
Abstract
Laryngeal squamous cell carcinoma (LSCC), one of the most common malignancies in the head and neck, has poor prognosis and high mortality. The need of novel and effective treatment for LSCC is urgent. Asparaginase, an enzyme-depriving asparagine, has been employed for the treatment of various cancers. In this study, we reported for the first time that asparaginase could induce remarkable cytotoxicity and caspase-dependent apoptosis in human LSCC Tu212 and Tu686 cells. Meanwhile, autophagy was triggered by asparaginase in LSCC cells, which was confirmed by accumulation of autophagosomes and the conversion of light chain 3-I (LC3-I) to LC3-II. Importantly, inhibition of autophagy by chloroquine (CQ) significantly enhanced asparaginase-induced cytotoxicity, indicating that autophagy has a cytoprotective role in asparaginase-treated LSCC cells. Meanwhile, we found that mitochondrial-originated reactive oxygen species (ROS) participated in asparaginase-induced autophagy and cytotoxicity. N-acetyl-L-cysteine (NAC), a common antioxidant, was employed to scavenge ROS, and our results demonstrated that NAC could significantly block asparaginase-induced autophagy and attenuate asparaginase-induced cytotoxicity, indicating that intracellular ROS played a crucial role in asparagine deprivation therapy. Furthermore, western blot analysis showed that asparaginase-induced autophagy was mediated by inactivation of Akt/mTOR and activation of the Erk signaling pathway in Tu212 and Tu686 cells. Therefore, these results indicated the protective role of autophagy in asparaginase-treated LSCC cells and provided a new attractive therapeutic strategy for LSCC by asparaginase alone or in combination with autophagic inhibitors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Laryngeal cancer, one of the most common malignancies in the head and neck, has been a significant health risk for its poor prognosis and high mortality (Unger et al. 2015). Approximately 90% of laryngeal cancers are laryngeal squamous cell carcinomas (LSCC) which usually originates from laryngeal epithelial cells (Piantelli et al. 2002; Surono et al. 2016). Despite the advances in LSCC therapies including surgery, chemotherapy, and radiotherapy, the overall 5-year survival of LSCC has not substantially improved in recent decades (Zhu et al. 2014). Therefore, the limited therapeutic options and dismal prognosis promote the searches for novel therapeutic strategies for LSCC.
As a semi-essential amino acid, asparagine plays an important role in mammalian cell metabolism (Wang et al. 2016). Meanwhile, for most tumor cells, the extracellular source of asparagine is essential for cell viability due to the lack of asparagine synthetase (ASNS) (Figueiredo et al. 2016; Kelo et al. 2009). Asparaginase, an enzyme which originates from Escherichia coli and Erwinia chrysanthemi, hydrolyzes asparagine into aspartic acid and ammonia, thereby suppressing the growth and inducing apoptosis of ASNS-deficient cells (Costa et al. 2016; Horvat et al. 2016). Based on asparagine deprivation, asparaginase has been employed in the therapy of acute lymphocytic leukemia, myelomonocytic leukemia, melanoma sarcoma, non-Hodgkin’s lymphoma, and pancreatic carcinoma (Abd El Baky and El Baroty 2016). Although asparaginase showed its broad spectrum and potent efficacy on tumors, its efficiency and the potential mechanism of asparagine-based therapy for LSCC have not been reported.
Long-term efficacy of tumor-targeted therapies is always challenged by therapy resistance. Autophagy is a conserved cellular catabolic pathway involving the orderly degradation and recycling of cellular proteins and organelles in eukaryotic cells (Casasampere et al. 2017; Cordani et al. 2016). Recently, autophagy has been widely reported to play a double-edged role in human health and disease (Fan et al. 2013, 2016a, b; Gozuacik and Kimchi 2004). Previous studies have demonstrated that antitumor drugs, especially chemotherapeutic agents and nutrient starvation, could trigger potent autophagy (Gozuacik and Kimchi 2004). With regard to the role of autophagy in tumor therapy, increasing lines of evidence inclined to suggest autophagy as a cytoprotective mechanism in cancer treatment, especially in amino acid deprivation-based therapies, which indicated that abolition of amino acid and autophagy might further enhanced the antitumor efficacy of amino acid deprivation alone (Zhang et al. 2016a). Notably, mitochondrial-originated ROS, Akt/mTOR, and Erk signaling pathway were also involved in the response to cancer therapeutics (Fan et al. 2013). In this study, we supposed that treatment with asparaginase alone or combined with autophagic inhibitors could be a potential therapeutic strategy for LSCC.
