
Abstract
Autophagy, a highly regulated cellular process, assumes a dual role in the context of cancer. On the one hand, it functions as a crucial homeostatic pathway, responsible for degrading malfunctioning molecules and organelles, thereby maintaining cellular health. On the other hand, its involvement in cancer development and regression is multifaceted, contingent upon a myriad of factors. This review meticulously examines the intricacies of autophagy, from its molecular machinery orchestrated by Autophagy-Related Genes (ATG) initially discovered in yeast to the various modes of autophagy operative within cells. Beyond its foundational role in cellular maintenance, autophagy reveals context-specific functions in processes like angiogenesis and inflammation. Our analysis delves into how autophagy-related factors directly impact inflammation, underscoring their profound implications for cancer dynamics. Additionally, we extend our inquiry to explore autophagy’s associations with cardiovascular conditions, neurodegenerative disorders, and autoimmune diseases, illuminating the broader medical relevance of this process. Furthermore, this review elucidates how autophagy contributes to sustaining hallmark cancer features, including stem cell maintenance, proliferation, angiogenesis, metastasis, and metabolic reprogramming. Autophagy emerges as a pivotal process that necessitates careful consideration in cancer treatment strategies. To this end, we investigate innovative approaches, ranging from enzyme-based therapies to MTOR inhibitors, lysosomal blockers, and nanoparticle-enabled interventions, all aimed at optimizing cancer treatment outcomes by targeting autophagy pathways. In summary, this comprehensive review provides a nuanced perspective on the intricate and context-dependent role of autophagy in cancer biology. Our exploration not only deepens our understanding of this fundamental process but also highlights its potential as a therapeutic target. By unraveling the complex interplay between autophagy and cancer, we pave the way for more precise and effective cancer treatments, promising better outcomes for patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autophagy is a highly regulated and essential intracellular pathway that plays a crucial role in maintaining cellular health and balance. It governs various cellular processes, such as cell survival, adaptability, and programmed cell death, and it is involved in the management of pathological conditions. This intricate process involves the coordination of multiple genes and proteins within the body (Chun and Kim 2018). At its core, autophagy is a catabolic process that facilitates the degradation of cellular components by delivering them to lysosomes. This degradation is achieved through the sequestration of these components within double-walled lipid structures known as autophagosomes, which subsequently merge with lysosomes containing hydrolytic enzymes. The substrates or materials targeted for autophagy can originate from within the cell (endogenous) or from outside the cell (exogenous). Endogenous substrates include intracellular pathogens, damaged mitochondria, aggregated non-functional proteins, and nuclear fragments. On the other hand, exogenous substrates encompass extracellular pathogens like viruses and bacteria, as well as other molecules or chemicals (Galluzzi, Baehrecke, et al. 2017). Autophagy plays a central role in regulating the normal and healthy state of cells. Any disruption or impairment of this process can have serious implications and contribute to the development of various pathologies, including different types of cancers. Therefore, autophagy has emerged as a potential target for therapeutic approaches, particularly in the field of cancer treatment. By understanding and manipulating the mechanisms of autophagy, researchers and medical professionals can explore novel strategies to modulate this process to promote cell survival or induce selective cell death in cancer cells. By targeting autophagy, it may be possible to develop therapies that specifically impact cancer cells while sparing normal healthy cells, offering a promising avenue for future treatments (Galluzzi, Bravo-San Pedro, et al. 2017). Overall, autophagy represents a complex and interconnected cellular pathway that influences cell fate and contributes to the maintenance of cellular equilibrium. Its dysregulation can have profound consequences, making it a focal point for studying and developing therapeutic interventions, particularly in the context of cancer (Galluzzi et al. 2015), neurodegenerative disorders (Frake et al. 2015), aging (Rubinsztein et al. 2011), inflammation (Netea-Maier et al. 2016), immunity (Gomes & Dikic 2014) and genome stability (Goldsmith et al. 2014), and cardiovascular abnormalities (Shirakabe et al. 2016). In the past, this process was commonly understood as a tumor suppressor mechanism. However, recent studies and findings have revealed a more complex picture, recognizing it as a “double-edged weapon” that can both impede and promote the onset and advancement of cancer in certain circumstances (Chavez-Dominguez et al. 2020). It is triggered by a range of stimuli or stresses, such as hypoxia (low oxygen levels), withdrawal of growth factors, nutrient deprivation, oxidative stress, and infection (Goldsmith et al. 2014). Autophagy, a highly complex cellular process responsible for the degradation and recycling of damaged or unnecessary cellular components, holds a multifaceted and intriguing role in the context of cancer. This intricate relationship between autophagy and cancer is characterized by a paradoxical duality, as it exhibits both tumor-suppressive and tumor-promoting effects depending upon the stage and context of the disease. In the early stages of cancer, autophagy often acts as a tumor suppressor, playing a vital role in maintaining cellular homeostasis by eliminating defective organelles, proteins, and mitigating oxidative stress. Key autophagy-related genes (ATGs), including BECN1, are essential for this tumor-suppressive function. However, as cancer progresses and becomes established, autophagy takes on a dual role (Verma et al. 2021; Yun and Lee 2018). It not only supports the survival and proliferation of cancer cells, helping them adapt to stressors like hypoxia and nutrient scarcity within the tumor microenvironment, but also fuels their metabolic demands by recycling intracellular components for energy production (Zaarour et al. 2021). Moreover, autophagy can contribute to various hallmarks of cancer, including promoting epithelial–mesenchymal transition (EMT), sustaining proliferation, facilitating angiogenesis, enhancing invasion and metastasis, maintaining cancer stem cell properties, and orchestrating tumor metabolism reprogramming (Gugnoni et al. 2016). Despite substantial advancements in understanding autophagy’s involvement in cancer, numerous enigmatic facets remain to be explored through rigorous scientific investigation, holding the potential to uncover novel therapeutic strategies and shed light on the complex mechanisms underpinning cancer progression. The complex interplay between autophagy, inflammation, and tumorigenesis is a subject of intense research and has far-reaching implications for understanding cancer biology and developing novel therapeutic strategies (Patergnani et al. 2021). In cancer development, inflammation can exert both promotive and inhibitory effects contingent upon contextual elements and molecular participants. Conversely, autophagy, a fundamental cellular process responsible for preserving cellular equilibrium through the degradation of damaged organelles and proteins, profoundly influences the inflammatory response (Yang et al. 2011). Dysregulated autophagy has intricate links to cancer progression. This connection pivots on various factors, chiefly the release of damage-associated molecular patterns (DAMPs) from stressed or damaged cells. DAMPs can initiate an innate immune response, recruiting immune cells like macrophages to sites of injury or tumors (Zhang et al. 2013). While a crucial part of the body’s defense, this response may foster chronic inflammation, a recognized cancer initiation and progression risk. Specialized autophagic processes, such as mitophagy targeting damaged mitochondria, can amplify the inflammatory response (Gkikas et al. 2018). Mitochondria, pivotal for energy generation and immune signaling regulation, release reactive oxygen species (ROS) and other pro-inflammatory molecules when dysfunctional. Impaired mitophagy sustains these damaged mitochondria, further fueling chronic inflammation and potentially supporting tumorigenesis (Tirichen et al. 2021). Molecular actors, including heat shock proteins (HSPs) and autophagy-related genes (ATGs), serve as vital modulators within this intricate interplay. HSPs have the capacity to activate the NF-κB pathway, prompting the elevation of inflammatory mediators like high-mobility group protein B1 (HMGB1) (Penke et al. 2018). Furthermore, autophagy can participate in the secretion of inflammatory cytokines such as IL-6 in response to certain mutations, intensifying inflammation and possibly enhancing cancer cell invasiveness.
Comprehending this multifaceted relationship between autophagy, inflammation, and tumorigenesis is critical for developing targeted therapies manipulating these processes for therapeutic advantage. Researchers are actively exploring autophagy modulators to enhance the immune response against cancer. Concurrently, strategies inhibiting autophagy in cancer cells to heighten treatment sensitivity are under investigation (Amaravadi et al. 2016). This ongoing exploration holds substantial promise for advancing cancer biology understanding and improving treatment outcomes. To gain a comprehensive grasp of autophagy and distinguish it from other cellular catabolic pathways, like proteasomal degradation, is fundamental. Proteasomal degradation involves the breakdown of short-lived, misfolded, damaged, or surplus proteins via the 26S proteasome, a complex proteolytic machinery, followed by engulfment by phagocytes like macrophages and dendritic cells. Conversely, autophagy predominantly revolves around lysosomal degradation, marked by sequestration of cellular components within autophagosomes that subsequently merge with lysosomes for degradation. A pivotal distinction lies in the fate of degraded material; proteasomal degradation yields short peptides for cellular repurposing, whereas autophagy leads to complete degradation into constituent molecules for recycling. These distinctions provide clearer insight into autophagy’s unique role, implications in cellular homeostasis, disease, and therapeutic strategies.
Sizeable literature suggests role of autophagy in cancer and inflammation. To talk about inflammation, this process is body’s response at injury site or any stimuli or pathogen invasion and is a complex signaling pathway involving whole lot of chemicals and receptors in case of continuous stimulus or chronic inflammation abnormal cell proliferation, damage, fibrosis etc. starts there is when autophagy come into play, and hence, a few factors that relate inflammation to autophagy are also briefly discussed. Literature guides that cancer is one of the most dreadful disease of mankind and we apply know it all theory for this disease and since autophagy is like cleaning the unwanted biomolecule system it is unlikely they remain uninteracted. Interestingly, their interaction is cancer stage dependent as autophagy suppresses tumor at early stages but promotes the same at advance stages. Autophagy is also assumed to play notable roles in various anomalies related with brain like neurodegeneration, Alzheimer’s disease (AD), and Parkinson disease (PD) which are still a battle to win for mankind. The absence of autophagy leads to abnormal or harmful protein accumulation that leads to aggregation leads to oxidative stress, tau protein hyperphosphorylation, and ER stress. Therefore, neurodegenerative disorders also have a direct connection to autophagy which is an area to be explored (F. Guo et al. 2018). Advances in bioelectronics has shifted cancer therapies to target-based therapies using compatible nanoparticles. Multiple types of nanoparticles are now synthesized in vitro for attacking specific targets in autophagic process or tumor biomarkers to diagnose and treat cancers using autophagy remodeling.
Different forms of autophagy
Autophagy can also occur in a selective manner, where it relies on specific autophagy receptors known as selective autophagy receptors (SARs) to facilitate the targeted degradation of specific autophagic substrates. These substrates can include protein aggregates, pathogens, and other cellular components that need to be eliminated. This form of autophagy is aptly named “selective autophagy” due to its ability to specifically target and degrade particular cellular materials, thereby contributing to the maintenance of cellular quality control and homeostasis (Galluzzi et al. 2017). Selective autophagy is a fundamental cellular process that operates in both normal and induced situations, playing a crucial role in maintaining cellular homeostasis. In non-induced or normal circumstances, selective autophagy serves as an ongoing mechanism for the clearance of specific cellular components, such as damaged organelles, misfolded proteins, and protein aggregates. This continuous clearance process helps prevent the accumulation of harmful materials and contributes to the overall health and functionality of the cell.
In induced situations, selective autophagy is triggered in response to specific signals or stresses, such as nutrient deprivation, oxidative stress, or pathogen invasion. These signals activate selective autophagy receptors (SARs), which act as molecular adapters, recognizing and binding to the targeted substrates to be degraded. The selective autophagy receptors then recruit the autophagic machinery, guiding the engulfment of the substrates into specialized vesicles called autophagosomes. Subsequently, the autophagosomes fuse with lysosomes, leading to the degradation of the captured substrates by lysosomal hydrolases (Zaffagnini and Martens 2016). By selectively targeting and eliminating specific substrates, selective autophagy contributes to cellular quality control, ensuring the removal of damaged or obsolete components. This process helps maintain cellular homeostasis by regulating organelle turnover, protein quality control, and defense against intracellular pathogens. Defects in selective autophagy have been implicated in various pathological conditions, including neurodegenerative diseases, cancer, and infections. Thus, understanding the intricate mechanisms underlying selective autophagy is of significant scientific interest, as it provides insights into the fundamental processes governing cellular health and disease (Farré and Subramani 2016).
Autophagy can also occur in a non-selective manner, known as non-selective autophagy, where the process operates without the involvement of selective autophagy receptors (SARs) and lacks specific targeting of substrates. In non-selective autophagy, the engulfment of substrates into autophagosomes happens randomly, without any specific recognition or selection of cellular components. Unlike selective autophagy, which selectively targets specific substrates such as protein aggregates or damaged organelles, non-selective autophagy is characterized by a more generalized and indiscriminate approach. It involves the engulfment of a broad range of cellular materials, including various cytoplasmic components and organelles, in a somewhat random manner. This random influx of substrates into autophagosomes distinguishes non-selective autophagy from its selective counterpart. Non-selective autophagy serves as a vital process in cellular maintenance and adaptation, particularly during times of cellular stress, nutrient deprivation, or energy imbalance. It plays a role in removing excess or damaged cellular components, recycling cellular building blocks, and generating energy. While non-selective autophagy lacks the specificity of selective autophagy, it still contributes to cellular homeostasis and supports the overall health and survival of the cell. Understanding the interplay between selective and non-selective autophagy is crucial for comprehending the diverse strategies employed by cells to maintain their functionality under various physiological and pathological conditions. Both selective and non-selective autophagy mechanisms work in concert to ensure cellular quality control and adaptability, highlighting the complexity and importance of autophagy as a fundamental cellular process (Gatica et al. 2018). Major forms of autophagic process seen inside cells are as follows:
Chaperone-mediated autophagy (CMA)
This unique form of autophagy, known as chaperone-mediated autophagy, represents a distinct variation in the autophagic process. Unlike the traditional pathways that involve autophagosome formation and vesicular engulfing, chaperone-mediated autophagy operates through a different mechanism. It does not require these conventional steps for the transfer of cargo or targets to the lysosomes for degradation. Instead, targets are directly translocated to the lumen of the lysosome utilizing a protein-translocation complex present on the lysosome membrane. Chaperone-mediated autophagy primarily focuses on targeting soluble proteins rather than other macromolecules or organelles. Within this process, heat shock proteins, specifically HSC70 and HSPA8, play a crucial role. They recognize and interact with a specific pentapeptide motif found on the target proteins, facilitating their translocation (Kaushik and Cuervo 2012), the KFERQ sequence in the target (Vakifahmetoglu-Norberg et al. 2013). According to Cuervo et al. (Cuervo and Dice 1996), interaction of target protein with HSC protein causes a little misfolding in the target protein before its direct transportation to the lysosome, and transport into lysosome is facilitated by lysosome-associated membrane protein 2A (LAMP2A). LAMP-2A levels and assembly/disassembly on the lysosomal membrane are rate-limiting steps for CMA. Several chaperones, including HSP90, HSP40, Hop, Hip, and BAG-1, facilitate substrate unfolding, enhance binding to LAMP2A, and stabilize LAMP2A during multimerization. CMA regulation involves phosphorylation and dephosphorylation events, with factors like AKT and PHLPP1 affecting CMA activity. Various transcription factors, such as NFAT, NRF2, and TPD52, can modulate CMA expression (Arias et al. 2015; Tekirdag and Cuervo 2018). Studies have shown that macroautophagy switch-off also serves as an inducer for CMA. Recent studies have proven that CMA might be a reason for some neurodegenerative disorders (Orenstein et al. 2013) and its progression, oncogenesis (Kon et al. 2011), and preservation of genome integrity (Park et al. 2015).
Macroautophagy
Macroautophagy is a distinct form of autophagy that differs from other types, such as chaperone-mediated autophagy (CMA) and microautophagy. It involves the formation of specialized lipid-based double-membrane vesicles known as “autophagosomes,” which serve as crucial structures for the process. The process involves the intricate coordination of proteins and lipids from various cellular membranes, including the endoplasmic reticulum (ER), ER/mitochondria contact sites (MAM), ER exit sites, recycling endosomes, Golgi, and the plasma membrane. Over 40 autophagy-related proteins (ATGs), initially discovered in yeast, play pivotal roles at different stages of macroautophagy. A crucial step involves the conjugation of ATG8 family members like LC3 and GABARAP to phosphatidylethanolamine in precursor membranes. Autophagosomes are sealed with the help of the ESCRT machinery and released, a process mediated by DNM2 (Kohler et al. 2020).
