
Abstract
Parkinson’s disease (PD) and l-DOPA-induced dyskinesia (LID) are motor disorders with significant impact on the patient’s quality of life. Unfortunately, pharmacological treatments that improve these disorders without causing severe side effects are not yet available. Delay in initiating l-DOPA is no longer recommended as LID development is a function of disease duration rather than cumulative l-DOPA exposure. Manipulation of the endocannabinoid system could be a promising therapy to control PD and LID symptoms. In this way, phytocannabinoids and synthetic cannabinoids, such as cannabidiol (CBD), the principal non-psychotomimetic constituent of the Cannabis sativa plant, have received considerable attention in the last decade. In this review, we present clinical and preclinical evidence suggesting CBD and other cannabinoids have therapeutic effects in PD and LID. Here, we discuss CBD pharmacology, as well as its neuroprotective effects and those of other cannabinoids. Finally, we discuss the modulation of several pro- or anti-inflammatory factors as possible mechanisms responsible for the therapeutic/neuroprotective potential of Cannabis-derived/cannabinoid synthetic compounds in motor disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Pharmacology of Cannabidiol
There are more than 500 phytochemical components in the Cannabis sativa plant, and at least 104 are cannabinoids (Elsohly and Slade 2005) Fig. 1). Cannabidiol (CBD), the major non-psychotropic component in Cannabis (Zuardi et al. 1982; McPartland et al. 2015), is a phytocannabinoid that comprises up to 40% of some plant extracts (Atakan 2012). CBD was isolated for the first time in 1940 by Adams and Madison (1940), and its structure was identified 23 years later by Mechoulam and Shvo (1963). CBD concentration is highly variable and depends on the plant phenotype, the cultivation conditions, and the part used to obtain the extract (Fischedick et al. 2010).

Chemical structures of three major phytocannabinoids: Δ9-tetrahydrocannabinol (Δ9-THC), cannabidiol (CBD), and Δ9-tetrahydrocannabivarin (Δ9-THCV)
In the mid-1980s, Howlett and colleagues suggested that Δ9-tetrahydrocannabinol (Δ9-THC), the primary psychoactive compound of the Cannabis plant, could act on a G protein-coupled receptor decreasing cAMP production (Howlett and Fleming 1984). In 1988, the first evidence indicating the existence of Δ9-THC receptors was described in the rat brain (Devane et al. 1988). Later, these sites were cloned and named cannabinoid receptor type 1 (CB1) (Matsuda et al. 1990) and cannabinoid receptor type 2 (CB2) (Devane et al. 1992; Munro et al. 1993; Mechoulam et al. 1995).
CBD is a pleiotropic compound with activity at multiple targets. It has low affinity for cannabinoid receptors, acting in CB1 and CB2 only at very high concentrations (≥ 10 μM) (Howlett and Fleming 1984; Lynn and Herkenham 1994; Jones et al. 2010; McPartland et al. 2015). CBD did not demonstrate any agonist activity for the CB1 receptor, seen through a [35S]GTPγS binding assay from cortical membranes (Jones et al. 2010), but could, in vitro, antagonize cannabinoid agonists, acting as an inverse agonist for both CB1 and CB2 receptors (Thomas et al. 2007). It also displays indirect agonist activity at CB1 receptors and increases endogenous levels of anandamide (AEA), by inhibiting the enzyme responsible for its degradation, fatty acid amide hydrolase (FAAH) (Bisogno et al. 2001; Rimmerman et al. 2011; Leweke et al. 2012).
CBD does not affect the metabolism of the endocannabinoid 2-arachidonoylglycerol (2-AG) (Rimmerman et al. 2011), consequently not influencing the action of 2-AG on CB1 or CB2 receptors (Piomelli 2003; Pertwee 2015). On the other hand, CBD reduces the efficacy and potency of 2-AG in CB1, thus acting as a negative allosteric modulator of CB1 receptors (Laprairie et al. 2015). CBD also inhibits psychoactive effects of Δ9-THC (Zuardi et al. 1982) by mechanisms not completely understood that could involve, for example, CB1 receptor antagonism (Zuardi et al. 1982; Pertwee 2008; Niesink and van Laar 2013) or antagonism of the orphan G protein-coupled receptor GPR55 (Pertwee 2008). Although CBD can impair Δ9-THC hydrolysis by CYP450 enzymes (Bornheim et al. 1993; Bornheim et al. 1995), no pharmacokinetic interaction between Δ9-THC and CBD has been found at clinically relevant doses (Pertwee 2008). Since Δ9-THC-like psychotropic effects depend on CB1 receptor activation (Mackie 2006), and CBD does not activate CB1 receptors in physiological concentrations, CBD is devoid of psychotomimetic properties.
CBD also binds to non-cannabinoid targets in low concentrations. It interacts with transient receptor potential ion channels such as the transient receptor potential vanilloid channel type 2 (TRPV-2) and transient receptor potential for melastatin, TRPM8 (Qin et al. 2008; De Petrocellis et al. 2011), respectively, as an agonist and antagonist. CBD can also directly stimulate potential vanilloid channel type 1 (TRPV-1) receptors (De Petrocellis et al. 2011) and acts as an indirect agonist of these receptors by enhancing the levels of AEA, an endogenous agonist of TRPV-1 (Bisogno et al. 2001; Rimmerman et al. 2011). CBD is an antagonist of the G-coupled receptors GPR18 (McHugh et al. 2012) and GPR55 (Ryberg et al. 2007) and binds to equilibrative nucleoside transporter (ENT) (Carrier et al. 2006), adenosine receptors (Carrier et al. 2006; Pandolfo et al. 2011), glycine receptors (Ahrens et al. 2009), serotonin 1A receptor (5-HT1A) (Russo et al. 2005), and peroxisome proliferator-activated receptor-γ (PPAR-γ) (O’Sullivan et al. 2009).
CBD influences dopaminergic neurotransmission by inhibiting the dopamine uptake transporter (Pandolfo et al. 2011), thus increasing the endogenous levels of dopamine (Murillo-Rodriguez et al. 2011). In mice striatal membrane assay, CBD acts as a negative allosteric modulator of dopamine D2 receptors (Bloom and Hillard 1985), suggesting that CBD can modulate the dopaminergic neurotransmission in basal ganglia. Moreover, CBD influences the activity of cyclooxygenases, thus reducing the production of prostaglandin E2 (Costa et al. 2004). CBD also decreases the expression of inducible nitric oxide synthase and the production of reactive oxygen species (Hampson et al. 1998; Iuvone et al. 2004; Lastres-Becker et al. 2005; Esposito et al. 2006), and influences the production of several pro-inflammatory factors/cytokines, such as NF-κB, TNF-α, INF-γ, IL-1β, IL-1α, and IL-6 in different experimental conditions (Watzl et al. 1991; Malfait et al. 2000; Kozela et al. 2010; Rimmerman et al. 2011; Li et al. 2013). These anti-inflammatory and antioxidant properties may help explain CBD’s neuroprotective action.
The Endocannabinoid System and Their Role in the Basal Ganglia
Endocannabinoids are derived from membrane phospholipids (Cadas et al. 1997). The most studied endocannabinoids are AEA and 2-AG (Di Marzo and De Petrocellis 2012). The enzyme N-acyl-phosphatidylethanolamine phospholipase (NAPE-PLD) synthesizes AEA, while the α and β isoforms of diacylglycerol lipase catalyze 2-AG formation (Cadas et al. 1997; Bisogno et al. 2001). The synthesis of these endocannabinoids occurs after cell depolarization or receptor stimulation (e.g., NMDA or mGlu5 activation) (Piomelli 2003). Therefore, endocannabinoids are synthesized “on-demand” after stimuli, thus acting as neuromodulators. The main enzymes that metabolize AEA and 2-AG are FAAH and monoacylglycerol lipase (MAGL), respectively (Desarnaud et al. 1995; Cravatt et al. 1996; Cadas et al. 1997). These enzymes are mostly responsible for ending endocannabinoid actions (Piomelli 2003).
2-AG is a full agonist of CB2 receptors (Sugiura et al. 2006) while AEA seems to be a partial agonist of CB1 receptors and does not bind significantly to CB2 receptors in normal conditions (Mechoulam and Hanus 2000; Sugiura et al. 2006). However, AEA was recently linked to activation of the CB2 receptor in pathological states (Eljaschewitsch et al. 2006). AEA also activates non-cannabinoid receptors, such as TRPV-1 and PPAR-γ, while 2-AG has been postulated to act as endogenous activators of TRPV-4.
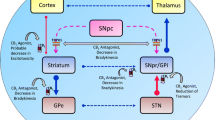
CB1 receptors are the most expressed G protein-coupled receptors in the central nervous system (Herkenham et al. 1991a, b; Howlett et al. 2010). In the basal ganglia, CB1 receptors are present in excitatory projections from the subthalamic nucleus to the internal globus pallidus and substantia nigra pars reticulata (SNpr) (Brotchie 2003; van der Stelt and Di Marzo 2003; Benarroch 2007). GABAergic medium spiny neurons also express CB1 receptors in their dendrites and presynaptic axon terminals, innervating the outer and inner segments of the globus pallidus and SNpr. Moreover, CB1 receptors are present at the level of corticostriatal excitatory glutamatergic terminals (Brotchie 2003; van der Stelt and Di Marzo 2003; Benarroch 2007), but not in dopaminergic presynaptic terminals from substantia nigra pars compacta (SNpc) (Herkenham et al. 1991a, b; Howlett et al. 2010).
Immunostaining for CB1 is widespread throughout the striatum in GABAergic, cholinergic, and NOS-positive neurons (Fernandez-Ruiz 2009; Fusco et al. 2004; Hohmann and Herkenham 2000). The higher expression of CB1 receptors is found on GABAergic spiny neurons projecting to the SNpc/internal globus pallidus (“direct” striatal efferent pathway) and the external globus pallidus (“indirect” striatal efferent pathway) (Herkenham et al. 1991a, b; Ameri 1999; Moldrich and Wenger 2000). GABAergic and cholinergic interneurons express CB1 receptors in a lesser extension (Marsicano and Lutz 1999; Hohmann and Herkenham 2000; Fusco et al. 2004).
CB2 receptors are discreetly expressed in specific brain areas such as the hypothalamus, brain stem, hippocampus, and SNpr (Onaivi et al. 2006), at much lower levels when compared to CB1. The functionality of CB2 receptors in neurons has also been controversial since their expression levels in central neurons are very low (Onaivi et al. 2006). CB2 is expressed primarily in the peripheral immune system, such as the marginal zone of the spleen (Onaivi et al. 2006). In the healthy brain, there is a discreet presence of CB2 receptors in glial cells (microglia and astrocytes), while its expression seems to be elevated in certain neurological disorders such as PD.
The endocannabinoid system can functionally interact with the dopaminergic neurotransmission in the striatum (Pisani et al. 2011). CB1 receptor stimulation reduces glutamate release in the striatum (Brotchie 2003), an effect that is frequency-dependent and involves dopamine D2 receptors (Giuffrida et al. 1999). Furthermore, CB1 is co-expressed with dopamine D1 receptors in the striatum of mice and humans (Glass et al. 2000; Hermann et al. 2002), where they can interact to form heterodimers (Hermann et al. 2002; Marcellino et al. 2008; Khan and Lee 2014), thus suggesting a functional crosstalk between cannabinoid and dopaminergic neurotransmission.
It has been proposed that AEA is synthesized in striatal postsynaptic GABAergic neurons acting on glutamatergic presynaptic terminals from cortical areas decreasing glutamate release (Gerdeman and Lovinger 2001). This CB1 stimulation by AEA is critical for long-term depression (LTD) in corticostriatal synapses (Gerdeman et al. 2002; Brotchie 2003), reducing glutamatergic synaptic effectiveness. Furthermore, CB1 receptor activation dampens the hyperlocomotion and the enhanced dopamine and glutamate release in the striatum induced by amphetamine (Polissidis et al. 2014) while reducing the GABAergic input to dopaminergic neurons of the SNpc, thereby modulating the firing activity of nigral neurons (Lovinger 2010). In this way, it has been usually accepted that the endocannabinoid system modifies the striatal functioning and interferes in movement control.
TRPV-1 is also highly expressed in basal ganglia (Kunert-Keil et al. 2006). AEA binds to and activates this receptor, behaving as a partial or full agonist, depending on the tissue receptor reserve or circumstances associated with some disease states (Ross 2003). In the striatum, TRPV-1 channels co-localize with CB1 receptors (Mezey et al. 2000; Cristino et al. 2006), suggesting a close functional interaction between these receptors. Pharmacological modulation of TRPV-1 has been reported to influence motor behaviors and nigrostriatal dopaminergic activity (de Lago et al. 2004) and to modulate motor symptoms originating from striatal dysfunction (Lastres-Becker et al. 2003; Tzavara et al. 2006). The inhibition of AEA degradation amplifies the tonic activation of TRPV-1 channels in the striatum, increasing glutamate but not GABA release (Musella et al. 2009), thus suggesting an essential role of TRPV-1 receptors in modulating glutamatergic activity in the striatum.
