
Abstract
Autophagy is an evolutionarily conserved process that delivers intracellular constituents to the lysosomes for degradation and recycling. Autophagy plays a central role in diverse physiological processes and has been implicated in the pathogenesis of various diseases including cancer. The role of autophagy in cancer is complex and largely context-dependent. Accumulating evidence indicates that autophagy facilitates tumorigenesis by enabling acquisition of cancer hallmarks. Autophagy manipulation has emerged as a promising strategy in cancer treatment. In this chapter, we provide an overview of the autophagic process, highlight the autophagy conundrum in cancer, examine the complex and conflicting reports on autophagy in tumour suppression and tumour promotion, as well as the role of autophagy in the acquisition of cancer hallmarks. Finally, from the clinical perspective, we summarise the evidence for autophagy-related genes and proteins as reliable markers of disease severity and prognosis and analyse the efficacy of autophagy manipulation in improving cancer treatment outcomes and circumventing chemoresistance.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
11.1 Introduction
Autophagy is an evolutionarily conserved process by which aberrant, unwanted proteins and damaged organelles are sequestered into double-membraned vesicles called autophagosomes and subsequently delivered to the lysosomes for degradation to maintain cellular homeostasis (Bishop and Bradshaw 2018). The term ‘autophagy’, coined by Christian de Duve in 1963, is derived from the Greek words, ‘auto’ meaning “self” and ‘phagein’ meaning “to eat” (Klionsky 2008).
Autophagy is categorised into three distinct types based on the mechanism of cargo delivery to the lysosomes for degradation, microautophagy, chaperone-mediated autophagy (CMA) and macroautophagy. Microautophagy seen in yeast involves the sequestration of small cargoes by protrusion or invagination of endolysosomal membranes. CMA mediates the degradation of soluble proteins in the lysosomes with the help of molecular chaperones and lysosome-associated membrane protein 2A (LAMP2A). Macroautophagy (henceforth referred to as autophagy), the best-characterised and evolutionarily conserved type of autophagy, requires the formation of double-membrane structures termed autophagosomes for the delivery of cargoes to the lysosomes. Macroautophagy may be further classified into selective autophagy, characterised by high cargo specificity, and non-selective (bulk) autophagy which lacks cargo specificity (Allen and Baehrecke 2020; Parzych and Klionsky 2014).
Autophagy is intricately involved in health and disease. It plays a vital role in cellular turnover, development, differentiation, tissue remodelling and cell death. Autophagy is believed to function as a double-edged sword in disease processes and may have a causative or protective role. Autophagy has been implicated in ageing, infections, neurodegenerative disorders and cancer (Shintani and Klionsky 2004). Yoshinori Ohsumi was awarded the Nobel Prize for Physiology or Medicine in 2016 for his seminal work on autophagy that led to a new paradigm in understanding physiological processes such as the adaptation to starvation as well as diseases such as cancer (https://www.nobelprize.org/prizes/medicine/2016/press-release/).
11.2 Physiological Functions of Autophagy
Autophagy is essential at every stage during the development of various organisms and mediates a plethora of diverse cellular processes. Autophagy plays a critical role in the maintenance of cellular homeostasis. Under basal conditions, autophagy is involved in housekeeping functions such as removal of damaged organelles, misfolded proteins and protein aggregates. On the other hand, during starvation, autophagy promotes bioenergetic homeostasis by breaking down cellular macromolecules to generate ATP for cellular functions (Klionsky 2020; Mowers et al. 2017). Besides nutrient deprivation, autophagy is also induced to mitigate stress due to hypoxia and reactive oxygen species (ROS). During embryogenesis, autophagy catalyses the removal of paternal mitochondria. Autophagy is required for mediating immune and inflammatory response, defence against microbial infections, cell-fate determination, tissue remodelling, preservation of organelle function, recycling of intracellular proteins, prevention of toxic build-up of waste products and gene silencing. Autophagy also protects cells from undergoing programmed cell death by apoptosis (Allen and Baehrecke 2020; Singh et al. 2018).
11.3 The Autophagic Process and Components
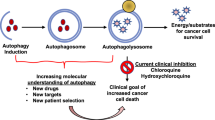
Autophagy occurs at a basal level in all cells and can be induced by various types of stress including nutrient deprivation, hypoxia, ROS, damaged cell organelles and as a part of the DNA damage response (DDR) (Singh et al. 2018). Autophagy is divided into five stages: initiation, nucleation of the initial sequestering compartment termed the phagophore, expansion and elongation of the phagophore to form the double-membrane structure called the autophagosome, fusion of the outer membrane of the autophagosome with the lysosome to form the autolysosome and cargo degradation and recycling (Hansen et al. 2018). Each stage of autophagy has potential therapeutic targets for intervention (Fig. 11.1) (Mulcahy Levy and Thorburn 2020).

Schematic representation of the mechanism and regulation of autophagy. The mTOR kinase is the key signalling molecule involved in the regulation of autophagy. In an unstressed state, activated mTORC1 phosphorylates and inactivates autophagy-related proteins and inhibits the ULK/FIP200/ATG13 complex with consequent inhibition of autophagy. Induction of autophagy by starvation, oxidative stress and hypoxia, inhibits mTORC1 that in turn releases and activates the ULK/FIP200/ATG13 complex. This leads to activation of the class III phosphoinositide 3-kinase (PI3K) complex comprised of Vps34, p150, Beclin-1, Atg14L and Autophagy and Beclin1 Regulator 1 (AMBRA1), which then drives the nucleation of the isolation membrane. Expansion and elongation of the isolation membrane involve conversion of cytoplasmic LC3-I to the lipidated LC3-II, followed by conjugation of phosphatidylethanolamine (PE) to LC3-II mediated by ATG4B and ATG7. Localisation of ATG5-ATG12/ATG16L complex helps in elongation by recruitment of LC3-II to the membrane. The ends of the isolation membrane fuse to form the autophagosome, which fuses with the lysosomes to form autolysosomes. The cargo is degraded in the autolysosomes by lysosomal enzymes and biomolecules recycled back to the cytoplasm
The process of autophagy is mediated by the highly conserved autophagy-related genes (ARGs), (Allen and Baehrecke 2020; Singh et al. 2018). Autophagy is initiated in response to various cellular signals by the Unc-51-like autophagy activating kinase (ULK1) complex comprising ULK1, ULK2, Atg13, Atg101 and the scaffolding protein RB1 inducible coiled-coil 1 (RBCC1) also known as FAK family kinase-interacting protein of 200 kDa (FIP200). This is followed by membrane nucleation and formation of the phagophore that requires synthesis of phosphatidylinositol-3-phosphate by activation of a class III phosphoinositide 3-kinase (PI3K) complex, composed of a PI3K, ATG14L, vacuolar protein sorting-associated proteins 15 and 34 (VPS15 and VPS34) and Beclin-1. The ATG9 trafficking system (ATG2A/ATG2B, WDR45/WIP14 and ATG9A) is responsible for elongation of the phagophore. The phagophore expands by acquisition of lipids promoted by two ubiquitin-like conjugation systems, the ATG5–ATG12–ATG16 complex and microtubule-associated protein light chain 3 (LC3) to form the autophagosome. Formation of the ATG5–ATG12–ATG16 complex is followed by conversion of the cytosolic LC3-I to the lipidated LC3-II that conjugates to phosphatidylethanolamine and incorporated into the phagophore membrane. The adaptor protein p62/sequestosome 1 (SQSTM1) binds to LC3-II during autophagosome formation and facilitates the degradation of ubiquitinated proteins (Bishop and Bradshaw 2018; Marinkovic et al. 2018). The autophagosome then fuses with a lysosome, to form an autolysosome in a process requiring small G-protein Rab7, soluble N-ethylmaleimide-sensitive factor attachment proteins (SNAREs), syntaxin17 (Stx17) and the membrane tethering complex HOPS (Dikic and Elazar 2018; Zhi et al. 2018). The autophagic process is completed within the autolysosomes by enzymatic degradation of the cargo and recycling of nutrients (Fig. 11.1).
Autophagy is regulated by the mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) signalling pathways. mTORC1 which is activated by nutrients and growth factors at the lysosomes induces phosphorylation of the ULK complex with repression of autophagy. On the other hand, nutrient deprivation inactivates mTORC1 leading to activation of the ULK complex and autophagy induction (Fig. 11.1). Recent evidence indicates that autophagy is also regulated by epigenetic mechanisms including histone modifications, DNA methylation and by noncoding RNAs (ncRNAs) (Baek and Kim 2017; Hu 2019). Epigenetic changes influence ARGs as well as the signalling molecules and pathways that regulate autophagy. While the co-activator-associated arginine methyltransferase 1 (CARM1) enhances transcriptional activation of ARGs, EZH2, a methyltransferase is reported to silence autophagy-activating promoters by methylation. Shin et al. (2016a) found CARM1-mediated arginine methylation (H3R17me2) as a critical epigenetic mark in autophagic induction.