In recent years, macroautophagy has become a subject of extensive scientific investigation due to its pivotal role in safeguarding cellular health. It functions as a quality control mechanism, specifically targeting and eliminating damaged or dysfunctional organelles, aggregated proteins, and other molecules that have the potential to induce severe pathological conditions, including cancer. Researchers are actively exploring the intricate workings of macroautophagy, seeking to unravel its molecular mechanisms and decipher the intricate interplay between ATG genes and protein products. By elucidating these processes, scientists aim to gain a deeper understanding of how macroautophagy can be harnessed to prevent and treat various diseases. The scientific community’s focus on macroautophagy underscores its significance in cellular homeostasis and disease prevention. The knowledge gained through ongoing research efforts holds promise for the development of innovative therapeutic strategies that leverage the potential of macroautophagy to combat a wide range of debilitating conditions, ultimately leading to improved health outcomes for individuals affected by such diseases (Kaur and Debnath 2015). Autophagy serves as a critical mechanism that helps cells maintain their internal balance, known as homeostasis. It plays a crucial role in enabling cells to adapt and withstand a wide range of stressors, including hypoxia (oxygen deprivation), intracellular or extracellular pathogens, starvation, deficiency in growth factors, and various other factors. By engaging in autophagy, cells are empowered to efficiently deal with these stressors, ensuring their survival and overall health (White 2015). Autophagy has a very controversial role when it comes to cancer as it has experimentally proved that it promotes tumor initiation and survival and metastasis depending on the context, but in some cases, at earlier stages, it also helps as tumor suppressor (Onorati et al. 2018).
Selective macroautophagy (mitophagy, ER-phagy, pexophagy, aggrephagy, and lysophagy)
Selective macroautophagy targets specific cellular components for degradation, including damaged organelles and aggregate-prone proteins. Adapter proteins bridge the targeted substrate, often ubiquitinated, and components of the nascent autophagosome, typically LC3 (Gatica et al. 2018). Macroautophagy can be induced by various stresses, including nutrient depletion, growth factor deprivation, oxidative stress, and protein aggregation. While macroautophagy studies have predominantly been conducted in fibroblasts and cancer cell lines, the fundamental principles likely apply to all cell types, with potential adaptations in neurons and glial cells.
Mitophagy
It is a special kind of autophagic process that maintains the pool of mitochondria that somehow became mangled and useless (Fivenson et al. 2017). Mitochondria also called the “powerhouse of the cell” maintains ATP level and is the location of oxidative phosphorylation inside a cell, but mitochondrial dysfunctioning causes some disorders–neurodegenerative disorders like Alzheimer’s disease (Ye et al. 2015), Parkinson’s disease (Ryan et al. 2015), Huntington’s disease (Khalil et al. 2015), and is considered to be one of the hallmarks of aging (López-Otín et al. 2013). PINK1/Parkin is a common and understood pathway for sweeping off damaged mitochondria in mammalian cells (Pickrell and Youle 2015). Intense studies on drosophila for PINK1/Parkin pathway have reported that knockout of Pink1 causes sterility, mitochondrial abnormalities, and stress sensitivity along with defects in locomotion and neuronal injury (Clark et al. 2006). Mitophagy could be PINK1-Parkin-dependent wherein a decline in mitochondrial membrane potential causes activation of protein kinase-ubiquitin kinase (PTEN)-induced kinase 1 (PINK1) due to damaged mitochondria. This activation of PINK1 then prompts ubiquitin ligase PARKIN; moreover, PARKIN polyubiquitinates other proteins of mitochondria. This process facilitates the interaction of the ubiquitin-binding domains of autophagy receptors and further initiation of the formation of phagophores followed by autophagosomes (Fig. 1). However, autophagy receptors can directly be recruited by PINK1 irrespective of PARKIN (Fivenson et al. 2017). Studies revealed that mutations in PINK and PARKIN also contribute to Parkinson’s disease (Pickrell and Youle 2015) and aging (Fivenson et al. 2017), the reason being mitochondrial dysfunctioning. Multiple mitophagy-related proteins are found to perform in a Parkin-independent manner, like Bcl2-L-13, FUNDC1, MUL1, and Nix/BNIP3L, AMBRA1 (Fivenson et al. 2017).Mitophagy, a process critical for maintaining healthy mitochondria, primarily relies on the recruitment of Parkin to damaged mitochondrial membranes by elevated PINK1 levels. This initiates the degradation of dysfunctional mitochondria through autophagy (Wang et al. 2019). Several receptors, including OPTN, NDP52, and TAX1BP1, aid in recruiting the autophagosome machinery to mitochondria.

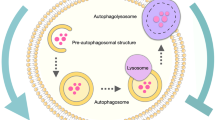
“Diverse Forms of Autophagy and Cargo Selectivity”. This figure illustrates the various forms of autophagy, each with distinct functions and mechanisms. The selectivity of autophagic cargos is regulated by specific cargo receptors or adaptors, which recognize and sequester target substrates for degradation
Macroautophagy also encompasses several distinct selective processes in cellular maintenance. ER-phagy involves receptors like FAM134B, which interact with LC3 or GABARAP, enabling the targeted degradation of specific regions within the Endoplasmic Reticulum (ER) (Ferro-Novick et al. 2021). Pexophagy, on the other hand, relies on the ubiquitination of peroxisomal proteins, recognized by autophagy adapters, such as P62 and NBR1, leading to the degradation of peroxisomes (Cho et al. 2018). In the context of neurodegenerative diseases, aggrephagy plays a crucial role by selectively eliminating misfolded, ubiquitinated proteins with the assistance of adapters like P62, NBR1, OPTN, and TAX1BP1 (Lamark and Johansen 2012). Additionally, lysophagy ensures the clearance of damaged lysosomes through ubiquitination and involves canonical autophagy receptors like TAX1BP1 and TBK1 (Hoyer et al. 2022). These diverse selective autophagic processes are vital for preserving cellular health by eliminating damaged or dysfunctional cellular components.
Microautophagy
Microautophagy, a lesser-explored cousin of macroautophagy, differs significantly in its mechanisms. While macroautophagy has garnered substantial attention, microautophagy remains relatively uncharted territory. One of the notable distinctions is the absence of double-walled autophagosome formation in microautophagy. Instead, it directly includes cargos, either selectively or non-selectively, into lysosomes or endosomes. Subsequently, autophagic bodies or luminal bodies are formed, which are then degraded by hydrolytic enzymes. The core autophagy machinery comprises a set of complex and conserved proteins involved in this process.
The first evidence of microautophagy emerged in the 1960s when rat liver analysis revealed invaginations of lysosomal membranes degrading soluble abnormal proteins (Ahlberg and Glaumann 1985). Since then, significant research has been conducted on microautophagy involving various cargos, including mitochondria, certain cytosolic enzymes, parts of the nucleus, pigment aggregates, and chloroplasts in plants (Ding et al. 2022; Stefaniak et al. 2020). It degrades the cellular components via membrane invaginations in compartments of the endolysosomal system. In mammals, microautophagy pathways target cargo to late endosomes/multivesicular bodies (LE/MVBs). Endosomal microautophagy (eMI) refers to the degradation of cytosolic proteins in LE/MVBs, and it can be selective or non-selective (Krause and Cuervo 2021; Mejlvang et al. 2018). Selective eMI involves recognition of a KFERQ-like motif by HSC70, similar to CMA, followed by internalization into LE/MVBs in an ESCRT-dependent manner. Non-selective eMI, induced by amino acid starvation, targets several macroautophagy receptors and contributes to the regulation of selective macroautophagy. eMI’s regulation and functional implications are still not fully understood in mammals, but it appears to respond to stress and nutrient availability (Gautreau et al. 2014).
Microautophagy occurs through several approaches:
(A) Lysosomal invagination: This process involves the canonical fusion by the invagination of the membrane of apical vacuoles (lysosome-like organelles) and endosomes. This fusion plays a crucial role in nutrient and signaling pathways, particularly in the presence of GTPase Rab7 (GTP-dependent rab7) (Huotari & Helenius 2011). A third pathway resembles the formation of tiny luminal vesicles in endosomes, similar to microautophagy. The Endosomal Sorting Complex Required for Transport (ESCRT) is a vital protein complex that plays a crucial role in organelle membrane remodeling, including invagination and budding. Some studies have shown promising binding between ESCRT proteins and ubiquitylated cargos for selective microautophagy (Gautreau et al. 2014; He et al. 2021).
(B) Endosomal invagination: Also known as endosomal microautophagy, this has been studied in Drosophila melanogaster and dendritic cell lines. This approach recruits ESCRT proteins and accessory proteins like Nbr1 and Hsc70, providing selectivity to the process (Schnebert et al. 2022). Multivesicular bodies or intraluminal vesicles are formed inside endosomes. In D. melanogaster, which lacks Chaperone-Mediated Autophagy (CMA), Hsc70 protein is recruited via electrostatic interactions with phosphatidylserine, deforming the endosomal membrane, a requirement for autophagy. Nbr1, in conjunction with ESCRT-0, aids cargo targeting and the ubiquitination of cytoplasmic proteins on the endosomal surface for delivery into the vacuole (Schnebert et al. 2022; Tekirdag and Cuervo 2018).
(C) Lysosomal protrusion: In this form of microautophagy, membranous or arm-like protrusions arise from lysosomes, as observed in mouse liver cells. These protrusions play a role in maintaining protein homeostasis. While the molecular understanding of this approach in mammals is still under research, in yeast, the extension of these protrusions involves Vac8 and various ATG proteins (Oku et al. 2018).
Various types of microautophagy are observed depending upon the cargo or organelle involved. One example is Micro-ER-phagy, which includes ESCRT-dependent microautophagy and the scission of ER stacks within the vacuole. In this process, excess or misfolded ER stacks and abnormal proteins are converted into spherical whorls, which are subsequently degraded in the vacuole (Schäfer et al. 2020).
Autophagic pathway
Autophagy operates through a highly efficient and tightly controlled pathway or mechanism, characterized by its robustness, sequential nature, and organized regulation. Each form of autophagy follows a multistep process, with multiple genes and their corresponding protein products orchestrating the intricate machinery responsible for degradation. These genes and proteins work in harmony, meticulously monitoring and maintaining the degradation mechanisms involved in autophagy. This well-coordinated process ensures the proper execution of autophagy, allowing cells to effectively eliminate unwanted or damaged components while upholding cellular health (Nakatogawa et al. 2009). CMA, mitophagy, and macroautophagy cycle through different paths, but they all reach a common destination of lysosomal degradation, where different cytotoxic substrates are fed, and after their digestion, the constituents are resupplied for energy replenishment. Term autophagic flux is defined as the rate at which the autophagic machinery spots, characterizes, separates, and dumps off the target (via lysosomal trashing).
Initiation and phagophore formation
Macroautophagy or autophagy pathway begins with the initiation and formation of autophagosome, a double-layer vesicle, and multiple ATG genes are needed for the origination of the autophagosome. In yeast, autophagy have initiation sites called phagophore assembly site (PAS). Stressors like (cytotoxic stressors, nutrient deprivation, hypoxia, oxidative stress, etc.) are responsible for initiating autophagy. Mechanistic target of rapamycin (mTOR) complex I (MTORCI) and AMP-activated protein kinase (AMPK) are two major modulators of the autophagic pathway, where inhibition of (MTORCI) and activation of (AMPK) are considered major stimulators of the process (Huang et al. 2018). Activators or triggering molecules of autophagy inactivate (MTORCI) by signal transduction. (MTORCI) and (AMPK) sense a relative difference in concentrations of AMP and ATP. This ratio gets imbalanced during starvation conditions (nutrient deficiency) (Inoki et al. 2012). (MTORCI) regulates a very crucial complex called unc-51-like autophagy-activating kinase 1 (ULK1, considered as a mammalian ortholog of gene Atg1 present in yeast), by phosphorylating the inhibitory sites of ULK complex. At the time of nutrient deficiency, (MTORCI) dissociates from inhibitory sites and this causes phosphorylation of the active sites and causes activation of the ULK complex (Egan et al. 2011). Moreover, AMPK also stimulates the ULK1 complex by phosphorylating variable locations in the core intrinsically disordered region (IDR), in autophagy and mitophagy Additionally, AMPK inhibits mTORC1 through phosphorylation of the regulatory-associated protein of mTOR (RAP-TOR). ULK1 complex now further activates phosphatidylinositol 3-kinase, catalytic subunit type 3 (PIK3C3, aka VPS34), and BECLIN 1 (Kim et al. 2013). These 2 proteins are part of a multi-protein complex called phosphatidylinositol 3-phosphate (PI3P); this complex synthesizes lipids and hence causes nucleation and biogenesis of autophagosomes (Zhao and Klionsky 2011). PI3P recruitment more of autophagy-specific PI3P effectors, like WD-repeat domain PI-interacting protein-2 (WIPI2). WIPI2 then interacts with ATG12–ATG16L complex to accomplish the expansion of the phagophore (Dooley et al. 2014) (Fig. 1).
Nucleation
The final formation of a mature autophagosome involves several distinct stages, with four functional units playing essential roles in these intricate processes. First, multiple Atg proteins converge at a site known as the pre-autophagosomal structure (PAS), kickstarting the formation of a phagophore, which is the initial isolation membrane (Parzych and Klionsky 2014). The PAS acts as a nucleating site, recruiting various Atg proteins to initiate this crucial step. The process is set in motion by the ULK1/Atg1 complex (Mizushima 2010). In response to nutrient deprivation, ULK1/Atg1 associates with Atg13, FIP200/Atg17, Atg29, and Atg31, forming a complex that acts as the foundation for the PAS scaffold complex. Subsequently, the PI3K complex is recruited to the PAS, facilitating phagophore formation through the interaction between ATG14L and ATG13. Additionally, ATG9A-positive membrane vesicles are brought to the PAS through their interaction with FIP200, anchoring them to this pivotal site. These concerted actions of multiple Atg proteins orchestrate the generation of the isolation membrane (Hitomi et al. 2023). The formation of autophagosomes, crucial components of the autophagy process, involves a series of intricate steps and regulatory complexes. The class III phosphatidylinositol 3-kinase (PtdIns3K) complex, featuring ATG14, is a pivotal player in this process, generating PtdIns3P, essential for macroautophagy in both yeast and mammals. In mammals, this complex can participate in macroautophagy or the endocytic pathway, depending upon its association with ATG14 or UVRAG, respectively (Itakura et al. 2008). Regulation of PtdIns3K complex activity is multifaceted, involving proteins that interact with BECN1, a critical component. BCL2 inhibits macroautophagy by binding BECN1, while KIAA0226/Rubicon suppresses PIK3C3 activity. Positive regulators include AMBRA1 and SH3GLB1/Bif-1. In yeast and mammalian cells, proteins like Atg18, WIPI1, WIPI2, and ZFYVE1 bind to PtdIns3P, playing roles in macroautophagy, though their precise functions are still under investigation (Polson et al. 2010). Understanding the complex regulation of these processes provides valuable insights into the intricacies of autophagosome formation.
This step requires the assembly of phosphatidylinositol 3-phosphate by the activity of class III phosphatidylinositol 3-kinase (PI3K), VPS34 (Goldsmith et al. 2014). After conjugating with VPS34, Beclin 1 complex associates with UV irradiation resistance-associated gene (UVRAG) (Itakura et al. 2008), ATG14L (Matsunaga et al. 2009), and AMBRA1 (Maria Fimia et al. 2007). VPS34 complex relocation is associated with phagophore nucleation. The flexibility of the phagophore is facilitated by the interaction between UVRAG and Bif1 (domain protein). Autophagy is repressed by BCL-2, BCL-xL, Rubicon, AKT, and EGFR by having a negative effect on BECLIN1/VPS34 autophagy-promoting complex (Wei et al. 2013), (Zhong et al. 2009), (Wang et al. 2012a, b).
Elongation and cargo targeting
Next comes the elongation of the phagophore before it matures into autophagosome and becomes fully functional which requires two conjugation systems called LC3 complex (microtubule-associated light-chain B LC3B, the mammalian orthologue of Atg8 gene in yeast) and the ATG16L complex (Goldsmith et al. 2014). ATG16L complex is a multimeric protein complex formed from the ATG5–ATG12 complex binding ATG16 complex. ATG5–ATG12 complex also consists of covalent binding between ATG5 and ATG12 aided by enzymes like ATG7 and ATG10 (Nakatogawa 2013). LC3 complex couples with lipid phosphatidylethanolamine (PE) in the presence of E2-like ATG3 enzyme and ATG7. LC3 along with Atg8 protein complexes facilitate tethering and fusion of the outer membrane followed by the inner one aka hemifusion, indicating importance in closing the phagophore membrane into a functional autophagosome (Fig. 1).