Parkinson’s Disease: a Brief Introduction
James Parkinson in “An essay on the shaking palsy” (Parkinson 2002) described for the first time the “paralysis agitans” or, as it is currently known, Parkinson’s disease (PD). In general, the disease is symptomatically characterized by abnormal posture, bradykinesia, resting tremor, and rigidity, among other motor and non-motor symptoms. PD is the second most frequent neurodegenerative disorder, and more than 90% of the cases are sporadic.
The pathogenesis of idiopathic PD is not fully understood. The disease is thought to be multifactorial, involving environmental and genetic factors (Lees et al. 2009; Nalls et al. 2014; Obeso et al. 2017). Studies have reported an association between PD and head injury, rural living, middle-age obesity, lack of exercise, and herbicide/insecticide exposure (paraquat, organophosphates, and rotenone) (Elbaz and Tranchant 2007; Thacker et al. 2008; Lees et al. 2009; Shih et al. 2017), while smokers and regular drinkers of coffee have lower risk to develop PD (Ascherio et al. 2003; Allam et al. 2004; Ascherio et al. 2004).
The pathophysiology of PD includes—but is not restricted to—several molecular and cellular malfunctions, such as mitochondrial dysfunction, oxidative stress, misfolding and aggregation of α-synuclein, dysregulation of calcium homeostasis, and neuroinflammation (Reglodi et al. 2017; Bortolanza et al. 2018). These alterations might lead to the ultimate aspect of PD, the loss of dopaminergic neurons in the nigrostriatal pathway (Michel et al. 2016). Therefore, more effective and tolerable treatments are necessary to retard the neurodegeneration and improve the patients’ quality of life.
Endocannabinoids and Cannabinoids in PD: Evidence from Patients and Animal Models
Targeting the Endocannabinoid System in PD
Studies investigating changes in the endocannabinoid signaling using experimental models of PD have produced contradictory results, with reports of reduction (Van Laere et al. 2012) or increase (Lastres-Becker et al. 2001) in CB1 binding within the striatum or other basal ganglia structures.
Parkinsonian patients present increased levels of AEA in the cerebrospinal fluid and l-DOPA treatment restores AEA to normal levels, which suggests that abnormal cerebrospinal AEA is a compensatory mechanism of dopamine depletion (Maccarrone et al. 2003; van der Stelt et al. 2005; Pisani et al. 2010). In rodents, 6-hydroxydopamine (6-OHDA) nigral lesions increase striatal levels of AEA, but not of 2-AG. They also decrease AMT and FAAH activity but did not affect AEA binding to cannabinoid receptors (Gubellini et al. 2002).
However, divergent data showed that the enhancement in AEA levels through chronic FAAH inhibition (using 1 mg/kg of URB597 into the midbrain and cerebellum of mice treated with MPTP over 5 weeks) prevents MPTP-induced motor impairment, without reducing glial activation or protecting the dopaminergic loss in the nigrostriatal pathway (Celorrio et al. 2016). In a different report, the indirect increase of AEA by the chronic administration of URB597 (0.2 mg/kg every 3 days for 30 days in MPTP-lesioned mice) decreased the dopaminergic cell loss and reduced glial activation (Viveros-Paredes et al. 2019). Chronic inhibition of MAGL with JZL184 (8 mg/kg), indirectly increasing the levels of 2-AG, also prevented MPTP-induced motor impairment, reduced astroglial activation, and preserved the nigrostriatal pathway (Fernandez-Suarez et al. 2014). Altogether, these data suggest that the modulation of endocannabinoid levels can modify the progression of PD.
Regarding cannabinoid receptors, MPTP-lesioned marmosets displayed an increased CB1 receptor binding in the striatum, an effect normalized by l-DOPA treatment (Lastres-Becker et al. 2005). On the other hand, the pharmacological blockade of CB1 receptors promoted anti-parkinsonian effects in rats with severe nigral lesions (> 95% cell loss), but not in rats with lesser lesions (85–95% cell loss). The authors suggest that this discrepancy is due to a nigral CB1-mediated stimulatory effect that counterbalances the striatal CB1-mediated inhibitory motor effect. Thus, after severe nigral degeneration, this CB1-mediated nigral stimulation disappears, and the anti-parkinsonian effects of CB1 antagonists via striatal CB1 become apparent (Fernandez-Espejo et al. 2005). Another study showed that a low dose (0.1 mg/kg) of the CB1 antagonist rimonabant partially reduced hypokinesia in animals lesioned with 6-OHDA injection, an effect that was not associated with dopamine depletion or changes in GABAergic and glutamatergic neurotransmissions in the striatum (Gonzalez et al. 2006). Together, these results indicate that nigrostriatal lesions are associated with changes in the endocannabinoid signaling in the basal ganglia (Lastres-Becker et al. 2001).
Synthetic cannabinoid agonists also interfere with experimentally induced parkinsonism. The non-selective cannabinoid receptor agonist WIN55,212-2 protected nigrostriatal neurons from MPTP-induced neurotoxicity and neuroinflammation. These effects were dependent on CB2 but not CB1 receptors (Price et al. 2009). HU-210, another non-selective cannabinoid agonist, displayed similar effects to WIN55,212-2 in MPTP-lesioned mice (Chung et al. 2011). Furthermore, CB2 receptors are elevated in microglial cells within the SN of PD patients, and both the striatum and SN of LPS-lesioned mice. The genetic ablation of CB2 receptors aggravated LPS-induced inflammation while the pharmacological activation of CB2 receptors diminished the pro-inflammatory response (Gomez-Galvez et al. 2016).
In MPTP-lesioned mice, WIN55,212-2 also increased the survival of dopaminergic neurons in the nigrostriatal pathway by reducing reactive oxygen species and pro-inflammatory cytokines production (Chung et al. 2011). In addition, WIN55,212-2 decreased cellular death via a reduction of extracellular magnesium concentration (Gilbert et al. 2007), increased the expression of growth factors such as FGF and BDNF (Galve-Roperh et al. 2013), and decreased glial activation and neuronal death in the presence of beta-amyloid protein (Ramirez et al. 2005).
The effects of the endocannabinoid system can also involve TRPV-1 receptors (Di Marzo et al. 2001; Ross 2003). MPTP treatment increases TRPV-1 expression in astrocytes of the SNpc, but not in microglial cells. This MPTP-evoked lesion also reduced the expression of TRPV-1 receptors in TH-positive neurons of the SNpc. The administration of a TRPV-1 agonist rescued the dopaminergic loss while the knockdown of TRPV-1 receptors exacerbated the MPTP-induced loss of dopaminergic neurons (Nam et al. 2015).
The role of PPAR-γ receptors in PD/parkinsonism state has also been studied (Liu et al. 2003; Rockwell and Kaminski 2004; O’Sullivan 2007). A preclinical study has demonstrated that a PPAR-γ agonist attenuated the MPTP-induced glial activation and prevented the SNpc dopaminergic cell loss (Breidert et al. 2002). PPAR-γ agonists have neuroprotective properties and anti-inflammatory actions that might help explain their promising actions in PD models (Carta et al. 2011b). For instance, the use of pioglitazone, a PPAR-γ agonist, protected rats against hypolocomotion and 6-OHDA-induced dopaminergic neurodegeneration. This effect was associated with a decrease in microglial activation and p65 subunit of NF-κB (Machado et al. 2019). The same agonist reduced mortality, prevented depressive-like behavior, and facilitated the neurogenesis in the hippocampus of the 6-OHDA rat model of PD, suggesting a generalized beneficial effect for PPAR-γ in experimental parkinsonian models. Pioglitazone also improved the symptomatic parkinsonian features of a PD genetic mouse model by reducing the neuroinflammation process in the striatum and midbrain of these animals (Pinto et al. 2016). Further studies exploring the AEA neuroprotective effect are necessary to unveil if it may produce its neuroprotection in PD via PPAR-γ receptor activation.
PD and Cannabis
In 1888, Gowers first noted benefits of the “Indian hemp” on a parkinsonian syndrome (Russo 2018). A century later, studies using Cannabis as a possible PD therapy have gained ground. An observational study showed that smoked Cannabis was well tolerated and improved tremor, rigidity, and bradykinesia in parkinsonian patients (Lotan et al. 2014). Five of nine patients using Cannabis reported significant improvement, particularly on mood and sleep (Finseth et al. 2015). A carefully crafted survey of 339 Czech patients using oral Cannabis leaves reported significant alleviations of multiple symptoms, particularly those using the treatment for 3 or more months, with improvement in general function, resting tremor, bradykinesia, and rigidity with few side effects (Venderova et al. 2004). An oral Δ9-THC:CBD extract, however, showed no significant benefits on dyskinesia or other signs in 17 patients (Carroll et al. 2004).
Neuroprotective Effect of Phytocannabinoids in Experimental Models of PD
Beyond endocannabinoids, works have demonstrated that phytocannabinoids such as Δ9-THC also have neuroprotective properties. Δ9-THC reduced the decrement in dopamine content and tyrosine hydroxylase activity evoked by 6-OHDA (Lastres-Becker et al. 2005) and inhibited the oxidative stress induced by 1-methyl-4-phenylpyridinium iodide (MPP+) in differentiated SH-SY5Y neuroblastoma cells, restoring the mitochondrial content via PPAR-γ receptor (Carroll et al. 2012; Zeissler et al. 2016).
Δ9-Tetrahydrocannabivarin (Δ9-THCV) is another phytocannabinoid with antioxidant properties (McPartland et al. 2015). Garcia and colleagues have shown that the Δ9-THCV decreased the 6-OHDA-evoked motor deficit and diminished the dopaminergic neuron loss in the SNpc in hemiparkinsonian rodents (Garcia et al. 2011). In the same way, the phytocannabinoid β-caryophyllene attenuated the oxidative stress, neuroinflammation, glial activation, and loss in nigrostriatal dopaminergic neurons in a rotenone animal model of PD (Ojha et al. 2016).
Preclinical and Clinical Evidence of the Neuroprotective Effect of CBD in PD
CBD is under intense preclinical and clinical research for numerous neurodegenerative disorders (Lopez-Sendon Moreno et al. 2016; Aymerich et al. 2018). The neuroprotective properties of CBD do not appear to depend on the direct activation of CB1 receptors (Fernandez-Ruiz et al. 2013), and even if the involvement of CB2 receptors has been documented in specific pathological conditions (Castillo et al. 2010), the direct activity of CBD at these cannabinoid receptors remains controversial (Bisogno et al. 2001; Pertwee 2008). However, an indirect mechanism could occur: by decreasing FAAH activity, CBD could facilitate AEA-mediated effects (Bisogno et al. 2001).
In a cellular model of PD using PC12 and SH-SY5Y cells treated with MPP(+), CBD increased the cell viability, differentiation, and expression of axonal (GAP-43) and synaptic proteins synaptophysin and synapsin I (Santos et al. 2015). CBD also increased cell viability and decreased microglia activation after incubation with LPS (Martin-Moreno et al. 2011; Janefjord et al. 2014). Since chronic inflammation is a significant feature in PD (McGeer and McGeer 2004; Herrero et al. 2015)—for instance, parkinsonian patients present augmented levels of pro-inflammatory cytokines (TNF-α, iNOS, IL-1β, etc.) (Reale et al. 2009; Herrero et al. 2015)—and dopaminergic neurons are particularly vulnerable to glial activation, CBD anti-inflammatory effects could contribute to its neuroprotective potential in PD.
In vivo studies using CBD in PD animal models have produced conflicting results. The administration of CBD (5 mg/kg) for 5 weeks did not reduce the loss of dopaminergic neurons or improve the motor deficits induced by MPTP in mice (Celorrio et al. 2016). Contrarily, a different study reported that a daily administration of CBD (3 mg/kg) for 14 days decreased the depletion of dopamine and tyrosine hydroxylase in the striatum of rats injected with 6-OHDA when administered immediately after the lesion (Lastres-Becker et al. 2005). No effect was found, however, when the treatment started 1 week later. CBD effects were associated with the upregulation of Cu2+/Zn superoxide dismutase mRNA levels, a key enzyme for endogenous defenses against oxidative stress (Garcia-Arencibia et al. 2007).