In this study, we investigated the potential effect of asparaginase on LSCC and reported for the first time that asparaginase could induce remarkable cytotoxicity and caspase-dependent apoptosis in LSCC Tu212 and Tu686 cells. Meanwhile, autophagy was triggered by asparaginase, which was confirmed by autophagosomes, autophagic flux, and conversion of LC3-I to LC3-II. Further exploration indicated that mitochondrial-originated ROS, Akt/mTOR, and Erk signaling pathway were also involved in the molecular mechanisms underlying the autophagy induction. Notably, inhibition of autophagy by chloroquine (CQ) could significantly enhance asparaginase-induced cytotoxicity and apoptosis, indicating that autophagy had a cytoprotective role in asparaginase-based therapy. Therefore, our results showed a novel viewpoint that autophagy had a cytoprotecitve role in asparaginase-induced LSCC cytotoxicity, and highlighted the novel approach for LSCC treatment by asparaginase alone or combined with autophagic inhibitors.
Materials and methods
Cell lines and culture
Human LSCC cell lines Tu212 and Tu686 were purchased from the Cell Bank of Xiangya Central Experiment Laboratory (Changsha, China). Cells were cultured in RPMI-1640 (San Diego, CA, USA) supplemented with 10% of heat-inactivated fetal bovine serum (San Diego, CA, USA) and maintained at 37 °C in humidified air with 5% CO2.
Reagents and antibodies
Asparaginase (derived from E. chrysanthemi) was obtained from Baiyunshan Mingxing Pharmaceutical Co., Ltd. (Guangzhou, China). The caspase inhibitor benzyloxycarbonyl Val-Ala-Asp (O-methyl)-fluoro-methylketone (z-VAD-fmk) and the antioxidant N-acetyl-L-cysteine (NAC) were acquired from Beyotime Institute of Biotechnology (Jiangsu, China). CQ and 3-(4,5-dimertrylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were obtained from Sigma-Aldrich (St. Louis, MO, USA). The primary antibodies were as follows: anti-tubulin, anti-β-actin, anti-caspase 3, anti-cleaved caspase 3, anti-PARP, anti-cleaved PARP, anti-LC3, anti-phospho-mTOR, anti-phospho-Akt, anti-phospho-p70 S6 kinase, anti-4EBP1 phospho (pT45), which were purchased from Cell Signaling Technology (Danvers, MA, USA). The secondary antibodies, including horseradish peroxidase-conjugated goat anti-mouse and anti-rabbit immunoglobulin G, were supplied by MR Biotech (Shanghai, China).
Cell viability assay
Cell viability was determined by the MTT assay. Cells were seeded into 96-well plates at a density of 1 × 104 cells/100 μL. Then, cells were incubated with different concentrations of asparaginase with or without CQ. After 48 h of incubation, cells were incubated with MTT (0.5 mg/mL) for 4 h at 37 °C. The optical density (OD) was measured at an absorbance wavelength of 590 nm.
Apoptosis assay
Annexin V-FITC/PI apoptosis detection kit (BD Bioscience, Franklin Lakes, NJ, USA) was employed to measure apoptosis. After treatment with asparaginase, cells were collected and washed with phosphate-buffered saline (PBS), then resuspended in 500 μL binding buffer at a concentration of 1 × 106 cells/mL. Subsequently, cells were incubated with Annexin V-FITC and PI for 15 min. Data was analyzed by FACS calibur flow cytometer.
Transmission electron microscopy
Tu212 and Tu686 cells were incubated with or without 0.5 IU/mL asparaginase for 24 h. Then, cells were harvested and fixed with glutaraldehyde in 0.1 M PBS (pH 7.4) for 2 h at 4 °C. After dehydration, samples were cut into ultrathin sections, then stained with uranyl acetate and lead citrate. Finally, samples were detected with a JEM 1410 transmission electron microscope (JEOL, Inc., USA).
Western blot analysis
To analyze protein expression levels, Tu212 and Tu686 laryngeal cancer cells were collected after treatment with different concentrations of asparaginase. Then, cells were washed with PBS and lysed in RIPA buffer (Beyotime Biotechnology, China) on ice for 30 min. The cell lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membrane. Then, the membrane was blocked with 3% bovine serum albumin (BSA) powder in 0.05% Tris-buffered saline and incubated with primary antibodies. After incubation with secondary antibodies, the membranes were observed with enhanced chemiluminescent reagents (Pierce, Rockford, IL, USA). Densitometry was determined by the ImageJ software.
Confocal immunofluorescence
Cyto-ID Autophagy Detection Kit was acquired from Enzo Life Sciences (Farmingdale, NY, USA), and LysoTracker Red was obtained from Invitrogen (San Diego, CA, USA). After treatment with or without asparaginase, LSCC cells were stained with Cyto-ID, Hoechst 33342 and LysoTracker Red DND-99 according to the manufacturer’s protocol. The samples were analyzed by laser confocal microscopy.
Statistical analysis
In this study, all experiments were repeated three times and the data were presented as the mean ± standard deviations (SD). Qualitative variables were analyzed by GraphPad Prism 5 under Student’s t test. P < 0.05 and P < 0.01 were indicated by * and **. P < 0.05 was considered as statistical significance of the differences.