Fusion and feed degradation
The outer membrane of a fully functional autophagosome is formed its merges with that of the lysosome. This fusion yields autolysosome and requires the interference of three families of protein namely—soluble N-ethyl maleimide-sensitive factor attachment proteins (SNAREs) and Rab GTPases, membrane-tethering factors (such as HOPS and EPG5) (Goldsmith et al. 2014). It is seen that autophagophores can either fuse with endosomes or produce an “amphisome”; this amphisome then merges with the lysosome to carry out degradation (Nakamura and Yoshimori 2017) or can directly fuse with the lysosome. After fusion, the feed is digested by the enzymes in lysosomes. Rab7 GTPase links mature autophagosomes to a microtubule motor through FYCO1 [FYVE and Coiled-Coil Domain Autophagy Adaptor 1] to facilitate kinesin protein-mediated movement to the periphery or boundary of the cell (Kriegenburg et al. 2018). Moreover, according to studies ATG8 has a special role in the planting of autophagosomes (Kriegenburg et al. 2018) (Fig. 1).
Role of autophagy
In angiogenesis
It is the process of the formation of novel vessels/capillaries out of existing vessels/capillaries. This process has a role to play in organ development, wound healing, and any pathological conditions (like cancer metastasis). In a similar process called vasculogenesis where endothelial precursor cells from bone marrow are responsible for vessel formation while in angiogenesis endothelial cells in the vicinity form the vessel, but the purpose remains the same supplying nutrients and oxygen. Angiogenesis supports cancer cells in surviving the problem of high energy demand, hypoxia, and nutrient deficiency (Kardideh et al. 2019). Equilibrium between anti- and pro-angiogenic factors drives the process of new vessel formation; moreover, around the tumor area, endothelial cells experience high Vascular Endothelial Growth factor A (VEGF-A), hypoxia, and nutrient deprivation (Schaaf et al. 2019). Hypoxic conditions stimulate cells in the TME to release (VEGF-A) in abundance and the receptor of EC cells after receiving it causes capillary formation in the presence of matrix metalloproteins in the matrix. In addition, hypoxic conditions stabilize HIF-1α which otherwise gets hydroxylated and degraded under normoxia (Masoud and Li 2015). Autophagy drives homeostasis in cells. Experiments suggest that rapamycin-induced autophagy promotes angiogenesis by altering AMPK/Akt/mTOR signaling. Autophagy can have an angiogenic effect as seen in the acute myocardial infarction (AMI) mice model through increased Vascular Endothelial Growth Factor (VEGFA) (Zou et al. 2019), although autophagy can have an anti-angiogenic effect also as seen by applying mebendazole in endothelial cell (Sung et al. 2019). Proteoglycan-like decorin has both pro-angiogenic and anti-angiogenic effects via interacting with different molecules and factors; likewise, another proteoglycan perlecan also exerts dual effects on endothelial cells (Kardideh et al. 2019). Tumor vasculature has a crucial part in two aspects of cancer cell growth that is hypoxia and invasive metastasis. Hypoxia promotes angiogenesis and provides chemoresistance; hence, controlling hypoxia or maintaining normoxia has proven effective in controlling cancer vasculature via autophagy according to recent studies. Example chloroquine not only controls lysosomal inhibition but also controls tumor vasculature. Another molecule triptolide is anti-cancerous against osteosarcoma cells and also restricts angiogenesis and promotes apoptosis through the Wnt/β-catenin pathway mediates processes like angiogenesis, cell proliferation, and cell death (Li et al. 2018).
In inflammation
Inflammation, a complex and vital response triggered by the body in the face of various stimuli, is influenced by a range of cellular and molecular factors. Immune cells, such as macrophages, monocytes, and neutrophils, are recruited to the site of injury or insult and initiate a sophisticated repair mechanism. Within this intricate process, autophagy plays a crucial role in regulating inflammation and promoting tissue homeostasis. Autophagy, the cellular process of self-digestion and recycling, intersects with inflammation at multiple levels. It influences the activation and function of immune cells involved in the inflammatory response. Autophagy promotes the clearance of intracellular pathogens and damaged organelles, reducing the burden on immune cells and preventing the release of pro-inflammatory signals. Furthermore, autophagy contributes to the regulation of cytokine production and secretion. It helps maintain a balanced cytokine profile by selectively degrading excessive or pro-inflammatory cytokines, thus preventing exaggerated immune responses. Autophagy also modulates the activation and function of immune cells by fine-tuning signaling pathways involved in inflammation. In addition to its role in immune cell regulation, autophagy aids in tissue repair and the resolution of inflammation. It facilitates the removal of cellular debris and damaged components, promoting the regeneration and restoration of healthy tissue. Autophagy also contributes to the clearance of apoptotic cells, preventing the release of inflammatory mediators from dying cells (Fig. 2).

The multistep process of autophagy and its regulatory components. Autophagy involves several stages: initiation, nucleation, elongation, fusion, and degradation. AMPK and mTOR serve as major modulators of autophagy, while ATGs (autophagy-related genes) are closely associated with the process. During initiation, ULK (Unc-51-like kinase) phosphorylates ATG13 and FIP200, leading to their activation. In nucleation, ULK1 phosphorylates Ambra1, which interacts with Beclin-1. Beclin-1 then forms a complex with other proteins, called the PI3KC3 complex. The phagophore is formed, enclosing cytoplasmic components during the elongation stage. The phagophore expands to form autophagosomes. Subsequently, autophagosomes fuse with lysosomes, forming autolysosomes. Within the autolysosomes, various cytoplasmic components are degraded by enzymes. AMPK AMP-activated protein kinase, mTOR mammalian target of rapamycin, ULK Unc-51-like kinase, FIP200 Focal adhesion kinase family interacting protein of 200-kDa, PI3KC3 Class III phosphatidylinositol 3-kinase. (The image legend clarifies that autophagic cargos are represented by purple and green colors, while red and yellow colors symbolize lysosomal enzymes. In the lower corner of the image, it is explained that the light green color corresponds to LC3.)
By orchestrating these diverse functions, autophagy acts as a critical modulator of inflammation. It helps maintain immune cell homeostasis, regulates cytokine production, promotes tissue repair, and aids in the resolution of inflammation. The dynamic interplay between autophagy and inflammation highlights the intricate nature of these processes and their significance in maintaining tissue integrity and overall immune function (Fig. 2) (Singh et al. 2019). Inflammation could be acute if the damage gets controlled or the stimulation stops after a time; otherwise, it could turn chronic if the damage is not controllable or stimulation persists. In that condition, inflammation becomes devastating and it initiates other processes like cell proliferation or tissue damage, mutation, and fibrosis (Germolec et al. 2018). Experiments have proved the involvement of chronic inflammation in multiple diseases, to name some are diabetes, atherosclerosis, arthritis, asthma, autoimmune diseases, cancer, and to aging (Monkkonen and Debnath 2018). There are a few factors that relate inflammation to autophagy and these are the following:
Innate immune receptors
Toll-like receptors—endosomal membrane, retinoic acid-inducible gene-I- (RIG-I-) like receptors (RLRs), nod like receptors—cytosol (Dambuza and Brown 2015), etc. The interaction between autophagy and pattern recognition receptors (PRRs) is a complex process that plays a crucial role in identifying and eliminating pathogens. PRRs recognize specific patterns or structures known as pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). When PRRs detect these patterns, they signal autophagic proteins to initiate the degradation of the foreign bodies through lysosomal pathways. This interplay is highly intricate and varies across different cell types. Autophagy enhances pathogen recognition, promotes immune activation, and contributes to pathogen clearance and tissue integrity. Dysregulation of this interaction can have detrimental effects on pathogen elimination and inflammation. Understanding the interplay between autophagy and PRRs is essential for comprehending innate immune responses and developing targeted interventions against infectious diseases (Lapaquette et al. 2015).
Cytokines
Cytokines, a class of chemical mediators, play a significant role in inflammation and autophagy modulation. Some cytokines, such as Interleukin-1, TGFβ, Interleukin-2, Interleukin-6, Tumor necrosis factor-α, and Interferon-γ, induce autophagy, promoting pathogen clearance and cellular remodeling. Conversely, Th2 cytokines like Interleukin-4, Interleukin-10, and Interleukin-5 have limited impact on autophagy or suppress its induction, focusing on immune regulation and tissue repair. Achieving a balanced cytokine profile is crucial for coordinated immune responses and inflammation resolution (Harris 2011). Research has demonstrated that autophagy plays a significant role in pathogen elimination, particularly in the cases of Mycobacterium tuberculosis and Chlamydia trachomatis. The induction of the cytokine IFN-γ has been associated with autophagy-driven killing of these pathogens. IFN-γ promotes the activation of autophagic pathways, enhancing the degradation of intracellular pathogens and contributing to their elimination. These findings highlight the importance of autophagy as a defense mechanism against specific pathogens, with IFN-γ playing a key role in inducing autophagy-mediated pathogen clearance (Al-Zeer et al. 2009). Pro-inflammatory cytokine TNF-α works as an autophagy inducer in T-lymphoblastic leukemic cells, osteoclasts bone cells, and Ewing sarcoma cells (Lin et al. 2013). IL-4 and IL-13, anti-Th2 and anti-inflammatory cytokines, negatively regulate autophagy by suppressing its induction during nutrient deprivation. They achieve this by activating the PI3K/Akt pathway, which inhibits the initiation of autophagy in response to cellular starvation. This mechanism helps maintain cellular resources and homeostasis under nutrient-deprived conditions. Moreover, IL-4 and IL-13 also function as inhibitors of IFN-γ-induced autophagy. IFN-γ is known to induce autophagy in different cellular contexts, but these cytokines act as counteractive factors, dampening the autophagic response triggered by IFN-γ. By inhibiting IFN-γ-induced autophagy, IL-4 and IL-13 contribute to immune regulation and the maintenance of immune homeostasis. (Lapaquette et al. 2015). Therefore, it is very clear that cytokine and cell type may drive the autophagic process.
Reactive oxygen species
Defined as unstable oxygen radicals or associated oxygen compounds when reacts with DNA, RNA, proteins damage their structure. Under nutrient deprivation, these species induce autophagy and this process is reversed in the presence of antioxidants as shown by several studies (Levonen et al. 2014). During nutrient deprivation, reduced glutathione (GSH), which serves as a potent antioxidant, is expelled from the cytosol of cells through a membrane translocator called ABCC1MRP1. This expulsion alters the cellular redox conditions, creating an oxidizing environment that can impact redox-sensitive proteins. Under starvation conditions, mitochondria generate reactive oxygen species (ROS), including superoxide and hydrogen peroxide. These ROS play a role in inducing autophagy through two distinct mechanisms. First, they oxidize cysteine residues in ATG4, an autophagic cysteine protease, which enhances overall autophagic activity. Second, the increased production of ROS can serve as a signaling cue for autophagy induction (Scherz-Shouval et al. 2007). AMPK, short for 5′-adenosine monophosphate-activated protein kinase, can be activated through a process known as S-glutathionylation. This activation occurs when cysteine residues within AMPK undergo post-translational modification. Upon activation, AMPK initiates a cascade of events that lead to the phosphorylation of TSC2, a gene associated with tumor suppression. Phosphorylation of TSC2 triggers the activation of ULK1 through phosphorylation and simultaneously inhibits mTORC1, a complex involved in regulating autophagy (Fig. 2).
Inflammation and transcriptional factors
Certain transcription factors, including HIF-1, NF-κB, STAT3, and STAT1, have been found to regulate the expression of genes associated with autophagy. HIF-1 is involved in sensing and responding to low oxygen levels and influences the cellular autophagic response. NF-κB, a central player in inflammation, also modulates autophagy through its transcriptional activity. Similarly, STAT3 and STAT1, known for their involvement in signaling pathways and inflammation, impact the expression of genes critical for autophagy. These transcriptional factors highlight the interconnectedness between inflammation and cellular self-degradation processes (Füllgrabe et al. 2014). Hypoxia-inducible factor-1 (HIF-1) and nuclear factor-κB (NF-κB) are transcription factors that regulate autophagy. HIF-1 activates autophagy by increasing the levels of pro-autophagic proteins and inhibiting anti-autophagic proteins. NF-κB activates autophagy by up-regulating the expression of two key genes: BECN1 and SQSTM1. BECN1 is involved in initiating autophagosome formation, while SQSTM1 facilitates the degradation of ubiquitinated proteins through autophagy. Both HIF-1 and NF-κB play important roles in the regulation of autophagy and the cellular response to stress (Bellot et al. 2009). STAT1 and STAT3 are transcription factors that negatively regulate autophagy. When these factors are repressed, it leads to an increase in autophagy through the stimulation of specific genes, such as Atg12 and Beclin1. Atg12 is involved in the formation of autophagosomes, while Beclin1 plays a critical role in initiating autophagy. The repression of STAT1 and STAT3 allows for the up-regulation of these autophagy-related genes, promoting the activation and progression of the autophagic pathway (Mccormick et al. 2012). FXR activation suppresses autophagy, while the inflammatory state can affect the activity of TFEB, a transcription factor responsible for regulating genes involved in lysosomal function and autophagy. This interplay between nuclear receptor proteins, inflammatory signals, and cellular self-degradation processes is complex and regulates the expression of genes related to autophagy and lysosomal function (Settembre et al. 2011).
Tumor microenvironment (tme)
The tumor microenvironment (TME) consists of a diverse range of cells, including fibroblasts, mesenchymal stem cells, immune cells, and endothelial cells. Within this complex environment, autophagy plays a crucial role in supporting tumor progression by facilitating the recycling of biomolecules. However, the interplay between autophagy and the tumor microenvironment is highly intricate and depends upon the specific context. Numerous interactions have been observed in the TME that involve autophagy. These interactions are influenced by various factors and can have diverse effects on tumor growth and survival. A typical niche or place hosting crosstalks among tumor cells and resident cells in that environment takes place (Folkerts et al. 2019).
Stromal fibroblast cells
Induction of autophagy in fibroblast cells has emerged in enhanced growth rates in numerous cancers like colon, breast carcinoma, head, and neck squamous-cell carcinoma (New et al. 2017). This was validated by observing enhanced LC3II vesicles and co-culturing of fibroblast cells with respective cancer cells (New et al. 2017) (Zhou et al. 2017). To confirm cells when treated with autophagy inhibitors like chloroquine and 3 methyl adenine resulted in reduced proliferation and growth in these cancer cells. Moreover, autophagy in fibroblasts cell variants also known as pancreatic stellate cells (PSC) also promotes protein catabolism the resultant alanine helps surrounding tumor cells to survive by pushing the TCA cycle, and lipid synthesis and also helps in overcoming chemotherapy in pancreatic ductal adenocarcinoma (PDAC) (Mowers et al. 2018), (Von Ahrens et al. 2017). Cancer cells co-cultured with fibroblasts depicted a reduction in apoptotic levels, study showed a 40% reduction in apoptotic levels when co-cultured as compared to their monocultures (Ko et al. 2011). Fibroblasts or MSCs also enhance oxidative stress resistance as seen in ovarian cancer cells, knockout of specific autophagy genes like Beclin1 or Atg5 brought back the sensitivity to stress (Wang et al. 2016).
Endothelial cells
Endothelial cells make the cellular bed in vessels and are primarily associated with the vascular repair, healing, and formation of new blood vessels. Tumor cells stimulate angiogenesis in endothelial cells to deal with multiple stressors. Hypoxic conditions stimulate tumor cells to release excess VEGF factor along with low glucose and increased blood flow around the tumor microenvironment, and endothelial cells are subjected to stimulate rigorous vessel sprouting, vessel maturation, and differentiation (Mowers et al. 2018). These tumor vessels seem to be structurally weak. Enhanced overall autophagic flux is seen in the tumor microenvironment due to stressful conditions as compared to normal endothelial cells. It is yet to be fully explored whether autophagic process in these cells should be promoted or repressed to stop angiogenesis. In human dermal endothelial cells, rapamycin activated angiogenesis, while autophagy blockers like chloroquine and 3-MA terminated angiogenesis (Sachdev et al. 2012).