An open-label pilot study in PD patients showed that CBD, when associated with the usual anti-Parkinson’s pharmacotherapy, reduced the psychotic symptoms without influencing the cognitive and motor signs (Zuardi et al. 2009). In a subsequent clinical trial, Chagas and colleagues treated PD patients for 6 weeks or more with increasing doses of CBD. In this work, CBD improved the mobility, emotional well-being, cognition, communication, and patients’ body discomfort, but failed to produce any difference in the total motor score compared to placebo-treated patients (Chagas et al. 2014b). High doses of CBD were also administered in five patients with dystonia, two of them with co-existing parkinsonian features. A dose-related improvement in dystonia was observed in all patients. In the two patients with co-existing parkinsonian symptoms, CBD in lower doses did not modify hypokinesia and resting tremor (Consroe et al. 1986). Since CBD is well tolerated in humans, these reported positive effects suggest that this drug could be a promising complementary treatment for PD. More preclinical and clinical studies, however, are needed to assess this possibility. The clinical trials and preclinical studies with CBD are summarized in Table 1.
l-DOPA-Induced Dyskinesia: an Introduction to Clinical and Pathophysiological Features
The use of the amino acid precursor of dopamine, l-3,4-dihydroxyphenylalanine (l-DOPA), remains the standard treatment for ameliorating PD motor symptoms. However, its long-term efficacy is limited by the development of disabling motor complications such as l-DOPA-induced dyskinesia (LID). LID is a set of disabling abnormal involuntary movements (Lundblad et al. 2002; Jenner 2008). It is expressed as a mixture of chorea, ballism and dystonia, and, to a lesser extent, myoclonus (a sudden and involuntary muscle contraction that occurs mainly in body extremities) (Nutt 1990).
l-DOPA treatment in the early stages of PD is responsible for “positive” plasticity, resulting in long-term symptomatic benefit (Cenci et al. 2009; Prescott et al. 2009). At later stages of the disease, however, the long-lasting symptomatic improvement triggered by l-DOPA disappears. In these late stages, l-DOPA worsens the striatum-dependent learning functions (Feigin et al. 2003; Cools et al. 2007) and negatively affects cortical plasticity (Cenci and Konradi 2010; Calabresi et al. 2015).
LID is associated with corticostriatal overactivation and molecular alterations in the basal ganglia. Striatal neurons integrate cortical and thalamic inputs to modify the output of the basal ganglia, playing a pivotal role in movement selection and adaptive motor control (Wiecki and Frank 2010). As described by Cenci and Konradi (2010), “the overactivation of dopamine type-1 receptor (D1R)-dependent signaling pathways [in LID] can cause large morphological and functional rearrangements in striatal neurons (“too much plasticity”), but also the molecular machinery by which these neurons normally respond to received stimuli.”
Increased glutamatergic neurotransmission has also been associated with the pathophysiology of LID (Papa and Chase 1996; Rylander et al. 2009; Ahmed et al. 2011; Huot et al. 2013; Morin and Di Paolo 2014; Solis et al. 2016). The non-competitive antagonist of the N-methyl-d-aspartate (NMDA)-type glutamate receptors amantadine is the only clinically available pharmacological treatment for LID (Metman et al. 1999; Wandinger et al. 1999; Luginger et al. 2000; Blanpied et al. 2005; Ossola et al. 2011). Nonetheless, pharmacotherapy with this drug is limited due to the development of central adverse effects including dizziness, confusion, and hallucinations (Shannon et al. 1987; Macchio et al. 1993; Thomas et al. 2004; Wolf et al. 2010). Preclinical studies show that LID could also be attenuated by drugs acting on other neurotransmitters including noradrenaline, acetylcholine, serotonin, adenosine, and nitric oxide (Blandini 2003; Colosimo and Craus 2003; Carta and Tronci 2014; Del-Bel et al. 2015; Perez et al. 2018).
Intervening in the Cannabinoid System to Treat LID
Preclinical Studies
Several studies have investigated whether the modulation of the cannabinoid system could represent a potential tool to alleviate l-DOPA-induced abnormal involuntary movements (AIMs). AEA and 2-AG levels were increased in the striatum and SN of MPTP-lesioned non-human primates, but 2-AG was decreased in the external globus pallidus. l-DOPA treatment restored endocannabinoid levels to those observed in non-dyskinetic animals, but not in dyskinetic ones (Maccarrone et al. 2003; van der Stelt et al. 2005). For AEA, the synthesizing/degrading enzymes were described as enhanced in the globus pallidus of untreated parkinsonian monkeys, but not of dyskinetic animals. A recent article confirmed the presence of several dysregulated metabolites in the striatum of dyskinetic rats, including AEA and 2-AG. The same work described that the intrastriatal administration of both endocannabinoids in hemiparkinsonian rats prior to l-DOPA treatment prevented the onset of LID (Wang et al. 2018).
CB1 and CB2 receptor modifications also indicate a potential therapeutic target for the active phase of LID (Rojo-Bustamante et al. 2018). The cannabinoid agonist R(+)-WIN55,212-2 also prevented l-DOPA-induced AIMs in two different reports (Morgese et al. 2007; Martinez et al. 2012). WIN dose dependently reduced l-DOPA-induced AIMs via CB1-mediated mechanisms (Morgese et al. 2007). This cannabinoid agonist also decreased protein kinase A (PKA) activity in the ipsilateral dorsal striatum of 6-OHDA-lesioned dyskinetic rats (Martinez et al. 2012). Supporting this proposal, CB1 agonist nabilone decreased LID in MPTP-lesioned non-human primates treated with l-DOPA (Fox et al. 2002). Small doses of the synthetic CB1 agonist HU-210 also substantially reduced l-DOPA- and apomorphine-induced contralateral rotations in hemiparkinsonian rats (Gilgun-Sherki et al. 2003).
However, there are conflicting results regarding CB1 activation on LID pathophysiology. Pérez-Rial and colleagues showed that hemiparkinsonian mice lacking CB1 receptors developed mild rather than severe dyskinesia when treated with l-DOPA (Perez-Rial et al. 2011). The co-administration of l-DOPA (8 mg/kg) and CB1 receptor antagonist rimonabant (1 and 3 mg/kg) in MPTP-lesioned marmosets decreased LID severity without affecting the anti-parkinsonian effect of l-DOPA (van der Stelt et al. 2005). In rats, this treatment also reduced LID and partially preserved the dopaminergic cells (Gutierrez-Valdez et al. 2013). The blockade of CB1 in MPTP-treated rhesus monkeys also augmented l-DOPA responses on motor disability (Cao et al. 2007).
In summary, the majority of reports have described LID mitigation after CB1 pharmacological manipulation. However, several studies have used WIN 55,212-2, a non-selective agonist of CB receptors, as a pharmacological tool to confirm the importance of CB1 in LID (Pertwee 2005), without mentioning the possible role of CB2 in the pathophysiology of this disorder. There is a lack of studies explicitly aimed at investigating the relationship between LID and CB2 receptors.
One recent work found that MPTP-evoked parkinsonism did not change CB2 expression in pallidothalamic projection neurons from macaques while l-DOPA-induced dyskinetic animals presented reduced neuronal pallidothalamic CB2 receptors. The presence of CB1–CB2 heteromers in basal ganglia output neurons has also been described. Their expression decreases in dyskinetic animals (Sierra et al. 2015). Further studies are needed to investigate the role of these receptors in LID attenuation.
LID beyond the Endocannabinoid System
Different cannabinoid-related compounds have also been studied in LID. The FAAH inhibitor URB597 increased AEA striatal concentrations but did not change LID manifestation (Morgese et al. 2007; Johnston et al. 2011). Nevertheless, URB597 significantly decreased AIMs when co-administered with TRPV-1 antagonist capsazepine (CPZ) (Morgese et al. 2007), suggesting that TRPV-1 receptors might also be essential regulators of LID display. The data concerning TRPV-1 involvement in LID remain contradictory (Morgese et al. 2007; Gonzalez-Aparicio and Moratalla 2014). In hemiparkinsonian l-DOPA-primed rodents, CPZ alone did not reduce LID (Morgese et al. 2007; Dos-Santos-Pereira et al. 2016). Nonetheless, the blockade of TRPV-1 using the endocannabinoid oleoylethanolamide prevented LID development after concomitant treatment with l-DOPA (Gonzalez-Aparicio and Moratalla 2014). The latter compound, however, also activates PPARα and the orphan receptor GPR119 (Overton et al. 2006).
Drugs that interfere with PPAR-γ receptors have also produced positive results in LID. These receptors are abundant in the basal ganglia, indicating that they participate in motor functions (Carta et al. 2011a). PPAR signaling was increased in the lesioned neostriatum of dyskinetic rats (Wang et al. 2014). The direct activation of these receptors by rosiglitazone alleviated l-DOPA-induced AIMs in 6-OHDA rats (Martinez et al. 2015). This effect was associated with a decrease of classic molecular markers for LID, such as dynorphin, zif-268, and extracellular signal-regulated kinase (ERK) phosphorylation (Martinez et al. 2015).
CBD on LID
Only one preclinical study has investigated the effects of CBD on LID (Dos-Santos-Pereira et al. 2016). In our work, CBD (10, 30, and 60 mg/kg) alone was not effective in eliciting an anti-dyskinetic effect in hemiparkinsonian mice chronically treated with l-DOPA. Nonetheless, the combined use of the TRPV-1 antagonist CPZ and the multifaceted compound CBD induced a significant anti-dyskinetic effect.
A mutual modulation of CB1 and TRPV-1 receptors is believed to occur when they are co-expressed in the same cell or in a close neuron-neuron/neuron-glia interaction (Fakhfouri et al. 2012; Citraro et al. 2013; Payandemehr et al. 2015). The stimulation of CB1 and TRPV-1 receptors produces opposing effects on excitatory and inhibitory neurotransmissions in principal neurons and interneurons of hippocampal cells, respectively (Cristino et al. 2006). Also, the concomitant activation of CB1 and TRPV-1 causes different effects on intracellular calcium concentrations (Szallasi and Di Marzo 2000).
One hypothesis to explain our findings is that TRPV-1 activation by either AEA (indirectly increased by CBD) or CBD itself facilitates LID or impairs the beneficial effects mediated by other mechanisms, such as the activation of CB1 and PPAR-γ receptors. The positive effects observed with the administration of a potent FAAH inhibitor and TRPV-1 antagonist (arachidonoyl serotonin) corroborates this hypothesis, once the specific increase of AEA levels (in conjunction with TRPV-1 receptor antagonism) reduced LID manifestation (Dos-Santos-Pereira et al. 2016). Corroborating these results, AM251 reversed the anti-dyskinetic effects of CPZ + CBD on limb and orofacial LID, whereas the PPAR-γ antagonist GW9662 inhibited only the anti-dyskinetic effect on axial AIMs. Despite reports of a reciprocal interaction between these receptors, this was the first time a complementary/selective effect of CB1 and PPAR-γ receptors has been suggested in LID. Therefore, CB1 and PPAR-γ direct (or indirect) activation (associated with TRPV-1 blockade) could be a promising mechanism to alleviate LID (Fig. 2).

Proposed mechanisms through which CPZ + CBD decrease LID. 1, one of the main pharmacological actions of CBD is to inhibit the enzyme FAAH; 2, indirectly increasing the levels of AEA; 3, CPZ antagonizes TRPV-1 receptor and thus counteracts the pro-dyskinetic effects of TRPV-1 in dyskinesia; 4, AEA can bind/activate cannabinoid (CB1) or cannabinoid-related receptors, such as PPAR-γ. CBD can also directly activate PPAR-γ. Their activation results in LID mitigation, reduction of classical LID markers (p-ERK and p-AcH3), pro-inflammatory markers (COX-2 and NF-κB), and glial activation. AEA, anandamide; CB1, cannabinoid receptor type 1; CBD, cannabidiol; CPZ, capsazepine; FAAH, fatty acid amide hydrolase; PPAR-γ, peroxisome proliferator-activated receptor gamma; TRPV-1, transient receptor potential vanilloid receptor 1
Clinical Studies with Cannabis, CBD, and Cannabinoid Compounds
No clinical study has been performed so far with the specific objective of observing CBD effects on LID mitigation in patients with PD. The few works that have examined the effects of Cannabis/cannabinoids on PD-associated motor dysfunctions (such as LID) have yielded conflicting results. Several single-case reports have associated the use of cannabinoids with positive and beneficial effects on motor symptoms derived from PD treatment. However, only four randomized placebo-controlled trials (RCTs) with 49 PD patients analyzed the effects of different cannabinoids—CBD, THC/CBD, nabilone, and rimonabant—on PD motor symptoms such as akinesia, tremor, or LID. From these, one presented a significant cannabinoid effect on parkinsonian motor symptoms or LID when used as an add-on therapy (Buhmann et al. 2019). This pilot study, performed in a mixed male/female population with different time of PD onset, showed that nabilone (a synthetic cannabinoid that mimics THC) improved up to 60% of patients with l-DOPA-induced AIMs (Sieradzan et al. 2001). In an exploratory, randomized, double-blind, placebo-controlled study, the CB1 antagonist rimonabant (SR141716, 20 mg) failed to reduce LID severity in PD patients (Mesnage et al. 2004). One possibility raised by the authors is that rimonabant failure was due to the high dose of l-DOPA, which would have masked rimonabant subtle benefic action on LID.
