given as mean ± SE (n = 8/group). e Immunoblot analysis of Nrf2 proteins in nuclear fraction of livers of the WT mice fed a control diet or an MCDD with or without for 2 days or 6 or 13 weeks. Data are given as mean ± SE (n = 8/group)
Next, we measured liver tissue neutral fat, MDA and non-heme iron levels in the livers with or without SFN treatment for 13 weeks (Fig. 5c). Neutral fat level generally decreased following SFN treatment, although this change was not statistically significant. The MDA level improved significantly following SFN treatment. Thus, oxidative stress was suppressed by SFN treatment. The non-heme iron level decreased slightly yet significantly following 13-week SFN treatment, thereby indicating significant suppression of iron accumulation. GSH levels showed no elevation following SFN treatment (data not shown). α-Sma expression was suppressed following 13-week SFN treatment, thus indicating the suppression of stellate cell activation (Fig. 5d).
Changes in Nrf2 expression level following SFN treatment were analyzed, and an increase in expression (activation) was noted early (2 days after the start of treatment) compared to that found in the group treated with MCDD alone (Fig. 5e). However, there was no difference in the Nrf2 expression level at 6 weeks after the start of treatment between the MCDD + SFN treatment group and the MCDD alone treatment group. At 13 weeks, Nrf2 expression was no longer apparent in both the MCDD + SFN treatment group and the MCDD alone treatment group. These results indicate that activation of Nrf2 by MCDD + SFN treatment occurs earlier than that following MCDD alone, but the difference in Nrf2 activation levels between these two groups eventually decreases. Changes in liver tissue Fpn1 expression following SFN treatment were analyzed using immunoblot analysis. Fpn1 expression increased both in the groups with and without SFN treatment, and did not differ depending on whether or not SFN was administered together with MCDD (data not shown).
Discussion
The results of this study have demonstrated the importance of hepatic Nrf2 and its downstream signaling in the development of nutritional steatohepatitis, using a mouse model induced by an MCDD. The sustained activation of Nrf2, which is found in the livers of Keap1-kd mice, exhibits a protective role against the exacerbation of the pathological state of the steatohepatitis. Moreover, the Nrf2 activation induced by a potent Nrf2 activator, SFN, partially prevents the progression of the pathological state of the steatohepatitis.
Nrf2 serves as an oxidative stress sensor and functions as a comprehensive host defense factor [11, 12, 14]. It has been reported that oxidative stress plays an important role in both the onset and progression of NASH [1, 2, 5–8]. Important sources of oxidative stress involved in NASH are thought to be ROS and radicals from fatty acid β-oxidation, inflammatory cytokines and/or iron accumulation [1, 2, 4, 20, 23]. In this study, increased accumulation of MDA and 4-HNE, i.e., increased oxidative stress in the livers, was noted in Nrf2-null mouse livers, while this accumulation did not occur in Keap1-kd mouse livers. γ-Gcs, Nqo1 and Gsta1, which are known as antioxidative stress response genes regulated by Nrf2 and factors involved in direct elimination of ROS [11, 12], were not induced in Nrf2-null mouse livers administered MCDD, while they were markedly induced in Keap1-kd mouse livers (Fig. 3b). The differences in these defense systems against oxidative stress should lead to the observed resistance in Keap1-kd mice, although, on the other hand, lead to the susceptibility in Nrf2-null mice in terms of the onset and progression of steatohepatitis induced by MCDD. Moreover, in this study, expression levels of factors involved in inflammation (inflammatory cytokines such as Tnf-α and Mcp-1) and factors involved in fibrosis (fiber growth factors such as α-Sma, Tgf-β1, and α1-procollagen) were upregulated in Nrf2-null mice, while they were down-regulated in Keap1-kd mice. Both experimental and clinical studies have shown that prolonged exposure to oxidative stress results in the progression of hepatic inflammation and fibrosis [3, 8]. To be consistent, this study has shown that the enhancement of the antioxidative defense systems induced by sustained Nrf2 activation leads to a marked suppression against the progression of hepatic inflammation and fibrosis.
In this study, excessive iron accumulated in Nrf2-null mouse livers; this is an interesting source of oxidative stress in mouse models of MCDD-induced steatohepatitis (Figs. 1a, 4a). Fpn1 is the only transporter known to be involved in the excretion of iron from liver tissue [24]. A recent study demonstrated that regulation of macrophages by Fpn1 involves Nrf2 [18]. The present study also examined the iron balance in liver tissue and hepatocytes. Iron staining using Berlin blue (Fig. 1a, b) and measurement of the non-heme iron level (Fig. 4a) in the mouse liver tissue following MCDD treatment revealed a significantly higher iron level in Nrf2-null mice liver tissue and a lower level in Keap1-kd mice. However, the expression levels of hepcidin 1 and Hamp, which are inhibitory molecules of Fpn1, were not significantly different between these two kinds of mice fed MCDD (Fig. 4b, d). Furthermore, primary hepatocyte cultures were examined, and we found that hepatocytes from Nrf2-null mice showed reduced excretion of radioisotope-labeled iron. Only hepatocytes from Nrf2-null mice showed iron accumulation following exposure to MCD medium (Fig. 4e, f), thereby suggesting that Nrf2 is an indispensable factor involved in excretion of iron from hepatocytes in mouse models of MCDD-induced steatohepatitis.