Immune cells
Autophagy plays a crucial and critical role in the development of adaptive immunity, antigenic presentation. During T-cell maturation into circulating T cells, this process drives mitochondrial abundance and checking self-reactive T cells (Mizushima and Levine 2010). Apart from this failure in the autophagic process resulted in a defective cross-presentation in ATG-5 deleted dendritic cells during HSV and Listeria infections in mice model, where mice were found to be more prone to lethal infection (Lee et al. 2010). Additionally, autophagy gifts cancer cells the power to evade lysis by T lymphocyte cytotoxicity and Natural killer cells in multiple ways playing a dual role in this context too. Autophagy regulates the connexin‐43 protein, necessary for forming a gap junction between target and effector cells also required for NK lysis (Tittarelli et al. 2014). Autophagy degrades the connexin‐43 protein which also affects granzyme B-mediated NK cell and CTL killing (Tittarelli et al. 2014). Autophagy degrades MHC-1 molecules resulting in decreased antigenic presentation; moreover, autophagy is up-regulated in both hypoxic and normoxic conditions overall decreasing the efficacy of CTL-NK cell-mediated execution of cancer cells (Khazen et al. 2016).
Role of autophagy in cancer
Autophagy, a cellular process involved in the degradation and recycling of damaged or unnecessary cellular components, possesses intriguing and enigmatic implications in the realm of cancer. Numerous protein kinases have been identified as key regulators of autophagy, dictating its contribution to cancer progression and regression. Extensive experimentation has consistently revealed that during the early stages of cancer, autophagy acts as a suppressor of cell proliferation, thereby facilitating cancer regression. However, as the disease advances, autophagy assumes a dual role, actively supporting cellular proliferation while simultaneously enabling tumor cells to withstand various stressors, thereby enhancing their malignant potential. Despite significant progress, there remain unexplored facets of autophagy’s intricate involvement in cancer, which warrant further scientific investigation.
Tumor suppression
Autophagy has an important part in clearing inoperative organelles, proteins, and regulating reactive oxygen species (ROS). For maintenance of homeostasis working of all ATG genes including BECN1 (encoding Beclin1 involved in phagophore formation) is required. It has been seen that depletion or deletion of this gene is associated with prostate, breast, and ovarian cancers in humans enlightening its role in tumor suppression. Decreased Beclin 1 levels have resulted in cervical squamous-cell and hepatocellular carcinomas (Yue et al. 2003). Factors like Bax-interacting factor-1 (BIF-1) and ultraviolet radiation resistance-associated gene (UVRAG) positively regulate BECLN 1 via increasing its interaction with VPS34 (Y. Takahashi et al. 2007). Negative effects on both UVRAG and BIF1 lead to cancer progression as evident in different cancers (Liang et al. 2006). Increased ROS and damaged mitochondria are prominent stimulators of autophagy and the reason for their increase could be the deletion or knocking out of autophagic core proteins like ATG5, ATG7, ATG3, ATG9, or induction of mitophagy by PINK1 and PARK2 pathway through mitochondrial damage. Abnormality in the PARK2 gene results in pathological conditions like hepatocellular carcinoma; therefore, both mitophagy and oxidative stress play important roles in tumor suppression (Goldsmith et al. 2014).
Tumor progression
Autophagy hampers tumor initiation and cancer development as evidenced by studies, but once a tumor has been established, autophagy supports cancer cell survival and proliferation by dodging stressors like hypoxia present in the central core of solid tumors, nutrient deprivation, and the reduced blood supply in solid tumors. Moreover, autophagy serves their metabolic and energetic requirements by recycling intracellular components to supply energy; in fact, cancer cells express autophagy more than normal cells (Huang et al. 2018). Induced deficiency of autophagy in tumor cells resulted in ATP limitations, toxic ROS accumulation, pile up of dysfunctional mitochondria, absence of TCA cycle intermediates, and overall metabolic crisis. Autophagic tumor cells overcome main metabolic and physiological stressors like hypoxia and nutrient limitation by entering the quiescent stage and thereby feeding on whatever glucose or other nutrients are available in the tumor microenvironment, this prolongs the survival and expansion of cancer cells. Apart from this autophagy maintains stem cell properties in cancer cells, it maintains a constant balance in cancer stem cells (White 2012). Oncogenes like RAS and BRAF when activated depend highly on autophagy to support, and sustain the expansion and progression of cancer cells. K-RAS is a protein member of the RAS/MAPK pathway encoded by the K-RAS gene when activated inhibits complex1 inside mitochondria. Moreover, it affects mitochondrial respiration in cancer cells and thus acetylcholine production, this pushes cancer cells to use autophagic mode to provide ingredients for acetyl-CoA biosynthesis and sustain the tricarboxylic acid cycle (Guo et al. 2011). Autophagy and combination with certain drugs in RAS-activated cancer cells could be very effective in tumor regression. The function of autophagy in cancer had been debatable for a long. Literature suggests that autophagy maintains all the hallmarks of cancer listed below.
Promoting epithelial–mesenchymal transition (EMT)
EMT transition occurs thrice in the body first during embryonic development, second during adult tissue regeneration, and third during cancer development. In this process, epithelial cells lose contact inhibition and become invasive mesenchymal cells. The relationship between autophagy and EMT transition is not clear, because their effect on each other is context-dependent, there are studies where autophagy promotes EMT transition as seen in hepatocellular carcinoma (HCC) at the same time autophagy inhibition by silencing ATG genes or using inhibitors (chloroquine) prevented EMT transition (Li et al. 2013a, b). In a similar experiment, the knockdown of the BECN-1 gene extinguished the EMT transition in colon cancer cells (Shen et al. 2018). In opposition, a few studies indicated toward the suppression of autophagy (silencing of BECLIN 1) have led to EMT activation by up-regulation of two regulators of EMT transition like zinc finger protein SNAI1 (SNAIL and SLUG). In addition, ATG5 and ATG7 deletion increased the metastatic nature of glioblastoma cells (Catalano et al. 2015). Multiple studies highlight direct crosstalk between autophagy and EMT transition. P62 an autophagy adaptor protein supports EMT transition by appearing in different roles like NF-κB transcription enhancement, thereby increasing p65 translocations. Moreover, p62 stabilizes junctional proteins like SMAD4 and TWIST required in EMT transition (Bertrand et al. 2015). Another important modulator is TGF-β (transforming growth factor beta), this factor promotes EMT transition by inducing autophagy in various human cancers like hepatocellular carcinoma (Li et al. 2013a, b), by activating autophagy proteins (like ATG5, ATG7, and BECLN1) (Kiyono et al. 2009) to support these experiments showed that autophagy blockage by knocking of ATG5 or treating with chloroquine repressed TGF-β-mediated EMT transition (Dash et al. 2018).
Sustained proliferation
Every normal cell favors controlled proliferation by passing through checkpoints of the cell cycle. TP53 and retinoblastoma are two well-studied tumor suppressor genes; any mutation in these genes would lead to disorders like cancers giving them the power of limitless replication. At the early stages of tumor initiation autophagy represses tumor growth. Abnormality in autophagy genes ATG5 and BECLN 1 resulted in increased chances of liver cancer (Takamura et al. 2011). Autophagy turn off by up-regulated mTOR signaling/AKT pathway, and PTEN deletions are some factors reflecting the role of autophagy in the initial restriction of tumors (Shen et al. 2008). In the case of mature tumors, role of autophagy is case-dependent it has been generalized to increase autophagic levels to meet increasing nutrient demands, overcome apoptosis, etc. BRAF and KRAS mutations are commonly observed mutations in carcinomas, ATG7 deletion prevented autophagy in the BRAF lung cancer model and reflected positive results by restricting the progression of the tumor (Singh et al. 2018). Therefore, autophagy process regulates this hallmark in a context-dependent manner.
Supporting angiogenesis
This process is defined as the sprouting of novel vessels from the existing vessel, this process is extremely important for tumor cells to grow and reach distant tissues, absorb maximum nutrition, and surpass hypoxic stress. Angiogenesis is needed for invasive and metastatic behavior. When such cells do not get the vascular supply they get into the hypoxic mode and starve; moreover, apoptosis starts within the cells. However, in this situation, they switch to autophagy via HIF-1α-mediated signaling to help them survive this starvation [91] and also adapt to the restricted blood supply and resistance to chemotherapy [91].
Invasion and metastasis
These are two lethal properties of cancer cells by which after entering blood or lymphatic vasculature they start invading new tissues and reach to multiple locations. Tumor cells face numerous challenges like facing immune cells, and fighting starvation, and hence, they have a tendency to leave the source tumor microenvironment. They form small colonies at multiple sites and remain dormant for little time. Autophagy helps them tackle all the stress in a new environment to maintain stemness. This activation is induced by a tumor suppressor gene called aplasia Ras homolog member I (ARHI gene), which increases dormancy during this period (Lu et al. 2008). To add to the survival and invasion of these cells autophagy prevents anoikis and suppresses macrophagic intrusion in the primary site to avoid inflammation, which further increases the metastatic abilities (Guadamillas et al. 2011).
Maintaining stemness in cancer stem cells
Like normal stem, tumor colony also contains cancer stem cells carrying the self-renewal capability, chemoresistance, irradiation resistance, and formation of heterogeneous lineage, and overall a resistant tumor core. Experiments support a strong relationship between ATG genes and cancer stem cell markers. Autophagy induction in glioma cells enhanced stem cells marker CD133 and in an experiment where autophagy was inhibited it resulted in reduced cancer stem cell population in breast cancer [33].
Tumor metabolism reprograming
Tumor cells tend to behave in a unique way when they divert the route from oxidative phosphorylation as an energy generation pathway to aerobic glycolysis, an effect known as the “Warburg effect’. This reprogramming of the metabolic pathway depends upon the cell’s microenvironment (Heiden et al. 2009). After effects of this change cause the conversion of pyruvate to lactic acid instead of acetyl-CoA, which further deficits TCA substrates to fuel ATP production. However, here comes the protective role of autophagy; here, autophagy recirculates biomolecules present in the cell to activate the TCA cycle and fuel ATP production. This enables cells to survive starvation, and due to mitochondrial dysfunction also helps them thrive with low blood and oxygen supply. RAS-mutated cancers increase glucose and glutamine intake by increasing receptors to run the TCA cycle (Huang et al. 2018), (Singh et al. 2018). Autophagy clearly has some role to play in cancer, but its role is very complicated.
Autophagy and its role in other diseases
Neurodegeneration
The autophagy process has a key role in maintaining proper functioning and homeostasis of the nervous system as clearly depicted by whole-body knockout mouse models. Gradual motor and behavioral abnormality were seen after 3 weeks of age in mouse pups delivered from mother mice having deleted autophagy-related 5 (Atg5) or Atg7 in the CNS during embryogenesis. Impairment of autophagic flux or any mutation in autophagy-related genes (ATGs) can result in deadly neurodegenerative disorders like Huntington’s disease (HD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), Parkinson disease (PD), dementia with Lewy bodies (DLB), and Lafora disease (Menzies et al. 2015). Neuronal function is hampered in the majority of these disorders, neurons exhibit an inflated requirement for ATP indicating the need for sustaining axonal transport, formation of synaptic vesicles, functioning and maintenance of ionic gradients, and balancing membrane potential. As a result, there should be a balance in mitophagy too. Dysfunctional mitophagy is responsible for various neurodegenerative disorders mentioned above. Moreover, mitophagy also affects aging (Fivenson et al. 2017).
Alzheimer’s disease (ad)
Most studied neurological disorder characterized by loss of cognitive functions, the presence of amyloid (Aβ) plaques, and neurofibrillary tangles caused by aggregation of the protein tau and shrinkage in brain tissues, Amyloid precursor proteins (APPs) are precursors of these plaques, APPs are cleaved by two enzymes initially by β-secretase called β-site APP-cleaving enzyme 1 (BACE1) and then by γ-secretase producing Aβ aggregation which causes plaque formation in the extracellular matrix. This aggregation leads to oxidative stress, tau protein hyperphosphorylation, and ER stress [94]. This cellular situation ultimately induces autophagy and mitophagy. As understood autophagy has a role in eliminating protein aggregates (here Aβ and tau aggregates), but unlike the early stage of AD at a later stage, autophagy fails to compensate for the disposal of aggregated protein and gets side-lined along with other cellular pathways like PI3K/Akt/AMPK and MAPK/ERK (Zare-Shahabadi et al. 2015). Accumulation of vesicular structures, Aβ toxicity, and plaque formation are very well seen in the later stage of AD along with dysfunctional regulatory mechanisms.
Parkinson’s disease (pd)
Another neurological abnormality involves cognitive loss. Seen more commonly among old age (80 years) as compared to younger group. The pathology behind PD is the loss of dopaminergic neurons (neurons or collection of neurons responsible for the synthesis of neurotransmitter dopamine) in the central nervous system (CNS), sometimes also seen inside the gastrointestinal tract of the patients and there is seen the presence of Lewy bodies (protein body inclusions comprised of aggregates of misfolded α-synuclein protein, ubiquitin, and other proteins) (Stefanoni et al. 2012). A PD patient experiences posture instability, muscular stiffness, distorted speech, loss of smell, frequent tremors, and bradykinesia (sedate movements) (Stefanoni et al. 2012). Impairment of autophagy, mitophagy, and CMA along with mutations in some genes like SNCA gene-encoding α-synuclein protein (failure of CMA is considered to be a major reason for stacking up of soluble, detergent-insoluble, high-molecular-weight α-synuclein protein, because it contains CMA targeting motifs for heat shock 70KD protein and this α-synuclein is one of prime component of Lewy bodies) (Xilouri et al. 2016), LRRK2 gene-encoding leucine-rich repeat kinase 2, recent studies have shown the effect of silencing of LRRK2 on increased macro-autophagy, but controversial results have been seen in fibroblasts of some PD patients showing lower autophagic induction (Manzoni et al. 2013), VPS35 gene-encoding vacuolar protein sorting 35 in endosomes and vesicles involved in protein trafficking in the cytoplasm), CHCHD2 (gene coding a mitochondrial protein) and DNAJC13 (gene-encoding REM-8, a chaperon involved in protein trafficking) leads to an autosomal dominant form of Parkinson’s Disease.
An autosomal recessive form of PD occurs due to the mutations of ubiquitin kinase (PTEN)-induced kinase 1 (PINK1), DJ-1 or PARK7 (codes a mitochondrial protein dealing with oxidative stress), and PARKIN (Phosphorylation of PINK1 activates Parkin). PARKIN/PINK1 is associated with ubiquitin-binding domains of autophagy or specifically mitophagy (Pickrell and Youle 2015). The frequency of mutation in the PARK2 gene responsible for coding PARKIN protein is very high. Mutations in both genes hamper mitochondrial protein function and cell response to oxidative stress and cause mitophagy failure. Other mutations include GBA (glucocerebrosidase β acid, an autophagy regulator), ATP13A2 gene (ATPase 13A), SCARB2 (coding lysosomal integral membrane protein-2 (LIMP-2), required for the functioning of lysosome/autophagosome) (Stefanoni et al. 2012). Both AD and PD are characterized by protein aggregation and plaque deposition which affects dopaminergic neuronal function; autophagy and mitophagy regulation might serve as effective solutions in clearing this aggregation.
Cardiovascular diseases
Autophagy is one process that affects every cell of the body including cardiac cells; this should be regulated to maintain cardiac homeostasis. It has been proved in mice models with deleted LAMP 2 gene (Bravo-San Pedro et al. 2017) which hampers CMA development cardio skeletal myopathy. Also, the deletion of some of ATGs leads to cardiac hypertrophy and the accumulation of atherosclerotic plaques. Specifically, Atg5 − / − mice lead to complications like contractile dysfunction, cardiac hypertrophy, dilatation in the left ventricle, and in some also caused premature death (Bhandari et al. 2014). In the park − / − flies (Drosophila) model, it caused Dilated cardiomyopathy (Taneike et al. 2010). The heart employs intracellular processes like autophagy and mitophagy (mitochondrial autophagy) as adaptive mechanisms to deal with situations like energetic stress, enhanced oxidative stress, and cell death. Earlier studies have directed that autophagy is activated during cardiac hypertrophy and heart failure, here also then autophagy plays a dual function of guarding and damaging (Bravo-San Pedro et al. 2017). However, although cardiac hypertrophy and HF are commonly accompanied by general and non-specific autophagy, the presence of organelle-specific mitochondrial autophagy and its role during pathological hypertrophy have not been well characterized. The heart experiences hypertrophy during hemodynamic overloads, such as pressure overload (PO), initially with the purpose of reducing wall stress.
Autoimmune disorders
Autophagy happens to interact with immune cells and inflammatory molecules leading to processes like lymphocyte development, secretory pathways, killing of intracellular pathogens, etc (Table 1).