11.4 Role of Autophagy in Cancer
The role of autophagy in cancer is complex and bidirectional. Autophagy has been documented to suppress or promote tumour development based on the context and the stage of tumorigenesis. Autophagy has been documented to be low in premalignant lesions and enhanced in advanced cancers (Galluzzi et al. 2015; Mulcahy Levy and Thorburn 2020).
11.4.1 Tumour Suppressive Effects of Autophagy
Autophagy prevents carcinogenesis by virtue of its ability to remove aggregated, misfolded and oncogenic proteins. Additionally, autophagy also exerts tumour-suppressive effects by stimulating the immune response. Decreased autophagy was shown to be associated with infiltration of regulatory T cells, leading to diminished immunosurveillance that facilitates tumour development (Parzych and Klionsky 2014). The tumour preventive role of autophagy has also been attributed to be mediated via scavenging endogenous sources of ROS and maintaining genomic stability (Galluzzi et al. 2015). Although genetic alterations in several ARGs have been extensively documented, a large-scale human genomic analysis of somatic mutations in ATG genes across 11 cancer types revealed that the core autophagy machinery, which plays a critical role in maintaining genomic stability does not undergo genetic alterations (Lebovitz et al. 2015).
Monoallelelic deletion of Beclin-1, a haploinsufficient tumour suppressor gene, has been reported in breast, ovarian and prostate cancers (Delaney et al. 2020; Qu et al. 2003). The loss of Beclin-1 was associated with reduced autophagy and increased proliferation (Lee and Wu 2012; Zhang et al. 2018). However, biallelic Beclin-1 mutations that could cause embryonic lethality do not occur in cancer. This implies that monoallelic Beclin-1 is adequate to facilitate the requirement of functional autophagy necessary for neoplastic transformation (Yue et al. 2003). The Vps34-binding domain of Beclin-1 was shown to be essential for its tumour suppressor activity (Furuya et al. 2005). The tumour suppressor functions of Beclin-1 are also mediated through UVRAG and Bax-interacting factor-1 (Bif-1), which increase binding of Beclin-1 to Vps34 (Takahashi et al. 2007). Monoallelic deletion or mutations of UVRAG as well as downregulation of Bif-1 have been documented in diverse malignancies (Kung et al. 2011).
In addition to Beclin-1, several components of the core autophagy machinery were also found to display tumour suppressor functions. Loss-of-function mutations in ATG2B, ATG5, ATG9B and ATG12 leading to truncated ATG proteins were identified in gastric and colorectal cancers with microsatellite instability (Kang et al. 2009). Mice with deficiency of Atg5 and Atg7 showed mitochondrial damage, oxidative stress and propensity to develop liver tumours (Takamura et al. 2011). Loss of ATG4C, involved in processing LC3/ATG8 during autophagosome formation, was reported in chemically induced murine fibrosarcomas (Kimmelman 2011). Somatic mutations of ATG5 coupled with overexpression of ATG16L2 observed in various tumours prevented the interaction of ATG5 with ATG16L1, with consequent proteasomal degradation of ATG12 and ATG16L1, resulting in inhibition of autophagy (Wible et al. 2019).
p62/SQSTM1, an autophagy receptor and selective substrate for autophagy, accumulates when autophagy is inhibited with fall in levels when autophagy is induced. It thus serves as a reliable marker of autophagic flux (Mathew et al. 2009). Aberrant accumulation of p62/SQSTM1 has been reported in gastrointestinal cancer (Su et al. 2005), prostate cancer (Kitamura et al. 2006), hepatocellular carcinoma (Umemura et al. 2016), breast cancer (Li et al. 2017) and lung adenocarcinoma (Inoue et al. 2012), suggesting that autophagy inhibits tumorigenesis by decreasing p62 accumulation (Li et al. 2020).
There is growing evidence to indicate that autophagy is stimulated by well-established tumour suppressors such as TP53 and phosphatase and tensin homolog (PTEN). In HT-29 colon cancer cells, PTEN was found to promote autophagy, whereas loss-of-function mutations in PTEN suppressed autophagy (Errafiy et al. 2013). Taken together, these findings underscore the anti-tumour effects of autophagy (Fig.11.2).

The dual role of autophagy in tumorigenesis
11.4.2 Tumour-Promoting Effects of Autophagy
Although autophagy is reported to suppress the development and progression of tumours, substantial evidence indicates that autophagy facilitates tumorigenesis. Autophagy is a strategy that enables acquisition of cancer hallmarks and survives tumour microenvironmental stress. Several studies have demonstrated the key role of autophagy in providing essential metabolites to meet the growing demands of proliferating tumour cells (Kocaturk et al. 2019; Mulcahy Levy and Thorburn 2020; Singh et al. 2018; Yang and Klionsky 2020). Autophagy fuels enhanced metabolic and energy needs of cancer cells by mediating the degradation of macromolecules to their constituent monomer units. In addition, autophagy promotes tumour survival by enhancing tolerance to oxidative and genotoxic stress as well as stress induced by increased metabolic rate and hypoxia (Fig.11.2).
RAS are small GTPases involved in important signal pathways for proliferation, survival and metabolism. Cancers driven by the K-Ras oncogene rely heavily on autophagy even in the absence of external stressors, a phenomenon known as ‘autophagy addiction’ that helps in evasion of metabolic stress and cell death (Kim et al. 2011b). Several studies have reported a correlation between RAS-mediated autophagy and the development of various human malignancies, including cancers of the lung, colon and pancreas, suggesting that autophagy plays an important role in survival and growth of various tumours that depend on RAS activation (Goel et al. 2015; Guo et al. 2011; Kim et al. 2011a). High rates of KRAS mutations are seen in pancreatic ductal adenocarcinomas (PDACs) that are believed to depend on autophagy to fuel tumour metabolism (Guo et al. 2011; Mulcahy Levy and Thorburn 2020; Yang et al. 2011). The tumour-promoting potential of autophagy is believed to be mediated by suppression of TP53 induction and by maintenance of mitochondrial function (Guo et al. 2013b; Mancias and Kimmelman 2011).
Cancer stem cells (CSCs) that display self-renewal and malignant transformation showed higher levels of autophagy (Nazio et al. 2019). The influence of autophagy on CSCs is rather complex and based on several factors such as origin and differentiation status. Inhibition of autophagy in CSCs induced death of CD34+ progenitor cells in chronic myeloid leukaemia, whereas in acute myeloid leukaemia, it caused expansion of progenitor cells in haematopoietic stem cells (Auberger and Puissant 2017). Conflicting findings have been reported on the effect of silencing ARGs on CSCs. While silencing of Beclin-1 or ATG genes such as ATG7, ATG12 or LC3 inhibited proliferation of CSCs, ATG7 deficiency in KRAS-driven tumours had no effect (Cufi et al. 2011; Eng et al. 2016; Gong et al. 2013).
11.5 Autophagy and Cancer Hallmarks
Tumorigenesis involves the acquisition of ten essential alterations that enable the growth and functional abilities of cancer cells to survive, proliferate, invade and disseminate, collectively denoted as hallmarks of cancer. These include self-sufficiency in growth signals, insensitivity to growth-inhibitory signals, evasion of programmed cell death, limitless replicative potential, sustained angiogenesis, tissue invasion and metastasis, reprogramming of energy metabolism, evading immune destruction, genome instability and inflammation (Sasahira and Kirita 2018). Several studies have unravelled the role of autophagy in the acquisition of cancer hallmarks, some of which (sustained cell proliferation, invasion, metastasis, apoptosis evasion and drug resistance) are discussed below.
11.5.1 Cell Proliferation and Autophagy
There are conflicting reports on the role of autophagy in tumour cell proliferation (Singh et al. 2018). High levels of autophagy have been documented to be essential for the growth of cancers with KRAS or BRAF mutations such as PDACs (Yang et al. 2011). In a BRAF-driven lung cancer model, Atg7 deletion resulted in tumour regression providing proof-of-concept for the involvement of autophagy in the proliferation of these tumours (Guo et al. 2013a). Other studies found a correlation between low levels of autophagy and high rate of proliferation in cancer that could be attributed to dysregulated PI3K/Akt/mTOR pathway and deletion of the tumour suppressor PTEN. Further, rapamycin, an mTOR inhibitor and autophagy inducer was shown to cause cell cycle arrest and inhibits proliferation of mantle cell lymphoma and MDA-MB-231 breast cancer cells (Chatterjee et al. 2015; Yazbeck et al. 2008). Collectively, these findings indicate that autophagy-mediated regulation of cell proliferation is context-dependent.
11.5.2 Interplay Between Autophagy and Apoptosis
Although apoptosis and autophagy are distinct forms of cell death that maintain cellular homeostasis, they are intricately interconnected by protein networks (Nikoletopoulou et al. 2013; Vijayarathna et al. 2015). Autophagy is a cytoprotective survival mechanism that tumour cells employ to evade apoptosis (Mulcahy Levy and Thorburn 2020). Understanding the mechanisms by which autophagy circumvents apoptosis in tumours will enable the development of successful therapeutic strategies. Inefficient mitochondrial outer membrane permeabilisation (MOMP) that enables tumours to recover from apoptosis and regain the ability to proliferate has been suggested as the mechanism underlying autophagy-mediated apoptosis avoidance (Ichim et al. 2015).