Results
Asparaginase elicited potent cytotoxicity in Tu212 and Tu686 cells
Mounting lines of evidence have shown that ASNS deficiency formed the basis in asparaginase treatment (Hurteau et al. 2005). To explore whether ASNS was deficient in LSCC, western blot analysis was performed to determine the expression of ASNS in Tu212 and Tu686 cells. As shown in Fig. 1a, K562 cells and NCI-H1975 cells were serviced as the positive and the negative control, respectively. ASNS was deficient in Tu212 and Tu686 cells indicating that Tu212 and Tu686 cells were sensitive to asparaginase treatment. Next, we analyzed asparaginase-induced cytotoxicity in Tu212 and Tu686 cells. After treatment with different concentrations of asparaginase for 48 h, significant cytotoxicity was induced by asparaginase in a dose-dependent manner in Tu212 and Tu686 cells (Fig. 1b).

Asparaginase induced growth inhibition and apoptosis in laryngocarcinoma cell lines. a The expression of ASNS in Tu212 and Tu686 cells was measured by western blot. K562 cells served as the positive control, while H1975 cells served as the negative one. Densitometric values were quantified by the ImageJ software, and the data represented the mean of three independent experiments. b Exposed to asparaginase for 48 h, the cell viability recorded a concentration-dependent decrease in Tu212 and Tu686 cells. Densitometric values were quantified by the ImageJ software, and the data represented the mean ± SD of three independent experiments. c Treated with different concentrations of asparaginase for 48 h, the apoptotic rate of the cells was analyzed by flow cytometry. d The percentage of Annexin V-positive Tu212 and Tu686 cells was presented in bar charts. Results were represented as mean ± SD and analyzed by using Student’s t test (two tailed) (*P < 0.05, **P < 0.01)
Altogether, these results indicated that asparaginase induced cytotoxicity in ASNS-deficient LSCC cells.
Caspase-dependent apoptosis was activated in asparaginase-treated Tu212 and Tu686 cells
Apoptosis plays vital roles in antitumor drug-induced cytotoxicity, and caspase-3 is found to be one of the key family members of caspases (Mili et al. 2016). First, Annexin V/PI-based FCM analysis was used to determine the apoptosis in asparaginase-treated Tu212 and Tu686 cells. As shown in Fig. 1c, d, treated with asparaginase for 48 h, apoptosis was remarkable induced in a dose-dependent manner, indicating that apoptosis was activated by asparaginase in Tu212 and Tu686 cells. Then, after treatment with asparaginase for 24 h, LSCC cells showed an increased level of cleaved-caspase 3 and cleaved-PARP in a dose- and time-dependent manner (Fig. 2a–d and Fig. S1A–D). Moreover, upon pretreatment with z-VAD-fmk, a high efficiency caspase blocker, asparaginase-induced cell cytotoxicity was significantly reversed (Fig. 2f), indicating that caspase-dependent pathways contributed to asparaginase-induced apoptosis in LSCC cells.

Apoptosis induced by asparaginase depended on caspase activation in Tu212 and Tu686 cells. a, b After treated with 0.125, 0.25, 0.5, and 1 IU/mL of asparaginase for 24 h, the level of caspase 3, cleaved-caspase 3, PARP, and cleaved-PARP was detected by western blot analysis. Densitometric values were quantified by ImageJ software, and the data represented the mean of three independent experiments. c, d After treatment with 0.25 IU/mL of asparaginase for different time, the level of caspase 3, cleaved-caspase 3, PARP, and cleaved-PARP was detected by western blot analysis. Densitometric values were quantified using ImageJ software, and the data represented the mean of three independent experiments. e Densitometric values of the level of cleaved-caspase 3 and cleaved-PARP in asparaginase-treated Tu212 cells were quantified by ImageJ software, and the data represented as the mean ± SD of three independent experiments. f Exposed to asparaginase (0.25 IU/mL) and/or 20 μM z-VAD-fmk for 48 h, cell viability was analyzed by MTT assay. Results were represented as mean ± SD of three independent experiments and analyzed by Student’s t test (two tailed) (**P < 0.01)
Collectively, our data demonstrated that asparaginase induced caspase-dependent apoptosis in Tu212 and Tu686 cells.