A survey of Colorado residents with PD using self-administered complementary therapies found that nine patients using medical Cannabis (4%) reported an improvement of mood and sleep, but only two showed improvement of motor symptoms, not specifically LID (Finseth et al. 2015). Venderova and colleagues found that the continuous use of Cannabis alleviated LID in 14% of PD patients (Venderova et al. 2004). On the other hand, Cannabis extract failed to improve parkinsonism or LID in a large double-blind, randomized placebo-controlled crossover trial (Carroll et al. 2004). As concluded in a systematic review conducted by Subcommittee on Developmental Guidelines by the American Academy of Neurology on the efficacy and safety of medical Cannabis in neurological disorders, using two controlled trials as reference, oral extract of Cannabis is probably ineffective for treating PD patients with LID (Gutierrez-Valdez et al. 2013; Catlow and Sanchez-Ramos 2015; Fernandez-Ruiz et al. 2015; Crippa et al. 2019).
LID as a Neuroimmune Disorder: How Could Cannabinoids Act?
Since parkinsonian patients are generally under l-DOPA or other anti-parkinsonian treatment, it cannot be ruled out that these drugs participate in neuroinflammatory reactions in PD (Del-Bel et al. 2016). l-DOPA administration increases extracellular dopamine and glutamate, creating a pro-inflammatory state (Farber and Kettenmann 2005), which could exacerbate any previous neuroinflammatory associated with the disorder (Gao et al. 2003; Cunningham et al. 2005). To date, the striatum remains the most studied region on neuroinflammatory processes possibly associated with LID. However, the involvement of other basal ganglia areas, such as the globus pallidus, has also been described (Bortolanza et al. 2015a).
Changes in the levels of inflammatory factors in the periphery could also be an important feature of LID pathophysiology. This chronic neuroinflammatory process leads to the production of pro-inflammatory factors, such as inducible nitric oxide synthase, cyclooxygenase-2, and cytokine IL-1β (Del-Bel et al. 2016; Carta et al. 2017). These factors can further boost the inflammatory response and even influence synaptic plasticity (Barnum et al. 2008; Bortolanza et al. 2015a, 2015b; Del-Bel et al. 2016; Dos-Santos-Pereira et al. 2016; Mulas et al. 2016; Carta et al. 2017). Anti-inflammatory drugs have beneficial effects on LID symptoms, reinforcing the proposal that neuroinflammatory changes are involved with the pathological mechanisms of the disorder (Tansey et al. 2007; Barnum et al. 2008; Teema et al. 2016).
The anti-inflammatory properties of CBD have been associated with its therapeutic effects in neurological disorders (Campos et al. 2016). The levels of the pro-inflammatory markers cyclooxygenase-2 (COX-2) and nuclear factor-κB (NF-κB) increased in dyskinetic animals after chronic treatment with l-DOPA, and the subchronic treatment with CPZ + CBD reduced the production of these markers (Dos-Santos-Pereira et al. 2016). By suppressing COX-2 overexpression, AEA and 2-AG can prevent neurotoxicity (Zhang and Chen 2008; Mounsey et al. 2015). This action appears to be mediated via CB1-dependent NF-κB signaling pathway. Furthermore, CB1-dependent PPAR-γ expression modulates the endocannabinoid-mediated decrease of the NF-κB-coupled COX-2 elevation in neuroinflammation (PERTWEE AND ROSS, 2002) (Du et al. 2011).
CBD + CPZ also decreased phospho-ERK levels (Dos-Santos-Pereira et al. 2016). This kinase has been associated with LID (Bezard et al. 2005; Pavon et al. 2006; Santini et al. 2009; Santini et al. 2010; Heiman et al. 2014; Cerovic et al. 2015) and can activate cAMP response element-binding (CREB) protein and NF-κB (Baldwin Jr 1996; Lawrence 2009; Kirschmann et al. 2014). At our knowledge, no report so far has demonstrated a straight link between ERK and inflammation using a LID experimental model. However, several papers have associated the increase of the pro-inflammatory transcription factor NF-κB (which is elevated in dyskinetic animals) to an ERK-dependent mechanism (Hiscott et al. 2001; Reber et al. 2009; Chiang et al. 2012).
The anti-inflammatory mechanism of CPZ + CBD in LID can also be receptor-dependent since CB1 and PPAR-γ receptors are important modulatory targets in neuroinflammation (Ashton 2007; O’Sullivan 2007). CB1 receptors and AEA levels are upregulated in brain cells in response to injury and inflammation (Sagredo et al. 2007; Di Filippo et al. 2008). The neuroprotector mechanism of CB1 receptor activation after a traumatic brain injury depends on the inhibition of NF-κB transactivation (Panikashvili et al. 2005), which is directly associated with the COX-2 activity (Tanabe and Tohnai 2002; Gasparini and Feldmann 2012).
As discussed above, PPAR receptors repress transcription factors such as NF-κB and suppress inflammatory pathways (Barish 2006). PPAR-γ negatively regulates the activation of glial cells by increasing anti-inflammatory-related gene expression and downregulation of pro-inflammatory mediators through their action on activated microglia and astrocytes (Villapol 2018). In pathological conditions, astrocytes and microglial cells release excessive amounts of cytokines, and this production might be a component of the neuroinflammatory response that can strongly modulate the neurotransmission, formation of long-term potentiation, and synaptic plasticity and can influence LID pathophysiology (Vesce et al. 2007; Domercq et al. 2013; Pisanu et al. 2018). Accordingly, the PPAR-γ agonists pioglitazone and rosiglitazone have anti-inflammatory properties. Pioglitazone inhibits dopaminergic nerve cell death, microglial activation, and production of pro-inflammatory mediators by blocking the translocation of the NF-κB subunit p65 to the nucleus in dopaminergic neurons, glial cells, and astrocytes (Breidert et al. 2002). Rosiglitazone modulates pro-inflammatory cytokines and inhibits the inflammatory phenotype of microglia (Pisanu et al. 2014). In summary, by activating CB1 and PPAR-γ receptors, cannabinoids could control inflammatory processes and improve LID (Barnum et al. 2008; Ossola et al. 2011; Bortolanza et al. 2015a, 2015b).
CPZ + CBD also reduced TNF-α levels in the lesioned striatum of dyskinetic mice (unpublished results from our laboratory). The increase of TNF-α in the injured striatum after treatment with l-DOPA has already been previously described (Mulas et al. 2016; Pisanu et al. 2018). In the healthy central nervous system, TNF-α works as a regulatory molecule in crucial physiological processes, such as synaptic plasticity, astrocyte-induced synaptic strengthening, and learning and memory mechanisms (Beattie et al. 2002; Baune et al. 2008; Santello et al. 2011; Kaneko and Stryker 2017). Nonetheless, this cytokine in unbalanced levels could produce deleterious effects on brain functions (Wyss-Coray and Mucke 2002; McCoy and Tansey 2008; Santello and Volterra 2012; Marin and Kipnis 2013).
One hypothesis that may explain how TNF-α influences LID is that this cytokine could increase glutamate concentrations, which in turn may further increase the release of this cytokine in a vicious circle (Sgambato-Faure and Cenci 2012; Olmos and Llado 2014). The physiological role played by TNF-α in controlling synaptic transmission and plasticity occurs through the modulation of ionotropic glutamate receptor traffic. However, excessive levels of TNF-α have an inhibitory effect on glutamate transporters, increasing the extracellular concentration of this neurotransmitter. Thus, cannabinoids such as CBD can influence the manifestation of LID not only by decreasing the production of TNF-α by glial cells but also by inhibiting the release of glutamate by neurons, as already demonstrated in other studies (Gerdeman and Lovinger 2001; Schlicker and Kathmann 2001).
Conclusion
The studies reviewed here indicate that cannabinoids could influence the development and manifestations of PD and LID. Several mechanisms, ranging from direct changes in critical neurotransmitters such as dopamine and glutamate to indirect anti-inflammatory effects, seem to be involved. Among the cannabinoids investigated so far, CBD appears one of the most promising drugs in preclinical trials. It is a “multi-targeted” compound, with an extensive range of biological effects in different neuropsychiatric disorders. The specific role of this compound in the treatment of these disorders, however, remains to be established by large and comparative clinical trials.
Abbreviations
- 2-AG:
-
2-Arachidonoylglicerol
- 6-OHDA:
-
6-Hydroxydopamine
- AEA:
-
Anandamide
- AIMs:
-
Abnormal involuntary movements
- AMT:
-
Anandamide membrane transporter
- cAMP:
-
Cyclic adenosine monophosphate
- CBD:
-
Cannabidiol
- COX-2:
-
Cyclooxygenase-2
- CPZ:
-
Capsazepine
- CREB:
-
cAMP response element-binding protein
- ERK:
-
Extracellular signal-regulated kinase
- FAAH:
-
Fatty acid amide hydrolase
- IL-1β:
-
Interleukin-1β
- LID:
-
l-DOPA-induced dyskinesia
- MAGL:
-
Monoacylglycerol lipase
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NF-κB:
-
Nuclear factor κB
- PD:
-
Parkinson’s disease
- PKA:
-
Protein kinase A
- PPAR:
-
Peroxisome proliferator-activated receptor
- SN:
-
Substantia nigra
- SNpc:
-
Substantia nigra pars compacta
- SNpr:
-
Substantia nigra pars reticulata
- THC:
-
Tetrahydrocannabinol
- THCV:
-
Tetrahydrocannabivarin
- TNF-α:
-
Tumor necrosis factor-α
- TRPV:
-
Transient receptor potential vanilloid receptor
References
Adams R, Madison H (1940) Structure of cannabidiol, a product isolated from the marihuana extract of Minnesota wild hemp. J Am Chem Soc 62:196–200
Ahmed I, Bose SK, Pavese N, Ramlackhansingh A, Turkheimer F, Hotton G, Hammers A, Brooks DJ (2011) Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain J Neurol 134:979–986
Ahrens J, Demir R, Leuwer M, de la Roche J, Krampfl K, Foadi N, Karst M, Haeseler G (2009) The nonpsychotropic cannabinoid cannabidiol modulates and directly activates alpha-1 and alpha-1-beta glycine receptor function. Pharmacology 83:217–222
Allam MF, Campbell MJ, Hofman A, Del Castillo AS, Fernandez-Crehuet Navajas R (2004) Smoking and Parkinson’s disease: systematic review of prospective studies. Mov Disord 19:614–621
Ameri A (1999) The effects of cannabinoids on the brain. Prog Neurobiol 58:315–348
Ascherio A, Chen H, Schwarzschild MA, Zhang SM, Colditz GA, Speizer FE (2003) Caffeine, postmenopausal estrogen, and risk of Parkinson’s disease. Neurology 60:790–795
Ascherio A, Weisskopf MG, O'Reilly EJ, McCullough ML, Calle EE, Rodriguez C, Thun MJ (2004) Coffee consumption, gender, and Parkinson’s disease mortality in the cancer prevention study II cohort: the modifying effects of estrogen. Am J Epidemiol 160:977–984
Ashton JC (2007) Cannabinoids for the treatment of inflammation. Curr Opin Investig Drugs 8:373–384
Atakan Z (2012) Cannabis, a complex plant: different compounds and different effects on individuals. Ther Adv Psychopharmacol 2:241–254
Aymerich MS, Aso E, Abellanas MA, Tolon RM, Ramos JA, Ferrer I, Romero J, Fernandez-Ruiz J (2018) Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem Pharmacol
Baldwin AS Jr (1996) The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 14:649–683
Barish GD (2006) Peroxisome proliferator-activated receptors and liver X receptors in atherosclerosis and immunity. J Nutr 136:690–694
Barnum CJ, Eskow KL, Dupre K, Blandino P Jr, Deak T, Bishop C (2008) Exogenous corticosterone reduces L-DOPA-induced dyskinesia in the hemi-parkinsonian rat: role for interleukin-1beta. Neuroscience 156:30–41