The BCL2 family proteins that regulate apoptosis are also involved in autophagy initiation (Fitzwalter and Thorburn 2015). The proapoptotic BH3-only proteins such as PUMA, NOXA, NIX, BID and BNIP3 disrupt the Beclin 1/BCL2 complex releasing Beclin 1 that complexes with VPS34 to stimulate autophagy. The anti-apoptotic BCL2 proteins on the other hand inhibit Beclin-1 by binding to its BH3 domain (Pattingre et al. 2005). Death-associated protein kinase (DAPK) has been shown to induce autophagy by phosphorylating Beclin 1. Upon phosphorylation, Beclin 1 dissociates from BCL-2 and binds to VPS34. In addition, DAPK also activates VPS34 via a second kinase, protein kinase D (PKD) (Eisenberg-Lerner and Kimchi 2012; Zalckvar et al. 2009).
c-jun N-terminal kinase (JNK), involved in a vast array of cellular processes, has been demonstrated to disrupt the Beclin 1-BCL-2 complex by phosphorylating BCL-2. This leads to release of Beclin 1 and formation of an active Beclin 1–VPS34 complex resulting in induction of autophagy. Wei et al. (2008) proposed a model on the dual role of JNK1-mediated BCL2 phosphorylation in regulating autophagy and apoptosis. They speculated that JNK1 initially phosphorylates BCl-2 to stimulate autophagy. However, once autophagy is unable to sustain cell survival, Bcl-2 phosphorylation inactivates its anti-apoptotic function and apoptosis is initiated.
The tumour suppressor protein TP53 also plays a dual role in autophagy based on its activation status and intracellular localisation. Cytosolic p53 inhibits autophagy by interacting with FIP200 and interfering with the ULK1 complex activity (Morselli et al. 2011; Tasdemir et al. 2008). However, under conditions of cellular stress, p53 localises to the nucleus and binds to the promoter region of multiple pro-autophagic genes, including AMPK, DRAM1, sestrin 1, sestrin 2 and PTEN, as well as pro-apoptotic genes of the BCL-2 family and p53 upregulated modulator of apoptosis (PUMA) (Budanov and Karin 2008; Gao et al. 2011; Kenzelmann Broz et al. 2013; Riley et al. 2008). Under certain conditions, p53 also induces both mitophagy and apoptosis by triggering MOMP (Youle and Narendra 2011).
The transcription factor FOXO3/FOXO3A (forkhead box O3), which confers apoptosis sensitisation by transactivating PUMA, reciprocally regulates autophagy (Warr et al. 2013). Elevated PUMA prevents the interaction between BCL2 and BAX/BAK with release of BAX/BAK MOMP and cell death by apoptosis. Fitzwalter and Thorburn (2018) postulated that FOXO3 functions as a cell surveillance mechanism to rectify perturbations in autophagy and induces apoptosis if autophagy regulation fails.
BH-3 only proteins that function at the crossroads of apoptosis and autophagy have emerged as attractive therapeutic targets in cancer. Several BH3 mimetics which are inhibitors of the anti-apoptotic BCL2 proteins have been developed. Venetoclax, a BH3-mimetic small-molecule inhibitor of BCL-2, is used in the treatment of chronic lymphocytic leukaemia (CLL) and small lymphocytic lymphoma. In acute myeloid leukaemia (AML), overexpression of vacuole membrane protein (VMP1) increased autophagic flux, protected against oxidative stress, reduced the response to venetoclax-induced MOMP and apoptotic cell death (Folkerts et al. 2019).
11.5.3 Angiogenesis and Autophagy
Angiogenesis, the formation of new blood vessels from existing vasculature, facilitates tumour invasion and metastasis. With increasing growth of a malignant tumour, the centre of the tumour is deprived of oxygen and nutrients due to decreased perfusion. Autophagy has been suggested to enable tumour cells to thrive under avascular and hypoxic conditions. In the tumour microenvironment (TME), autophagy flux induces migration of ECs and angiogenesis (Vion et al. 2017). Resistance to anti-angiogenic therapy has been attributed to high levels of autophagy in tumours. Anti-angiogenesis treatment in concert with administration of an autophagy inhibitor was found to exhibit greater efficacy besides stimulating apoptosis of tumours (Ramakrishnan et al. 2007). However, enhanced autophagy in neuroblastomas was demonstrated to block angiogenesis via degradation of pro-angiogenic gastrin-releasing peptide (GRP) (Kim et al. 2013).
Matrix glycoproteins that regulate the interplay between autophagy and angiogenesis in the tumour microenvironment are considered to be critical determinants of the fate of cancer cells. Decorin and Perlecan, matrix proteoglycans have been envisaged to influence the crosstalk between angiogenesis and autophagy signalling in endothelial cells. In a recent study, decorin, a small leucine-rich proteoglycan, was demonstrated to evoke the autophagic clearance of vascular endothelial growth factor A (VEGFA) by functioning as a partial agonist of vascular endothelial growth factor 2 (VEGFR2) in a process that requires the energy-sensing protein, AMPK and the autophagic regulator, paternally expressed gene 3 (PEG3). Further, pharmacological depletion of ATG5 led to intracellular accumulation of VEGFA, indicating that VEGFA is a substrate for autophagy. These findings underscore the therapeutic potential of decorin as a next-generation anticancer agent (Neill et al. 2020).
11.5.4 Tissue Invasion, Metastasis and Autophagy
Autophagy has a complex role in tumour invasion. In a primary tumour, autophagy prevents tissue necrosis and inflammation, thereby preventing invasion (Kenific et al. 2010). Autophagy also inhibits epithelial–mesenchymal transition (EMT) by degradation of p62/SQSTM1 as well as its cargo TWIST1 that is known to stimulate EMT (Qiang et al. 2014). However, once the tumour becomes invasive and progresses, autophagy affords protection against apoptosis and facilitates tumour dormancy. Autophagy has been implicated in various features of invasion such as cell motility, epithelial–mesenchymal transition (EMT), quiescence, stem cell phenotype and drug resistance (Mowers et al. 2017). Autophagy was found to be essential for secretion of factors critical for tumour invasion such as interleukin-6, matrix metalloproteinase-2 (MMP-2) and WNT-5A (Lock et al. 2014). Interestingly, hypoxia and transforming growth factor beta (TGFβ) are known to induce EMT also induce autophagy (Kiyono et al. 2009; Li et al. 2013). MicroRNA-mediated suppression of Smad2 was found to interrupt autophagy, resulting in inhibition of cell survival and invasive potential (Zhai et al. 2015). Conversely, ULK2, which promotes autophagy enhanced EMT and invasiveness (Kim et al. 2016). Autophagy was reported to induce EMT via SPHK1-TRAF2-Beclin-1-CDH1 signal cascades in hepatocellular carcinoma cells (Liu et al. 2017a).
Emerging evidence indicates the involvement of autophagy in metastasis (Dower et al. 2018). Several steps in the metastatic cascade are believed to be autophagy-dependent, including establishment of a pre-metastatic niche, tumour cell dormancy, resistance to anoikis and escape from immune surveillance (Kenific et al. 2010; Mowers et al. 2017). Autophagy also plays an important role in preventing tumour cells that detach from the ECM from dying by the process of anoikis, thereby promoting metastasis (Lock and Debnath 2008). Autophagy is induced by the same factors that promote metastasis such as hypoxia. Interestingly, several features of autophagy, such as mesenchymal characteristics, escape from immune surveillance and stem cell-like phenotype, are shared by metastasis. Increased staining for the autophagy marker, microtubule-associated light chain B (LC3B), is a common feature in solid tumours that is associated with metastasis (Lazova et al. 2012). Increased autophagy and EMT promote the cancer stem cell (CSC) phenotype that drives metastasis (May et al. 2011). In breast ductal carcinoma in situ (DCIS), high levels of autophagy were observed in subpopulations of cells that displayed tumour-invasive potential and stem cell phenotype (Espina et al. 2010).
The tumour microenvironment (TME), which interacts with the malignant tumour, profoundly influences tumour progression as well as therapeutic response. Autophagy is documented to promote migration and invasion of tumour cells, maintain tumour cell stemness and drug-resistance phenotypes and influence the crosstalk between the tumour and the TME (Mowers et al. 2018). In the TME, autophagy facilitates polarisation of macrophages into tumour-associated macrophages (TAMs) (Chen et al. 2014; Wen et al. 2018), and differentiation of fibroblasts into cancer-associated fibroblasts (CAFs) (Ngabire and Kim 2017; Peiris-Pages et al. 2015; Wang et al. 2017) and myeloid-derived suppressor cells (MDSCs) (Dong et al. 2017; Ostrand-Rosenberg et al. 2020).