Autophagy was induced by asparaginase in Tu212 and Tu686 cells
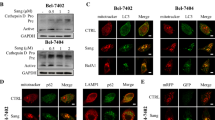
Recent studies have identified that autophagy could be induced by amino acid depletion to maintain cell homeostasis (Chen et al. 2016). To determine whether autophagy participated in asparagine deprivation-based LSCC treatment, three well-established methods including transmission electron microscopy, fluorescence staining, and immunoblotting were employed in this study. First, after treatment with asparaginase in LSCC cells for 24 h, an increased accumulation of double-membrane autophagosomes was observed when compared with negative controls (Fig. 3a). Then, autophagic vesicles were reported by the Cyto-ID Green dye with fluorescence microscopy. Similar to the positive control rapamycin-treated cells, asparaginase-treated cells exhibited increased green fluorescent dots (Fig. 3b). Furthermore, LC3-II, a reliable biomarker of autophagic level, was detected in asparaginase-treated LSCC cells. As shown in Fig. 3c, d and Fig. S2A–D, asparaginase induced LC3-II overexpression and the transformation from LC3-I to LC3-II in Tu212 and Tu686 cells.

Asparaginase triggered autophagy in Tu212 and Tu686 cells. a Exposed to asparaginase (0.5 IU/mL) for 24 h, the autophagosomes in Tu212 and Tu686 cells were examined by TEM. Red arrows labeled autophagic vacuoles. b After treated with asparaginase (0.5 IU/mL) for 24 h in Tu212 and Tu686 cells, autophagy stained with Cyto-ID Green dye was detected by confocal fluorescent microscopy. Rapamycin (50 nM) was serviced as a positive control. c, d Tu212 and Tu686 cells were treated with asparaginase in a dose- and time-dependent manner, and the expression of LC3-I/II was measured by western blot analysis
Taken together, these data forcefully suggested that asparaginase induced autophagy in Tu212 and Tu686 cells.
Autophagic flux was elicited in asparaginase-treated LSCC cells
Autophagy is a vital process involved in autophagosome formation, fusion of autophagosomes to lysosomes, and degradation of cellular contents in lysosomes (Zhang et al. 2016b). To further elucidate the dynamic process of autophagy, autophagic flux was evaluated in asparaginase-treated cells. Western blot analysis showed that cotreatment with CQ, a lysosomal pH neutralizing agent that inhibited autophagosome-lysosome fusion to restrain autophagy flux, further improved the expression of LC3-II in asparaginase-treated cells (Fig. 4a, b). Moreover, Cyto-ID and LysoTracker staining were used to examine autophagic flux by confocal fluorescent microscopy. Three stages of autophagic flux were distinctly observed in asparaginase-treated cells (Fig. 4c, d): the formation and accumulation of autophagosomes at 12 h (green fluorescence), fusion of autophagosomes to lysosomes at 24 h (yellow fluorescence), and then finally degradation of autophagosomes by lysosomes at 48 h (orange fluorescence).

Asparaginase triggered autophagic flux in Tu212 and Tu686 cells. a, b Tu212 and Tu686 cells were treated with 0.25 IU/mL of asparaginase and/or CQ (5 μM) for different times, then western blot analysis was performed to assess the expression level of LC3-II. Densitometric values were quantified by ImageJ software, and the data were presented as means ± SD of three independent experiments. c, d After treated with asparaginase (0.25 IU/mL) for different times, autophagic flux was measured in Tu212 and Tu686 cells by a confocal microscope
Briefly, these results strongly demonstrated that autophagic flux was activated by asparaginase in Tu212 and Tu686 cells.
Blocking autophagy enhanced asparaginase-induced cytotoxicity in Tu212 and Tu686 cells
To explore the role of autophagy in asparaginase-treated LSCC cells, CQ, an autophagosome-lysosome fusion blocker, was used to abolish autophagy. As shown in Fig. 5a, asparaginase-triggered autophagy was successfully inhibited by CQ in Tu212 and Tu686 cells, while cotreatment with asparaginase and CQ induced enhanced cytotoxicity in Tu212 and Tu686 cells compared with asparaginase alone (Fig. 5b). Western blot analysis exhibited a significant increase of cleaved-PARP in the cells cotreated with asparaginase and CQ (Fig. 5c). To further elucidate the biological role of autophagy in asparaginase-induced cytotoxicity, asparaginase-induced apoptosis was also detected. Figure 5d, e shows that cotreatment with asparaginase and CQ induced an enhanced apoptosis in LSCC cells compared with asparaginase treatment alone (Fig. 5d, e).

Inhibiting autophagy enhanced asparaginase-induced apoptosis and cytotoxicity in Tu212 and Tu686 cells. a Tu212 and Tu686 cells were treated with asparaginase (0.25 IU/mL) and/or CQ (5 μM) for 24 h. Autophagy-associated protein LC3-I/II was measured by western blot analysis. b Tu212 and Tu686 cells were incubated with 0.25 IU/mL of asparaginase in the absence or presence of CQ (5 μM) for 48 h, then cell viability was measured by MTT. Densitometric values were quantified by the ImageJ software, and the data represented mean ± SD of three independent experiments. c Tu212 and Tu686 cells were treated with asparaginase (0.25 IU/mL) and/or CQ (5 μM) for 24 h. The levels of PARP and cleaved-PARP were detected by western blot analysis. d Tu212 and Tu686 cells were treated with asparaginase (0.25 IU/mL) and/or CQ (5 μM) for 48 h, then flow cytometry was performed to measure cell apoptosis. e The percentage of Annexin V-positive/PI-negative cells was presented in bar charts. Results were represented as mean ± SD (**P < 0.01)
These data indicated that asparaginase-induced autophagy represented a cytoprotective mechanism in Tu212 and Tu686 cells, and blocking autophagy could potentiate asparaginase-induced cytotoxicity and caspase-dependent apoptosis in LSCC cells.