Killing of intracellular pathogens
Autophagy performs pathogen degradation by two processes (Fig. 3): (a) Xenophagy—autophagy machinery specifically degrading bacteria or other pathogens is termed as xenophagy, wherein the pathogen is targeted and after trapping in autophagolysosomes gets dumped in the lysosome (Levine 2005). (b) LC3-associated phagocytosis (LAP)—sometimes, pathogens get trapped and killed in a single-walled phagophore coated with a Toll-like receptor (TLR).

“The Intricate Interplay of Autophagy, Inflammasome Activation, and Cancer”.The figure illustrate the intricate relationship between autophagy, inflammasome activation, and cancer. Dysfunctional autophagy and mitochondrial impairment culminate in heightened oxidative stress, triggering autophagic cell death, inflammasome activation. Elevated levels of mitochondrial reactive oxygen species (ROS) further promote oncogenesis, chemoresistance, and metastasis. Autophagy serves a dual role in cancer and inflammation, both supporting tumor growth and orchestrating immune responses. This reciprocal interaction between cancer, inflammation, and autophagy involves the activation of oncogenic pathways, metabolic alterations, and the modulation of the tumor microenvironment. This complex crosstalk holds significant implications for disease progression and potential therapeutic strategies
Lymphocyte development
Apart from innate immune response autophagy also mediates adaptive immune response by regulating T and B cells. During viral infection, autophagy enhances MHC class I and II presentation. Studies have shown its value in generating MHC class II specific CD4 ( +) T cells, failure of which might cause self-reactive T cells (Nedjic et al. 2008). Moreover, autophagy is sincerely required in plasma B-cell homeostasis and protects antibodies from apoptotic damage and for their sustained production (Pengo et al. 2013).
Secretory pathways
As discussed earlier autophagy abnormalities increase pro-inflammatory cytokines. Experiments, where autophagy deficiency was produced, resulted in the up-regulation of cytokines like IL-1α in macrophages causing increased inflammatory response (Castillo et al. 2012) and increased IL-1β secretion (Harris et al. 2011) (Fig. 3).
Unraveling the impact: impaired crosstalk between autophagy and immune cells fuels the onset of multifarious diseases
Autophagy is a crucial process that safeguards cellular health and sustains life by selectively packaging and degrading components of the cytoplasm within double-membrane vesicles called autophagosomes. This cellular self-digestion mechanism acts as a protective response against stress, ensuring the removal of damaged organelles and proteins that could jeopardize the cell’s survival. A key benefit of autophagy is the generation of energy and the recycling of metabolic precursors. In the realm of the immune system, autophagy plays a pivotal role in regulating various processes. It oversees the uptake and presentation of antigens, the elimination of pathogens, and the survival of both short- and long-lived immune cells. Additionally, autophagy influences cytokine-dependent inflammation, acting as a crucial mediator. Maintaining an optimal level of autophagic activity is vital to strike a balance between catabolic processes, cellular repair, and the induction of inflammation. However, when autophagosome formation and the subsequent flux of autophagic activity become dysregulated, it can lead to detrimental consequences. Such dysregulation may result in a failure to efficiently "clean house," leaving damaged components unaddressed. Conversely, it can also trigger autophagy-induced cell death, a process in which excessive autophagy contributes to cell demise. The autophagic pathway’s abnormalities have been implicated in numerous autoimmune diseases, highlighting its significance in maintaining immune system homeostasis. These conditions arise when the delicate equilibrium of autophagy is disrupted, further emphasizing the crucial role of autophagy in ensuring cellular well-being and defending against autoimmune disorders. The table below covers four autoimmune disorders, pathology, the role of autophagy, and related workout with resultant outcomes (Table 1).
Novel approaches to target autophagy
Enzyme-based autophagy modulation
L-asparaginase (l-asp) and chloroquine (CQ)-based therapy
Multiple studies showed that L-Asp causes deamination of L-asparagine (Asn) dedicated amino acid deprivation. L-asp resulted in mitochondrial dysfunctioning and ROS generation evolving in cytoprotective autophagy. At this point, treatment with chloroquine (CQ) seized autophagy and increased ROS levels, DNA damage, and p53 expression leading to up-regulation of pro-apoptotic proteins and down-regulation of hexokinase 2 resulting in apoptosis-driven lymphoblastic leukemia tumor cell death (Takahashi et al. 2017). FDA has approved L-asparaginase in the treatment of leukemia and other cancers. The use of L-asparaginase and autophagy blockers (like chloroquine) can modulate autophagy by regulating AKT/mTORC1 in laryngeal squamous-cell carcinoma (LSCC) (Ji et al. 2017).
Arginase-based therapy
Arginine deficiency due to hydrolysis by arginase, its deprivation causes autophagy induction. Recombinant human arginase (RhArg) is one approach that is used in different lymphoma, melanomas and is currently under clinical trials. In a study carried out on non-small-cell lung cancer H1975 cells, recombinant human arginase was served that induced cytoprotective autophagy via phosphorylating Mtorc1/AKT pathway, ROS generation and ERK1/2 (extracellular signal‑regulated protein kinase). Here also the use of autophagy blockers enhanced tumor cell death by apoptosis (Shen et al. 2017).
Arginine deiminase-based therapy
This enzyme converts arginine to citrulline, arginine deiminase (ADI) along with autophagy inhibitors has shown positive results in Argininosuccinate Synthase 1 (ASS1)-deficient sarcoma cells by enhancing cytotoxicity (Bean et al. 2016). Prostate cancer cells (CWR22Rv1) found to be deficient in ASS1, showed ADI-dependent autophagic cytotoxicity, and this activation was regulated through the Mtorc1/AKT pathway and activation of the AMPK, ERK pathway. Autophagy inhibitors multiplied cytotoxicity due to apoptosis (Kim et al. 2009).
MTOR inhibitors
The regulation of autophagy is a highly complex and finely tuned process involving intricate interactions between key proteins and cellular signaling pathways. One critical player in this regulation is the mammalian target of rapamycin complex 1 (mTORC1), which exerts both positive and negative control over autophagy. AMP-activated protein kinase (AMPK) negatively regulates mTORC1, leading to the inhibition of mTORC1 and the promotion of autophagy (Dossou and Basu 2019). AMPK also directly interacts with and phosphorylates Unc-51-like kinase 1 (ULK1), a key initiator of autophagy. However, there are controversies regarding how mTORC1 influences the interaction between ULK1 and AMPK, with some studies, suggesting that mTORC1 can either promote or prevent this interaction (Alers et al. 2012). Various factors, including differences in nutrient availability and experimental conditions, can influence the outcomes of these interactions. Additionally, mTORC1 and AMPK can also regulate autophagy initiation by phosphorylating other autophagy-related proteins, such as Atg13 (S. Wang et al. 2022). Moreover, mTORC1 plays a role in the nucleation and expansion of autophagosomes by phosphorylating proteins like Atg14 and p300 acetyltransferase. Furthermore, mTORC1 regulates the transcription of autophagy-related genes through its control of transcription factors, such as TFEB, TFE3, and MITF. These transcription factors can modulate autophagy by promoting the expression of genes involved in autophagosome formation, lysosome biogenesis, and nutrient sensing. This intricates interplay between mTORC1 and various autophagy regulators underscores the complexity of autophagy regulation and its importance in cellular homeostasis (Di Malta et al. 2019).
Nutrient deprivation, such as amino acid depletion, inhibits MTORC1 and induces autophagy. Small molecules like rapamycin, Torin 1, and niclosamide are known to inhibit MTORC1 and induce autophagy (Tan & Miyamoto 2016).
Lysosomal inhibitors
Autophagy, a crucial cellular process, facilitates the transport and degradation of various materials in lysosomes. It plays a vital role in nutrient provision during cellular stress and the removal of unnecessary cellular components (Murrow and Debnath 2013). Recently, the lysosome has been found to play a significant role in MTORC1 activation, particularly through the Rag/RRAG GTPase family located on the lysosomal membrane (Li et al. 2013a). Rag proteins, when GTP-loaded, recruit MTORC1 to the lysosome, enabling its activation. Inhibition of lysosome function, either by interfering with v-ATPase, LAMTOR, or Rag interactions, or using lysosome inhibitors like bafilomycin A1 and concanomycin A, leads to MTORC1 inhibition and autophagy activation(Sancak et al. 2010). The link between lysosome functionality and the signaling pathway involving Rag GTPases underscores the lysosome’s involvement in both the activation of autophagy and its conventional role in the degradation process (Li et al. 2013b).
Nanoparticle-based autophagy targeting for cancer therapy
Nanosciences (especially nanobiotechnology) are currently treated as the best alternative therapeutic approach as compared to the conventional/traditional methods for drug delivery, tissue remodeling, and cytotoxicity. It deals with engineered devices designed in nanodimension with special chemical, mechanical, biological, and physical properties. Nanobiotechnology in combination with chemistry and material sciences offers efficient and safe solutions to deal with diseases even at complex locations of the body in addition to decreasing side effects and increasing the efficacy of standard chemotherapies. Unique biological, physical, chemical, optical, mechanical, and properties make them novel and powerful therapeutic tools. Characteristic properties of nanocarriers include higher surface-to-volume ratios, high bioavailability, appropriate drug release capability, and biodegradability along with flexibility in designing and targeting (Sinha et al. 2006). Nanocarriers show increased permeability and retention effect making them suitable for controlled drug release (Albanese et al. 2012).
Nanoparticles can be categorized into: Lipid-based NP (E.g., Liposome, Solid lipid NP), Polymer-based (E.g., Polymeric NP, Polymeric Miscelle), Inorganic NP (E.g., Metallic NP, Silica NP), and Conjugated NP (E.g., Antibody–Drug conjugate, Polymer–Drug conjugate).
Microenvironmental stimuli can trigger the release ensuring toxicity to the target tissue, and keeping the healthy tissue safe. Internal stimuli include alterations in pH, ionic strength, redox state, and other stress around target sites that serve as internal stimuli. External stimuli are temperature, ultrasound, radiation, magnetic, and electric forces (Wicki et al. 2015). Magnetic hyperthermia is one approach where local heating is used to achieve desired chemotherapeutics, and local heating is given by radiation, laser (Bañobre-López et al. 2013).
Silver-based nanoparticles
Silver-based nanoparticles used in combination with different agents has shown promising results by inducing autophagic and apoptotic pathways, thereby killing cancer cells by cellular damage. Ag-NP along with radiotherapy showed toxic effects on mouse brain tumor model and U251 glioblastoma cells, results of LC3-II protein level, staining by nucleic acid binding dye acridine orange and monodansyl cadaverine (MDC) showed enhanced autophagy (Liu et al. 2016). The reduced form of graphene oxide–silver nanocomposite and cisplatin combination (rGO–Ag-NPs) proves effective against cervical cancer cells in combination with chemotherapy as experiments direct that this composite induces cytotoxicity by apoptosis and autophagy evident by increased expression of ATG genes (Yuan and Gurunathan 2017). Salymicin (Sal) in combination with Ag-NPs displayed a significant collaborative cell killing and collection of autophagolysosomes in A2780 ovarian cancer cells (Zhang and Gurunathan 2016), elevated levels of LC3-II (protein complex) were seen in adenocarcinoma pancreatic cancer cells when these tumor cells were treated with Ag-NPs, reduced tumor viability, and increased autophagy and apoptosis were observed (Zielinska et al. 2018). SKBR3 cells experienced high ROS levels, increased autophagy, and apoptosis when cells were treated with Ag-NPs, enclosed into a specific exopolysaccharide (EPS), validation of the study was done by western blot and fluorescence microscopy, and this depicted increased cell death/necrotic cell death (Buttacavoli et al. 2018).
Gold-based nanoparticles
There are multiple studies that conclude that Gold nanoparticles (Au-NP) can be conjugated with different molecules like drugs, proteins, different chemicals, oligonucleotide sequences, etc. Au-NP is a successful drug carrier because of properties like good biocompatibility and, low toxicity and immunogenicity. Au-NP when assembled with SMI-9 in SUM1315 triple-negative breast cancer (TNBC) cell, using the approach for its slow release inside the cells. This SMI-9 acts as an inhibitor of Rad6; this protein has a role to play in mitochondrial stability, DNA damage maintenance, and DNA repair mechanism. Researchers found enhanced autophagy and autophagy markers (Haynes et al. 2016).
Snake venom toxin (NKCT1) when conjugated with Au-NP and tested in K562 and human leukemic U937 cell lines. There was an increase in apoptotic levels and autophagy levels because of alteration in mtorc1 and AKT signaling pathways; this could be an effective approach as therapy (Bhowmik and Gomes 2016).
Tethering of (TRAIL—tumor necrosis factor (TNF)-related apoptosis-inducing ligand) with Au-NP prompted a Drp1-mediated mitochondrial fault causing induction of mitophagy, along with apoptosis and autophagy (Ke et al. 2017). Activation of autophagy and mitophagy (evident by PINK1 and PARKIN markers) helps to overcome TRAIL resistance often seen in tumors (Ke et al. 2017). Another natural compound called Quercetin, a type of flavonoid found in plants, is a molecule that restricts cellular growth and tumor progression, Au-NP and Quercetin when stabilized by PLGA resulted in autophagy, apoptosis, and deregulation of many signaling pathways in cancer cells (Ren et al. 2017).
Silica-based nanoparticles
Silica-based nanoparticles are popular therapeutic nanoparticles because of their established role in cancer treatment through autophagy induction. Studies suggest that silica nanoparticles (Si-NPs) activate this self-degradative process by Reactive Oxygen Species (ROS) accumulation, generating ER stress, and creating oxidative stress; moreover, Si-NPs display positive properties with high surface area, biocompatibility, and flexible pore size making them idle drug delivery candidate, gene transfection agent, suitable for bio-imaging. Si-NPs showed anticancer properties by activating autophagy along with mitophagy and apoptosis in target LBC3 and LN-18 glioblastoma (brain tumor) cells. These highly aggressive and invasive tumor cells were exposed to silver nanoparticles and research finding through quantitative RT-PCR showed higher expressions of apoptotic (like Bax, BIM) and autophagy-related genes (ATG5, high LC3-II/LC3-I ratio) (Krętowski et al. 2017). In another study where colorectal cancer cells in humans were exposed to Genistein corporated with PEGylated silica hybrid nanomaterials results favored that genistein imposed anti-proliferative and antioxidant actions on colorectal cells (HT29 colon cancer cells); moreover, it activated apoptotic and autophagic pathways to stop cancer growth (Pool et al. 2018). Si-NPs have also been found to cause endoplasmic reticulum (ER) stress-mediated autophagy in colon cancer HCT-116 cells (Wei et al. 2017). Si-NP sometimes also cause autophagy failure by down-regulating lysosomal protease enzyme, but the dosage remains a key factor (Wang et al. 2017). Therefore, usage of silica nanoparticle in cancer treatment through the autophagy process is worth exploring.
Metal oxide-based nanoparticles
There are a lot of metal oxides that have found their way into medical and research fields:
Iron oxide nanoparticles
These nanoparticles are widely researched nanoparticles with the capability of treating multiple types of cancers by inducing autophagy and producing endoplasmic reticulum stress. Iron adds super-paramagnetism to the nanoparticle that makes them a bio-imaging tool and biocompatibility makes them a drug carrier. Multi-drug resistant gastric cancer cell lines were targeted using chitosan chloride alginate encapsulated magnetite nanoparticles (HTCC–MNPs) in an experiment. Results revealed autophagy-mediated cell cytotoxicity in both gastric cancer cell line and variant one (SGC7901 & SGC7901/ADR, respectively); this was proved by increased LC3 localization with LAMP 2 protein, high LC3-II/LC3-I ratio. Therefore, these nanoparticles were effective in cancer regulation (Li et al. 2016). In research carried out using iron oxide nanoparticles on MCF-7 cancer cells and the xenograft model, the photothermal effect of NP created an autophagic response in dose dependence fashion, and in the same model, inhibition of autophagy resulted in apoptotic cell killing by photothermal effect (Li et al. 2016).