In macrophages reported by Harada et al. [18], activation of Nrf2 resulted in increased expression of Fpn1 and enhanced excretion of radioisotope-labeled iron. In the liver tissue, however, an increase in Fpn1 expression was not potent even in Keap1-kd mice in which Nrf2 was permanently highly expressed, and no increase in RI excretion was noted in Keap1-kd mouse hepatocytes as compared to that in WT mouse hepatocytes (Fig. 4c, d, f). To date, only Fpn1 has been identified as a transporter for excreting iron from hepatocytes, but we cannot rule out the presence of unknown factor(s) involved in this process.
Initial treatment of NASH uses diet and exercise therapy; no first-line drug therapy for NASH has been established. Recently, large-scale interventional studies have demonstrated the effectiveness of vitamin E known as an antioxidant in adults with NASH [25]. In the present study, SFN (an Nrf2 activator) was administered together with MCDD to evaluate its effect in alleviating steatohepatitis, with an expectation of steatohepatitis alleviation through drug-induced Nrf2 activation. SFN is known to be contained in broccoli sprouts and other plant foods, and was recently shown to serve as a potent Nrf2 activator [21]. Treatment with SFN did not alleviate fat droplet deposition in histopathological specimens, but did suppress inflammation and fibrosis (Fig. 5a, b). Additionally, oxidative stress was suppressed by SFN treatment (Fig. 5a, c). These effects may be attributable to the enhanced oxidative stress scavenging potential following hepatic Nrf2 activation. The effect of SFN in suppressing liver tissue iron accumulation was not as evident as that seen in Keap1-kd mice, which express excessive Nrf2. SFN rapidly induced Nrf2 activation over approximately 2 days, in contrast to activation by MCDD alone, which occurred over approximately 1 week (Fig. 5d). Considering that Nrf2 expression permanently occurs in excess in Keap1-kd mice, prior activation of Nrf2 may be important in suppressing the progression of steatohepatitis. Thus, SFN is promising for preventing the progression of steatohepatitis.
In conclusion, this study using mouse models of MCDD-induced steatohepatitis revealed that Nrf2 plays an important role in regulating the onset and progression of this disease. Examination of the pathophysiological background for these effects of Nrf2 suggests that Nrf2 is a comprehensive factor that defends the host from oxidative stress in various aspects and stages (Fig. 6). The alleviation of steatohepatitis following treatment with SFN suggests the feasibility of drug therapy using Nrf2 activators. Nrf2 activation by pharmaceutical intervention could be a new option for prevention and treatment of steatohepatitis.

Schematic summary for the protective role of Nrf2 against oxidative stress-induced liver injury in steatohepatitis induced by an MCDD. Examination of the pathophysiological background suggests that Nrf2 is a comprehensive factor that defends the host from oxidative stress in various aspects and stages. These effects include not only host defense against oxidative stress by direct elimination of active oxygen through stimulation of antioxidative stress response genes (a primary role of Nrf2 according to a conventional view), but also regulation of iron metabolism in new hepatocytes (shown in the present study), among other effects. It appears likely that suppression of oxidative stress by Nrf2 leads to suppression of stellate cell activation, thus allowing suppression of hepatic fibrosis progression
Abbreviations
- α-Sma:
-
Alpha-smooth muscle actin
- ALP:
-
Alkaline phosphatase
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- Fpn1:
-
Ferroportin-1
- γ-Gcs:
-
γ-Glutamylcysteine synthetase
- GSH:
-
Glutathione
- Gst:
-
Glutathione S-transferase
- Hamp:
-
Hepcidin gene
- 4-HNE:
-
4-Hydroxy-2-nonenal
- Keap1:
-
Kelch-like Ech-associated protein 1
- MCDD:
-
Methionine- and choline-deficient diet
- MDA:
-
Malondialdehyde
- NASH:
-
Non-alcoholic steatohepatitis
- Nrf2:
-
Nuclear factor-E2-related factor-2
- Nqo1:
-
NAD(P)H: quinone oxidoreductase 1
- ROS:
-
Reactive oxygen species
- SFN:
-
Sulforaphane
- TfR:
-
Transferrin receptor
- Tgf:
-
Transforming growth factor
- WT:
-
Wild type
References
Torres DM, Harrison SA. Diagnosis and therapy of nonalcoholic steatohepatitis. Gastroenterology. 2008;134:1682–98.