ROS was involved in asparaginase-induced autophagy and cytotoxicity in Tu212 and Tu686 cells
Previous studies have reported that ROS was involved in various antitumor treatment-induced autophagy (Poillet-Perez et al. 2015). First, confocal microscopy was employed to detect whether ROS participated in asparaginase-induced autophagy in LSCC cells. As shown in Fig. 6a, treatment with asparaginase induced the generation of ROS and autophagy (red and green fluorescence) in LSCC cells, while incubation with asparaginase and NAC, a free radical scavenging agent, led to a severe reduction of autophagy specific fluorescence, further suggesting that inhibition of ROS attenuated asparaginase-induced autophagy. We also found that inhibition of ROS by NAC could partially rescue the cytotoxicity and apoptosis in asparaginase-treated LSCC cells (Fig. 6b–d).

ROS generation played an important role in asparaginase-induced autophagy and cytotoxicity in cells. a Tu212 and Tu686 cells were incubated with asparaginase and NAC for 48 h, and autophagy and ROS were determined by confocal microscopy. b After treated with asparaginase (0.5 IU/mL) and/or NAC (20 μM) for 48 h, cell viability was determined by MTT assay. Results were expressed as mean ± SD of three independent experiments and analyzed by using Student’s t test (two tailed). c, d Tu212 and Tu686 cells were treated with asparaginase (0.5 IU/mL) and/or NAC (20 μM) for 48 h and detected by flow cytometry. The percentages of Annexin V-positive/PI-negative cells were presented in bar charts (**P < 0.01)
Collectively, these data demonstrated that intracellular ROS had a crucial role in asparaginase-induced autophagy and cytotoxicity in Tu212 and Tu686 cells.
The Akt/mTOR and Erk signaling pathways were implicated in asparaginase-induced autophagy in Tu212 and Tu686 cells
Akt/mTOR signaling pathway is a classic negative regulatory pathway for autophagy (Piccione et al. 2016; Zhou et al. 2016). To investigate whether Akt/mTOR signaling pathway was involved in asparaginase-induced autophagy, the phosphorylated level of Akt and mTOR in Tu212 and Tu686 cells was assessed. Figure 7a. b shows that the phosphorylated level of mTOR and Akt significantly decreased in a dose-dependent manner in Tu212 and Tu686 cells after asparaginase treatment for 24 h. Furthermore, the phosphorylated level of p70S6K and 4EBP1, two downstream substrates of Akt/mTOR signaling pathway, was also downregulated (Fig. 7a, b).

Akt/mTOR and Erk signaling pathways were implicated in autophagy triggered by asparaginase in Tu212 and Tu686. a–d Tu212 and Tu686 cells were exposed to different concentrations of asparaginase (0, 0.125, 0.25, 0.5, 1 IU/mL). a, b The level of p-Akt, p-mTOR, p-p70S6K, and p-4EBP1 was determined by western blot analysis. c, d Western blot was performed to analyze the protein Erk1/2 and p-Erk1/2. Densitometric values were quantified using the ImageJ software, and the data are presented as means ± SD of three independent experiments
Previous studies have shown that extracellular-signal-regulated kinase 1/2 (Erk1/2) is another important pathway in amino acid deprivation-induced autophagy (Song et al. 2015). Upon exposure to different concentrations of asparaginase, western blot analysis showed an increased phosphorylation level of Erk1/2 in TU212 and TU686 cells (Fig. 7c, d).
These results demonstrated that inactivation of Akt/mTOR and activation of the Erk signaling pathway were involved in asparaginase-induced autophagy in Tu212 and Tu686 cells.
Discussion
According to the data published in 2014 by the American Cancer Society, the 5-year survival rate of LSCC patients is still decreasing in recent years (Lian et al. 2016; Lin et al. 2015; Lu et al. 2016). Targeting cellular metabolism has been regarded as a promising approach for the therapy of malignancies (Zhu et al. 2016). Asparaginase, an asparagine-depleting enzyme, has been approved for the therapy of acute lymphoblastic leukemia (Kelo et al. 2009). Moreover, previous studies have demonstrated that ASNS-deficient cancer cells were susceptible to asparaginase treatment (Rizzari 2014). In this study, our results demonstrated that ASNS was deficient in LSCC TU212 and TU686 cells and asparaginase could significantly induce cytotoxicity and caspase-dependent apoptosis in LSCC for the first time.