The interplay between autophagy and exosomes is increasingly recognised to influence the TME. Exosomes, cargo-laden vesicles secreted by various cell types, establish intercellular communication to transfer their contents such as RNA and proteins to other cells, which may impact autophagy. Both exosomes and autophagy exert influence on the TME and metastasis and reciprocally regulate each other (Lin et al. 2019; Ruivo et al. 2017). The interaction of autophagy and exosomes is also mediated by autophagy-related proteins. ATG5 silencing significantly attenuated the release of exosomes as well as exosome-mediated lipidation of LC3B, a central protein of the autophagy pathway (Xu et al. 2018a).
11.5.5 Drug Resistance and Autophagy
There is growing evidence to indicate the involvement of autophagy in resistance to chemotherapeutic agents. The anticancer drug 5-fluorouracil (5-FU) used in the treatment of solid tumours such as breast, pancreatic and colorectal cancers, inhibits thymidylate synthetase, an enzyme essential for DNA synthesis. Induction of cytoprotective autophagy that results in chemoresistance is a major limitation of this drug. Autophagy induction by 5-FU has been attributed to overexpression of beclin-1, followed by conversion of LC3I to LC3II, JNK-mediated protective autophagy and BCL2-mediated autophagic flux. The DNA-damaging chemotherapeutic drug, cisplatin, is also documented to induce autophagy and chemoresistance. Several mechanisms have been suggested for enhanced autophagy induced by cisplatin. These include modulation of ERK pathway and upregulation of beclin 1 with consequent conversion of LC3 proteins, increase of ATG7 expression and downregulation of miR-199a-5p (Xu et al. 2012). However, combined administration of cisplatin and an autophagy inhibitor induced tumour cell death. In mitoxantone-resistant breast cancer cells, miR-181a targets Atg5 and impedes autophagy by targeting breast cancer–resistance protein (Jiao et al. 2013). Likewise, miR-874 inhibits autophagy and sensitises gastric cancer cells to chemotherapy via the target gene ATG16L1 (Huang et al. 2018). Shuhua et al. (2015) observed a positive correlation between the expressions of the ARGs Raptor, Rictor and Beclin1 and the multidrug resistance (MDR) gene in colorectal cancer (CRC) patients. Targeting autophagy by modulating Atgs such as Beclin1 (Eum and Lee 2011), Atg5 (Ge et al. 2014), Atg7 (Singh et al. 2012) and Atg12 (An et al. 2015) sensitised MDR cells to therapeutic agents. Taken together, these findings imply that chemoresistance can be circumvented by targeting autophagy.
11.6 ARGs as Prognostic Markers
There is substantial evidence to indicate that ARGs are reliable markers of disease severity and prognosis (Bortnik and Gorski 2017; Yang and Klionsky 2020). The expression levels of ATG genes vary based on the site of the tumour and stage of the disease. In colon cancer, ATG16L2, CAPN2 and TP63 were upregulated, whereas SIRT1, RPS6KB1, PEX3, UVRAG and NAF1 were downregulated and associated with disease recurrence (Mo et al. 2019). On the other hand, in gastric cancer, ULK1, Beclin-1, ATG3 and ATG10 were identified as favourable prognostic markers (Cao et al. 2016). An eight-gene autophagy-related signature (BLOC1S1, IL24, NRG4, PDK4, PEX3, PRKG1, SIRT2 and WDR45L) was identified as an independent and accurate predictor for the prognosis of serous ovarian cancer (An et al. 2018). Recently, Mao et al. (2020) showed that ATGs are crucial factors in the progression of HCC and could serve as potential prognostic markers for diagnosis and treatment. An autophagy score signature was validated to classify CRC patients into low and high risk of early relapse to predict post-operative survival (Zhou et al. 2019). Gene expression microarray data obtained from TCGA was used to develop ARG expression signature as a predictive tool for overall survival (OS) and disease-free survival (DFS) in prostate cancer patients. Five OS-related and 22 DFS-related ARG signatures were identified that could function as promising prognostic biomarkers of prostate cancer (Hu et al. 2020). Despite these studies, correlation between the ARG signature and the cancer type still remains obscure. The moonlighting functions of ATG proteins are believed to be responsible for the lack of correlation. Many ATG proteins are multifunctional and exert their influence beyond autophagy on diverse signalling pathways and cellular processes.
11.7 Autophagy Manipulation in Cancer Therapeutics
Autophagy manipulation has emerged as a promising strategy in cancer treatment. However, the paradoxical role of autophagy in cancer merits attention while designing therapeutic strategies. While enhancing autophagy is an option in premalignant lesions, and in some malignant tumours, inhibiting autophagy appears to be effective in many tumours, especially in advanced cancers. Several clinical trials are underway to target autophagy in cancer with more emphasis on the discovery and development of drugs that inhibit autophagy (Towers and Thorburn 2016).
11.7.1 Autophagy Induction
Several chemotherapeutic drugs are known to induce autophagy. The mToR inhibitor rapamycin has been successfully used to inhibit angiogenesis by preventing the synthesis of VEGF and downstream signalling events. Temsirolimus and everolimus, water-soluble analogues of rapamycin administered alone or in combination with chemotherapeutic drugs inhibited proliferation and induced autophagic cell death in mantle cell lymphoma and acute lymphoblastic leukaemia (Crazzolara et al. 2009; Yazbeck et al. 2008). Significant improvement in progression-free survival (PFS) was evident with everolimus treatment in patients with advanced neuroendocrine tumours in the Phase III RAD001 in Advanced Neuroendocrine Tumours (RADIANT)-3 and RADIANT-4 studies, respectively (Gajate et al. 2017). Everolimus in combination with exemestane, an aromatase inhibitor was found to be an important treatment option for patients with hormone receptor-positive (HR+) and human epidermal growth factor receptor 2- (HER2-) metastatic breast cancer (Riccardi et al. 2018). Combination chemotherapy with the autophagy inducers temozolomide and dasatinib was effective in killing glioblastoma cells resistant to apoptosis (Milano et al. 2009).
11.7.2 Autophagy Inhibition
There is substantial evidence to indicate that autophagy enhances tumour development and progression as well as chemoresistance in a wide variety of neoplasms. Ample evidence from cell-based in vitro studies, genetically engineered mouse models (GEMMs) and patient-derived xenograft (PDX) mouse models demonstrate that autophagy inhibition by anti-cancer drugs enhances tumour cell death (Levy and Thorburn 2011; Mulcahy Levy and Thorburn 2020). Autophagy inhibition has been demonstrated to sensitise tumour cells to chemotherapeutic agents and potentiate apoptosis (Amaravadi et al. 2016).
Autophagy inhibition as a treatment modality for cancer may be tumour-specific or systemic. Tumour-specific autophagy inhibition causes perturbations in tumour cell metabolism, impairment in redox and energy homeostasis, mitochondrial dysfunction and reduced nucleotide pools eventually leading to tumour cell death. Systemic inhibition of autophagy, on the other hand, causes changes in the tumour microenvironment (Kimmelman and White 2017).
Several strategies have been used to inhibit autophagy in malignant tumours including small molecule inhibitors, genetic ablation of ARGs such as beclin-1, ATG5 or ATG7, and repurposed drugs such as chloroquine (Mulcahy Levy and Thorburn 2020). Current clinical efforts have explored the different stages of autophagy as potential therapeutic targets to maximise benefit in cancer treatment (Mulcahy Levy and Thorburn 2020). The serine/threonine kinases ULK1 and ULK2 are prime targets to block autophagy in the early stages. The selective ATP competitive inhibitor of ULK1 kinase, SBI-0206965 (SBI) was found to induce apoptosis in lung cancer during nutrient deprivation (Egan et al. 2015). Preclinical results using inhibitors of VPS34 (VPS34-IN1 and SB02024), ATG4B (NSC185058, UAMC-2526 and S130) are encouraging (Dyczynski et al. 2018; Fu et al. 2019).
Autophagy inhibition both alone and in combination with anticancer drugs is emerging as a promising option in cancer therapy. The autophagy inhibitor 3-methyladenine (3-MA) when used in concert with tratsuzumab increased chemotherapeutic efficacy in HER2-positive breast cancer cells (Jain et al. 2013). Treatment with 3-MA or deletion of beclin-1 induced chemosensitisation of hepatocellular carcinoma cells (Song et al. 2009). Knockdown of ARGs was found to overcome resistance to tamoxifen in ER-positive breast cancer cells (Cook et al. 2011). In cisplatin-resistant ovarian cancer cells, Atg5 deletion induced apoptosis (Wang and Wu 2014).