Cuprous and copper oxide nanoparticle (Cu-NP and C0-NP) are another class of autophagy-promoting metal oxide nanoparticles. CO-NP is reported to stimulate autophagy in the breast cancer cell (MCF7 cell lines) by switching off apoptotic response and vice versa (Laha et al. 2014), and also Cu-NP seems to work in cervical cancer cell line by activating autophagy by reduced phosphorylation of mTOR and AKT (Xia et al. 2017). Cuprous nanoparticles have been found to be effective in treating leukemia, and lung and breast cancer (Wang et al. 2012a, b)
Zinc oxide (ZnO) is a widely studied metal oxide that is found to be effective in multidisciplinary areas of science. Some of the useful properties include antibacterial, ability to perform photocatalytic degradation, UV absorber, and photoluminescence. Zinc oxide nanoparticles can stimulate mitophagy (PINK/Parkin-mediated) in tongue squamous cell carcinoma (CAL 27 human tongue cancer cell) (Wang et al. 2018) and targeted cytotoxicity was produced in SKOV3 ovarian cancer cells using autophagy (Bai et al. 2017). ZnO-NP conjugated with meso-tetra (4-carboxyphenyl) porphyrin (MTCP) showed autophagy-mediated cytotoxicity inside MDA-MB-468 and MCF-7 breast cancer cells (Mozdoori et al. 2017).
Crosstalk between autophagy, inflammation, and tumorigenesis
Inflammation poses a high risk of cancer initiation and progression. The relationship between autophagy and inflammation is quite complex. Any stress or trauma causes the release of DAMPs (damage-associated molecular patterns), a signal of innate immune response. These signals are released from damaged or affected cells which attracts macrophages inviting an inflammatory response for repair and digesting dead cells. However, an increased inflammatory response may pose complications of hyperplasia and tumor. To regulate this mitophagy comes into the picture. This interaction causes mitochondrial stress, and mitophagy releases mitochondrial reactive oxygen species and other immunopathies.
Heat shock proteins (HSP70) were used to give extracellular stress to tumor cells, causing NF-κB pathway activation resulting in up-regulation of high-mobility group protein B1 (HMGB1) expression (refer Fig. 2). In addition, HSP70 enhanced the sustainability of H22 hepatocarcinoma cells by activating Jun N-terminal kinase (JNK), thereby increasing BECLN1 expression and ejection of HMGB1 protein. HMGB1 phosphorylates NF-κB and further activates TLR-2 and TLR-4 receptor signaling to promote invasive behavior of hepatocarcinoma cells (Gong et al. 2013). ATG5, ATG7–ATG12-mediated release of HMGB1 protein in tumor cells killed by Epidermal Growth Factor Receptor (EGFR)-targeted diphtheria toxin (DT-EGF) (refer Fig. 2). (Dupont et al. 2011). Autophagy guided the secretion of inflammatory cytokine IL6 inside cells hit by RAS mutation. Mutated epithelial cells showed enhanced invasiveness due to autophagy-backed IL6 secretion, and to support this, reverse behavior was seen in autophagy-deficient cells (Lock et al. 2014). Tumor cells having Ras mutation lead to (OIC) oncogene-induced senescence (Fig. 4). Measured by increased LC3-II, ULK, and CXCL8 expression in fibroblast co-cultured with breast cancer cells (Young et al. 2009) (Fig. 5).

Autophagy and its role in different aspects of the immune system. Autophagy serves as a crucial mechanism in various facets of the immune system, contributing to immune cell development, function, and regulation, while also playing a pivotal role in host defense against intracellular pathogens and maintaining immune system balance

Crosstalk between autophagy, inflammation, and tumorigenesis in the context of RAS-mutated and tumor cells. This diagram illustrates the intricate interplay between autophagy, inflammation, and tumorigenesis, with a particular focus on the role of high-mobility group protein B1 (HMGB1), interleukin-6 (IL-6), and interleukin-8 (IL-8) in promoting increased invasion and tumorigenesis. Additionally, these factors contribute to heightened inflammation, a phenomenon prominently observed in RAS-mutated cells and various tumor cell types
Conclusion
Autophagy is a cellular process that plays a vital role in maintaining normal cell functioning and overall cellular balance, known as homeostasis. It involves the degradation and recycling of cellular components to provide the cell with the necessary nutrients and energy. Extensive research has been conducted on autophagy, leading to the identification of various substrates and protein complexes involved in this pathway. Disruptions in autophagy can lead to various abnormalities, including cancer and inflammation. In the context of cancer, autophagy can confer several advantages to mature cancer cells. It enables these cells to obtain nutrients, survive chemotherapy, and adapt to low oxygen conditions, thereby enhancing their ability to invade multiple tissues and metastasize. Different protein complexes, such as MTORC1, BECLN1, ULK1, and AMBRA1, are potential targets for manipulating autophagy through appropriate therapeutic interventions. By modulating the activity of these protein complexes, it is possible to regulate autophagy and potentially impact cancer progression. Furthermore, autophagy assists cancer cells in performing critical processes like angiogenesis (formation of new blood vessels), metabolic reprogramming, epithelial–mesenchymal transition (a process involved in metastasis), and overall immune evasion. These functions support cancer cells’ ability to adapt to their environment and evade immune responses. Autophagy’s role in cancer progression can be assessed through various direct and indirect pathways. Researchers use these pathways to measure autophagic activity and evaluate its impact on cancer development and treatment outcomes. In recent years, novel approaches utilizing nanoparticles and enzymes have emerged as potential strategies for targeting autophagy. These approaches are currently being evaluated in different phases of clinical trials, aiming to develop therapies that can selectively manipulate autophagy in cancer cells. It is important to note that autophagy’s impact on cancer is highly context-dependent. While it can promote cancer progression in mature cancers, its effects may vary depending upon the type and stage of cancer. Therefore, the manipulation of autophagy in cancer therapy requires careful consideration and a personalized approach to ensure optimal outcomes while avoiding potential risks.
Data availability
Not applicable.
References
Ahlberg J, Glaumann H (1985) Uptake-microautophagy-and degradation of exogenous proteins by isolated rat liver lysosomes. Effects of pH, ATP, and inhibitors of proteolysis. Experim Mol Pathol 42(1):78–88. https://doi.org/10.1016/0014-4800(85)90020-6
Albanese A, Tang PS, Chan WCW (2012) The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng 14:1–16. https://doi.org/10.1146/annurev-bioeng-071811-150124
Alers S, Löffler AS, Wesselborg S, Stork B (2012) Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 32(1):2. https://doi.org/10.1128/MCB.06159-11
Alirezaei M, Fox HS, Flynn CT, Moore CS, Hebb AL, Frausto RF, Bhan V, Kiosses WB, Whitton JL, Robertson GS, Crocker SJ (2009) Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 5(2):152–158. https://doi.org/10.4161/auto.5.2.7348
Al-Zeer MA, Al-Younes HM, Braun PR, Zerrahn J, Meyer TF (2009) IFN-γ-inducible Irga6 mediates host resistance against chlamydia trachomatis via autophagy. PLoS One. https://doi.org/10.1371/journal.pone.0004588
Amaravadi R, Kimmelman AC, White E (2016) Recent insights into the function of autophagy in cancer. Genes Dev 30(17):1913. https://doi.org/10.1101/GAD.287524.116
Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM (2015) Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol Cell 59(2):270. https://doi.org/10.1016/J.MOLCEL.2015.05.030
Bai DP, Zhang XF, Zhang GL, Huang YF, Gurunathan S (2017) Zinc oxide nanoparticles induce apoptosis and autophagy in human ovarian cancer cells. Int J Nanomed 12:6521–6535. https://doi.org/10.2147/IJN.S140071
Bañobre-López M, Teijeiro A, Rivas J (2013) Magnetic nanoparticle-based hyperthermia for cancer treatment. Reports Practi Oncol Radiother 18(6):397–400. https://doi.org/10.1016/j.rpor.2013.09.011
Bean GR, Kremer JC, Prudner BC, Schenone AD, Yao JC, Schultze MB, Chen DY, Tanas MR, Adkins DR, Bomalaski J, Rubin BP, Michel LS, Van Tine BA (2016) A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death Dis 7(10):e2406–e2406. https://doi.org/10.1038/cddis.2016.232
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM (2009) Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29(10):2570–2581. https://doi.org/10.1128/mcb.00166-09
Bertrand M, Petit V, Jain A, Amsellem R, Johansen T, Larue L, Codogno P, Beau I (2015) SQSTM1/p62 regulates the expression of junctional proteins through epithelial–mesenchymal transition factors. Cell Cycle 14(3):364–374. https://doi.org/10.4161/15384101.2014.987619
Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW (2014) Mitochondrial contagion induced by parkin deficiency in drosophila hearts and its containment by suppressing mitofusin. Circ Res 114(2):257–265. https://doi.org/10.1161/CIRCRESAHA.114.302734
Bhowmik T, Gomes A (2016) NKCT1 (purified Naja kaouthia protein toxin) conjugated gold nanoparticles induced Akt/mTOR inactivation mediated autophagic and caspase 3 activated apoptotic cell death in leukemic cell. Toxicon 121:86–97. https://doi.org/10.1016/j.toxicon.2016.08.004
Bottini N, Firestein GS (2013) Epigenetics in rheumatoid arthritis: a primer for rheumatologists. Curr Rheumatol Rep 15(11):372. https://doi.org/10.1007/s11926-013-0372-9
Bravo-San Pedro JM, Kroemer G, Galluzzi L (2017) Autophagy and mitophagy in cardiovascular disease. Circul Res Lippincott Williams Wilkins 120(11):1812–1824. https://doi.org/10.1161/CIRCRESAHA.117.311082
Buttacavoli M, Albanese NN, Di Cara G, Alduina R, Faleri C, Gallo M, Pizzolanti G, Gallo G, Feo S, Baldi F, Cancemi P (2018) Anticancer activity of biogenerated silver nanoparticles: an integrated proteomic investigation. Oncotarget 9(11):9685–9705. https://doi.org/10.18632/oncotarget.23859
Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, Hutt J, Lyons CR, Dobos KM, Deretic V (2012) Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1210500109
Catalano M, D’Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, Faienza F, Esposito V, Limatola C, Cecconi F, Di Bartolomeo S (2015) Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol 9(8):1612–1625. https://doi.org/10.1016/j.molonc.2015.04.016
Chavez-Dominguez R, Perez-Medina M, Lopez-Gonzalez JS, Galicia-Velasco M, Aguilar-Cazares D (2020) The double-edge sword of autophagy in cancer: from tumor suppression to pro-tumor activity. Front Oncol 10:578418. https://doi.org/10.3389/FONC.2020.578418/BIBTEX
Cho DH, Kim YS, Jo DS, Choe SK, Jo EK (2018) Pexophagy: molecular mechanisms and implications for health and diseases. Mol Cells 41(1):55. https://doi.org/10.14348/MOLCELLS.2018.2245
Chun Y, Kim J (2018) Autophagy: an essential degradation program for cellular homeostasis and life. Cells. https://doi.org/10.3390/CELLS7120278
Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441(7097):1162–1166. https://doi.org/10.1038/nature04779
Clarke AJ, Ellinghaus U, Cortini A, Stranks A, Simon AK, Botto M, Vyse TJ (2015) Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis 74(5):912–920. https://doi.org/10.1136/annrheumdis-2013-204343
Compston A, Coles A (2008) Multiple sclerosis. Lancet 372(9648):1502–1517. https://doi.org/10.1016/S0140-6736(08)61620-7
Cuervo AM, Dice JF (1996) A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273(5274):501–503. https://doi.org/10.1126/science.273.5274.501
Dambuza IM, Brown GD (2015) C-type lectins in immunity: Recent developments. Curr Opin Immunol 32:21–27. https://doi.org/10.1016/j.coi.2014.12.002
Dash S, Sarashetti PM, Rajashekar B, Chowdhury R, Mukherjee S (2018) TGF-β2-induced EMT is dampened by inhibition of autophagy and TNF-α treatment. Oncotarget 9(5):6433–6449. https://doi.org/10.18632/oncotarget.23942
Di Malta C, Cinque L, Settembre C (2019) Transcriptional regulation of autophagy: mechanisms and diseases. Front Cell Develop Biol 7:456488. https://doi.org/10.3389/FCELL.2019.00114/BIBTEX
Ding X, Zhang X, Paez-Valencia J, McLoughlin F, Reyes FC, Morohashi K, Grotewold E, Vierstra RD, Otegui MS (2022) Microautophagy mediates vacuolar delivery of storage proteins in maize aleurone cells. Front Plant Sci. https://doi.org/10.3389/FPLS.2022.833612/FULL
Dooley HC, Razi M, Polson HEJ, Girardin SE, Wilson MI, Tooze SA (2014) WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell 55(2):238–252. https://doi.org/10.1016/j.molcel.2014.05.021
Dossou AS, Basu A (2019) The emerging roles of mTORC1 in macromanaging autophagy. Cancers. https://doi.org/10.3390/CANCERS11101422
Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V (2011) Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J 30(23):4701–4711. https://doi.org/10.1038/emboj.2011.398
Egan D, Kim J, Shaw RJ, Guan KL (2011) The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7(6):643–644. https://doi.org/10.4161/auto.7.6.15123
Farré JC, Subramani S (2016) Mechanistic insights into selective autophagy pathways: lessons from yeast. In Nat Rev Mol Cell Biol 17(9):537–552. https://doi.org/10.1038/nrm.2016.74
Ferro-Novick S, Reggiori F, Brodsky JL (2021) ER-phagy, ER homeostasis, and ER quality control: Implications for disease. Trends Biochem Sci 46(8):630. https://doi.org/10.1016/J.TIBS.2020.12.013
Fivenson EM, Lautrup S, Sun N, Scheibye-Knudsen M, Stevnsner T, Nilsen H, Bohr VA, Fang EF (2017) Mitophagy in neurodegeneration and aging. Neurochem Int 109:202–209. https://doi.org/10.1016/j.neuint.2017.02.007
Folkerts H, Hilgendorf S, Vellenga E, Bremer E, Wiersma VR (2019) The multifaceted role of autophagy in cancer and the microenvironment. Med Res Rev 39(2):517–560. https://doi.org/10.1002/med.21531
Frake RA, Ricketts T, Menzies FM, Rubinsztein DC (2015) Autophagy and neurodegeneration. J Clin Investig 125(1):65–74. https://doi.org/10.1172/JCI73944
Füllgrabe J, Klionsky DJ, Joseph B (2014) The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol 15(1): 65–74). https://doi.org/10.1038/nrm3716
Galluzzi L, Pietrocola F, Bravo‐San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, Kimmelman A, Kumar S, Levine B, Maiuri MC, Martin SJ, Penninger J, Piacentini M, Rubinsztein DC, Simon H, Kroemer G (2015) Autophagy in malignant transformation and cancer progression. EMBO J 34(7):856–880. https://doi.org/10.15252/embj.201490784
Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G (2017) Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov 16(7):487–511. https://doi.org/10.1038/NRD.2017.22
Galluzzi L, Baehrecke EH, Ballabio A. Boya P, Pedro JMB, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, Cuervo AM, Debnath J, Deretic V, Dikic I, Eskelinen E, Fimia GM, Fulda S, Gewirtz DA, Green DR, Kroemer G (2017) Molecular definitions of autophagy and related processes. EMBO J 36(13):1811. https://doi.org/10.15252/EMBJ.201796697
Gatica D, Lahiri V, Klionsky DJ (2018) Cargo recognition and degradation by selective autophagy. Nat Cell Biol 20(3):233–242. https://doi.org/10.1038/s41556-018-0037-z
Gautreau A, Oguievetskaia K, Ungermann C (2014) Function and regulation of the endosomal fusion and fission machineries. Cold Spring Harbor Perspect Biol. https://doi.org/10.1101/CSHPERSPECT.A016832