Autophagy, a conserved cellular catabolic pathway, has been triggered in environmental stimulus such as nutrient starvation and chemotherapeutic agents (Gomes et al. 2016; Wang et al. 2014). It has been reported that autophagy played a dual role in cancer therapy (Gomes et al. 2016). In this study, we found that autophagy was induced in LSCC cells after asparaginase treatment. The appearance of autophagy was validated by the accumulation of autophagic vacuoles, autophagy-specific fluorescence, and the conversion of LC3-I to LC3-II in Tu212 and Tu686 cells. Previous studies showed that abolishing autophagy could enhance the antitumor efficacy of asparaginase in human pulmonary adenocarcinoma and acute myeloid leukemia (Zhang et al. 2016a). The biological function of autophagy in asparaginase-treated Tu212 and Tu686 cells was explored to determine its potential effect for antitumor therapy. Autophagolysosome inhibitor CQ was employed to block asparaginase-induced autophagy. Our results demonstrated that the combined treatment with asparaginase and CQ induced a significant enhanced cytotoxicity, indicating that autophagy was a cytoprotective mechanism in asparaginase-induced cytotoxicity. Further experiment clarified the relation between autophagy and apoptosis in asparaginase-treated cells. After inhibiting autophagy, the percentage of Annexin V-positive cells and cleaved-PARP was remarkably increased in asparaginase-treated cells, which strongly demonstrated that autophagy played a cytoprotective role in asparaginase-induced cytotoxicity and inhibiting autophagy could be a therapeutic approach to enhance the antitumor efficacy in laryngocarcinoma.
We explored the probable underlying molecular mechanisms in asparaginase-induced autophagy. Previous studied have demonstrated that ROS, Akt/mTOR, and Erk signaling pathway played a key role in autophagy induction (Yang et al. 2015). Thus, we first detected whether ROS was accumulated in asparaginase-treated LSCC cells. We found that ROS was notably accumulated and scavenging ROS by NAC could reduce asparaginase-induced autophagy and cytotoxicity in LSCC cells indicating that ROS was essential in asparaginase-induced autophagy and cytotoxicity in laryngocarcinoma cells. Then, the protein expression level of phosphorylated mTOR, Akt, p70S6K, and p-4EBP1 in asparaginase-treated cells was determined. Our results demonstrated that the expression of phosphorylated mTOR, Akt, p70S6K, and p-4EBP1 was significantly decreased, indicating that asparaginase induced autophagy via inactivation of the Akt/mTOR signaling pathway. Meanwhile, we observed the activation of phosphorylation of Erk1/2 in asparaginase-treated cells, suggesting that the Erk pathway was also involved in asparaginase-induced autophagy. Therefore, our data demonstrated that ROS, inactivation of Akt/mTOR, and stimulation of the Erk signaling pathway played key roles in asparaginase-induced autophagy, providing the feasible underlying molecular mechanisms of asparaginase treatment in LSCC.
In conclusion, deprivation of asparagine by asparaginase induced cytotoxicity and caspase-dependent apoptosis in Tu212 and Tu686 cells. Meanwhile, the cytoprotective autophagy was triggered by asparaginase. Inhibiting autophagy by CQ could remarkably enhance asparaginase-induced cytotoxicity and apoptosis. Moreover, our results revealed that asparaginase-induced autophagy depended on ROS production and inactivation of Akt/mTOR and activation of the Erk signaling pathway. Therefore, our results demonstrated that asparaginase alone or in combination with autophagy inhibitors could serve as an attractive therapeutic strategy for LSCC.