Baune BT, Wiede F, Braun A, Golledge J, Arolt V, Koerner H (2008) Cognitive dysfunction in mice deficient for TNF- and its receptors. Am J Med Genet B Neuropsychiatr Genet 147B:1056–1064
Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC (2002) Control of synaptic strength by glial TNFalpha. Science 295:2282–2285
Benarroch E (2007) Endocannabinoids in basal ganglia circuits: implications for Parkinson disease. Neurology 69:306–309
Bezard E, Gross CE, Qin L, Gurevich VV, Benovic JL, Gurevich EV (2005) L-DOPA reverses the MPTP-induced elevation of the arrestin2 and GRK6 expression and enhanced ERK activation in monkey brain. Neurobiol Dis 18:323–335
Bisogno T, Hanus L, De Petrocellis L, Tchilibon S, Ponde DE, Brandi I, Moriello AS, Davis JB, Mechoulam R, Di Marzo V (2001) Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol 134:845–852
Blandini F (2003) Adenosine receptors and L-DOPA-induced dyskinesia in Parkinson’s disease: potential targets for a new therapeutic approach. Exp Neurol 184:556–560
Blanpied TA, Clarke RJ, Johnson JW (2005) Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J Neurosci 25:3312–3322
Bloom AS, Hillard CJ (1985) Cannabinoids, neurotransmitter receptors and brain membranes. In: Harvey DJ (ed) Marihuana ‘84. IRL Press, Oxford, pp 217–231
Bornheim LM, Everhart ET, Li J, Correia MA (1993) Characterization of cannabidiol-mediated cytochrome P450 inactivation. Biochem Pharmacol 45:1323–1331
Bornheim LM, Kim KY, Li J, Perotti BY, Benet LZ (1995) Effect of cannabidiol pretreatment on the kinetics of tetrahydrocannabinol metabolites in mouse brain. Drug Metab Dispos 23:825–831
Bortolanza M, Cavalcanti-Kiwiatkoski R, Padovan-Neto FE, da-Silva CA, Mitkovski M, Raisman-Vozari R, Del-Bel E (2015a) Glial activation is associated with l-DOPA induced dyskinesia and blocked by a nitric oxide synthase inhibitor in a rat model of Parkinson’s disease. Neurobiol Dis 73:377–387
Bortolanza M, Padovan-Neto FE, Cavalcanti-Kiwiatkoski R, Dos Santos-Pereira M, Mitkovski M, Raisman-Vozari R, Del-Bel E (2015b) Are cyclooxygenase-2 and nitric oxide involved in the dyskinesia of Parkinson’s disease induced by L-DOPA? Philos Trans R Soc Lond Ser B Biol Sci 370
Bortolanza M, Nascimento GC, Socias SB, Ploper D, Chehin RN, Raisman-Vozari R, Del-Bel E (2018) Tetracycline repurposing in neurodegeneration: focus on Parkinson’s disease. J Neural Transm (Vienna) 125:1403–1415
Breidert T, Callebert J, Heneka MT, Landreth G, Launay JM, Hirsch EC (2002) Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson’s disease. J Neurochem 82:615–624
Brotchie JM (2003) CB1 cannabinoid receptor signalling in Parkinson’s disease. Curr Opin Pharmacol 3:54–61
Buhmann C, Mainka T, Ebersbach G, Gandor F (2019) Evidence for the use of cannabinoids in Parkinson’s disease. J Neural Transm
Cadas H, di Tomaso E, Piomelli D (1997) Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. J Neurosci 17:1226–1242
Calabresi P, Ghiglieri V, Mazzocchetti P, Corbelli I, Picconi B (2015) Levodopa-induced plasticity: a double-edged sword in Parkinson’s disease? Philos Trans R Soc Lond Ser B Biol Sci 370
Campos AC, Fogaca MV, Sonego AB, Guimaraes FS (2016) Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharmacol Res 112:119–127
Cao X, Liang L, Hadcock JR, Iredale PA, Griffith DA, Menniti FS, Factor S, Greenamyre JT, Papa SM (2007) Blockade of cannabinoid type 1 receptors augments the antiparkinsonian action of levodopa without affecting dyskinesias in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated rhesus monkeys. J Pharmacol Exp Ther 323:318–326
Carrier EJ, Auchampach JA, Hillard CJ (2006) Inhibition of an equilibrative nucleoside transporter by cannabidiol: a mechanism of cannabinoid immunosuppression. Proc Natl Acad Sci U S A 103:7895–7900
Carroll CB, Bain PG, Teare L, Liu X, Joint C, Wroath C, Parkin SG, Fox P, Wright D, Hobart J, Zajicek JP (2004) Cannabis for dyskinesia in Parkinson disease: a randomized double-blind crossover study. Neurology 63:1245–1250
Carroll CB, Zeissler ML, Hanemann CO, Zajicek JP (2012) Delta(9)-tetrahydrocannabinol (delta(9)-THC) exerts a direct neuroprotective effect in a human cell culture model of Parkinson’s disease. Neuropathol Appl Neurobiol 38:535–547
Carta M, Tronci E (2014) Serotonin system implication in l-DOPA-induced dyskinesia: from animal models to clinical investigations. Front Neurol 5:78
Carta AR, Frau L, Pisanu A, Wardas J, Spiga S, Carboni E (2011a) Rosiglitazone decreases peroxisome proliferator receptor-gamma levels in microglia and inhibits TNF-alpha production: new evidences on neuroprotection in a progressive Parkinson’s disease model. Neuroscience 194:250–261
Carta AR, Pisanu A, Carboni E (2011b) Do PPAR-gamma agonists have a future in Parkinson’s disease therapy? Parkinsons Dis 2011:689181
Carta AR, Mulas G, Bortolanza M, Duarte T, Pillai E, Fisone G, Vozari RR, Del-Bel E (2017) L-DOPA-induced dyskinesia and neuroinflammation: do microglia and astrocytes play a role? Eur J Neurosci 45:73–91
Castillo A, Tolon MR, Fernandez-Ruiz J, Romero J, Martinez-Orgado J (2010) The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol Dis 37:434–440
Catlow B, Sanchez-Ramos J (2015) Cannabinoids for the treatment of movement disorders. Curr Treat Options Neurol 17:370
Celorrio M, Fernandez-Suarez D, Rojo-Bustamante E, Echeverry-Alzate V, Ramirez MJ, Hillard CJ, Lopez-Moreno JA, Maldonado R, Oyarzabal J, Franco R, Aymerich MS (2016) Fatty acid amide hydrolase inhibition for the symptomatic relief of Parkinson’s disease. Brain Behav Immun 57:94–105
Cenci MA, Konradi C (2010) Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog Brain Res 183:209–233
Cenci MA, Ohlin KE, Rylander D (2009) Plastic effects of L-DOPA treatment in the basal ganglia and their relevance to the development of dyskinesia. Parkinsonism Relat Disord 15(Suppl 3):S59–S63
Cerovic M, Bagetta V, Pendolino V, Ghiglieri V, Fasano S, Morella I, Hardingham N, Heuer A, Papale A, Marchisella F, Giampa C, Calabresi P, Picconi B, Brambilla R (2015) Derangement of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and extracellular signal-regulated kinase (ERK) dependent striatal plasticity in L-DOPA-induced dyskinesia. Biol Psychiatry 77:106–115
Chagas MH, Eckeli AL, Zuardi AW, Pena-Pereira MA, Sobreira-Neto MA, Sobreira ET, Camilo MR, Bergamaschi MM, Schenck CH, Hallak JE, Tumas V, Crippa JA (2014a) Cannabidiol can improve complex sleep-related behaviours associated with rapid eye movement sleep behaviour disorder in Parkinson’s disease patients: a case series. J Clin Pharm Ther 39:564–566
Chagas MH, Zuardi AW, Tumas V, Pena-Pereira MA, Sobreira ET, Bergamaschi MM, dos Santos AC, Teixeira AL, Hallak JE, Crippa JA (2014b) Effects of cannabidiol in the treatment of patients with Parkinson’s disease: an exploratory double-blind trial. J Psychopharmacol 28:1088–1098
Chiang IT, Liu YC, Wang WH, Hsu FT, Chen HW, Lin WJ, Chang WY, Hwang JJ (2012) Sorafenib inhibits TPA-induced MMP-9 and VEGF expression via suppression of ERK/NF-kappaB pathway in hepatocellular carcinoma cells. In vivo 26:671–681
Chung YC, Bok E, Huh SH, Park JY, Yoon SH, Kim SR, Kim YS, Maeng S, Park SH, Jin BK (2011) Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J Immunol 187:6508–6517
Citraro R, Russo E, Scicchitano F, van Rijn CM, Cosco D, Avagliano C, Russo R, D’Agostino G, Petrosino S, Guida F, Gatta L, van Luijtelaar G, Maione S, Di Marzo V, Calignano A, De Sarro G (2013) Antiepileptic action of N-palmitoylethanolamine through CB1 and PPAR-alpha receptor activation in a genetic model of absence epilepsy. Neuropharmacology 69:115–126
Colosimo C, Craus A (2003) Noradrenergic drugs for levodopa-induced dyskinesia. Clin Neuropharmacol 26:299–305
Consroe P, Sandyk R, Snider SR (1986) Open label evaluation of cannabidiol in dystonic movement disorders. Int J Neurosci 30:277–282
Cools R, Lewis SJ, Clark L, Barker RA, Robbins TW (2007) L-DOPA disrupts activity in the nucleus accumbens during reversal learning in Parkinson’s disease. Neuropsychopharmacology 32:180–189
Costa B, Colleoni M, Conti S, Parolaro D, Franke C, Trovato AE, Giagnoni G (2004) Oral anti-inflammatory activity of cannabidiol, a non-psychoactive constituent of cannabis, in acute carrageenan-induced inflammation in the rat paw. Naunyn Schmiedeberg's Arch Pharmacol 369:294–299
Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83–87
Crippa JAS, Hallak JEC, Zuardi AW, Guimaraes FS, Tumas V, Dos Santos RG (2019) Is cannabidiol the ideal drug to treat non-motor Parkinson’s disease symptoms? Eur Arch Psychiatry Clin Neurosci 269:121–133
Cristino L, de Petrocellis L, Pryce G, Baker D, Guglielmotti V, Di Marzo V (2006) Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 139:1405–1415
Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH (2005) Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 25:9275–9284
de Lago E, de Miguel R, Lastres-Becker I, Ramos JA, Fernandez-Ruiz J (2004) Involvement of vanilloid-like receptors in the effects of anandamide on motor behavior and nigrostriatal dopaminergic activity: in vivo and in vitro evidence. Brain Res 1007:152–159
De Petrocellis L, Ligresti A, Moriello AS, Allara M, Bisogno T, Petrosino S, Stott CG, Di Marzo V (2011) Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol 163:1479–1494
Del-Bel E, Padovan-Neto FE, Bortolanza M, Tumas V, Aguiar AS Jr, Raisman-Vozari R, Prediger RD (2015) Nitric oxide, a new player in L-DOPA-induced dyskinesia. Front Biosci 7:168–192
Del-Bel E, Bortolanza M, Dos-Santos-Pereira M, Bariotto K, Raisman-Vozari R (2016) L-DOPA-induced dyskinesia in Parkinson’s disease: are neuroinflammation and astrocytes key elements? Synapse 70:479–500
Desarnaud F, Cadas H, Piomelli D (1995) Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J Biol Chem 270:6030–6035
Devane WA, Dysarz FA 3rd, Johnson MR, Melvin LS, Howlett AC (1988) Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol 34:605–613
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949
Di Filippo M, Pini LA, Pelliccioli GP, Calabresi P, Sarchielli P (2008) Abnormalities in the cerebrospinal fluid levels of endocannabinoids in multiple sclerosis. J Neurol Neurosurg Psychiatry 79:1224–1229
Di Marzo V, De Petrocellis L (2012) Why do cannabinoid receptors have more than one endogenous ligand? Philos Trans R Soc Lond Ser B Biol Sci 367:3216–3228
Di Marzo V, Bisogno T, De Petrocellis L (2001) Anandamide: some like it hot. Trends Pharmacol Sci 22:346–349
Domercq M, Vazquez-Villoldo N, Matute C (2013) Neurotransmitter signaling in the pathophysiology of microglia. Front Cell Neurosci 7:49
Dos-Santos-Pereira M, da-Silva CA, Guimaraes FS, Del-Bel E (2016) Co-administration of cannabidiol and capsazepine reduces L-DOPA-induced dyskinesia in mice: possible mechanism of action. Neurobiol Dis 94:179–195
Du H, Chen X, Zhang J, Chen C (2011) Inhibition of COX-2 expression by endocannabinoid 2-arachidonoylglycerol is mediated via PPAR-gamma. Br J Pharmacol 163:1533–1549
Elbaz A, Tranchant C (2007) Epidemiologic studies of environmental exposures in Parkinson’s disease. J Neurol Sci 262:37–44
Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S, Hoertnagl H, Raine CS, Schneider-Stock R, Nitsch R, Ullrich O (2006) The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 49:67–79
Elsohly MA, Slade D (2005) Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sci 78:539–548