11.7.2.1 Chloroquine and Hydroxycloroquine
The antimalarial drug chloroquine (7-chloro-4-(4-diethylamino-1-methylbutylamino)-quinoline, CQ) has attracted significant attention as a promising anticancer agent, a classic example of drug repurposing. Both CQ and hydroxychloroquine (HCQ) have been approved by the Food and Drug Administration (FDA) for clinical trials in cancer. CQ is a small molecule that is unprotonated at physiological pH. Being lipophilic, it traverses the cell membrane and accumulates in acidic compartments such as the lysosomes (Weyerhauser et al. 2018). CQ inhibits autophagy by preventing the fusion of autophagosome with the lysososome (Yang et al. 2013). CQ treatment reverted resistance to chemotherapeutic and anti-angiogenesis drugs (Selvakumaran et al. 2013).
Addition of a hydroxyl group to CQ lowered toxicity of CQ while retaining the efficacy. A large number of clinical trials have revealed the adjuvant effects of CQ/HCQ for diverse neoplasms. Following the identification of CQ as an autophagy inhibitor by Murakami et al. (1998), CQ was demonstrated to significantly improve clinical outcomes in patients with glioblastoma (Briceno et al. 2003). Subsequently, CQ/HCQ was reported to exhibit anti-neoplastic properties on a wide range of tumours (Xu et al. 2018b). CQ treatment is recognised to sensitise colorectal cancer cells to anti-angiogenesis treatment, DNA damaging chemotherapeutic drugs and photosan-II-mediated photodynamic therapy (PS-PDT) (Xiong et al. 2017). The ability of CQ to sensitise malignant tumours to radiation and chemotherapy was impaired by pharmacological inhibition or siRNA ablation of Beclin-1. CQ acts on a wide spectrum of molecular targets such as p53, NF–κB and ATM kinase, reflecting its functional pleiotropy. CQ has been hypothesised to play a dual role by activating DNA damage response (DDR) and suppressing DNA repair, thereby shifting the balance towards cell death (Weyerhauser et al. 2018). Recent research has provided evidence that CQ exerts anticancer effects independent of its ability to inhibit autophagy (Eng et al. 2016).
11.7.2.2 Lysosome-Targeted Inhibitors
Although CQ/HCQ showed positive results in GBM and pancreatic tumours, clinical efficacy was not encouraging in other tumours. Several lysosomal targeted inhibitors that are potent and selective are being developed as potential alternatives to CQ/HCQ (Mulcahy Levy and Thorburn 2020). Lys05, a bisaminoquinoline and DQ661, a dimeric quinacrine that concurrently inhibits lysosomes by deacidification and impairs lysosomal recruitment of mTOR were successful as single agents in mouse models of melanoma and CRC. DQ661 displayed greater efficacy relative to HCQ and Lys05 especially in acidic tumours, because it is able to maintain its activity in acidic media. Additionally, DQ661 was also found to be promising in combination with gemcitabine in PDAC (McAfee et al. 2012; Pellegrini et al. 2014; Rebecca et al. 2017).
11.7.2.3 Epigenetic Modulation of Autophagy
Given the importance of epigenetic players in regulating autophagy, epigenetic modifiers that influence autophagy through histone acetylation, methylation of CpG islands and by ncRNAs have been used to manipulate autophagy. Several natural products have been documented to target autophagy via epigenetic modification (Vidoni et al. 2019). Curcumin was demonstrated to inhibit autophagy by restoring the expression of miR-143 and induce apoptosis of prostate cancer cells exposed to radiation (Liu et al. 2017b). Ellagic acid, a naturally occurring polyphenol abundantly found in fruits and vegetables that exerts antiproliferative effects has been reported to inhibit CARM1-mediated H3R17 methylation, thereby suppressing autophagy (Shin et al. 2016b). Studies from this laboratory demonstrated that gedunin and nimbolide, limonoids from the neem tree (Azadirachta indica) exert their antiproliferative effects by inhibiting cytoprotective autophagy and inducing apoptosis in oral cancer cell lines and in the hamster buccal pouch model of oral oncogenesis (Sophia et al. 2018; Tanagala et al. 2018). While gedunin mediated its effects via downregulation of the oncomiR, miR-21, nimbolide augmented apoptosis by overcoming the shielding effects of autophagy through modulation of the PI3K/Akt/GSK-3β signalling axis as well as the ncRNAs miR-126 and HOTAIR. Autophagy modulators are thus a valuable addition to the armamentarium of compounds that offer promise in cancer therapeutics.
11.7.2.4 Pitfalls of Autophagy Inhibition
There are several concerns in using autophagy manipulation as a therapeutic strategy in cancer. Many of the autophagy inhibitors including CQ/HCQ are not autophagy specific and affect other essential signalling pathways. For instance, in dormant murine breast cancer stem cells autophagy inhibition induced aberrant expression of 6-phosphofructo2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) leading to proliferation and recurrent metastatic disease (Yang et al. 2013). The cytotoxicity due to global autophagy inhibition induced by some compounds is another concern although it may be circumvented by therapy breaks or by using agents that cause incomplete autophagy inhibition such as CQ. The uptake of HCQ is pH-dependent which limits its effectiveness in solid tumours that show differences in pH between central and peripheral regions (Pellegrini et al. 2014).
Autophagy inhibition has been reported to cause side effects such as inflammation and tissue damage. This can be overcome by intermittent dosing of autophagy inhibitors. An inducible dominant-negative ATG4BC74A mutant mouse model that mimics a pharmacological inhibitor by reversibly manipulating autophagy without a complete blockade has been developed (Yang et al. 2018). The interplay between autophagy and apoptosis lends credence to the development of intermittent autophagy inhibitors. However, the appropriate dose of autophagy inhibitors remains to be standardised.
Treatment outcomes may also depend on the concept of autophagy addiction. RAS-driven tumours such as PDACs may respond better to autophagy inhibition compared to autophagy-independent tumours providing a rationale for initiating clinical trials targeting autophagy addiction. Autophagy inhibition decreased tumour growth in xenograft models of PDAC and improved surgical outcomes in PDAC patients who were pre-operatively treated with gemcitabine, nab-paclitaxel and HCQ (Boone et al. 2015; La Belle Flynn et al. 2019). Autophagy inhibition in combination with direct targeting of MEK or ERK was found to be beneficial and clinical trials have been developed for NRAS melanoma and PDAC respectively (Kinsey et al. 2019). In addition to RAS, mutations in other genes have also been used to identify autophagy-dependence as well as to predict response to autophagy inhibition such as the epidermal growth factor receptor (EGFR) that regulates pathways influencing autophagy. GBM tumours expressing EGFR variant III (EGFRvIII), as well as head and neck squamous cell carcinoma (HNSCC) are autophagy-dependent and respond to autophagy inhibition (Jutten et al. 2018). Clinical trials have been carried out on autophagy inhibition in NSCLC and GBM patients with overexpressed or mutant EGFR (Massachusetts General Hospital 2019, https://ClinicalTrials.gov/show/NCT00977470; Maastricht Radiation Oncology 2020, https://ClinicalTrials.gov/show/NCT02378532).
11.8 Conclusion
The role of autophagy in cancer is highly complex and paradoxical. While autophagy has suppressive effects on some tumours, in most cases, autophagy is a survival pathway that enables tumour proliferation and progression. In particular, the interplay between autophagy and apoptosis is intriguing and has implications for cancer therapy. Autophagy is a therapeutically targetable process, although there are many factors that need to be considered to maximise benefit. It is increasingly important to weigh options such as targeting the early or late stages of the pathway, stage of the disease that will respond best to intervention, whether to use an autophagy inducer or inhibitor and whether to administer the autophagy modulator as a single agent or in combination. Patient selection is critical in delineating positive findings as well as to identify non-responders. Rationally based interventions are therefore essential to effectively maximise therapeutic benefit and minimise adverse outcomes.