Germolec DR, Shipkowski KA, Frawley RP, Evans E (2018) Markers of inflammation. Methods Mol Biol 1803:57–79. https://doi.org/10.1007/978-1-4939-8549-4_5
Gkikas I, Palikaras K, Tavernarakis N (2018) The role of mitophagy in innate immunity. Front Immunol. https://doi.org/10.3389/FIMMU.2018.01283
Goldsmith J, Levine B, Debnath J (2014) Autophagy and cancer metabolism. Methods Enzymol 542:25–57. https://doi.org/10.1016/B978-0-12-416618-9.00002-9
Gomes LC, Dikic I (2014) Autophagy in antimicrobial immunity. Mol Cell 54(2):224–233. https://doi.org/10.1016/j.molcel.2014.03.009
Gong W, Wang ZY, Chen GX, Liu YQ, Gu XY, Liu WW (2013) Invasion potential of H22 hepatocarcinoma cells is increased by HMGB1-induced tumor NF-κB signaling via initiation of HSP70. Oncol Rep 30(3):1249–1256. https://doi.org/10.3892/or.2013.2595
Guadamillas MC, Cerezo A, del Pozo MA (2011) Overcoming anoikis - pathways to anchorageindependent growth in cancer. J Cell Sci 14(19):3189–3197. https://doi.org/10.1242/jcs.072165
Gualtierotti R, Biggioggero M, Penatti AE, Meroni PL (2010) Updating on the pathogenesis of systemic lupus erythematosus. Autoimmun Rev 10(1):3–7. https://doi.org/10.1016/j.autrev.2010.09.007
Gugnoni M, Sancisi V, Manzotti G, Gandolfi G, Ciarrocchi A (2016) Autophagy and epithelial–mesenchymal transition: an intricate interplay in cancer. Cell Death Dis 7(12):e2520–e2520. https://doi.org/10.1038/cddis.2016.415
Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JMS, Karantza V, Coller HA, DiPaola RS, Gelinas C, Rabinowitz JD, White E (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25(5):460–470. https://doi.org/10.1101/gad.2016311
Guo F, Liu X, Cai H, Le W (2018) Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol 28(1):3. https://doi.org/10.1111/BPA.12545
Harris J (2011) Autophagy and cytokines. Cytokine. https://doi.org/10.1016/j.cyto.2011.08.022
Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H (2011) Autophagy controls IL-1β secretion by targeting Pro-IL-1β for degradation. J Biol Chem 286(11):9587–9597. https://doi.org/10.1074/jbc.M110.202911
Haynes B, Zhang Y, Liu F, Li J, Petit S, Kothayer H, Bao X, Westwell AD, Mao G, Shekhar MPV (2016) Gold nanoparticle conjugated Rad6 inhibitor induces cell death in triple negative breast cancer cells by inducing mitochondrial dysfunction and PARP-1 hyperactivation: synthesis and characterization. Nanomed Nanotechnol Biol Med 12(3):745–757. https://doi.org/10.1016/j.nano.2015.10.010
He CW, Cui XF, Ma SJ, Xu Q, Ran YP, Chen WZ, Mu JX, Li H, Zhu J, Gong Q, Xie Z (2021) Membrane recruitment of Atg8 by Hfl1 facilitates turnover of vacuolar membrane proteins in yeast cells approaching stationary phase. BMC Biol. https://doi.org/10.1186/S12915-021-01048-7
Heiden MGV, Cantley LC, Thompson CB (2009) Understanding the warburg effect: The metabolic requirements of cell proliferation. Science. https://doi.org/10.1126/science.1160809
Hitomi K, Kotani T, Noda NN, Kimura Y, Nakatogawa H (2023) The Atg1 complex, Atg9, and Vac8 recruit PI3K complex I to the pre-autophagosomal structure. J Cell Biol. https://doi.org/10.1083/JCB.202210017/214230
Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C (2010) ATG16L1 and NOD2 interact in an autophagydependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 139(5):1630–1641. https://doi.org/10.1053/j.gastro.2010.07.006
Hoyer MJ, Swarup S, Wade Harper J (2022) Mechanisms controlling selective elimination of damaged lysosomes. Curr Opin Physiol. https://doi.org/10.1016/J.COPHYS.2022.100590
Huang T, Song X, Yang Y, Wan X, Alvarez AA, Sastry N, Feng H, Hu B, Cheng SY (2018) Autophagy and hallmarks of cancer. Crit Rev Oncog 23(5–6):247–267. https://doi.org/10.1615/CritRevOncog.2018027913
Huotari J, Helenius A (2011) Endosome maturation. EMBO J 30(17):3481. https://doi.org/10.1038/EMBOJ.2011.286
Inoki K, Kim J, Guan KL (2012) AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol 52:381–400. https://doi.org/10.1146/annurev-pharmtox-010611-134537
Itakura E, Kishi C, Inoue K, Mizushima N (2008) Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19(12):5360–5372. https://doi.org/10.1091/MBC.E08-01-0080
Ji Y, Li L, Tao Q, Zhang X, Luan J, Zhao S, Liu H, Ju D (2017) Deprivation of asparagine triggers cytoprotective autophagy in laryngeal squamous cell carcinoma. Appl Microbiol Biotechnol 101(12):4951–4961. https://doi.org/10.1007/s00253-017-8221-9
Kardideh B, Samimi Z, Norooznezhad F, Kiani S, Mansouri K (2019) Autophagy, cancer and angiogenesis: Where is the link? Cell Biosci. https://doi.org/10.1186/s13578-019-0327-6
Kaur J, Debnath J (2015) Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. https://doi.org/10.1038/nrm4024
Kaushik S, Cuervo AM (2012) Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. https://doi.org/10.1016/j.tcb.2012.05.006
Ke S, Zhou T, Yang P, Wang Y, Zhang P, Chen K, Ren L, Ye S (2017) Gold nanoparticles enhance TRAIL sensitivity through Drp1-mediated apoptotic and autophagic mitochondrial fission in NSCLC cells. Int J Nanomed 12:2531–2551. https://doi.org/10.2147/IJN.S129274
Khalil B, El Fissi N, Aouane A, Cabirol-Pol MJ, Rival T, Liévens JC (2015) PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis 6(1):e1617. https://doi.org/10.1038/cddis.2014.581
Khazen R, Müller S, Gaudenzio N, Espinosa E, Puissegur MP, Valitutti S (2016) Melanoma cell lysosome secretory burst neutralizes the CTL-mediated cytotoxicity at the lytic synapse. Nat Commun. https://doi.org/10.1038/ncomms10823
Kim RH, Bold RJ, Kung HJ (2009) ADI, autophagy and apoptosis: Metabolic stress as a therapeutic option for prostate cancer. Autophagy 5(4):567–568. https://doi.org/10.4161/auto.5.4.8252
Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152(1–2):290–303. https://doi.org/10.1016/j.cell.2012.12.016
Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, Sugimoto K, Miyazono K (2009) Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Can Res 69(23):8844–8852. https://doi.org/10.1158/0008-5472.CAN-08-4401
Ko YH, Lin Z, Flomenberg N, Pestell RG, Howell A, Sotgia F, Lisanti MP, Martinez-Outschoorn UE (2011) Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: Implications for preventing chemotherapy resistance. Cancer Biol Ther 12(12):1085–1097. https://doi.org/10.4161/cbt.12.12.18671
Kohler V, Aufschnaiter A, Büttner S (2020) Closing the gap: membrane contact sites in the regulation of autophagy. Cells. https://doi.org/10.3390/CELLS9051184
Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM (2011) Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. https://doi.org/10.1126/scitranslmed.3003182
Krause GJ, Cuervo AM (2021) Assessment of mammalian endosomal microautophagy. Methods Cell Biol 164:167. https://doi.org/10.1016/BS.MCB.2020.10.009
Krętowski R, Kusaczuk M, Naumowicz M, Kotyńska J, Szynaka B, Cechowska-Pasko M (2017) The effects of silica nanoparticles on apoptosis and autophagy of glioblastoma cell lines. Nanomaterials. https://doi.org/10.3390/nano7080230
Kriegenburg F, Ungermann C, Reggiori F (2018) Coordination of autophagosome-lysosome fusion by Atg8 family members. Curr Biol. https://doi.org/10.1016/j.cub.2018.02.034
Laha D, Pramanik A, Maity J, Mukherjee A, Pramanik P, Laskar A, Karmakar P (2014) Interplay between autophagy and apoptosis mediated by copper oxide nanoparticles in human breast cancer cells MCF7. Biochim Biophys Acta Gen Subj 1840(1):1–9. https://doi.org/10.1016/j.bbagen.2013.08.011
Lamark T, Johansen T (2012) Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. https://doi.org/10.1155/2012/736905
Lapaquette P, Guzzo J, Bretillon L, Bringer MA (2015) Cellular and molecular connections between autophagy and inflammation. Mediat Inflamm. https://doi.org/10.1155/2015/398483
Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, Mizushima N, Grinstein S, Iwasaki A (2010) In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 32(2):227–239. https://doi.org/10.1016/j.immuni.2009.12.006
Levine B (2005) Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120(2):159–162. https://doi.org/10.1016/J.CELL.2005.01.005
Levonen AL, Hill BG, Kansanen E, Zhang J, Darley-Usmar VM (2014) Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radical Biol Med. https://doi.org/10.1016/j.freeradbiomed.2014.03.025
Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y, Song Z, Zheng Q, Xiong J (2013a) Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 34(6):1343–1351. https://doi.org/10.1093/carcin/bgt063
Li M, Khambu B, Zhang H, Kang JH, Chen X, Chen D, Vollmer L, Liu PQ, Vogt A, Yin XM (2013b) Suppression of lysosome function induces autophagy via a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J Biol Chem 288(50):35769. https://doi.org/10.1074/JBC.M113.511212
Li Y, Zhao T, Liu B, Halaweish I, Mazitschek R, Duan X, Alam HB (2015) Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic model. J Trauma Acute Care Surg 78(2):378–385. https://doi.org/10.1097/TA.0000000000000510
Li X, Feng J, Zhang R, Wang J, Su T, Tian Z, Han D, Zhao C, Fan M, Li C, Liu B, Feng X, Nie Y, Wu K, Chen Y, Deng H, Feng C (2016) Quaternized chitosan/alginate-fe3o4 magnetic nanoparticles enhance the chemosensitization of multidrug-resistant gastric carcinoma by regulating cell autophagy activity in mice. J Biomed Nanotechnol 12(5):948–961. https://doi.org/10.1166/jbn.2016.2232
Li X, Lu Q, Xie W, Wang Y, Wang G (2018) Anti-tumor effects of triptolide on angiogenesis and cell apoptosis in osteosarcoma cells by inducing autophagy via repressing Wnt/β-Catenin signaling. Biochem Biophys Res Commun 496(2):443–449. https://doi.org/10.1016/j.bbrc.2018.01.052
Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU (2006) Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 8(7):688–698. https://doi.org/10.1038/ncb1426
Lin NY, Beyer C, Gießl A, Kireva T, Scholtysek C, Uderhardt S, Enrique Munoz L, Dees C, Distler A, Wirtz S, Krönke G, Spencer B, Distler O, Schett G, Distler JHW (2013) Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann Rheum Dis 72(5):761–768. https://doi.org/10.1136/annrheumdis-2012-201671
Lisnevskaia L, Murphy G, Isenberg D (2014) Systemic lupus erythematosus. Lancet 384(9957):1878–1888. https://doi.org/10.1016/S0140-6736(14)60128-8
Liu P, Jin H, Guo Z, Ma J, Zhao J, Li D, Wu H, Gu N (2016) Silver nanoparticles outperform gold nanoparticles in radiosensitizing U251 cells in vitro and in an intracranial mouse model of glioma. Int J Nanomed 11:5003–5014. https://doi.org/10.2147/IJN.S115473
Lock R, Kenific CM, Leidal AM, Salas E, Debnath J (2014) Autophagy-dependent production of secreted factors facilitates oncogenic RAS-Driven invasion. Cancer Discov 4(4):466–479. https://doi.org/10.1158/2159-8290.CD-13-0841
López P, Alonso-Pérez E, Rodríguez-Carrio J, Suárez A (2013) Influence of Atg5 mutation in SLE depends on functional IL-10 genotype. PLoS One 8(10):e78756. https://doi.org/10.1371/journal.pone.0078756
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153(6):1194–1217. https://doi.org/10.1016/j.cell.2013.05.039
Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, Kondo S, Kondo Y, Yu Y, Mills GB, Liao WSL, Bast RC (2008) The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Investig 118(12):3917–3929. https://doi.org/10.1172/JCI35512
Manzoni C, Mamais A, Dihanich S, McGoldrick P, Devine MJ, Zerle J, Kara E, Taanman JW, Healy DG, Marti-Masso JF, Schapira AH, Plun-Favreau H, Tooze S, Hardy J, Bandopadhyay R, Lewis PA (2013) Pathogenic parkinson’s disease mutations across the functional domains of LRRK2 alter the autophagic/lysosomal response to starvation. Biochem Biophys Res Commun 441(4):862–866. https://doi.org/10.1016/j.bbrc.2013.10.159
Maria Fimia G, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F (2007) Ambra1 regulates autophagy and development of the nervous system. Nature 447(7148):1121–1125. https://doi.org/10.1038/nature05925
Masoud GN, Li W (2015) HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharmaceutica Sinica B. https://doi.org/10.1016/j.apsb.2015.05.007
Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T (2009) Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11(4):385–396. https://doi.org/10.1038/ncb1846
McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, Duerr RH, Silverberg MS, Taylor KD, Rioux JD, Altshuler D, Daly MJ, Xavier RJ (2008) Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet 40(9):1107–1112. https://doi.org/10.1038/ng.215
Mccormick J, Suleman N, Scarabelli TM, Knight RA, Latchman DS, Stephanou A (2012) STAT1 deficiency in the heart protects against myocardial infarction by enhancing autophagy. J Cell Mol Med 16(2):386–393. https://doi.org/10.1111/j.1582-4934.2011.01323.x
Mejlvang J, Olsvik H, Svenning S, Bruun JA, Abudu YP, Larsen KB, Brech A, Hansen TE, Brenne H, Hansen T, Stenmark H, Johansen T (2018) Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J Cell Biol 217(10):3640. https://doi.org/10.1083/JCB.201711002
Menzies FM, Fleming A, Rubinsztein DC (2015) Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. https://doi.org/10.1038/nrn3961
Mizushima N (2010) The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 22(2):132–139. https://doi.org/10.1016/J.CEB.2009.12.004
Mizushima N, Levine B (2010) Autophagy in mammalian development and differentiation. Nat Cell Biol. https://doi.org/10.1038/ncb0910-823
Monkkonen T (2017) Debnath J (2018) Inflammatory signaling cascades and autophagy in cancer. Autophagy 10(1080/15548627):1345412
Mowers EE, Sharifi MN, Macleod KF (2018) Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. https://doi.org/10.1111/febs.14388
Mozdoori N, Safarian S, Sheibani N (2017) Augmentation of the cytotoxic effects of zinc oxide nanoparticles by MTCP conjugation: Non-canonical apoptosis and autophagy induction in human adenocarcinoma breast cancer cell lines. Mater Sci Eng, C 78:949–959. https://doi.org/10.1016/j.msec.2017.03.300
Murrow L, Debnath J (2013) Autophagy as a stress response and quality control mechanism—implications for cell injury and human disease. Annu Rev Pathol 8:105. https://doi.org/10.1146/ANNUREV-PATHOL-020712-163918
Nakamura S, Yoshimori T (2017) New insights into autophagosome-lysosome fusion. J Cell Sci. https://doi.org/10.1242/jcs.196352
Nakatogawa H (2013) Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem 55(1):39–50. https://doi.org/10.1042/BSE0550039
Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y (2009) Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat Rev Mol Cell Biol. https://doi.org/10.1038/nrm2708
Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L (2008) Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 455(7211):396–400. https://doi.org/10.1038/nature07208
Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW (2015) Netea MG (2016) Modulation of inflammation by autophagy: Consequences for human disease. Autophagy. https://doi.org/10.1080/15548627.2015.1071759
New J, Arnold L, Ananth M, Alvi S, Thornton M, Werner L, Tawfik O, Dai H, Shnayder Y, Kakarala K, Tsue TT, Girod DA, Ding WX, Anant S, Thomas SM (2017) Secretory autophagy in cancer-associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Can Res 77(23):6679–6691. https://doi.org/10.1158/0008-5472.CAN-17-1077
Oku Y, Oku M, Sakai Y (2018) Three distinct types of microautophagy based on membrane dynamics and molecular machineries. BioEssays 2018(6):1800008–1802018. https://doi.org/10.1002/bies.201800008
Onorati AV, Dyczynski M, Ojha R, Amaravadi RK (2018) Targeting autophagy in cancer. Cancer. https://doi.org/10.1002/cncr.31335
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16(4):394–406. https://doi.org/10.1038/nn.3350
Park C, Suh Y, Cuervo AM (2015) Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun. https://doi.org/10.1038/ncomms7823