Esposito G, De Filippis D, Maiuri MC, De Stefano D, Carnuccio R, Iuvone T (2006) Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci Lett 399:91–95
Fakhfouri G, Ahmadiani A, Rahimian R, Grolla AA, Moradi F, Haeri A (2012) WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-gamma pathway. Neuropharmacology 63:653–666
Farber K, Kettenmann H (2005) Physiology of microglial cells. Brain Res Brain Res Rev 48:133–143
Feigin A, Ghilardi MF, Carbon M, Edwards C, Fukuda M, Dhawan V, Margouleff C, Ghez C, Eidelberg D (2003) Effects of levodopa on motor sequence learning in Parkinson’s disease. Neurology 60:1744–1749
Fernandez-Espejo E, Caraballo I, de Fonseca FR, El Banoua F, Ferrer B, Flores JA, Galan-Rodriguez B (2005) Cannabinoid CB1 antagonists possess antiparkinsonian efficacy only in rats with very severe nigral lesion in experimental parkinsonism. Neurobiol Dis 18:591–601
zcccxcx
Fernandez-Ruiz J, Sagredo O, Pazos MR, Garcia C, Pertwee R, Mechoulam R, Martinez-Orgado J (2013) Cannabidiol for neurodegenerative disorders: important new clinical applications for this phytocannabinoid? Br J Clin Pharmacol 75:323–333
Fernandez-Ruiz J, Romero J, Ramos JA (2015) Endocannabinoids and neurodegenerative disorders: Parkinson’s disease, Huntington’s chorea, Alzheimer’s disease, and others. Handb Exp Pharmacol 231:233–259
Fernandez-Suarez D, Celorrio M, Riezu-Boj JI, Ugarte A, Pacheco R, Gonzalez H, Oyarzabal J, Hillard CJ, Franco R, Aymerich MS (2014) Monoacylglycerol lipase inhibitor JZL184 is neuroprotective and alters glial cell phenotype in the chronic MPTP mouse model. Neurobiol Aging 35:2603–2616
Finseth TA, Hedeman JL, Brown RP 2nd, Johnson KI, Binder MS, Kluger BM (2015) Self-reported efficacy of cannabis and other complementary medicine modalities by Parkinson’s disease patients in Colorado. Evid Based Complement Alternat Med 2015:874849
Fischedick JT, Hazekamp A, Erkelens T, Choi YH, Verpoorte R (2010) Metabolic fingerprinting of Cannabis sativa L., cannabinoids and terpenoids for chemotaxonomic and drug standardization purposes. Phytochemistry 71:2058–2073
Fox SH, Henry B, Hill M, Crossman A, Brotchie J (2002) Stimulation of cannabinoid receptors reduces levodopa-induced dyskinesia in the MPTP-lesioned nonhuman primate model of Parkinson’s disease. Mov Disord 17:1180–1187
Fusco FR, Martorana A, Giampa C, De March Z, Farini D, D'Angelo V, Sancesario G, Bernardi G (2004) Immunolocalization of CB1 receptor in rat striatal neurons: a confocal microscopy study. Synapse 53:159–167
Galve-Roperh I, Chiurchiu V, Diaz-Alonso J, Bari M, Guzman M, Maccarrone M (2013) Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog Lipid Res 52:633–650
Gao HM, Liu B, Zhang W, Hong JS (2003) Novel anti-inflammatory therapy for Parkinson’s disease. Trends Pharmacol Sci 24:395–401
Garcia C, Palomo-Garo C, Garcia-Arencibia M, Ramos J, Pertwee R, Fernandez-Ruiz J (2011) Symptom-relieving and neuroprotective effects of the phytocannabinoid Delta(9)-THCV in animal models of Parkinson’s disease. Br J Pharmacol 163:1495–1506
Garcia-Arencibia M, Gonzalez S, de Lago E, Ramos JA, Mechoulam R, Fernandez-Ruiz J (2007) Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson’s disease: importance of antioxidant and cannabinoid receptor-independent properties. Brain Res 1134:162–170
Gasparini C, Feldmann M (2012) NF-kappaB as a target for modulating inflammatory responses. Curr Pharm Des 18:5735–5745
Gerdeman G, Lovinger DM (2001) CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol 85:468–471
Gerdeman GL, Ronesi J, Lovinger DM (2002) Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5:446–451
Gilbert GL, Kim HJ, Waataja JJ, Thayer SA (2007) Delta9-tetrahydrocannabinol protects hippocampal neurons from excitotoxicity. Brain Res 1128:61–69
Gilgun-Sherki Y, Melamed E, Mechoulam R, Offen D (2003) The CB1 cannabinoid receptor agonist, HU-210, reduces levodopa-induced rotations in 6-hydroxydopamine-lesioned rats. Pharmacol Toxicol 93:66–70
Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D (1999) Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci 2:358–363
Glass M, Dragunow M, Faull RL (2000) The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington's disease. Neuroscience 97:505–519
Gomez-Galvez Y, Palomo-Garo C, Fernandez-Ruiz J, Garcia C (2016) Potential of the cannabinoid CB(2) receptor as a pharmacological target against inflammation in Parkinson’s disease. Prog Neuro-Psychopharmacol Biol Psychiatry 64:200–208
Gonzalez S, Scorticati C, Garcia-Arencibia M, de Miguel R, Ramos JA, Fernandez-Ruiz J (2006) Effects of rimonabant, a selective cannabinoid CB1 receptor antagonist, in a rat model of Parkinson’s disease. Brain Res 1073-1074:209–219
Gonzalez-Aparicio R, Moratalla R (2014) Oleoylethanolamide reduces L-DOPA-induced dyskinesia via TRPV1 receptor in a mouse model of Parkinson’s disease. Neurobiol Dis 62:416–425
Gubellini P, Picconi B, Bari M, Battista N, Calabresi P, Centonze D, Bernardi G, Finazzi-Agro A, Maccarrone M (2002) Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci 22:6900–6907
Gutierrez-Valdez AL, Garcia-Ruiz R, Anaya-Martinez V, Torres-Esquivel C, Espinosa-Villanueva J, Reynoso-Erazo L, Tron-Alvarez R, Aley-Medina P, Sanchez-Betancourt J, Montiel-Flores E, Avila-Costa MR (2013) The combination of oral L-DOPA/rimonabant for effective dyskinesia treatment and cytological preservation in a rat model of Parkinson's disease and L-DOPA-induced dyskinesia. Behav Pharmacol 24:640–652
Hampson AJ, Grimaldi M, Axelrod J, Wink D (1998) Cannabidiol and (-)delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci U S A 95:8268–8273
Heiman M, Heilbut A, Francardo V, Kulicke R, Fenster RJ, Kolaczyk ED, Mesirov JP, Surmeier DJ, Cenci MA, Greengard P (2014) Molecular adaptations of striatal spiny projection neurons during levodopa-induced dyskinesia. Proc Natl Acad Sci U S A 111:4578–4583
Herkenham M, Lynn AB, de Costa BR, Richfield EK (1991a) Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res 547:267–274
Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC (1991b) Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci 11:563–583
Hermann H, Marsicano G, Lutz B (2002) Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience 109:451–460
Herrero MT, Estrada C, Maatouk L, Vyas S (2015) Inflammation in Parkinson’s disease: role of glucocorticoids. Front Neuroanat 9:32
Hiscott J, Kwon H, Genin P (2001) Hostile takeovers: viral appropriation of the NF-kappaB pathway. J Clin Invest 107:143–151
Hohmann AG, Herkenham M (2000) Localization of cannabinoid CB(1) receptor mRNA in neuronal subpopulations of rat striatum: a double-label in situ hybridization study. Synapse 37:71–80
Howlett AC, Fleming RM (1984) Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol Pharmacol 26:532–538
Howlett AC, Blume LC, Dalton GD (2010) CB(1) cannabinoid receptors and their associated proteins. Curr Med Chem 17:1382–1393
Huot P, Johnston TH, Koprich JB, Fox SH, Brotchie JM (2013) The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol Rev 65:171–222
Iuvone T, Esposito G, Esposito R, Santamaria R, Di Rosa M, Izzo AA (2004) Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J Neurochem 89:134–141
Janefjord E, Maag JL, Harvey BS, Smid SD (2014) Cannabinoid effects on beta amyloid fibril and aggregate formation, neuronal and microglial-activated neurotoxicity in vitro. Cell Mol Neurobiol 34:31–42
Jenner P (2008) Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci 9:665–677
Johnston TH, Huot P, Fox SH, Wakefield JD, Sykes KA, Bartolini WP, Milne GT, Pearson JP, Brotchie JM (2011) Fatty acid amide hydrolase (FAAH) inhibition reduces L-3,4-dihydroxyphenylalanine-induced hyperactivity in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned non-human primate model of Parkinson’s disease. J Pharmacol Exp Ther 336:423–430
Jones NA, Hill AJ, Smith I, Bevan SA, Williams CM, Whalley BJ, Stephens GJ (2010) Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. J Pharmacol Exp Ther 332:569–577
Kaneko M, Stryker MP (2017) Homeostatic plasticity mechanisms in mouse V1. Philos Trans R Soc Lond Ser B Biol Sci 372
Khan SS, Lee FJ (2014) Delineation of domains within the cannabinoid CB1 and dopamine D2 receptors that mediate the formation of the heterodimer complex. J Mol Neurosci 53:10–21
Kirschmann EK, Mauna JC, Willis CM, Foster RL, Chipman AM, Thiels E (2014) Appetitive cue-evoked ERK signaling in the nucleus accumbens requires NMDA and D1 dopamine receptor activation and regulates CREB phosphorylation. Learn Mem 21:606–615
Kozela E, Pietr M, Juknat A, Rimmerman N, Levy R, Vogel Z (2010) Cannabinoids delta(9)-tetrahydrocannabinol and cannabidiol differentially inhibit the lipopolysaccharide-activated NF-kappaB and interferon-beta/STAT proinflammatory pathways in BV-2 microglial cells. J Biol Chem 285:1616–1626
Kunert-Keil C, Bisping F, Kruger J, Brinkmeier H (2006) Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genomics 7:159
Laprairie RB, Bagher AM, Kelly ME, Denovan-Wright EM (2015) Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol 172:4790–4805
Lastres-Becker I, Cebeira M, de Ceballos ML, Zeng BY, Jenner P, Ramos JA, Fernandez-Ruiz JJ (2001) Increased cannabinoid CB1 receptor binding and activation of GTP-binding proteins in the basal ganglia of patients with Parkinson’s syndrome and of MPTP-treated marmosets. Eur J Neurosci 14:1827–1832
Lastres-Becker I, de Miguel R, De Petrocellis L, Makriyannis A, Di Marzo V, Fernandez-Ruiz J (2003) Compounds acting at the endocannabinoid and/or endovanilloid systems reduce hyperkinesia in a rat model of Huntington’s disease. J Neurochem 84:1097–1109
Lastres-Becker I, Molina-Holgado F, Ramos JA, Mechoulam R, Fernandez-Ruiz J (2005) Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: relevance to Parkinson’s disease. Neurobiol Dis 19:96–107
Lawrence T (2009) The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1:a001651
Lees AJ, Hardy J, Revesz T (2009) Parkinson’s disease. Lancet 373:2055–2066
Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C, Klosterkotter J, Hellmich M, Koethe D (2012) Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry 2:e94
Li K, Feng JY, Li YY, Yuece B, Lin XH, Yu LY, Li YN, Feng YJ, Storr M (2013) Anti-inflammatory role of cannabidiol and O-1602 in cerulein-induced acute pancreatitis in mice. Pancreas 42:123–129
Liu J, Li H, Burstein SH, Zurier RB, Chen JD (2003) Activation and binding of peroxisome proliferator-activated receptor gamma by synthetic cannabinoid ajulemic acid. Mol Pharmacol 63:983–992
Lopez-Sendon Moreno JL, Garcia Caldentey J, Trigo Cubillo P, Ruiz Romero C, Garcia Ribas G, Alonso Arias MA, Garcia de Yebenes MJ, Tolon RM, Galve-Roperh I, Sagredo O, Valdeolivas S, Resel E, Ortega-Gutierrez S, Garcia-Bermejo ML, Fernandez Ruiz J, Guzman M, Garcia de Yebenes Prous J (2016) A double-blind, randomized, cross-over, placebo-controlled, pilot trial with Sativex in Huntington’s disease. J Neurol 263:1390–1400
Lotan I, Treves TA, Roditi Y, Djaldetti R (2014) Cannabis (medical marijuana) treatment for motor and non-motor symptoms of Parkinson disease: an open-label observational study. Clin Neuropharmacol 37:41–44
Lovinger DM (2010) Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology 58:951–961
Luginger E, Wenning GK, Bosch S, Poewe W (2000) Beneficial effects of amantadine on L-dopa-induced dyskinesias in Parkinson’s disease. Mov Disord 15:873–878
Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA (2002) Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson’s disease. Eur J Neurosci 15:120–132