References
Allen EA, Baehrecke EH (2020) Autophagy in animal development. Cell Death Differ 27:903–918
Amaravadi R, Kimmelman AC, White E (2016) Recent insights into the function of autophagy in cancer. Genes Dev 30:1913–1930
An Y, Zhang Z, Shang Y, Jiang X, Dong J, Yu P, Nie Y, Zhao Q (2015) miR-23b-3p regulates the chemoresistance of gastric cancer cells by targeting ATG12 and HMGB2. Cell Death Dis 6:e1766
An Y, Bi F, You Y, Liu X, Yang Q (2018) Development of a novel autophagy-related prognostic signature for serous ovarian cancer. J Cancer 9:4058–4071
Auberger P, Puissant A (2017) Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood 129:547–552
Baek SH, Kim KI (2017) Epigenetic control of autophagy: nuclear events gain more attention. Mol Cell 65:781–785
Bishop E, Bradshaw TD (2018) Autophagy modulation: a prudent approach in cancer treatment? Cancer Chemother Pharmacol 82:913–922
Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, Wu WC, Singhi AD, Bao P, Bartlett DL, Liotta LA, Espina V, Loughran P, Lotze MT, Zeh HJ 3rd (2015) Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol 22:4402–4410
Bortnik S, Gorski SM (2017) Clinical applications of autophagy proteins in cancer: from potential targets to biomarkers. Int J Mol Sci 18:1496
Briceno E, Reyes S, Sotelo J (2003) Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg Focus 14:e3
Budanov AV, Karin M (2008) p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134:451–460
Cao QH, Liu F, Yang ZL, Fu XH, Yang ZH, Liu Q, Wang L, Wan XB, Fan XJ (2016) Prognostic value of autophagy related proteins ULK1, Beclin 1, ATG3, ATG5, ATG7, ATG9, ATG10, ATG12, LC3B and p62/SQSTM1 in gastric cancer. Am J Transl Res 8:3831–3847
Chatterjee A, Mukhopadhyay S, Tung K, Patel D, Foster DA (2015) Rapamycin-induced G1 cell cycle arrest employs both TGF-beta and Rb pathways. Cancer Lett 360:134–140
Chen P, Cescon M, Bonaldo P (2014) Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy 10:192–200
Cook KL, Shajahan AN, Clarke R (2011) Autophagy and endocrine resistance in breast cancer. Expert Rev Anticancer Ther 11:1283–1294
Crazzolara R, Cisterne A, Thien M, Hewson J, Baraz R, Bradstock KF, Bendall LJ (2009) Potentiating effects of RAD001 (Everolimus) on vincristine therapy in childhood acute lymphoblastic leukemia. Blood 113:3297–3306
Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA (2011) Autophagy positively regulates the CD44(+) CD24(−/low) breast cancer stem-like phenotype. Cell Cycle 10:3871–3885
Delaney JR, Patel CB, Bapat J, Jones CM, Ramos-Zapatero M, Ortell KK, Tanios R, Haghighiabyaneh M, Axelrod J, DeStefano JW, Tancioni I, Schlaepfer DD, Harismendy O, La Spada AR, Stupack DG (2020) Autophagy gene haploinsufficiency drives chromosome instability, increases migration, and promotes early ovarian tumors. PLoS Genet 16:e1008558
Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19:349–364
Dong G, Si C, Zhang Q, Yan F, Li C, Zhang H, Ma Q, Dai J, Li Z, Shi H, Wang B, Zhang J, Ming J, Hu Y, Geng S, Zhang Y, Li L, Xiong H (2017) Autophagy regulates accumulation and functional activity of granulocytic myeloid-derived suppressor cells via STAT3 signaling in endotoxin shock. Biochim Biophys Acta Mol Basis Dis 1863:2796–2807
Dower CM, Wills CA, Frisch SM, Wang HG (2018) Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy 14:1110–1128
Dyczynski M, Yu Y, Otrocka M, Parpal S, Braga T, Henley AB, Zazzi H, Lerner M, Wennerberg K, Viklund J, Martinsson J, Grander D, De Milito A, Pokrovskaja Tamm K (2018) Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett 435:32–43
Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, Teriete P, Asara JM, Turk BE, Cosford ND, Shaw RJ (2015) Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell 59:285–297
Eisenberg-Lerner A, Kimchi A (2012) PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ 19:788–797
Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, Fitzgerald SL, George E, Frias E, Cochran N, De Jesus R, McAllister G, Hoffman GR, Bray K, Lemon L, Lucas J, Fantin VR, Abraham RT, Murphy LO, Nyfeler B (2016) Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A 113:182–187
Errafiy R, Aguado C, Ghislat G, Esteve JM, Gil A, Loutfi M, Knecht E (2013) PTEN increases autophagy and inhibits the ubiquitin-proteasome pathway in glioma cells independently of its lipid phosphatase activity. PLoS One 8:e83318
Espina V, Mariani BD, Gallagher RI, Tran K, Banks S, Wiedemann J, Huryk H, Mueller C, Adamo L, Deng J, Petricoin EF, Pastore L, Zaman S, Menezes G, Mize J, Johal J, Edmiston K, Liotta LA (2010) Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PLoS One 5:e10240
Eum KH, Lee M (2011) Targeting the autophagy pathway using ectopic expression of Beclin 1 in combination with rapamycin in drug-resistant v-Ha-ras-transformed NIH 3T3 cells. Mol Cells 31:231–238
Fitzwalter BE, Thorburn A (2015) Recent insights into cell death and autophagy. FEBS J 282:4279–4288
Fitzwalter BE, Thorburn A (2018) FOXO3 links autophagy to apoptosis. Autophagy 14:1467–1468
Folkerts H, Wierenga AT, van den Heuvel FA, Woldhuis RR, Kluit DS, Jaques J, Schuringa JJ, Vellenga E (2019) Elevated VMP1 expression in acute myeloid leukemia amplifies autophagy and is protective against venetoclax-induced apoptosis. Cell Death Dis 10:421
Fu Y, Hong L, Xu J, Zhong G, Gu Q, Gu Q, Guan Y, Zheng X, Dai Q, Luo X, Liu C, Huang Z, Yin XM, Liu P, Li M (2019) Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 15:295–311
Furuya N, Yu J, Byfield M, Pattingre S, Levine B (2005) The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 1:46–52
Gajate P, Martinez-Saez O, Alonso-Gordoa T, Grande E (2017) Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manag Res 9:215–224
Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, Kimmelman A, Kumar S, Levine B, Maiuri MC, Martin SJ, Penninger J, Piacentini M, Rubinsztein DC, Simon HU, Simonsen A, Thorburn AM, Velasco G, Ryan KM, Kroemer G (2015) Autophagy in malignant transformation and cancer progression. EMBO J 34:856–880
Gao W, Shen Z, Shang L, Wang X (2011) Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ 18:1598–1607
Ge J, Chen Z, Huang J, Chen J, Yuan W, Deng Z, Chen Z (2014) Upregulation of autophagy-related gene-5 (ATG-5) is associated with chemoresistance in human gastric cancer. PLoS One 9:e110293
Goel S, Huang J, Klampfer L (2015) K-Ras, intestinal homeostasis and colon cancer. Curr Clin Pharmacol 10:73–81
Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V, Tharinger H, Delbos L, Gary-Gouy H, Morel AP, Ghavami S, Song E, Codogno P, Mehrpour M (2013) Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 32:2261–2272, 2272e.1–11
Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25:460–470
Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, Snyder E, Santanam U, Dipaola RS, Jacks T, Rabinowitz JD, White E (2013a) Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 27:1447–1461
Guo JY, Xia B, White E (2013b) Autophagy-mediated tumor promotion. Cell 155:1216–1219
Hansen M, Rubinsztein DC, Walker DW (2018) Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol 19:579–593
Hu LF (2019) Epigenetic regulation of autophagy. Adv Exp Med Biol 1206:221–236
Hu D, Jiang L, Luo S, Zhao X, Hu H, Zhao G, Tang W (2020) Development of an autophagy-related gene expression signature for prognosis prediction in prostate cancer patients. J Transl Med 18:160
Huang H, Tang J, Zhang L, Bu Y, Zhang X (2018) miR-874 regulates multiple-drug resistance in gastric cancer by targeting ATG16L1. Int J Oncol 53:2769–2779
Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, Haller M, Riley JS, Mason SM, Athineos D, Parsons MJ, van de Kooij B, Bouchier-Hayes L, Chalmers AJ, Rooswinkel RW, Oberst A, Blyth K, Rehm M, Murphy DJ, Tait SWG (2015) Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell 57:860–872
Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S, Watanabe M, Sakurada A, Endo C, Uruno A, Sasano H, Nakagawa T, Satoh K, Tanaka N, Kubo H, Motohashi H, Yamamoto M (2012) Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci 103:760–766
Jain K, Paranandi KS, Sridharan S, Basu A (2013) Autophagy in breast cancer and its implications for therapy. Am J Cancer Res 3:251–265
Jiao X, Zhao L, Ma M, Bai X, He M, Yan Y, Wang Y, Chen Q, Zhao X, Zhou M, Cui Z, Zheng Z, Wang E, Wei M (2013) MiR-181a enhances drug sensitivity in mitoxantone-resistant breast cancer cells by targeting breast cancer resistance protein (BCRP/ABCG2). Breast Cancer Res Treat 139:717–730
Jutten B, Keulers TG, Peeters HJM, Schaaf MBE, Savelkouls KGM, Compter I, Clarijs R, Schijns O, Ackermans L, Teernstra OPM, Zonneveld MI, Colaris RME, Dubois L, Vooijs MA, Bussink J, Sotelo J, Theys J, Lammering G, Rouschop KMA (2018) EGFRvIII expression triggers a metabolic dependency and therapeutic vulnerability sensitive to autophagy inhibition. Autophagy 14:283–295
Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS, Ahn CH, Yoo NJ, Lee SH (2009) Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol 217:702–706
Kenific CM, Thorburn A, Debnath J (2010) Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol 22:241–245
Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD (2013) Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 27:1016–1031
Kim JH, Kim HY, Lee YK, Yoon YS, Xu WG, Yoon JK, Choi SE, Ko YG, Kim MJ, Lee SJ, Wang HJ, Yoon G (2011a) Involvement of mitophagy in oncogenic K-Ras-induced transformation: overcoming a cellular energy deficit from glucose deficiency. Autophagy 7:1187–1198
Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, Hwang SG, Yoon G, Lee SJ (2011b) Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem 286:12924–12932
Kim KW, Paul P, Qiao J, Lee S, Chung DH (2013) Enhanced autophagy blocks angiogenesis via degradation of gastrin-releasing peptide in neuroblastoma cells. Autophagy 9:1579–1590
Kim YH, Baek SH, Kim EK, Ha JM, Jin SY, Lee HS, Ha HK, Song SH, Kim SJ, Shin HK, Yong J, Kim DH, Kim CD, Bae SS (2016) Uncoordinated 51-like kinase 2 signaling pathway regulates epithelial-mesenchymal transition in A549 lung cancer cells. FEBS Lett 590:1365–1374
Kimmelman AC (2011) The dynamic nature of autophagy in cancer. Genes Dev 25:1999–2010
Kimmelman AC, White E (2017) Autophagy and tumor metabolism. Cell Metab 25:1037–1043
Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT, Burrell LD, Lum DH, Whisenant JR, Gilcrease GW 3rd, Cavalieri CC, Rehbein KM, Cutler SL, Affolter KE, Welm AL, Welm BE, Scaife CL, Snyder EL, McMahon M (2019) Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 25:620–627
Kitamura H, Torigoe T, Asanuma H, Hisasue SI, Suzuki K, Tsukamoto T, Satoh M, Sato N (2006) Cytosolic overexpression of p62 sequestosome 1 in neoplastic prostate tissue. Histopathology 48:157–161
Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, Sugimoto K, Miyazono K (2009) Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res 69:8844–8852
Klionsky DJ (2008) Autophagy revisited: a conversation with Christian de Duve. Autophagy 4:740–743
Klionsky DJ (2020) Autophagy participates in, well, just about everything. Cell Death Differ 27:831–832
Kocaturk NM, Akkoc Y, Kig C, Bayraktar O, Gozuacik D, Kutlu O (2019) Autophagy as a molecular target for cancer treatment. Eur J Pharm Sci 134:116–137
Kung CP, Budina A, Balaburski G, Bergenstock MK, Murphy M (2011) Autophagy in tumor suppression and cancer therapy. Crit Rev Eukaryot Gene Expr 21:71–100
La Belle Flynn A, Calhoun BC, Sharma A, Chang JC, Almasan A, Schiemann WP (2019) Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun 10:3668
Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM (2012) Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res 18:370–379
Lebovitz CB, Robertson AG, Goya R, Jones SJ, Morin RD, Marra MA, Gorski SM (2015) Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 11:1668–1687
Lee JG, Wu R (2012) Combination erlotinib-cisplatin and Atg3-mediated autophagy in erlotinib resistant lung cancer. PLoS One 7:e48532
Levy JM, Thorburn A (2011) Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Ther 131:130–141
Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y, Song Z, Zheng Q, Xiong J (2013) Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 34:1343–1351
Li SS, Xu LZ, Zhou W, Yao S, Wang CL, Xia JL, Wang HF, Kamran M, Xue XY, Dong L, Wang J, Ding XD, Bella L, Bugeon L, Xu J, Zheng FM, Dallman MJ, Lam EWF, Liu Q (2017) p62/SQSTM1 interacts with vimentin to enhance breast cancer metastasis. Carcinogenesis 38:1092–1103
Li X, He S, Ma B (2020) Autophagy and autophagy-related proteins in cancer. Mol Cancer 19:12
Lin J, Lu X, Liao S, Chen X, Wang S, Zhao C, Li X, Xu YZ, Liu HF, Pan Q (2019) Cross-regulation between exosomal and autophagic pathways: promising therapy targets in disease. Discov Med 27:201–210
Liu H, Ma Y, He HW, Zhao WL, Shao RG (2017a) SPHK1 (sphingosine kinase 1) induces epithelial-mesenchymal transition by promoting the autophagy-linked lysosomal degradation of CDH1/E-cadherin in hepatoma cells. Autophagy 13:900–913
Liu J, Li M, Wang Y, Luo J (2017b) Curcumin sensitizes prostate cancer cells to radiation partly via epigenetic activation of miR-143 and miR-143 mediated autophagy inhibition. J Drug Target 25:645–652
Lock R, Debnath J (2008) Extracellular matrix regulation of autophagy. Curr Opin Cell Biol 20:583–588
Lock R, Kenific CM, Leidal AM, Salas E, Debnath J (2014) Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov 4:466–479
Maastricht Radiation Oncology (2020) The addition of chloroquine to chemoradiation for glioblastoma. https://ClinicalTrials.gov/show/NCT02378532
Mancias JD, Kimmelman AC (2011) Targeting autophagy addiction in cancer. Oncotarget 2:1302–1306
Mao D, Zhang Z, Zhao X, Dong X (2020) Autophagy-related genes prognosis signature as potential predictive markers for immunotherapy in hepatocellular carcinoma. Peer J 8:e8383
Marinkovic M, Sprung M, Buljubasic M, Novak I (2018) Autophagy modulation in cancer: current knowledge on action and therapy. Oxidative Med Cell Longev 2018:8023821
Massachusetts General Hospital (2019) Erlotinib with or without hydroxychloroquine in chemo-naive advanced NSCLC and (EGFR) mutations. https://ClinicalTrials.gov/show/NCT00977470
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E (2009) Autophagy suppresses tumorigenesis through elimination of p62. Cell 137:1062–1075
May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA (2011) Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res 13:202
McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, Lynch JP, Uehara T, Sepulveda AR, Davis LE, Winkler JD, Amaravadi RK (2012) Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A 109:8253–8258
Milano V, Piao Y, LaFortune T, de Groot J (2009) Dasatinib-induced autophagy is enhanced in combination with temozolomide in glioma. Mol Cancer Ther 8:394–406
Mo S, Dai W, Xiang W, Li Y, Feng Y, Zhang L, Li Q, Cai G (2019) Prognostic and predictive value of an autophagy-related signature for early relapse in stages I-III colon cancer. Carcinogenesis 40:861–870
Morselli E, Shen S, Ruckenstuhl C, Bauer MA, Marino G, Galluzzi L, Criollo A, Michaud M, Maiuri MC, Chano T, Madeo F, Kroemer G (2011) p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 10:2763–2769
Mowers EE, Sharifi MN, Macleod KF (2017) Autophagy in cancer metastasis. Oncogene 36:1619–1630
Mowers EE, Sharifi MN, Macleod KF (2018) Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J 285:1751–1766
Mulcahy Levy JM, Thorburn A (2020) Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ 27:843–857
Murakami N, Oyama F, Gu Y, McLennan IS, Nonaka I, Ihara Y (1998) Accumulation of tau in autophagic vacuoles in chloroquine myopathy. J Neuropathol Exp Neurol 57:664–673
Nazio F, Bordi M, Cianfanelli V, Locatelli F, Cecconi F (2019) Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ 26:690–702
Neill T, Chen CG, Buraschi S, Iozzo RV (2020) Catabolic degradation of endothelial VEGFA via autophagy. J Biol Chem 295:6064–6079
Ngabire D, Kim GD (2017) Autophagy and inflammatory response in the tumor microenvironment. Int J Mol Sci 18:2016
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N (2013) Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 1833:3448–3459
Ohsumi Y (2016) MLA style: press release. NobelPrize.org. Nobel Media AB 2020. 11 May 2020. https://www.nobelprize.org/prizes/medicine/2016/press-release/
Ostrand-Rosenberg S, Beury DW, Parker KH, Horn LA (2020) Survival of the fittest: how myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment. Cancer Immunol Immunother 69:215–221
Parzych KR, Klionsky DJ (2014) An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20:460–473
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939
Peiris-Pages M, Smith DL, Gyorffy B, Sotgia F, Lisanti MP (2015) Proteomic identification of prognostic tumour biomarkers, using chemotherapy-induced cancer-associated fibroblasts. Aging (Albany NY) 7:816–838
Pellegrini P, Strambi A, Zipoli C, Hagg-Olofsson M, Buoncervello M, Linder S, De Milito A (2014) Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies. Autophagy 10:562–571
Qiang L, Zhao B, Ming M, Wang N, He TC, Hwang S, Thorburn A, He YY (2014) Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci U S A 111:9241–9246
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112:1809–1820
Ramakrishnan S, Nguyen TM, Subramanian IV, Kelekar A (2007) Autophagy and angiogenesis inhibition. Autophagy 3:512–515