Parzych KR, Klionsky DJ (2014) An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20(3):460. https://doi.org/10.1089/ARS.2013.5371
Patergnani S, Missiroli S, Morciano G, Perrone M, Mantovani CM, Anania G, Fiorica F, Pinton P, Giorgi C (2021) Understanding the role of autophagy in cancer formation and progression is a real opportunity to treat and cure human cancers. Cancers 13(22):5622. https://doi.org/10.3390/cancers13225622
Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, Fagioli C, Merlini A, Mariani E, Pasqualetto E, Orfanelli U, Ponzoni M, Sitia R, Casola S, Cenci S (2013) Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol 14(3):298–305. https://doi.org/10.1038/ni.2524
Penke B, Bogár F, Crul T, Sántha M, Tóth ME, Vígh L (2018) Heat shock proteins and autophagy pathways in neuroprotection: from molecular bases to pharmacological interventions. Int J Mol Sci. https://doi.org/10.3390/IJMS19010325
Pickrell AM, Youle RJ (2015) The roles of PINK1, Parkin, and mitochondrial fidelity in parkinson’s disease. Neuron. https://doi.org/10.1016/j.neuron.2014.12.007
Polson HEJ, De Lartigue J, Rigden DJ, Reedijk M, Urbé S, Clague MJ, Tooze SA (2010) Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 6(4):506–522. https://doi.org/10.4161/AUTO.6.4.11863
Pool H, Campos-Vega R, Herrera-Hernández MG, García-Solis P, García-Gasca T, Sánchez IC, Luna-Bárcenas G, Vergara-Castañeda H (2018) Development of genistein-PEGylated silica hybrid nanomaterials with enhanced antioxidant and antiproliferative properties on HT29 human colon cancer cells. Am J Transl Res 10(8):2306–2323
Ren KW, Li YH, Wu G, Ren JZ, Lu HB, Li ZM, Han XW (2017) Quercetin nanoparticles display antitumor activity via proliferation inhibition and apoptosis induction in liver cancer cells. Int J Oncol 50(4):1299–1311. https://doi.org/10.3892/ijo.2017.3886
Rubinsztein DC, Mariño G, Kroemer G (2011) Autophagy and aging. Cell. https://doi.org/10.1016/j.cell.2011.07.030
Ryan BJ, Hoek S, Fon EA, Wade-Martins R (2015) Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci 40(4):200–210. https://doi.org/10.1016/j.tibs.2015.02.003
Sachdev U, Cui X, Hong G, Namkoong S, Karlsson JM, Baty CJ, Tzeng E (2012) High mobility group box 1 promotes endothelial cell angiogenic behavior in vitro and improves muscle perfusion in vivo in response to ischemic injury. J Vasc Surg 55(1):180–191. https://doi.org/10.1016/j.jvs.2011.07.072
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456(7219):264–268. https://doi.org/10.1038/nature07383
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141(2):290–303. https://doi.org/10.1016/J.CELL.2010.02.024
Schaaf MB, Houbaert D, Meçe O, Agostinis P (2019) Autophagy in endothelial cells and tumor angiogenesis. Cell Death Different. https://doi.org/10.1038/s41418-019-0287-8
Schäfer JA, Schessner JP, Bircham PW, Tsuji T, Funaya C, Pajonk O, Schaeff K, Ruffini G, Papagiannidis D, Knop M, Fujimoto T, Schuck S (2020) ESCRT machinery mediates selective microautophagy of endoplasmic reticulum in yeast. EMBO J https://doi.org/10.15252/EMBJ.2019102586
Scherz-Shouval R, Shvets E, Elazar Z (2007) Oxidation as a post-translational modification that regulates autophagy. Autophagy 3(4):371–373. https://doi.org/10.4161/auto.4214
Schnebert S, Goguet M, Vélez EJ, Depincé A, Beaumatin F, Herpin A, Seiliez I (2022) Diving into the evolutionary history of HSC70-linked selective autophagy pathways: endosomal microautophagy and chaperone-mediated autophagy. Cells. https://doi.org/10.3390/cells11121945ï
Settembre C, Di Malta C, Polito VA, Arencibia MG, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332(6036):1429–1433. https://doi.org/10.1126/science.1204592
Shen Y, Li DD, Wang LL, Deng R, Zhu XF (2008) Decreased expression of autophagy-related proteins in malignant epithelial ovarian cancer. Autophagy 4(8):1067–1068. https://doi.org/10.4161/auto.6827
Shen W, Zhang X, Fu X, Fan J, Luan J, Cao Z, Yang P, Xu Z, Ju D (2017) A novel and promising therapeutic approach for NSCLC: Recombinant human arginase alone or combined with autophagy inhibitor. Cell Death Dis 8(3):e2720–e2720. https://doi.org/10.1038/cddis.2017.137
Shen H, Yin L, Deng G, Guo C, Han Y, Li Y, Cai C, Fu Y, Liu S, Zeng S (2018) Knockdown of Beclin-1 impairs epithelial-mesenchymal transition of colon cancer cells. J Cell Biochem 119(8):7022–7031. https://doi.org/10.1002/jcb.26912
Shen D, Liu K, Wang H, Wang H (2022) Autophagy modulation in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol 209(2):140–150. https://doi.org/10.1093/cei/uxac017
Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J (2016) Aging and autophagy in the heart. Circul Res. https://doi.org/10.1161/CIRCRESAHA.116.307474
Singh SS, Vats S, Chia AYQ, Tan TZ, Deng S, Ong MS, Arfuso F, Yap CT, Goh BC, Sethi G, Huang RYJ, Shen HM, Manjithaya R, Kumar AP (2018) Dual role of autophagy in hallmarks of cancer. Oncogene 37(9):1142–1158. https://doi.org/10.1038/s41388-017-0046-6
Singh N, Baby D, Rajguru J, Patil P, Thakkannavar S, Pujari V (2019) Inflammation and cancer. Ann Afr Med 18(3):121–126. https://doi.org/10.4103/aam.aam_56_18
Sinha R, Kim GJ, Nie S, Shin DM (2006) Nanotechnology in cancer therapeutics: bioconjugated nanoparticles for drug delivery. Mol Cancer Ther. https://doi.org/10.1158/1535-7163.MCT-06-0141
Stefaniak S, Wojtyla Ł, Pietrowska-Borek M, Borek S (2020) Completing autophagy: formation and degradation of the autophagic body and metabolite salvage in plants. Int J Mol Sci. https://doi.org/10.3390/IJMS21062205
Stefanoni G, Sala G, Tremolizzo L, Brighina L, Ferrarese C (2012) Role of autophagy in Parkinson’s disease. Autophagy. https://doi.org/10.2174/0929867325666180226094351
Sung SJ, Kim HK, Hong YK, Joe YA (2019) Autophagy is a potential target for enhancing the anti-angiogenic effect of mebendazole in endothelial cells. Biomol Ther 27(1):117–125. https://doi.org/10.4062/biomolther.2018.222
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mulé JJ, Pledger WJ, Wang HG (2007) Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9(10):1142–1151. https://doi.org/10.1038/ncb1634
Takahashi H, Inoue J, Sakaguchi K, Takagi M, Mizutani S, Inazawa J (2017) Autophagy is required for cell survival under L-asparaginase-induced metabolic stress in acute lymphoblastic leukemia cells. Oncogene 36(30):4267–4276. https://doi.org/10.1038/onc.2017.59
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N (2011) Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25(8):795–800. https://doi.org/10.1101/gad.2016211
Tan VP, Miyamoto S (2016) Nutrient-sensing mTORC1: integration of metabolic and autophagic signals. J Mol Cell Cardiol 95:31. https://doi.org/10.1016/J.YJMCC.2016.01.005
Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, Nishida K, Shimizu T, Hori M, Komuro I, Shirasawa T, Mizushima N, Otsu K (2010) Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 6(5):600–606. https://doi.org/10.4161/auto.6.5.11947
Tekirdag K, Cuervo AM (2018) Chaperone-mediated autophagy and endosomal microautophagy: Jointed by a chaperone. J Biol Chem 293(15):5414. https://doi.org/10.1074/JBC.R117.818237
Tirichen H, Yaigoub H, Xu W, Wu C, Li R, Li Y (2021) Mitochondrial reactive oxygen species and their contribution in chronic kidney disease progression through oxidative stress. Front Physiol 12:627837. https://doi.org/10.3389/FPHYS.2021.627837/BIBTEX
Tittarelli A, Mendoza-Naranjo A, Farías M, Guerrero I, Ihara F, Wennerberg E, Riquelme S, Gleisner A, Kalergis A, Lundqvist A, López MN, Chambers BJ, Salazar-Onfray F (2014) Gap Junction intercellular communications regulate NK cell activation and modulate NK cytotoxic capacity. J Immunol 192(3):1313–1319. https://doi.org/10.4049/jimmunol.1301297
Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, Pan L, Ince TA, Kroemer G, Brugge JS, Yuan J (2013) Chaperone-mediated autophagy degrades mutant p53. Genes Develop 27(15):1718–1730. https://doi.org/10.1101/gad.220897.113
Verma AK, Bharti PS, Rafat S, Bhatt D, Goyal Y, Pandey KK, Ranjan S, Almatroodi SA, Alsahli MA, Rahmani AH, Almatroudi A, Dev K (2021) Autophagy paradox of cancer: role, regulation, and duality. Oxid Med Cell Longev 2021:8832541. https://doi.org/10.1155/2021/8832541
Von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A (2017) The role of stromal cancer-associated fibroblasts in pancreatic cancer. J Hematol Oncol. https://doi.org/10.1186/s13045-017-0448-5
Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B (2012a) Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 338(6109):956–959. https://doi.org/10.1126/science.1225967
Wang Y, Zi XY, Su J, Zhang HX, Zhang XR, Zhu HY, Li JX, Yin M, Yang F, Hu YP (2012b) Cuprous oxide nanoparticles selectively induce apoptosis of tumor cells. Int J Nanomed 7:2641–2652. https://doi.org/10.2147/IJN.S31133
Wang Q, Xue L, Zhang X, Bu S, Zhu X, Lai D (2016) Autophagy protects ovarian cancer-associated fibroblasts against oxidative stress. Cell Cycle 15(10):1376–1385. https://doi.org/10.1080/15384101.2016.1170269
Wang J, Yu Y, Lu K, Yang M, Li Y, Zhou X, Sun Z (2017) Silica nanoparticles induce autophagy dysfunction via lysosomal impairment and inhibition of autophagosome degradation in hepatocytes. Int J Nanomed 12:809–825. https://doi.org/10.2147/IJN.S123596
Wang J, Gao S, Wang S, Xu Z, Wei L (2018) Zinc oxide nanoparticles induce toxicity in CAL 27 oral cancer cell lines by activating PINK1/Parkin-mediated mitophagy. Int J Nanomed 13:3441–3450. https://doi.org/10.2147/IJN.S165699
Wang Y, Liu N, Lu B (2019) Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci Ther 25(7):859. https://doi.org/10.1111/CNS.13140
Wang S, Li H, Yuan M, Fan H, Cai Z (2022) Role of AMPK in autophagy. Front Physiol 13:1015500. https://doi.org/10.3389/FPHYS.2022.1015500/BIBTEX
Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, Grishin NV, Peyton M, Minna J, Bhagat G, Levine B (2013) XEGFR-mediated beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 154(6):1269. https://doi.org/10.1016/j.cell.2013.08.015
Wei F, Wang Y, Luo Z, Li Y, Duan Y (2017) New findings of silica nanoparticles induced ER autophagy in human colon cancer cell. Scient Rep. https://doi.org/10.1038/srep42591
White E (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. https://doi.org/10.1038/nrc3262
White E (2015) The role for autophagy in cancer. J Clin Invest. https://doi.org/10.1172/JCI73941
Wicki A, Witzigmann D, Balasubramanian V, Huwyler J (2015) Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Controll Release. https://doi.org/10.1016/j.jconrel.2014.12.030
Wu DJ, Adamopoulos IE (2017) Autophagy and autoimmunity. Clin Immunol 176:55–62. https://doi.org/10.1016/j.clim.2017.01.007
Xia L, Wang Y, Chen Y, Yan J, Hao F, Su X, Zhang C, Xu M (2017) Cuprous oxide nanoparticles inhibit the growth of cervical carcinoma by inducing autophagy. Oncotarget 8(37):61083–61092. https://doi.org/10.18632/oncotarget.17854
Xilouri M, Brekk OR, Stefanis L (2016) Autophagy and alpha-synuclein: relevance to Parkinson’s disease and related synucleopathies. Mov Dis. https://doi.org/10.1002/mds.26477
Yang ZJ, Chee CE, Huang S, Sinicrope FA (2011) The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 10(9):1533. https://doi.org/10.1158/1535-7163.MCT-11-0047
Ye X, Sun X, Starovoytov V, Cai Q (2015) Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum Mol Genet 24(10):2938–2951. https://doi.org/10.1093/hmg/ddv056
Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, Tavaré S, Arakawa S, Shimizu S, Watt FM, Narita M (2009) Autophagy mediates the mitotic senescence transition. Genes Dev 23(7):798–803. https://doi.org/10.1101/gad.519709
Yuan YG, Gurunathan S (2017) Combination of graphene oxide-silver nanoparticle nanocomposites and cisplatin enhances apoptosis and autophagy in human cervical cancer cells. Int J Nanomed 12:6537–6558. https://doi.org/10.2147/IJN.S125281
Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 100(25):15077–15082. https://doi.org/10.1073/pnas.2436255100
Yun CW, Lee SH (2018) The Roles of Autophagy in Cancer. Int J Mol Sci. https://doi.org/10.3390/IJMS19113466
Zaarour RF, Azakir B, Hajam EY, Nawafleh H, Zeinelabdin NA, Engelsen AST, Thiery J, Jamora C, Chouaib S (2021) Role of hypoxia-mediated autophagy in tumor cell death and survival. Cancers. https://doi.org/10.3390/CANCERS13030533
Zaffagnini G, Martens S (2016) Mechanisms of selective autophagy. J Mol Biol 428(9):1714. https://doi.org/10.1016/J.JMB.2016.02.004
Zare-Shahabadi A, Masliah E, Johnson GVW, Rezaei N (2015) Autophagy in Alzheimer’s disease. Rev Neurosci 26(4):385–395. https://doi.org/10.1515/revneuro-2014-0076
Zhang XF, Gurunathan S (2016) Combination of salinomycin and silver nanoparticles enhances apoptosis and autophagy in human ovarian cancer cells: An effective anticancer therapy. Int J Nanomed 11:3655–3675. https://doi.org/10.2147/IJN.S111279
Zhang Q, Kang R, Zeh HJ, Lotze MT, Tang D (2013) DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy 9(4):451. https://doi.org/10.4161/AUTO.23691
Zhao M, Klionsky DJ (2011) AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metabol. https://doi.org/10.1016/j.cmet.2011.01.009
Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 11(4):468–476. https://doi.org/10.1038/ncb1854
Zhou XJ, Lu XL, Lv JC, Yang HZ, Qin LX, Zhao MH, Su Y, Li ZG, Zhang H (2011) Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis 70(7):1330–1337. https://doi.org/10.1136/ard.2010.140111
Zhou W, Xu G, Wang Y, Xu Z, Liu X, Xu X, Ren G, Tian K (2017) Oxidative stress induced autophagy in cancer associated fibroblast enhances proliferation and metabolism of colorectal cancer cells. Cell Cycle 16(1):73–81. https://doi.org/10.1080/15384101.2016.1252882
Zielinska E, Zauszkiewicz-Pawlak A, Wojcik M, Inkielewicz-Stepniak I (2018) Silver nanoparticles of different sizes induce a mixed type of programmed cell death in human pancreatic ductal adenocarcinoma. Oncotarget 9(4):4675–4697. https://doi.org/10.18632/oncotarget.22563
Zou J, Fei Q, Xiao H, Wang H, Liu K, Liu M, Zhang H, Xiao X, Wang K, Wang N (2019) VEGF-A promotes angiogenesis after acute myocardial infarction through increasing ROS production and enhancing ER stress-mediated autophagy. J Cell Physiol 234(10):17690–17703. https://doi.org/10.1002/jcp.28395
Acknowledgements
It is with gratitude that the authors affirm the assistance of Delhi Technological University for the completion of this study.
Author information
Authors and Affiliations
Contributions
The study was designed and conceived by all authors. Data analysis and paper writing were done by the first and second authors. Each author made revisions to the manuscript. It is understood that all authors have reviewed the final manuscript version and agreed to be held accountable for its content.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethics approval
The study conducted at the Department of Biotechnology at Delhi Technological University in India was an observational study. According to the ethical guidelines of Delhi Technological University, ethical approval was not required for this study as it did not involve any humans or animals. The study was conducted under the supervision of Dr. Asmita Das and Dr. Prakash Chandra, who ensured compliance with the university’s ethical guidelines.
Consent for publication
Not applicable.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mathur, A., Ritu, Chandra, P. et al. Autophagy: a necessary evil in cancer and inflammation. 3 Biotech 14, 87 (2024). https://doi.org/10.1007/s13205-023-03864-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-023-03864-w