Lynn AB, Herkenham M (1994) Localization of cannabinoid receptors and nonsaturable high-density cannabinoid binding sites in peripheral tissues of the rat: implications for receptor-mediated immune modulation by cannabinoids. J Pharmacol Exp Ther 268:1612–1623
Maccarrone M, Gubellini P, Bari M, Picconi B, Battista N, Centonze D, Bernardi G, Finazzi-Agro A, Calabresi P (2003) Levodopa treatment reverses endocannabinoid system abnormalities in experimental parkinsonism. J Neurochem 85:1018–1025
Macchio GJ, Ito V, Sahgal V (1993) Amantadine-induced coma. Arch Phys Med Rehabil 74:1119–1120
Machado MMF, Bassani TB, Coppola-Segovia V, Moura ELR, Zanata SM, Andreatini R, Vital M (2019) PPAR-gamma agonist pioglitazone reduces microglial proliferation and NF-kappaB activation in the substantia nigra in the 6-hydroxydopamine model of Parkinson's disease. Pharmacol Rep 71:556–564
Mackie K (2006) Mechanisms of CB1 receptor signaling: endocannabinoid modulation of synaptic strength. Int J Obes 30(Suppl 1):S19–S23
Malfait AM, Gallily R, Sumariwalla PF, Malik AS, Andreakos E, Mechoulam R, Feldmann M (2000) The nonpsychoactive cannabis constituent cannabidiol is an oral anti-arthritic therapeutic in murine collagen-induced arthritis. Proc Natl Acad Sci U S A 97:9561–9566
Marcellino D, Carriba P, Filip M, Borgkvist A, Frankowska M, Bellido I, Tanganelli S, Muller CE, Fisone G, Lluis C, Agnati LF, Franco R, Fuxe K (2008) Antagonistic cannabinoid CB1/dopamine D2 receptor interactions in striatal CB1/D2 heteromers. A combined neurochemical and behavioral analysis. Neuropharmacology 54:815–823
Marin I, Kipnis J (2013) Learning and memory ... and the immune system. Learn Mem 20:601–606
Marsicano G, Lutz B (1999) Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci 11:4213–4225
Martinez A, Macheda T, Morgese MG, Trabace L, Giuffrida A (2012) The cannabinoid agonist WIN55212-2 decreases L-DOPA-induced PKA activation and dyskinetic behavior in 6-OHDA-treated rats. Neurosci Res 72:236–242
Martinez AA, Morgese MG, Pisanu A, Macheda T, Paquette MA, Seillier A, Cassano T, Carta AR, Giuffrida A (2015) Activation of PPAR gamma receptors reduces levodopa-induced dyskinesias in 6-OHDA-lesioned rats. Neurobiol Dis 74:295–304
Martin-Moreno AM, Reigada D, Ramirez BG, Mechoulam R, Innamorato N, Cuadrado A, de Ceballos ML (2011) Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: relevance to Alzheimer’s disease. Mol Pharmacol 79:964–973
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564
McCoy MK, Tansey MG (2008) TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation 5:45
McGeer PL, McGeer EG (2004) Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Relat Disord 10(Suppl 1):S3–S7
McHugh D, Page J, Dunn E, Bradshaw HB (2012) Delta(9) -tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells. Br J Pharmacol 165:2414–2424
McPartland JM, Duncan M, Di Marzo V, Pertwee RG (2015) Are cannabidiol and delta(9) -tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol 172:737–753
Mechoulam R, Hanus L (2000) A historical overview of chemical research on cannabinoids. Chem Phys Lipids 108:1–13
Mechoulam R, Shvo Y (1963) Hashish. I. The structure of cannabidiol. Tetrahedron 19:2073–2078
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR et al (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50:83–90
Mesnage V, Houeto JL, Bonnet AM, Clavier I, Arnulf I, Cattelin F, Le Fur G, Damier P, Welter ML, Agid Y (2004) Neurokinin B, neurotensin, and cannabinoid receptor antagonists and Parkinson disease. Clin Neuropharmacol 27:108–110
Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN (1999) Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 56:1383–1386
Mezey E, Toth ZE, Cortright DN, Arzubi MK, Krause JE, Elde R, Guo A, Blumberg PM, Szallasi A (2000) Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci U S A 97:3655–3660
Michel PP, Hirsch EC, Hunot S (2016) Understanding dopaminergic cell death pathways in Parkinson disease. Neuron 90:675–691
Moldrich G, Wenger T (2000) Localization of the CB1 cannabinoid receptor in the rat brain. An immunohistochemical study. Peptides 21:1735–1742
Moldzio R, Pacher T, Krewenka C, Kranner B, Novak J, Duvigneau JC, Rausch WD (2012) Effects of cannabinoids delta(9)-tetrahydrocannabinol, delta(9)-tetrahydrocannabinolic acid and cannabidiol in MPP+ affected murine mesencephalic cultures. Phytomedicine 19:819–824
Morgese MG, Cassano T, Cuomo V, Giuffrida A (2007) Anti-dyskinetic effects of cannabinoids in a rat model of Parkinson’s disease: role of CB(1) and TRPV1 receptors. Exp Neurol 208:110–119
Morin N, Di Paolo T (2014) Pharmacological treatments inhibiting levodopa-induced dyskinesias in MPTP-lesioned monkeys: brain glutamate biochemical correlates. Front Neurol 5:144
Mounsey RB, Mustafa S, Robinson L, Ross RA, Riedel G, Pertwee RG, Teismann P (2015) Increasing levels of the endocannabinoid 2-AG is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Exp Neurol 273:36–44
Mulas G, Espa E, Fenu S, Spiga S, Cossu G, Pillai E, Carboni E, Simbula G, Jadzic D, Angius F, Spolitu S, Batetta B, Lecca D, Giuffrida A, Carta AR (2016) Differential induction of dyskinesia and neuroinflammation by pulsatile versus continuous l-DOPA delivery in the 6-OHDA model of Parkinson’s disease. Exp Neurol 286:83–92
Munro S, Thomas KL, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65
Murillo-Rodriguez E, Palomero-Rivero M, Millan-Aldaco D, Mechoulam R, Drucker-Colin R (2011) Effects on sleep and dopamine levels of microdialysis perfusion of cannabidiol into the lateral hypothalamus of rats. Life Sci 88:504–511
Musella A, De Chiara V, Rossi S, Prosperetti C, Bernardi G, Maccarrone M, Centonze D (2009) TRPV1 channels facilitate glutamate transmission in the striatum. Mol Cell Neurosci 40:89–97
Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, International Parkinson’s Disease Genomics C, Parkinson’s Study Group Parkinson’s Research: The Organized GI, andMe, GenePd, NeuroGenetics Research C, Hussman Institute of Human G, Ashkenazi Jewish Dataset I, Cohorts for H, Aging Research in Genetic E, North American Brain Expression C, United Kingdom Brain Expression C, Greek Parkinson’s Disease C, Alzheimer Genetic Analysis G, Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46:989–993
Nam JH, Park ES, Won SY, Lee YA, Kim KI, Jeong JY, Baek JY, Cho EJ, Jin M, Chung YC, Lee BD, Kim SH, Kim EG, Byun K, Lee B, Woo DH, Lee CJ, Kim SR, Bok E, Kim YS, Ahn TB, Ko HW, Brahmachari S, Pletinkova O, Troconso JC, Dawson VL, Dawson TM, Jin BK (2015) TRPV1 on astrocytes rescues nigral dopamine neurons in Parkinson’s disease via CNTF. Brain J Neurol 138:3610–3622
Niesink RJ, van Laar MW (2013) Does cannabidiol protect against adverse psychological effects of THC? Front Psychiatry 4:130
Nutt JG (1990) Levodopa-induced dyskinesia: review, observations, and speculations. Neurology 40:340–345
O’Sullivan SE (2007) Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol 152:576–582
O’Sullivan SE, Sun Y, Bennett AJ, Randall MD, Kendall DA (2009) Time-dependent vascular actions of cannabidiol in the rat aorta. Eur J Pharmacol 612:61–68
Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, Burn D, Halliday GM, Bezard E, Przedborski S, Lehericy S, Brooks DJ, Rothwell JC, Hallett M, DeLong MR, Marras C, Tanner CM, Ross GW, Langston JW, Klein C, Bonifati V, Jankovic J, Lozano AM, Deuschl G, Bergman H, Tolosa E, Rodriguez-Violante M, Fahn S, Postuma RB, Berg D, Marek K, Standaert DG, Surmeier DJ, Olanow CW, Kordower JH, Calabresi P, Schapira AHV, Stoessl AJ (2017) Past, present, and future of Parkinson’s disease: a special essay on the 200th anniversary of the shaking palsy. Mov Disord 32:1264–1310
Ojha S, Javed H, Azimullah S, Haque ME (2016) Beta-caryophyllene, a phytocannabinoid attenuates oxidative stress, neuroinflammation, glial activation, and salvages dopaminergic neurons in a rat model of Parkinson disease. Mol Cell Biochem 418:59–70
Olmos G, Llado J (2014) Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediat Inflamm 2014:861231
Onaivi ES, Ishiguro H, Gong JP, Patel S, Perchuk A, Meozzi PA, Myers L, Mora Z, Tagliaferro P, Gardner E, Brusco A, Akinshola BE, Liu QR, Hope B, Iwasaki S, Arinami T, Teasenfitz L, Uhl GR (2006) Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann N Y Acad Sci 1074:514–536
Ossola B, Schendzielorz N, Chen SH, Bird GS, Tuominen RK, Mannisto PT, Hong JS (2011) Amantadine protects dopamine neurons by a dual action: reducing activation of microglia and inducing expression of GDNF in astroglia [corrected]. Neuropharmacology 61:574–582
Overton HA, Babbs AJ, Doel SM, Fyfe MC, Gardner LS, Griffin G, Jackson HC, Procter MJ, Rasamison CM, Tang-Christensen M, Widdowson PS, Williams GM, Reynet C (2006) Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab 3:167–175
Pandolfo P, Silveirinha V, dos Santos-Rodrigues A, Venance L, Ledent C, Takahashi RN, Cunha RA, Kofalvi A (2011) Cannabinoids inhibit the synaptic uptake of adenosine and dopamine in the rat and mouse striatum. Eur J Pharmacol 655:38–45
Panikashvili D, Mechoulam R, Beni SM, Alexandrovich A, Shohami E (2005) CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J Cereb Blood Flow Metab 25:477–484
Papa SM, Chase TN (1996) Levodopa-induced dyskinesias improved by a glutamate antagonist in Parkinsonian monkeys. Ann Neurol 39:574–578
Parkinson J (2002) An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci 14:223–236 discussion 222
Pavon N, Martin AB, Mendialdua A, Moratalla R (2006) ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol Psychiatry 59:64–74
Payandemehr B, Ebrahimi A, Gholizadeh R, Rahimian R, Varastehmoradi B, Gooshe M, Aghaei HN, Mousavizadeh K, Dehpour AR (2015) Involvement of PPAR receptors in the anticonvulsant effects of a cannabinoid agonist, WIN 55,212-2. Prog Neuro-Psychopharmacol Biol Psychiatry 57:140–145
Perez XA, Bordia T, Quik M (2018) The striatal cholinergic system in L-dopa-induced dyskinesias. J Neural Transm 125:1251–1262
Perez-Rial S, Garcia-Gutierrez MS, Molina JA, Perez-Nievas BG, Ledent C, Leiva C, Leza JC, Manzanares J (2011) Increased vulnerability to 6-hydroxydopamine lesion and reduced development of dyskinesias in mice lacking CB1 cannabinoid receptors. Neurobiol Aging 32:631–645
Pertwee RG (2005) Pharmacological actions of cannabinoids. Handb Exp Pharmacol:1–51
Pertwee RG (2008) The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol 153:199–215
Pertwee RG (2015) Endocannabinoids and their pharmacological actions. Handb Exp Pharmacol 231:1–37
Pertwee RG, Ross RA (2002) Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fat Acids 66:101–121
Pinto M, Nissanka N, Peralta S, Brambilla R, Diaz F, Moraes CT (2016) Pioglitazone ameliorates the phenotype of a novel Parkinson’s disease mouse model by reducing neuroinflammation. Mol Neurodegener 11:25
Piomelli D (2003) The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4:873–884
Pisani V, Moschella V, Bari M, Fezza F, Galati S, Bernardi G, Stanzione P, Pisani A, Maccarrone M (2010) Dynamic changes of anandamide in the cerebrospinal fluid of Parkinson’s disease patients. Mov Disord 25:920–924
Pisani V, Madeo G, Tassone A, Sciamanna G, Maccarrone M, Stanzione P, Pisani A (2011) Homeostatic changes of the endocannabinoid system in Parkinson’s disease. Mov Disord 26:216–222
Pisanu A, Lecca D, Mulas G, Wardas J, Simbula G, Spiga S, Carta AR (2014) Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol Dis 71:280–291