References
Abd El Baky HH, El Baroty GS (2016) Optimization of growth conditions for purification and production of L-asparaginase by Spirulina maxima. Evid Based Complemen Alterna Med 2016:1785938. doi:10.1155/2016/1785938
Casasampere M, Ordóñez YF, Casas J, Fabrias G (2017) Dihydroceramide desaturase inhibitors induce autophagy via dihydroceramide-dependent and independent mechanisms. Biochim Biophys Acta 1861(2):264–275. doi:10.1016/j.bbagen.2016.11.033
Chen R, Wang H, Liang B, Liu G, Tang M, Jia R, Fan X, Jing W, Zhou X, Wang H, Yang Y, Wei H, Li B, Zhao J (2016) Downregulation of ASPP2 improves hepatocellular carcinoma cells survival via promoting BECN1-dependent autophagy initiation. Cell Death Dis 7(12):e2512. doi:10.1038/cddis.2016.407
Cordani M, Butera G, Pacchiana R, Donadelli M (2016) Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim Biophys Acta 1867(1):19–28. doi:10.1016/j.bbcan.2016.11.003
Costa IM, Schultz L, de Araujo Bianchi Pedra B, Leite MS, Farsky SH, de Oliveira MA, Pessoa A, Monteiro G (2016) Recombinant L-asparaginase 1 from Saccharomyces cerevisiae: an allosteric enzyme with antineoplastic activity. Sci Rep 6:36239. doi:10.1038/srep36239
Fan J, Sun Y, Wang S, Li Y, Zeng X, Cao Z, Yang P, Song P, Wang Z, Xian Z, Gao H, Chen Q, Cui D, Ju D (2016a) Inhibition of autophagy overcomes the nanotoxicity elicited by cadmium-based quantum dots. Biomaterials 78:102–114. doi:10.1016/j.biomaterials.2015.11.029
Fan J, Zeng X, Li Y, Wang S, Wang Z, Sun Y, Gao H, Zhang G, Feng M, Ju D (2013) Autophagy plays a critical role in ChLym-1-induced cytotoxicity of non-Hodgkin’s lymphoma cells. PLoS One 8(8):e72478. doi:10.1371/journal.pone.0072478
Fan J, Zeng X, Li Y, Wang S, Yang P, Cao Z, Wang Z, Song P, Mei X, Ju D (2016b) A novel therapeutic approach against B-cell non-Hodgkin’s lymphoma through co-inhibition of Hedgehog signaling pathway and autophagy. Tumour Biol 37(6):7305–7314. doi:10.1007/s13277-015-4614-5
Figueiredo L, Cole PD, Drachtman RA (2016) Asparaginase Erwinia chrysanthemi as a component of a multi-agent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia who have developed hypersensitivity to E. coli-derived asparaginase. Expert Rev Hematol 9(3):227–234. doi:10.1586/17474086.2016.1142370
Gomes LC, Odedra D, Dikic I, Pohl C (2016) Autophagy and modular restructuring of metabolism control germline tumor differentiation and proliferation in C. elegans. Autophagy 12(3):529–546. doi:10.1080/15548627.2015.1136771
Gozuacik D, Kimchi A (2004) Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23(16):2891–2906. doi:10.1038/sj.onc.1207521
Horvat TZ, Pecoraro JJ, Daley RJ, Buie LW, King AC, Rampal RK, Tallman MS, Park JH, Douer D (2016) The use of Erwinia asparaginase for adult patients with acute lymphoblastic leukemia after pegaspargase intolerance. Leuk Res 50:17–20. doi:10.1016/j.leukres.2016.08.014
Hurteau GJ, Broome JD, Brock GJ (2005) Accurate detection of asparagine synthetase (ASNS) using quantitative real-time PCR (qRT-PCR), without requiring DNaseI treatment. Leukemia 19(12):2368–2370. doi:10.1038/sj.leu.2403969
Kelo E, Noronkoski T, Mononen I (2009) Depletion of L-asparagine supply and apoptosis of leukemia cells induced by human glycosylasparaginase. Leukemia 23(6):1167–1171. doi:10.1038/leu.2008.387
Lian R, Lu B, Jiao L, Li S, Wang H, Miao W, Yu W (2016) MiR-132 plays an oncogenic role in laryngeal squamous cell carcinoma by targeting FOXO1 and activating the PI3K/AKT pathway. Eur J Pharmacol 792:1–6. doi:10.1016/j.ejphar.2016.10.015
Lin C, Wang Z, Li L, He Y, Fan J, Liu Z, Zhao S, Ju D (2015) The role of autophagy in the cytotoxicity induced by recombinant human arginase in laryngeal squamous cell carcinoma. Appl Microbiol Biotechnol 99(20):8487–8494. doi:10.1007/s00253-015-6565-6
Lu M, Zhu H, Wang X, Zhang D, Xiong L, Zhu J, Mao Y, Qiang J (2016) LAMP1 expression is associated with malignant behaviours and predicts unfavourable prognosis in laryngeal squamous cell carcinoma. Pathology 48(7):684–690. doi:10.1016/j.pathol.2016.08.001
Mili D, Abid K, Rjiba I, Kenani A (2016) Effect of SP600125 on the mitotic spindle in HeLa cells, leading to mitotic arrest, endoreduplication and apoptosis. Mol Cytogenet 9:86. doi:10.1186/s13039-016-0296-y