Rebecca VW, Nicastri MC, McLaughlin N, Fennelly C, McAfee Q, Ronghe A, Nofal M, Lim CY, Witze E, Chude CI, Zhang G, Alicea GM, Piao S, Murugan S, Ojha R, Levi SM, Wei Z, Barber-Rotenberg JS, Murphy ME, Mills GB, Lu Y, Rabinowitz J, Marmorstein R, Liu Q, Liu S, Xu X, Herlyn M, Zoncu R, Brady DC, Speicher DW, Winkler JD, Amaravadi RK (2017) A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov 7:1266–1283
Riccardi F, Colantuoni G, Diana A, Mocerino C, Carteni G, Lauria R, Febbraro A, Nuzzo F, Addeo R, Marano O, Incoronato P, De Placido S, Ciardiello F, Orditura M (2018) Exemestane and Everolimus combination treatment of hormone receptor positive, HER2 negative metastatic breast cancer: a retrospective study of 9 cancer centers in the Campania Region (Southern Italy) focused on activity, efficacy and safety. Mol Clin Oncol 9:255–263
Riley T, Sontag E, Chen P, Levine A (2008) Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9:402–412
Ruivo CF, Adem B, Silva M, Melo SA (2017) The biology of cancer exosomes: insights and new perspectives. Cancer Res 77:6480–6488
Sasahira T, Kirita T (2018) Hallmarks of cancer-related newly prognostic factors of oral squamous cell carcinoma. Int J Mol Sci 19:2413
Selvakumaran M, Amaravadi RK, Vasilevskaya IA, O’Dwyer PJ (2013) Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin Cancer Res 19:2995–3007
Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko HJ, Kweon MN, Won KJ, Baek SH (2016a) AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 534:553–557
Shin HR, Kim H, Kim KI, Baek SH (2016b) Epigenetic and transcriptional regulation of autophagy. Autophagy 12:2248–2249
Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306:990–995
Shuhua W, Chenbo S, Yangyang L, Xiangqian G, Shuang H, Tangyue L, Dong T (2015) Autophagy-related genes Raptor, Rictor, and Beclin1 expression and relationship with multidrug resistance in colorectal carcinoma. Hum Pathol 46:1752–1759
Singh BN, Kumar D, Shankar S, Srivastava RK (2012) Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochem Pharmacol 84:1154–1163
Singh SS, Vats S, Chia AY, Tan TZ, Deng S, Ong MS, Arfuso F, Yap CT, Goh BC, Sethi G, Huang RY, Shen HM, Manjithaya R, Kumar AP (2018) Dual role of autophagy in hallmarks of cancer. Oncogene 37:1142–1158
Song J, Qu Z, Guo X, Zhao Q, Zhao X, Gao L, Sun K, Shen F, Wu M, Wei L (2009) Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy 5:1131–1144
Sophia J, Kowshik J, Dwivedi A, Bhutia SK, Manavathi B, Mishra R, Nagini S (2018) Nimbolide, a neem limonoid inhibits cytoprotective autophagy to activate apoptosis via modulation of the PI3K/Akt/GSK-3beta signalling pathway in oral cancer. Cell Death Dis 9:1087
Su Y, Qian H, Zhang J, Wang S, Shi P, Peng X (2005) The diversity expression of p62 in digestive system cancers. Clin Immunol 116:118–123
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ, Wang HG (2007) Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9:1142–1151
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N (2011) Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25:795–800
Tanagala KKK, Baba AB, Kowshik J, Reddy GB, Nagini S (2018) Gedunin, a neem limonoid in combination with epalrestat inhibits cancer hallmarks by attenuating aldose reductase-driven oncogenic signaling in SCC131 oral cancer cells. Anti Cancer Agents Med Chem 18:2042–2052
Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G (2008) Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10:676–687
Towers CG, Thorburn A (2016) Therapeutic targeting of autophagy. EBioMedicine 14:15–23
Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, Zhong Z, Subramaniam S, Raghunandan S, Duran A, Linares JF, Reina-Campos M, Umemura S, Valasek MA, Seki E, Yamaguchi K, Koike K, Itoh Y, Diaz-Meco MT, Moscat J, Karin M (2016) p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 29:935–948
Vidoni C, Ferraresi A, Secomandi E, Vallino L, Dhanasekaran DN, Isidoro C (2019) Epigenetic targeting of autophagy for cancer prevention and treatment by natural compounds. Semin Cancer Biol. https://doi.org/10.1016/j.semcancer.2019.04.006
Vijayarathna S, Gothai S, Jothy SL, Chen Y, Kanwar JR, Sasidharan S (2015) Can cancer therapy be achieved by bridging apoptosis and autophagy: a method based on microRNA-dependent gene therapy and phytochemical targets. Asian Pac J Cancer Prev 16:7435–7439
Vion AC, Kheloufi M, Hammoutene A, Poisson J, Lasselin J, Devue C, Pic I, Dupont N, Busse J, Stark K, Lafaurie-Janvore J, Barakat AI, Loyer X, Souyri M, Viollet B, Julia P, Tedgui A, Codogno P, Boulanger CM, Rautou PE (2017) Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc Natl Acad Sci U S A 114:E8675–E8684
Wang J, Wu GS (2014) Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem 289:17163–17173
Wang Y, Gan G, Wang B, Wu J, Cao Y, Zhu D, Xu Y, Wang X, Han H, Li X, Ye M, Zhao J, Mi J (2017) Cancer-associated fibroblasts promote irradiated cancer cell recovery through autophagy. EBioMedicine 17:45–56
Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegue E (2013) FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494:323–327
Wei Y, Sinha S, Levine B (2008) Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 7:949–951
Wen ZF, Liu H, Gao R, Zhou M, Ma J, Zhang Y, Zhao J, Chen Y, Zhang T, Huang F, Pan N, Zhang J, Fox BA, Hu HM, Wang LX (2018) Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J Immunother Cancer 6:151
Weyerhauser P, Kantelhardt SR, Kim EL (2018) Re-purposing chloroquine for glioblastoma: potential merits and confounding variables. Front Oncol 8:335
Wible DJ, Chao HP, Tang DG, Bratton SB (2019) ATG5 cancer mutations and alternative mRNA splicing reveal a conjugation switch that regulates ATG12-ATG5-ATG16L1 complex assembly and autophagy. Cell Discov 5:42
Xiong L, Liu Z, Ouyang G, Lin L, Huang H, Kang H, Chen W, Miao X, Wen Y (2017) Autophagy inhibition enhances photocytotoxicity of Photosan-II in human colorectal cancer cells. Oncotarget 8:6419–6432
Xu N, Zhang J, Shen C, Luo Y, Xia L, Xue F, Xia Q (2012) Cisplatin-induced downregulation of miR-199a-5p increases drug resistance by activating autophagy in HCC cell. Biochem Biophys Res Commun 423:826–831
Xu J, Camfield R, Gorski SM (2018a) The interplay between exosomes and autophagy—partners in crime. J Cell Sci 131:jcs215210
Xu R, Ji Z, Xu C, Zhu J (2018b) The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: a systematic review and meta-analysis. Medicine (Baltimore) 97:e12912
Yang Y, Klionsky DJ (2020) Autophagy and disease: unanswered questions. Cell Death Differ 27:858–871
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, Kimmelman AC (2011) Pancreatic cancers require autophagy for tumor growth. Genes Dev 25:717–729
Yang YP, Hu LF, Zheng HF, Mao CJ, Hu WD, Xiong KP, Wang F, Liu CF (2013) Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin 34:625–635
Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, Deng J, Hai J, Yang S, Wong KK, Kimmelman AC (2018) Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov 8:276–287
Yazbeck VY, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, Kondo S, Younes A (2008) Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol 36:443–450
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12:9–14
Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 100:15077–15082
Zalckvar E, Berissi H, Eisenstein M, Kimchi A (2009) Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 5:720–722
Zhai H, Fesler A, Ba Y, Wu S, Ju J (2015) Inhibition of colorectal cancer stem cell survival and invasive potential by hsa-miR-140-5p mediated suppression of Smad2 and autophagy. Oncotarget 6:19735–19746
Zhang Y, Li F, Liu L, Jiang H, Jiang X, Ge X, Cao J, Wang Z, Zhang L, Wang Y (2018) Salinomycin-induced autophagy blocks apoptosis via the ATG3/AKT/mTOR signaling axis in PC-3 cells. Life Sci 207:451–460
Zhi X, Feng W, Rong Y, Liu R (2018) Anatomy of autophagy: from the beginning to the end. Cell Mol Life Sci 75:815–831
Zhou Z, Mo S, Dai W, Ying Z, Zhang L, Xiang W, Han L, Wang Z, Li Q, Wang R, Cai G (2019) Development and validation of an autophagy score signature for the prediction of post-operative survival in colorectal cancer. Front Oncol 9:878
Acknowledgements
Financial support from the University Grants Commission, Basic Science Research (No.F.18-1/2011(BSR)) and the Science and Engineering Research Board (# EMR/2016/001984) of Department of Science and Technology New Delhi, India to Siddavaram Nagini is gratefully acknowledged.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Nagini, S., Manikandan, P., Malla, R.R. (2020). The Autophagy Conundrum in Cancer Development, Progression and Therapeutics. In: Bhutia, S.K. (eds) Autophagy in tumor and tumor microenvironment . Springer, Singapore. https://doi.org/10.1007/978-981-15-6930-2_11
Download citation
DOI: https://doi.org/10.1007/978-981-15-6930-2_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-6929-6
Online ISBN: 978-981-15-6930-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)