Pisanu A, Boi L, Mulas G, Spiga S, Fenu S, Carta AR (2018) Neuroinflammation in L-DOPA-induced dyskinesia: beyond the immune function. J Neural Transm
Polissidis A, Chouliara O, Galanopoulos A, Naxakis G, Papahatjis D, Papadopoulou-Daifoti Z, Antoniou K (2014) Cannabinoids negatively modulate striatal glutamate and dopamine release and behavioural output of acute D-amphetamine. Behav Brain Res 270:261–269
Prescott IA, Dostrovsky JO, Moro E, Hodaie M, Lozano AM, Hutchison WD (2009) Levodopa enhances synaptic plasticity in the substantia nigra pars reticulata of Parkinson’s disease patients. Brain J Neurol 132:309–318
Price DA, Martinez AA, Seillier A, Koek W, Acosta Y, Fernandez E, Strong R, Lutz B, Marsicano G, Roberts JL, Giuffrida A (2009) WIN55,212-2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Eur J Neurosci 29:2177–2186
Qin N, Neeper MP, Liu Y, Hutchinson TL, Lubin ML, Flores CM (2008) TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J Neurosci 28:6231–6238
Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de Ceballos ML (2005) Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci 25:1904–1913
Reale M, Iarlori C, Thomas A, Gambi D, Perfetti B, Di Nicola M, Onofrj M (2009) Peripheral cytokines profile in Parkinson’s disease. Brain Behav Immun 23:55–63
Reber L, Vermeulen L, Haegeman G, Frossard N (2009) Ser276 phosphorylation of NF-kB p65 by MSK1 controls SCF expression in inflammation. PLoS One 4:e4393
Reglodi D, Renaud J, Tamas A, Tizabi Y, Socias SB, Del-Bel E, Raisman-Vozari R (2017) Novel tactics for neuroprotection in Parkinson’s disease: role of antibiotics, polyphenols and neuropeptides. Prog Neurobiol 155:120–148
Rimmerman N, Juknat A, Kozela E, Levy R, Bradshaw HB, Vogel Z (2011) The non-psychoactive plant cannabinoid, cannabidiol affects cholesterol metabolism-related genes in microglial cells. Cell Mol Neurobiol 31:921–930
Rockwell CE, Kaminski NE (2004) A cyclooxygenase metabolite of anandamide causes inhibition of interleukin-2 secretion in murine splenocytes. J Pharmacol Exp Ther 311:683–690
Rojo-Bustamante E, Abellanas MA, Clavero P, Thiolat ML, Li Q, Luquin MR, Bezard E, Aymerich MS (2018) The expression of cannabinoid type 1 receptor and 2-arachidonoyl glycerol synthesizing/degrading enzymes is altered in basal ganglia during the active phase of levodopa-induced dyskinesia. Neurobiol Dis 118:64–75
Ross RA (2003) Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol 140:790–801
Russo EB (2018) Cannabis therapeutics and the future of neurology. Front Integr Neurosci 12:51
Russo EB, Burnett A, Hall B, Parker KK (2005) Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem Res 30:1037–1043
Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, Elebring T, Nilsson K, Drmota T, Greasley PJ (2007) The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152:1092–1101
Rylander D, Recchia A, Mela F, Dekundy A, Danysz W, Cenci MA (2009) Pharmacological modulation of glutamate transmission in a rat model of L-DOPA-induced dyskinesia: effects on motor behavior and striatal nuclear signaling. J Pharmacol Exp Ther 330:227–235
Sagredo O, Garcia-Arencibia M, de Lago E, Finetti S, Decio A, Fernandez-Ruiz J (2007) Cannabinoids and neuroprotection in basal ganglia disorders. Mol Neurobiol 36:82–91
Santello M, Volterra A (2012) TNFalpha in synaptic function: switching gears. Trends Neurosci 35:638–647
Santello M, Bezzi P, Volterra A (2011) TNFalpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 69:988–1001
Santini E, Alcacer C, Cacciatore S, Heiman M, Herve D, Greengard P, Girault JA, Valjent E, Fisone G (2009) L-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J Neurochem 108:621–633
Santini E, Sgambato-Faure V, Li Q, Savasta M, Dovero S, Fisone G, Bezard E (2010) Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in L-DOPA-induced dyskinesia. PLoS One 5:e12322
Santos NA, Martins NM, Sisti FM, Fernandes LS, Ferreira RS, Queiroz RH, Santos AC (2015) The neuroprotection of cannabidiol against MPP(+)-induced toxicity in PC12 cells involves trkA receptors, upregulation of axonal and synaptic proteins, neuritogenesis, and might be relevant to Parkinson’s disease. Toxicol In Vitro 30:231–240
Schlicker E, Kathmann M (2001) Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci 22:565–572
Sgambato-Faure V, Cenci MA (2012) Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog Neurobiol 96:69–86
Shannon KM, Goetz CG, Carroll VS, Tanner CM, Klawans HL (1987) Amantadine and motor fluctuations in chronic Parkinson’s disease. Clin Neuropharmacol 10:522–526
Shih IF, Starhof C, Lassen CF, Hansen J, Liew Z, Ritz B (2017) Occupational and recreational physical activity and Parkinson’s disease in Denmark. Scand J Work Environ Health 43:210–216
Sieradzan KA, Fox SH, Hill M, Dick JP, Crossman AR, Brotchie JM (2001) Cannabinoids reduce levodopa-induced dyskinesia in Parkinson’s disease: a pilot study. Neurology 57:2108–2111
Sierra S, Luquin N, Rico AJ, Gomez-Bautista V, Roda E, Dopeso-Reyes IG, Vazquez A, Martinez-Pinilla E, Labandeira-Garcia JL, Franco R, Lanciego JL (2015) Detection of cannabinoid receptors CB1 and CB2 within basal ganglia output neurons in macaques: changes following experimental parkinsonism. Brain Struct Funct 220:2721–2738
Solis O, Garcia-Sanz P, Herranz AS, Asensio MJ, Moratalla R (2016) L-DOPA reverses the increased free amino acids tissue levels induced by dopamine depletion and rises GABA and tyrosine in the striatum. Neurotox Res 30:67–75
Sugiura T, Kishimoto S, Oka S, Gokoh M (2006) Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog Lipid Res 45:405–446
Szallasi A, Di Marzo V (2000) New perspectives on enigmatic vanilloid receptors. Trends Neurosci 23:491–497
Tanabe T, Tohnai N (2002) Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat 68-69:95–114
Tansey MG, McCoy MK, Frank-Cannon TC (2007) Neuroinflammatory mechanisms in Parkinson's disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 208:1–25
Teema AM, Zaitone SA, Moustafa YM (2016) Ibuprofen or piroxicam protects nigral neurons and delays the development of l-dopa induced dyskinesia in rats with experimental Parkinsonism: influence on angiogenesis. Neuropharmacology 107:432–450
Thacker EL, Chen H, Patel AV, McCullough ML, Calle EE, Thun MJ, Schwarzschild MA, Ascherio A (2008) Recreational physical activity and risk of Parkinson’s disease. Mov Disord 23:69–74
Thomas A, Iacono D, Luciano AL, Armellino K, Di Iorio A, Onofrj M (2004) Duration of amantadine benefit on dyskinesia of severe Parkinson’s disease. J Neurol Neurosurg Psychiatry 75:141–143
Thomas A, Baillie GL, Phillips AM, Razdan RK, Ross RA, Pertwee RG (2007) Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol 150:613–623
Tzavara ET, Li DL, Moutsimilli L, Bisogno T, Di Marzo V, Phebus LA, Nomikos GG, Giros B (2006) Endocannabinoids activate transient receptor potential vanilloid 1 receptors to reduce hyperdopaminergia-related hyperactivity: therapeutic implications. Biol Psychiatry 59:508–515
van der Stelt M, Di Marzo V (2003) The endocannabinoid system in the basal ganglia and in the mesolimbic reward system: implications for neurological and psychiatric disorders. Eur J Pharmacol 480:133–150
van der Stelt M, Fox SH, Hill M, Crossman AR, Petrosino S, Di Marzo V, Brotchie JM (2005) A role for endocannabinoids in the generation of parkinsonism and levodopa-induced dyskinesia in MPTP-lesioned non-human primate models of Parkinson's disease. FASEB J 19:1140–1142
Van Laere K, Casteels C, Lunskens S, Goffin K, Grachev ID, Bormans G, Vandenberghe W (2012) Regional changes in type 1 cannabinoid receptor availability in Parkinson’s disease in vivo. Neurobiol Aging 33:620 e621–620 e628
Venderova K, Ruzicka E, Vorisek V, Visnovsky P (2004) Survey on cannabis use in Parkinson’s disease: subjective improvement of motor symptoms. Mov Disord 19:1102–1106
Vesce S, Rossi D, Brambilla L, Volterra A (2007) Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int Rev Neurobiol 82:57–71
Villapol S (2018) Roles of peroxisome proliferator-activated receptor gamma on brain and peripheral inflammation. Cell Mol Neurobiol 38:121–132
Viveros-Paredes JM, Gonzalez-Castaneda RE, Escalante-Castaneda A, Tejeda-Martinez AR, Castaneda-Achutigui F, Flores-Soto ME (2019) Effect of inhibition of fatty acid amide hydrolase on MPTP-induced dopaminergic neuronal damage. Neurologia 34:143–152
Wandinger KP, Hagenah JM, Kluter H, Rothermundt M, Peters M, Vieregge P (1999) Effects of amantadine treatment on in vitro production of interleukin-2 in de-novo patients with idiopathic Parkinson’s disease. J Neuroimmunol 98:214–220
Wang Y, Zhang QJ, Wang HS, Wang T, Liu J (2014) Genome-wide microarray analysis identifies a potential role for striatal retrograde endocannabinoid signaling in the pathogenesis of experimental L-DOPA-induced dyskinesia. Synapse 68:332–343
Wang Y, Zhang GJ, Sun YN, Yao L, Wang HS, Du CX, Zhang L, Liu J (2018) Identification of metabolite biomarkers for L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease by metabolomic technology. Behav Brain Res 347:175–183
Watzl B, Scuderi P, Watson RR (1991) Marijuana components stimulate human peripheral blood mononuclear cell secretion of interferon-gamma and suppress interleukin-1 alpha in vitro. Int J Immunopharmacol 13:1091–1097
Wiecki TV, Frank MJ (2010) Neurocomputational models of motor and cognitive deficits in Parkinson's disease. Prog Brain Res 183:275–297
Wolf E, Seppi K, Katzenschlager R, Hochschorner G, Ransmayr G, Schwingenschuh P, Ott E, Kloiber I, Haubenberger D, Auff E, Poewe W (2010) Long-term antidyskinetic efficacy of amantadine in Parkinson's disease. Mov Disord 25:1357–1363
Wyss-Coray T, Mucke L (2002) Inflammation in neurodegenerative disease--a double-edged sword. Neuron 35:419–432
Zeissler ML, Eastwood J, McCorry K, Hanemann CO, Zajicek JP, Carroll CB (2016) Delta-9-tetrahydrocannabinol protects against MPP+ toxicity in SH-SY5Y cells by restoring proteins involved in mitochondrial biogenesis. Oncotarget 7:46603–46614
Zhang J, Chen C (2008) Endocannabinoid 2-arachidonoylglycerol protects neurons by limiting COX-2 elevation. J Biol Chem 283:22601–22611
Zuardi AW, Shirakawa I, Finkelfarb E, Karniol IG (1982) Action of cannabidiol on the anxiety and other effects produced by delta 9-THC in normal subjects. Psychopharmacology 76:245–250
Zuardi AW, Crippa JA, Hallak JE, Pinto JP, Chagas MH, Rodrigues GG, Dursun SM, Tumas V (2009) Cannabidiol for the treatment of psychosis in Parkinson’s disease. J Psychopharmacol 23:979–983
Funding
This work was supported by Fundação de Apoio ao Ensino, Pesquisa e Assistência da Universidade de São Paulo (FAEPA USP/RP 98/16); Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2014/25029-4; 2017/14207-7; 12/17626-7); Conselho nacional de Desenvolvimento Científico e Tecnológico (CNPq 201187/2016-7); and Coordenação de Aperfeiçoamento de pessoal de Nível Superior (CAPES PROEX-USP PROEX0051047).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• Investigation of cannabidiol as a possible treatment for motor dysfunctions.
• Compilation of evidence on how cannabinoids can treat PD and LID.
• Preclinical and clinical works show an altered cannabinoid signaling in PD and LID.
Rights and permissions
About this article

Cite this article
Junior, N.C.F., dos- Santos-Pereira, M., Guimarães, F.S. et al. Cannabidiol and Cannabinoid Compounds as Potential Strategies for Treating Parkinson’s Disease and l-DOPA-Induced Dyskinesia. Neurotox Res 37, 12–29 (2020). https://doi.org/10.1007/s12640-019-00109-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-019-00109-8