Piantelli M, Iacobelli S, Almadori G, Iezzi M, Tinari N, Natoli C, Cadoni G, Lauriola L, Ranelletti FO (2002) Lack of expression of galectin-3 is associated with a poor outcome in node-negative patients with laryngeal squamous-cell carcinoma. J Clin Oncol 20(18):3850–3856. doi:10.1200/JCO.2002.01.078
Piccione EC, Juarez S, Tseng S, Liu J, Stafford M, Narayanan C, Wang L, Weiskopf K, Majeti R (2016) SIRPalpha-antibody fusion proteins selectively bind and eliminate dual antigen-expressing tumor cells. Clin Cancer Res 22(20):5109–5119. doi:10.1158/1078-0432.CCR-15-2503
Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M (2015) Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol 4:184–192. doi:10.1016/j.redox.2014.12.003
Rizzari C (2014) Shedding light on the asparaginase galaxy. Blood 123(13):1976. doi:10.1182/blood-2014-02-553040
Song P, Ye L, Fan J, Li Y, Zeng X, Wang Z, Wang S, Zhang G, Yang P, Cao Z, Ju D (2015) Asparaginase induces apoptosis and cytoprotective autophagy in chronic myeloid leukemia cells. Oncotarget 6(6):3861–3873. doi:10.18632/oncotarget.2869
Surono A, Priyanto P, Indrasari SR (2016) Hypoxia-inducible factor-1alpha expression in Indonesian laryngeal squamous cell carcinoma patients. J Oncol 2016:3215463. doi:10.1155/2016/3215463
Unger J, Lohscheller J, Reiter M, Eder K, Betz CS, Schuster M (2015) A noninvasive procedure for early-stage discrimination of malignant and precancerous vocal fold lesions based on laryngeal dynamics analysis. Cancer Res 75(1):31–39. doi:10.1158/0008-5472.CAN-14-1458
Wang S, Li Y, Fan J, Wang Z, Zeng X, Sun Y, Song P, Ju D (2014) The role of autophagy in the neurotoxicity of cationic PAMAM dendrimers. Biomaterials 35(26):7588–7597. doi:10.1016/j.biomaterials.2014.05.029
Wang X, Liu Y, Wang S, Pi D, Leng W, Zhu H, Zhang J, Shi H, Li S, Lin X, Odle J (2016) Asparagine reduces the mRNA expression of muscle atrophy markers via regulating protein kinase B (Akt), AMP-activated protein kinase alpha, toll-like receptor 4 and nucleotide-binding oligomerisation domain protein signalling in weaning piglets after lipopolysaccharide challenge. Br J Nutr 116(7):1188–1198. doi:10.1017/S000711451600297X
Yang J, Chen Q, Tian S, Song S, Liu F, Wang Q, Fu Z (2015) The role of 1,25-dyhydroxyvitamin D3 in mouse liver ischemia reperfusion injury: regulation of autophagy through activation of MEK/ERK signaling and PTEN/PI3K/Akt/mTORC1 signaling. Am J Transl Res 7(12):2630–2645
Zhang B, Fan J, Zhang X, Shen W, Cao Z, Yang P, Xu Z, Ju D (2016a) Targeting asparagine and autophagy for pulmonary adenocarcinoma therapy. Appl Microbiol Biotechnol 100(21):9145–9161. doi:10.1007/s00253-016-7640-3
Zhang C, Cai Z, Liang Q, Wang Q, Lu Y, Hu L, Hu G (2016b) RLIP76 depletion enhances autophagic flux in U251 cells. Cell Mol Neurobiol. doi:10.1007/s10571-016-0410-z
Zhou Q, Chen B, Wang X, Wu L, Yang Y, Cheng X, Hu Z, Cai X, Yang J, Sun X, Lu W, Yan H, Chen J, Ye J, Shen J, Cao P (2016) Sulforaphane protects against rotenone-induced neurotoxicity in vivo: involvement of the mTOR, Nrf2, and autophagy pathways. Sci Rep 6:32206. doi:10.1038/srep32206
Zhu J, Zheng Y, Zhang H, Sun H (2016) Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose regulates apoptosis in ovarian cancer cells via p38 MAPK/JNK signaling pathway. Am J Transl Res 8(11):4812–4821
Zhu M, Yin F, Yang L, Chen S, Chen R, Zhou X, Jing W, Fan X, Jia R, Wang H, Zheng H, Zhao J, Guo Y (2014) Contribution of TIP30 to chemoresistance in laryngeal carcinoma. Cell Death Dis 5:e1468. doi:10.1038/cddis.2014.424
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
The study was funded by the National Key Basic Research Program of China (2015CB931800, 2013CB932502), the Key Project for the Shanghai Health and Family Planning Commission (2013012, 201444), the National Natural Science Foundation of China (81573332), Shanghai Science and Technology Funds (14431900200), and Special Research Foundation of State Key Laboratory of Medical Genomics and Collaborative Innovation Center of Systems Biomedicine.
Conflict of interest
The authors declare that they have no competing interest.
Electronic supplementary material
ESM 1
(PDF 437 kb)
Rights and permissions
About this article

Cite this article
Ji, Y., Li, L., Tao, Q. et al. Deprivation of asparagine triggers cytoprotective autophagy in laryngeal squamous cell carcinoma. Appl Microbiol Biotechnol 101, 4951–4961 (2017). https://doi.org/10.1007/s00253-017-8221-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8221-9