Vuppalanchi R, Naga C. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: selected practical issues in their evaluation and management. Hepatology. 2009;49:306–17.
Falch-Ytter Y, Younossi ZM, Marchesini G, McCullough AJ. Clinical features and natural history of nonalcoholic steatosis syndromes. Semin Liver Dis. 2001;21:17–26.
Adams LA, Lymp JF, Sauver JS, Sanderson SO, Lindor KD, Feldstein A, et al. The natural history of nonalcoholic fatty liver disease: a population based cohort study. Gastroenterology. 2005;129:113–21.
Malaguarnera L, Madeddu R, Palio E, Arena N, Malaguarnera M. Heme oxygenase-1 levels and oxidative stress-related parameters in non-alcoholic fatty liver disease patients. J Hepatol. 2005;42:585–91.
Sumida Y, Nakashima T, Yoh T, Furutani M, Hirohama A, Kakisaka Y, et al. Serum thioredoxin levels as a predictor of steatohepatitis in patients with nonalcoholic fatty liver disease. J Hepatol. 2003;38:32–8.
Tomita K, Oike Y, Teratani T, Taguchi T, Noguchi M, Suzuki T, et al. Hepatic AdipoR2 signaling plays a protective role against progression of nonalcoholic steatohepatitis in mice. Hepatology. 2008;48:458–73.
Yu J, Chu ES, Wang R, Wang S, Wu CW, Wong VWS, et al. Heme oxygenase-1 protects against steatohepatitis in both cultured hepatocyte and mice. Gastroenterology. 2010;138:694–704.
Leonarduzz G, Scavazza A, Biasi F, Chiarpotto E, Camandola S, Vogel S, et al. The lipid peroxidation end product 4-hydroxy-2,3-nonenal up-regulates transforming growth factor beta expression in the macrophages lineage: a link between oxidative injury and fibrosclerosis. FASEB J. 1997;11:851–7.
Matsuzawa N, Takamura T, Kurita S, Nisu H, Ota T, Ando H, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46:1392–403.
Zhang DD, Hannink M. Distinct cysteine in Keap1 are required for Keap1-dependent ubiquitination on Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–51.
Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dithiole-3-thione. Mol Med. 2001;7:135–45.
Okada K, Shoda J, Taguchi K, Maher JM, Ishizaki K, Inoue Y, et al. Ursodeoxycholic acid stimulates Nrf2-mediated hepatocellular transport, detoxification, and antioxidative stress systems in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G735–47.
Okada K, Shoda J, Taguchi K, Maher JM, Ishizaki K, Inoue Y, et al. Nrf2 counteracts cholestatic liver injury via stimulation of hepatic defense systems. Biochem Biophys Res Commun. 2009;389:431–6.
Sugimoto H, Okada K, Shoda J, Warabi E, Ishige K, Ueda T, et al. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2010;298:G283–94.
Chowdhry S, Nazmy MH, Meakin PJ, Dinkova-Kostova AT, Walsh SV, Tsujita T, et al. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic Biol Med. 2010;48:357–71.
Zhang YKJ, Yeager RL, Tanaka Y, Klaassen CD. Enhanced expression of Nrf2 in mice attenuated the fatty liver produced by a methionine- and choline-deficient diet. Toxicol Appl Pharmacol. 2010;245:326–34.
Harada N, Kanayama M, Maruyama A, Yoshida A, Tazumi K, Hosoya T, et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch Biochem Biophys. 2011;508:101–9.
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3.
George DK, Goldwurm S, Macdonald GA, Cowley LL, Walker NI, Ward PJ, et al. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology. 1998;114:311–8.
Shinkai Y, Sumi D, Fukami I, Ishii T, Kumagai Y. Sulforaphane, an activator of Nrf2, suppresses cellular accumulation of arsenic and its cytotoxicity in primary mouse hepatocytes. FEBS Lett. 2006;580:1771–4.
Foy AL, Williams HL, Cortell S, Conrad ME. A modified procedure for the determination of non-heme iron in tissue. Anal Biochem. 1967;18:559–63.
Imeryuz N, Tahan V, Sonsuz A, Eren F, Uraz S, Yuksel M, et al. Iron preloading aggravates nutritional steatohepatitis in rats by increasing apoptotic cell death. J Hepatol. 2007;47:851–9.
Garrick MD. Human iron transporters. Genes Nutr. 2011;6:45–54.
Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85.
Acknowledgments
This work was supported in part by a Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (19791054) and Grants-in-Aid from Nakayama Cancer Research Institute (Tokyo, Japan).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Okada, K., Warabi, E., Sugimoto, H. et al. Nrf2 inhibits hepatic iron accumulation and counteracts oxidative stress-induced liver injury in nutritional steatohepatitis. J Gastroenterol 47, 924–935 (2012). https://doi.org/10.1007/s00535-012-0552-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-012-0552-9